
Remodeling of Cardiac Gap Junctional Cell–Cell Coupling

Abstract
:1. Introduction and General Considerations
2. Regulation of the Acute Opening and Closure of Gap Junction Channels
3. Regulation of the Expression of Gap Junction Proteins
4. Regulation of the Localization of Gap Junction Channels
5. Gap Junction Remodeling in Atrial Fibrillation (AF)
6. Gap Junction Remodeling in Ischemic Heart Disease
7. Gap Junction Remodeling in Cardiomyopathy
8. Concluding Remarks
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| DCM | dilative cardiomyopathy |
| ECM | extracellular matrix |
| EHD1/Eps15 | endocytic adaptor epidermal growth factor receptor substrate 15 |
| Erk | extracellular signal regulated kinase |
| FAK | focal adhesion kinase |
| GATA4 | transcription factor binding to the DNA sequence “GATA” |
| gGJ | gap junction conductance |
| GJ | gap junction |
| HOCM | hypertrophic obstructive cardiomyopathy |
| IL-1ß | interleukin-1ß |
| IL-6 | interleukin-6 |
| JNK | c-Jun-N-terminal kinase |
| MAPK | mitogen activated protein kinase |
| MEK | Mitogen-activated protein kinase kinase |
| MTOC | microtubule organizing center |
| NFAT | nuclear factor of activated T-cells |
| PKA | protein kinase A |
| PKC | protein kinase C |
| Rac1 | Rac Family Small GTPase 1 |
| rER | rough endoplasmic reticulum |
| Ry2R | ryanodine receptor |
| SF | safety factor |
| SR | sinus rhythm |
| θL | longitudinal propagation velocity |
| θT | transverse propagation velocity |
| TGFß | transforming growth factor ß |
| VEGF | vascular endothelial growth factor |
References
- Spach, M.S.; Dolber, P.C. Discontinuous anisotropic propagation. In Cardiac Electrophysiology: A Textbook; Rosen, M.R., Janse, M.J., Wit, A.L., Eds.; Futura Publishing Company Inc: Mount Kisco, NY, USA, 1990; pp. 517–534. [Google Scholar]
- Dhein, S.; Hammerrath, S.B. Aspects of the intercellular communication in aged hearts. Effects of the gap junction uncopler palmitoleic acid. Naunyn Schmiedeberg’s Arch. Pharmacol. 2001, 364, 397–408. [Google Scholar]
- Dolber, P.C.; Beyer, E.C.; Junker, J.L.; Spach, M.S. Distribution of gap junctions in dog and rat ventricle studied with a double-label technique. J. Mol. Cell. Cardiol. 1992, 24, 1443–1457. [Google Scholar] [CrossRef]
- Weingart, R.; Maurer, P. Action potential transfer in cell pairs isolated from adult rat and guinea pig ventricles. Circ. Res. 1988, 63, 72–80. [Google Scholar] [CrossRef] [Green Version]
- Veeraraghavan, R.; Lin, J.; Hoeker, G.S.; Keener, J.P.; Gourdie, R.G.; Poelzing, S. Sodium channels in the Cx43 gap junction perinexus may constitute a cardiac ephapse: An experimental and modeling study. Pflugers Arch. 2015, 467, 2093–2105. [Google Scholar] [CrossRef] [Green Version]
- George, S.A.; Hoeker, G.; Calhoun, P.J.; Entz, M., 2nd; Raisch, T.B.; King, D.R.; Khan, M.; Baker, C.; Gourdie, R.G.; Smyth, J.W.; et al. Modulating cardiac conduction during metabolic ischemia with perfusate sodium and calcium in guinea pig hearts. Am. J. Physiol. Heart Circ. Physiol. 2019, 316, H849–H861. [Google Scholar] [CrossRef]
- Gourdie, R.G. The cardiac gap junction has discrete functions in electrotonic and ephaptic coupling. Anat. Rec. 2019, 302, 93–100. [Google Scholar] [CrossRef] [Green Version]
- Hichri, E.; Abriel, H.; Kucera, J.P. Distribution of cardiac sodium channels in clusters potentiates ephaptic interactions in the intercalated disc. J. Physiol. 2018, 596, 563–589. [Google Scholar] [CrossRef] [PubMed]
- Carmeliet, E. Conduction in cardiac tissue. Historical reflections. Physiol. Rep. 2019, 7, e13860. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Seidel, T.; Salameh, A.; Dhein, S. A simulation study of cellular hypertrophy and connexin lateralization in cardiac tissue. Biophys. J. 2010, 99, 2821–2830. [Google Scholar] [CrossRef] [Green Version]
- Jongsma, H.J.; Wilders, R. Gap junctions in cardiovascular disease. Circ. Res. 2000, 86, 1193–1197. [Google Scholar] [CrossRef] [PubMed]
- Rohr, S. Role of gap junctions in the propagation of the cardiac action potential. Cardiovasc. Res. 2004, 62, 309–322. [Google Scholar] [CrossRef] [PubMed]
- Rohr, S.; Kucera, J.P.; Fast, V.G.; Kléber, A.G. Paradoxical improvement of impulse conduction in cardiac tissue by partial cellular uncoupling. Science 1997, 275, 841–844. [Google Scholar] [CrossRef] [PubMed]
- Rohr, S. Arrhythmogenic implications of fibroblast-myocyte interactions. Circ. Arrhythm. Electrophysiol. 2012, 5, 442–452. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, P.J.; Pogwizd, S.M. Micropatterns of propagation. Adv. Cardiol. 2006, 42, 86–106. [Google Scholar] [CrossRef] [PubMed]
- Joyner, R.W.; van Capelle, F.J. Propagation through electrically coupled cells. How a small SA node drives a large atrium. Biophys. J. 1986, 50, 1157–1164. [Google Scholar] [CrossRef] [Green Version]
- Boyett, M.R.; Inada, S.; Yoo, S.; Li, J.; Liu, J.; Tellez, J.; Greener, I.D.; Honjo, H.; Billeter, R.; Lei, M.; et al. Connexins in the sinoatrial and atrioventricular nodes. Adv. Cardiol. 2006, 42, 175–197. [Google Scholar]
- Shaw, R.M.; Rudy, Y. Electrophysiologic effects of acute myocardial ischemia. A mechanistic investigation of action potential conduction and conduction failure. Circ. Res. 1997, 80, 124–138. [Google Scholar] [CrossRef]
- Shaw, R.M.; Rudy, Y. Ionic mechanisms of propagation in cardiac tissue. Roles of the sodium and L-type calcium currents during reduced excitability and decreased gap junction coupling. Circ. Res. 1997, 81, 727–741. [Google Scholar] [CrossRef]
- Dhein, S.; Seidel, T.; Salameh, A.; Jozwiak, J.; Hagen, A.; Kostelka, M.; Hindricks, G.; Mohr, F.W. Remodeling of cardiac passive electrical properties and susceptibility to ventricular and atrial arrhythmias. Front. Physiol. 2014, 5, 424. [Google Scholar] [CrossRef]
- Schrickel, J.W.; Kreuzberg, M.M.; Ghanem, A.; Kim, J.S.; Linhart, M.; Andrié, R.; Tiemann, K.; Nickenig, G.; Lewalter, T.; Willecke, K. Normal impulse propagation in the atrioventricular conduction system of Cx30.2/Cx40 double deficient mice. J. Mol. Cell. Cardiol. 2009, 46, 644–652. [Google Scholar] [CrossRef]
- Kwak, B.R.; Hermans, M.M.; De Jonge, H.R.; Lohmann, S.M.; Jongsma, H.J.; Chanson, M. Differential regulation of distinct types of gap junction channels by similar phosphorylating conditions. Mol. Biol. Cell. 1995, 6, 1707–1719. [Google Scholar] [CrossRef] [Green Version]
- Valiunas, V.; Bukauskas, F.F.; Weingart, R. Conductances and selective permeability of connexin43 gap junction channels examined in neonatal rat heart cells. Circ. Res. 1997, 80, 708–719. [Google Scholar] [CrossRef] [PubMed]
- Bukauskas, F.F.; Verselis, V.K. Gap junction channel gating. Biochim. Biophys. Acta 2004, 1662, 42–60. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brink, P.R.; Ramanan, S.V.; Christ, G.J. Human connexin 43 gap junction channel gating: Evidence for mode shifts and/or heterogeneity. Am. J. Physiol. 1996, 271, C321–C331. [Google Scholar] [CrossRef] [PubMed]
- Bukauskas, F.F.; Peracchia, C. Two distinct gating mechanisms in gap junction channels: CO2-sensitive and voltage-sensitive. Biophys. J. 1997, 72, 2137–2142. [Google Scholar] [CrossRef] [Green Version]
- Ek-Vitorín, J.F.; Pontifex, T.K.; Burt, J.M. Determinants of Cx43 Channel Gating and Permeation: The Amino Terminus. Biophys. J. 2016, 110, 127–140. [Google Scholar] [CrossRef] [Green Version]
- Peracchia, C. Chemical gating of gap junction channels; roles of calcium, pH and calmodulin. Biochim. Biophys. Acta 2004, 1662, 61–80. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Verselis, V.K.; Ginter, C.S.; Bargiello, T.A. Opposite voltage gating polarities of two closely related connexins. Nature 1994, 368, 348–351. [Google Scholar] [CrossRef]
- Moreno, A.P.; Lau, A.F. Gap junction channel gating modulated through protein phosphorylation. Prog. Biophys. Mol. Biol. 2007, 94, 107–119. [Google Scholar] [CrossRef] [Green Version]
- Lampe, P.D.; TenBroek, E.M.; Burt, J.M.; Kurata, W.E.; Johnson, R.G.; Lau, A.F. Phosphorylation of connexin43 on serine368 by protein kinase C regulates gap junctional communication. J. Cell Biol. 2000, 149, 1503–1512. [Google Scholar] [CrossRef]
- Kwak, B.R.; van Veen, T.A.; Analbers, L.J.; Jongsma, H.J. TPA increases conductance but decreases permeability in neonatal rat cardiomyocyte gap junction channels. Exp. Cell Res. 1995, 220, 456–463. [Google Scholar] [CrossRef]
- Kwak, B.R.; Jongsma, H.J. Regulation of cardiac gap junction channel permeability and conductance by several phosphorylating conditions. Mol. Cell Biochem. 1996, 157, 93–99. [Google Scholar] [CrossRef] [PubMed]
- Leybaert, L.; Lampe, P.D.; Dhein, S.; Kwak, B.R.; Ferdinandy, P.; Beyer, E.C.; Laird, D.W.; Naus, C.C.; Green, C.R.; Schulz, R. Connexins in cardiovascular and neurovascular health and disease: Pharmacological implications. Pharmacol. Rev. 2017, 69, 396–478. [Google Scholar] [CrossRef] [PubMed]
- Dhein, S. Role of connexins in atrial fibrillation. Adv. Cardiol. 2006, 42, 161–174. [Google Scholar] [CrossRef] [PubMed]
- Jozwiak, J.; Dhein, S. Local effects and mechanisms of antiarrhythmic peptide AAP10 in acute regional myocardial ischemia: Electrophysiological and molecular findings. Naunyn Schmiedebergs Arch. Pharmacol. 2008, 378, 459–470. [Google Scholar] [CrossRef]
- De Groot, J.R.; Coronel, R. Acute ischemia-induced gap junctional uncoupling and arrhythmogenesis. Cardiovasc. Res. 2004, 62, 323–334. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Li, C.; Liang, D.; Lv, F.; Yuan, T.; The, E.; Ma, X.; Wu, Y.; Zhen, L.; Xie, D.; et al. LRP6 acts as a scaffold protein in cardiac gap junction assembly. Nat. Commun. 2016, 7, 11775. [Google Scholar] [CrossRef]
- Musil, L.S.; Goodenough, D.A. Multisubunit assembly of an integral plasma membrane channel protein, gap junction connexin43, occurs after exit from the ER. Cell 1993, 74, 1065–1077. [Google Scholar] [CrossRef]
- Musil, L.S.; Goodenough, D.A. Biochemical analysis of connexon assembly. In Intercellular Communication through Gap Junctions: Progress in Cell Research; Kanno, Y., Kataoka, K., Shiba, Y., Shibata, Y., Shimazu, T., Eds.; Elsevier: Amsterdam, The Netherlands, 1995; Volume 4, pp. 327–330. [Google Scholar]
- Falk, M.M. Connexin-specific distribution within gap junctions revealed in living cells. J. Cell Sci. 2000, 113, 4109–4120. [Google Scholar] [CrossRef]
- Giepmans, B.N. Gap junctions and connexin-interacting proteins. Cardiovasc. Res. 2004, 62, 233–245. [Google Scholar] [CrossRef] [Green Version]
- Berthoud, V.M.; Minogue, P.J.; Laing, J.G.; Beyer, E.C. Pathways for degradation of connexins and gap junctions. Cardiovasc. Res. 2004, 62, 256–267. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Salameh, A. Life cycle of connexins: Regulation of connexin synthesis and degradation. Adv. Cardiol. 2006, 42, 57–70. [Google Scholar] [PubMed]
- Olejnickova, V.; Kocka, M.; Kvasilova, A.; Kolesova, H.; Dziacky, A.; Gidor, T.; Gidor, L.; Sankova, B.; Gregorovicova, M.; Gourdie, R.G.; et al. Gap junctional communication via connexin43 between Purkinje fibers and working myocytes explains the epicardial activation pattern in the postnatal mouse left ventricle. Int. J. Mol. Sci. 2021, 22, 2475. [Google Scholar] [CrossRef]
- Bukauskas, F.F.; Jordan, K.; Bukauskiene, A.; Bennett, M.V.; Lampe, P.D.; Laird, D.W.; Verselis, V.K. Clustering of connexin 43-enhanced green fluorescent protein gap junction channels and functional coupling in living cells. Proc. Natl. Acad. Sci. USA 2000, 97, 2556–2561. [Google Scholar] [CrossRef] [Green Version]
- Moise, N.; Struckman, H.L.; Dagher, C.; Veeraraghavan, R.; Weinberg, S.H. Intercalated disk nanoscale structure regulates cardiac conduction. J. Gen. Physiol. 2021, 153, e202112897. [Google Scholar] [CrossRef]
- De Jong, A.M.; Maass, A.H.; Oberdorf-Maass, S.U.; De Boer, R.A.; Van Gilst, W.H.; Van Gelder, I.C. Cyclical stretch induces structural changes in atrial myocytes. J. Cell Mol. Med. 2013, 17, 743–753. [Google Scholar] [CrossRef] [Green Version]
- Salameh, A.; Wustmann, A.; Karl, S.; Blanke, K.; Apel, D.; Rojas-Gomez, D.; Franke, H.; Mohr, F.W.; Janousek, J.; Dhein, S. Cyclic mechanical stretch induces cardiomyocytes orientation and polarization of the gap junction protein connexin43. Circ. Res. 2010, 106, 1592–1602. [Google Scholar] [CrossRef] [Green Version]
- Dhein, S.; Schreiber, A.; Steinbach, S.; Apel, D.; Salameh, A.; Schlegel, F.; Kostelka, M.; Dohmen, P.M.; Mohr, F.W. Mechanical control of cell biology. Effects of cyclic mechanical stretch on cardiomyocyte cellular organization. Prog. Biophys. Mol. Biol. 2014, 115, 93–102. [Google Scholar] [CrossRef] [PubMed]
- Salameh, A.; Apel, D.; Gonzalez Casanova, J.; von Salisch, S.; Mohr, F.W.; Daehnert, I.; Dhein, S. On the different roles of AT1 and AT2 receptors in stretch-induced changes of connexin43 expression and localisation. Pflugers Arch. 2012, 464, 535–547. [Google Scholar] [CrossRef] [PubMed]
- Salameh, A.; Karl, S.; Djilali, H.; Dhein, S.; Janousek, J.; Daehnert, I. Opposing and synergistic effects of cyclic mechanical stretch and α- or β-adrenergic stimulation on the cardiac gap junction protein Cx43. Pharmacol. Res. 2010, 62, 506–513. [Google Scholar] [CrossRef] [PubMed]
- Ninio, D.M.; Saint, D.A. The role of stretch-activated channels in atrial fibrillation and the impact of intracellular acidosis. Prog Biophys. Mol. Biol. 2008, 97, 401–416. [Google Scholar] [CrossRef] [PubMed]
- Hocini, M.; Ho, S.Y.; Kawara, T.; Linnenbank, A.C.; Potse, M.; Shah, D.; Jaïs, P.; Janse, M.J.; Haïssaguerre, M.; De Bakker, J.M. Electrical conduction in canine pulmonary veins: Electrophysiological and anatomic correlation. Circulation 2002, 105, 2442–2448. [Google Scholar] [CrossRef] [Green Version]
- Po, S.S.; Li, Y.; Tang, D.; Liu, H.; Geng, N.; Jackman, W.M.; Scherlag, B.; Lazzara, R.; Patterson, E. Rapid and stable re-entry within the pulmonary vein as a mechanism initiating paroxysmal atrial fibrillation. J. Am. Coll. Cardiol. 2005, 45, 1871–1877. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- The, A.W.; Kistler, P.M.; Lee, G.; Medi, C.; Heck, P.M.; Spence, S.; Morton, J.B.; Sanders, P.; Kalman, J.M. Electroanatomic properties of the pulmonary veins: Slowed conduction, low voltage and altered refractoriness in AF patients. J. Cardiovasc. Electrophysiol. 2011, 22, 1083–1091. [Google Scholar] [CrossRef]
- Verheule, S.; Wilson, E.E.; Arora, R.; Engle, S.K.; Scott, L.R.; Olgin, J.E. Tissue structure and connexin expression of canine pulmonary veins. Cardiovasc. Res. 2002, 55, 727–738. [Google Scholar] [CrossRef] [Green Version]
- Franz, M.R.; Jamal, S.M.; Narayan, S.M. The role of action potential alternans in the initiation of atrial fibrillation in humans: A review and future directions. Europace 2012, 14 (Suppl. S5), v58–v64. [Google Scholar] [CrossRef] [Green Version]
- Harada, M.; Nattel, S. Implications of Inflammation and Fibrosis in Atrial Fibrillation Pathophysiology. Card Electrophysiol. Clin. 2021, 13, 25–35. [Google Scholar] [CrossRef] [PubMed]
- Nattel, S.; Heijman, J.; Zhou, L.; Dobrev, D. Molecular basis of atrial fibrillation pathophysiology and therapy: A translational perspective. Circ. Res. 2020, 127, 51–72. [Google Scholar] [CrossRef]
- Linz, D.; Hohl, M.; Dhein, S.; Ruf, S.; Reil, J.C.; Kabiri, M.; Wohlfart, P.; Verheule, S.; Böhm, M.; Sadowski, T.; et al. Cathepsin A mediates susceptibility to atrial tachyarrhythmia and impairment of atrial emptying function in Zucker diabetic fatty rats. Cardiovasc. Res. 2016, 110, 371–380. [Google Scholar] [CrossRef] [Green Version]
- Hohl, M.; Erb, K.; Lang, L.; Ruf, S.; Hübschle, T.; Dhein, S.; Linz, W.; Elliott, A.D.; Sanders, P.; Zamyatkin, O.; et al. Cathepsin A mediates ventricular remote remodeling and atrial cardiomyopathy in rats with ventricular ischemia/reperfusion. JACC Basic Transl. Sci. 2019, 4, 332–344. [Google Scholar] [CrossRef] [PubMed]
- Boldt, A.; Wetzel, U.; Lauschke, J.; Weigl, J.; Gummert, J.; Hindricks, G.; Kottkamp, H.; Dhein, S. Fibrosis in left atrial tissue of patients with atrial fibrillation with and without underlying mitral valve disease. Heart 2004, 90, 400–405. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Angel, N.; Li, L.I.; Macleod, R.S.; Marrouche, N.; Ranjan, R.; Dosdall, D.J. Diverse fibrosis architecture and premature stimulation facilitate initiation of reentrant activity following chronic atrial fibrillation. J. Cardiovasc. Electrophysiol. 2015, 26, 1352–1360. [Google Scholar] [CrossRef] [Green Version]
- Krul, S.P.; Berger, W.R.; Smit, N.W.; van Amersfoorth, S.C.; Driessen, A.H.; van Boven, W.J.; Fiolet, J.W.; van Ginneken, A.C.; van der Wal, A.C.; de Bakker, J.M.; et al. Atrial fibrosis and conduction slowing in the left atrial appendage of patients undergoing thoracoscopic surgical pulmonary vein isolation for atrial fibrillation. Circ. Arrhythm. Electrophysiol. 2015, 8, 288–295. [Google Scholar] [CrossRef] [Green Version]
- Wijffels, M.C.; Kirchhof, C.J.; Dorland, R.; Allessie, M.A. Atrial fibrillation begets atrial fibrillation. A study in awake chronically instrumented goats. Circulation 1995, 92, 1954–1968. [Google Scholar] [CrossRef]
- Polontchouk, L.; Haefliger, J.A.; Ebelt, B.; Schaefer, T.; Stuhlmann, D.; Mehlhorn, U.; Kuhn-Regnier, F.; De Vivie, E.R.; Dhein, S. Effects of chronic atrial fibrillation on gap junction distribution in human and rat atria. J. Am. Coll. Cardiol. 2001, 38, 883–891. [Google Scholar] [CrossRef] [Green Version]
- Wetzel, U.; Boldt, A.; Lauschke, J.; Weigl, J.; Schirdewahn, P.; Dorszewski, A.; Doll, N.; Hindricks, G.; Dhein, S.; Kottkamp, H. Expression of connexins 40 and 43 in human left atrium in atrial fibrillation of different aetiologies. Heart 2005, 91, 166–170. [Google Scholar] [CrossRef] [Green Version]
- Kostin, S.; Klein, G.; Szalay, Z.; Hein, S.; Bauer, E.P.; Schaper, J. Structural correlate of atrial fibrillation in human patients. Cardiovasc. Res. 2002, 54, 361–379. [Google Scholar] [CrossRef] [Green Version]
- Li, D.Q.; Feng, Y.B.; Zhang, H.Q. The relationship between gap junctional remodeling and human atrial fibrillation. Chin. Med. J. 2004, 117, 1256–1258. [Google Scholar] [PubMed]
- Dupont, E.M.; Ko, Y.; Rothery, S.; Coppen, S.R.; Baghai, M.; Haw, M.; Severs, N.J. The gap-junctional protein connexin40 is elevated in patients susceptible to postoperative atrial fibrillation. Circulation 2001, 103, 842–849. [Google Scholar] [CrossRef] [PubMed]
- Pfannmüller, B.; Boldt, A.; Reutemann, A.; Duerrschmidt, N.; Krabbes-Graube, S.; Mohr, F.W.; Dhein, S. Gender-specific remodeling in atrial fibrillation? Thorac. Cardiovasc. Surg. 2013, 61, 66–73. [Google Scholar] [CrossRef]
- Neuberger, H.R.; Schotten, U.; Verheule, S.; Eijsbouts, S.; Blaauw, Y.; van Hunnik, A.; Allessie, M. Development of a substrate of atrial fibrillation during chronic atrioventricular block in the goat. Circulation 2005, 111, 30–37. [Google Scholar] [CrossRef] [Green Version]
- Dhein, S.; Rothe, S.; Busch, A.; Rojas-Gomez, D.M.; Boldt, A.; Reutemann, A.; Seidel, T.; Salameh, A.; Pfannmüller, B.; Rastan, A.; et al. Effects of metoprolol therapy on cardiac gap junction remodelling and conduction in human chronic atrial fibrillation. Br. J. Pharmacol. 2011, 164, 607–616. [Google Scholar] [CrossRef] [Green Version]
- Colman, M.A.; Varela, M.; Hancox, J.C.; Zhang, H.; Aslanidi, O.V. Evolution and pharmacological modulation of the arrhythmogenic wave dynamics in canine pulmonary vein model. Europace 2014, 16, 416–423. [Google Scholar] [CrossRef]
- Jungk, L.; Franke, H.; Salameh, A.; Dhein, S. Golgi Fragmentation in human patients with chronic atrial fibrillation: A new aspect of remodeling. Thorac. Cardiovasc. Surg. 2019, 67, 98–106. [Google Scholar] [CrossRef]
- Bellouze, S.; Baillat, G.; Buttigieg, D.; de la Grange, P.; Rabouille, C.; Haase, G. Stathmin 1/2-triggered microtubule loss mediates Golgi fragmentation in mutant SOD1 motor neurons. Mol. Neurodegener. 2016, 11, 43. [Google Scholar] [CrossRef]
- Sun, K.H.; de Pablo, Y.; Vincent, F.; Johnson, E.O.; Chavers, A.K.; Shah, K. Novel genetic tools reveal Cdk5′s major role in Golgi fragmentation in Alzheimer’s disease. Mol. Biol. Cell 2008, 19, 3052–3069. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Boldt, A.; Wetzel, U.; Weigl, J.; Garbade, J.; Lauschke, J.; Hindricks, G.; Kottkamp, H.; Gummert, J.F.; Dhein, S. Expression of angiotensin II receptors in human left and right atrial tissue in atrial fibrillation with and without underlying mitral valve disease. J. Am. Coll Cardiol. 2003, 42, 1785–1792. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Boldt, A.; Scholl, A.; Garbade, J.; Resetar, M.E.; Mohr, F.W.; Gummert, J.F.; Dhein, S. ACE-inhibitor treatment attenuates atrial structural remodeling in patients with lone chronic atrial fibrillation. Basic Res. Cardiol. 2006, 101, 261–267. [Google Scholar] [CrossRef] [PubMed]
- Adam, O.; Lavall, D.; Theobald, K.; Hohl, M.; Grube, M.; Ameling, S.; Sussman, M.A.; Rosenkranz, S.; Kroemer, H.K.; Schäfers, H.J.; et al. Rac1-induced connective tissue growth factor regulates connexin 43 and N-cadherin expression in atrial fibrillation. J. Am. Coll. Cardiol. 2010, 55, 469–480. [Google Scholar] [CrossRef] [Green Version]
- Salameh, A.; Frenzel, C.; Boldt, A.; Rassler, B.; Glawe, I.; Schulte, J.; Mühlberg, K.; Zimmer, H.G.; Pfeiffer, D.; Dhein, S. Subchronic alpha- and beta-adrenergic regulation of cardiac gap junction protein expression. FASEB J. 2006, 20, 365–367. [Google Scholar] [CrossRef] [PubMed]
- Salameh, A.; Krautblatter, S.; Karl, S.; Blanke, K.; Rojas-Gomez, D.; Dhein, S.; Pfeiffer, D.; Janousek, J. The signal transduction cascade regulating the expression of the gap junction protein connexin43 by beta-adrenoceptors. Br. J. Pharmacol. 2009, 158, 198–208. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, Y.; Hou, M.C.; Li, J.J.; Qi, Y.; Zhang, Y.; She, G.; Ren, Y.J.; Wu, W.; Pang, Z.D.; Xie, W.; et al. Cardiac β-adrenergic receptor activation mediates distinct and cell type-dependent changes in the expression and distribution of connexin 43. J. Cell Mol. Med. 2020, 24, 8505–8517. [Google Scholar] [CrossRef]
- Salameh, A.; Djilali, H.; Blanke, K.; Gonzalez Casanova, J.; von Salisch, S.; Savtschenko, A.; Dhein, S.; Dähnert, I. Cardiac fibroblasts inhibit β-adrenoceptor-dependent connexin43 expression in neonatal rat cardiomyocytes. Naunyn Schmiedebergs Arch. Pharmacol. 2013, 386, 421–433. [Google Scholar] [CrossRef]
- Hagen, A.; Dietze, A.; Dhein, S. Human cardiac gap-junction coupling: Effects of antiarrhythmic peptide AAP10. Cardiovasc. Res. 2009, 83, 405–415. [Google Scholar] [CrossRef] [Green Version]
- Jeyaraman, M.; Tanguy, S.; Fandrich, R.R.; Lukas, A.; Kardami, E. Ischemia-induced dephosphorylation of cardiomyocyte connexin-43 is reduced by okadaic acid and calyculin A but not fostriecin. Mol. Cell Biochem. 2003, 242, 129–134. [Google Scholar] [CrossRef]
- Xue, J.; Yan, X.; Yang, Y.; Chen, M.; Wu, L.; Gou, Z.; Sun, Z.; Talabieke, S.; Zheng, Y.; Luo, D. Connexin 43 dephosphorylation contributes to arrhythmias and cardiomyocyte apoptosis in ischemia/reperfusion hearts. Basic Res. Cardiol. 2019, 114, 40. [Google Scholar] [CrossRef]
- Martins-Marques, T.; Catarino, S.; Gonçalves, A.; Miranda-Silva, D.; Gonçalves, L.; Antunes, P.; Coutinho, G.; Leite Moreira, A.; Falcão Pires, I.; Girão, H. EHD1 modulates Cx43 gap junction remodeling associated with cardiac diseases. Circ. Res. 2020, 126, e97–e113. [Google Scholar] [CrossRef] [PubMed]
- Frangogiannis, N.G.; Kovacic, J.C. Extracellular matrix in ischemic heart disease, Part 4/4: JACC Focus Seminar. J. Am. Coll. Cardiol. 2020, 75, 2219–2235. [Google Scholar] [CrossRef] [PubMed]
- Peet, C.; Ivetic, A.; Bromage, D.I.; Shah, A.M. Cardiac monocytes and macrophages after myocardial infarction. Cardiovasc. Res. 2020, 116, 1101–1112. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Camelliti, P.; Green, C.R.; Kohl, P. Structural and functional coupling of cardiac myocytes and fibroblasts. Adv. Cardiol. 2006, 42, 132–149. [Google Scholar] [CrossRef]
- Mouton, A.J.; Rivera, O.J.; Lindsey, M.L. Myocardial infarction remodeling that progresses to heart failure: A signaling misunderstanding. Am. J. Physiol. Heart Circ. Physiol. 2018, 315, H71–H79. [Google Scholar] [CrossRef]
- De Jesus, N.M.; Wang, L.; Lai, J.; Rigor, R.R.; Francis Stuart, S.D.; Bers, D.M.; Lindsey, M.L.; Ripplinger, C.M. Antiarrhythmic effects of interleukin 1 inhibition after myocardial infarction. Heart Rhythm 2017, 14, 727–736. [Google Scholar] [CrossRef] [Green Version]
- Camelliti, P.; Devlin, G.P.; Matthews, K.G.; Kohl, P.; Green, C.R. Spatially and temporally distinct expression of fibroblast connexins after sheep ventricular infarction. Cardiovasc. Res. 2004, 62, 415–425. [Google Scholar] [CrossRef] [PubMed]
- Quinn, T.A.; Camelliti, P.; Rog-Zielinska, E.A.; Siedlecka, U.; Poggioli, T.; O’Toole, E.T.; Knöpfel, T.; Kohl, P. Electrotonic coupling of excitable and nonexcitable cells in the heart revealed by optogenetics. Proc. Natl. Acad. Sci. USA 2016, 113, 14852–14857. [Google Scholar] [CrossRef] [Green Version]
- Schultz, F.; Swiatlowska, P.; Alvarez-Laviada, A.; Sanchez-Alonso, J.L.; Song, Q.; de Vries, A.A.F.; Pijnappels, D.A.; Ongstad, E.; Braga, V.M.M.; Entcheva, E.; et al. Cardiomyocyte-myofibroblast contact dynamism is modulated by connexin-43. FASEB J. 2019, 33, 10453–10468. [Google Scholar] [CrossRef] [PubMed]
- Severs, N.J.; Dupont, E.; Thomas, N.; Kaba, R.; Rothery, S.; Jain, R.; Sharpey, K.; Fry, C.H. Alterations in cardiac connexin expression in cardiomyopathies. Adv. Cardiol. 2006, 42, 228–242. [Google Scholar] [CrossRef]
- Gottwald, E.; Gottwald, M.; Dhein, S. Enhanced dispersion of epicardial activation-recovery intervals at sites of histological inhomogeneity during regional cardiac ischaemia and reperfusion. Heart 1998, 79, 474–480. [Google Scholar] [CrossRef]
- Müller, A.; Dhein, S. Sodium channel blockade enhances dispersion of the cardiac action potential duration. A computer simulation study. Basic Res. Cardiol. 1993, 88, 11–22. [Google Scholar] [CrossRef]
- Kucera, J.P.; Rohr, S.; Kleber, A.G. Microstructure, cell-to-cell coupling, and ion currents as determinants of electrical propagation and arrhythmogenesis. Circ. Arrhythm. Electrophysiol. 2017, 10, e004665. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Spach, M.S.; Miller, W.T., 3rd; Dolber, P.C.; Kootsey, J.M.; Sommer, J.R.; Mosher, C.E., Jr. The functional role of structural complexities in the propagation of depolarization in the atrium of the dog. Cardiac conduction disturbances due to discontinuities of effective axial resistivity. Circ. Res. 1982, 50, 175–191. [Google Scholar] [CrossRef] [Green Version]
- Miragoli, M.; Gaudesius, G.; Rohr, S. Electrotonic modulation of cardiac impulse conduction by myofibroblasts. Circ. Res. 2006, 98, 801–810. [Google Scholar] [CrossRef] [Green Version]
- Miragoli, M.; Salvarani, N.; Rohr, S. Myofibroblasts induce ectopic activity in cardiac tissue. Circ. Res. 2007, 101, 755–758. [Google Scholar] [CrossRef] [Green Version]
- Brown, T.R.; Krogh-Madsen, T.; Christini, D.J. Illuminating myocyte-fibroblast homotypic and heterotypic gap junction dynamics using dynamic clamp. Biophys. J. 2016, 111, 785–797. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kostin, S.; Dammer, S.; Hein, S.; Klovekorn, W.P.; Bauer, E.P.; Schaper, J. Connexin 43 expression and distribution in compensated and decompensated cardiac hypertrophy in patients with aortic stenosis. Cardiovasc. Res. 2004, 62, 426–436. [Google Scholar] [CrossRef]
- Kostin, S. Zonula occludens-1 and connexin 43 expression in the failing human heart. J. Cell Mol. Med. 2007, 11, 892–895. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Glukhov, A.V.; Fedorov, V.V.; Kalish, P.W.; Ravikumar, V.K.; Lou, Q.; Janks, D.; Schuessler, R.B.; Moazami, N.; Efimov, I.R. Conduction remodeling in human end-stage nonischemic left ventricular cardiomyopathy. Circulation 2012, 125, 1835–1847. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Akar, F.G.; Spragg, D.D.; Tunin, R.S.; Kass, D.A.; Tomaselli, G.F. Mechanisms underlying conduction slowing and arrhythmogenesis in nonischemic dilated cardiomyopathy. Circ. Res. 2004, 95, 717–725. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Macquart, C.; Jüttner, R.; Morales Rodriguez, B.; Le Dour, C.; Lefebvre, F.; Chatzifrangkeskou, M.; Schmitt, A.; Gotthardt, M.; Bonne, G.; Muchir, A. Microtubule cytoskeleton regulates Connexin 43 localization and cardiac conduction in cardiomyopathy caused by mutation in A-type lamins gene. Hum. Mol. Genet. 2019, 28, 4043–4052. [Google Scholar] [CrossRef] [PubMed]
- Trembley, M.A.; Quijada, P.; Agullo-Pascual, E.; Tylock, K.M.; Colpan, M.; Dirkx, R.A., Jr.; Myers, J.R.; Mickelsen, D.M.; de Mesy Bentley, K.; Rothenberg, E.; et al. Mechanosensitive gene regulation by myocardin-related transcription factors is required for cardiomyocyte integrity in load-induced ventricular hypertrophy. Circulation 2018, 138, 1864–1878. [Google Scholar] [CrossRef]
- De Smet, M.A.; Lissoni, A.; Nezlobinsky, T.; Wang, N.; Dries, E.; Pérez-Hernández, M.; Lin, X.; Amoni, M.; Vervliet, T.; Witschas, K.; et al. Cx43 hemichannel microdomain signaling at the intercalated disc enhances cardiac excitability. J. Clin. Investig. 2021, 131, e137752. [Google Scholar] [CrossRef]
- Lissoni, A.; Hulpiau, P.; Martins-Marques, T.; Wang, N.; Bultynck, G.; Schulz, R.; Witschas, K.; Girao, H.; De Smet, M.; Leybaert, L. RyR2 regulates Cx43 hemichannel intracellular Ca2+-dependent activation in cardiomyocytes. Cardiovasc. Res. 2021, 117, 123–136. [Google Scholar] [CrossRef] [PubMed]
- Sugita, J.; Fujiu, K.; Nakayama, Y.; Matsubara, T.; Matsuda, J.; Oshima, T.; Liu, Y.; Maru, Y.; Hasumi, E.; Kojima, T.; et al. Cardiac macrophages prevent sudden death during heart stress. Nat. Commun. 2021, 12, 1910. [Google Scholar] [CrossRef] [PubMed]
- Himelman, E.; Lillo, M.A.; Nouet, J.; Gonzalez, J.P.; Zhao, Q.; Xie, L.H.; Li, H.; Liu, T.; Wehrens, X.H.; Lampe, P.D.; et al. Prevention of connexin-43 remodeling protects against Duchenne muscular dystrophy cardiomyopathy. J. Clin. Investig. 2020, 130, 1713–1727. [Google Scholar] [CrossRef] [PubMed]
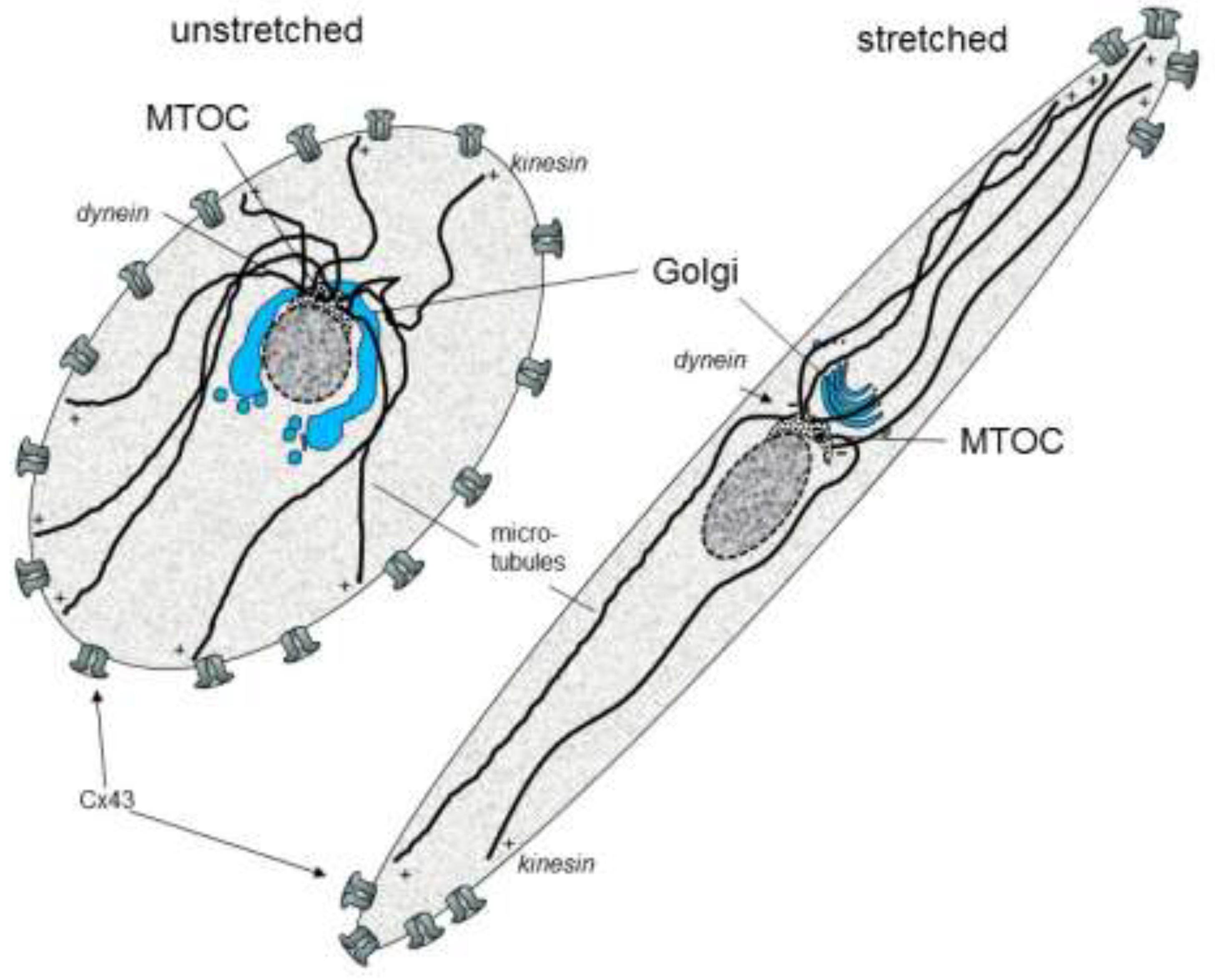


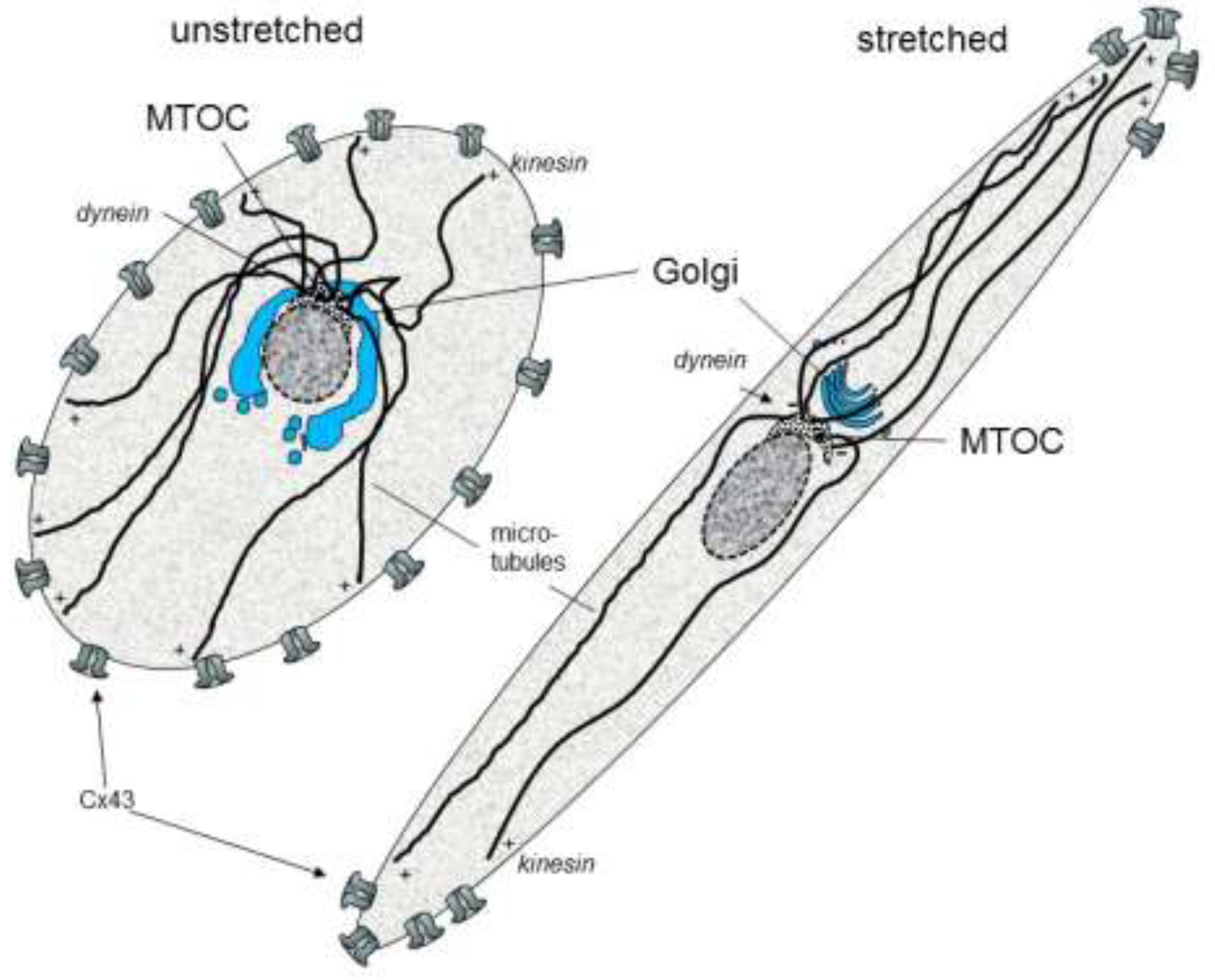{kind=link}
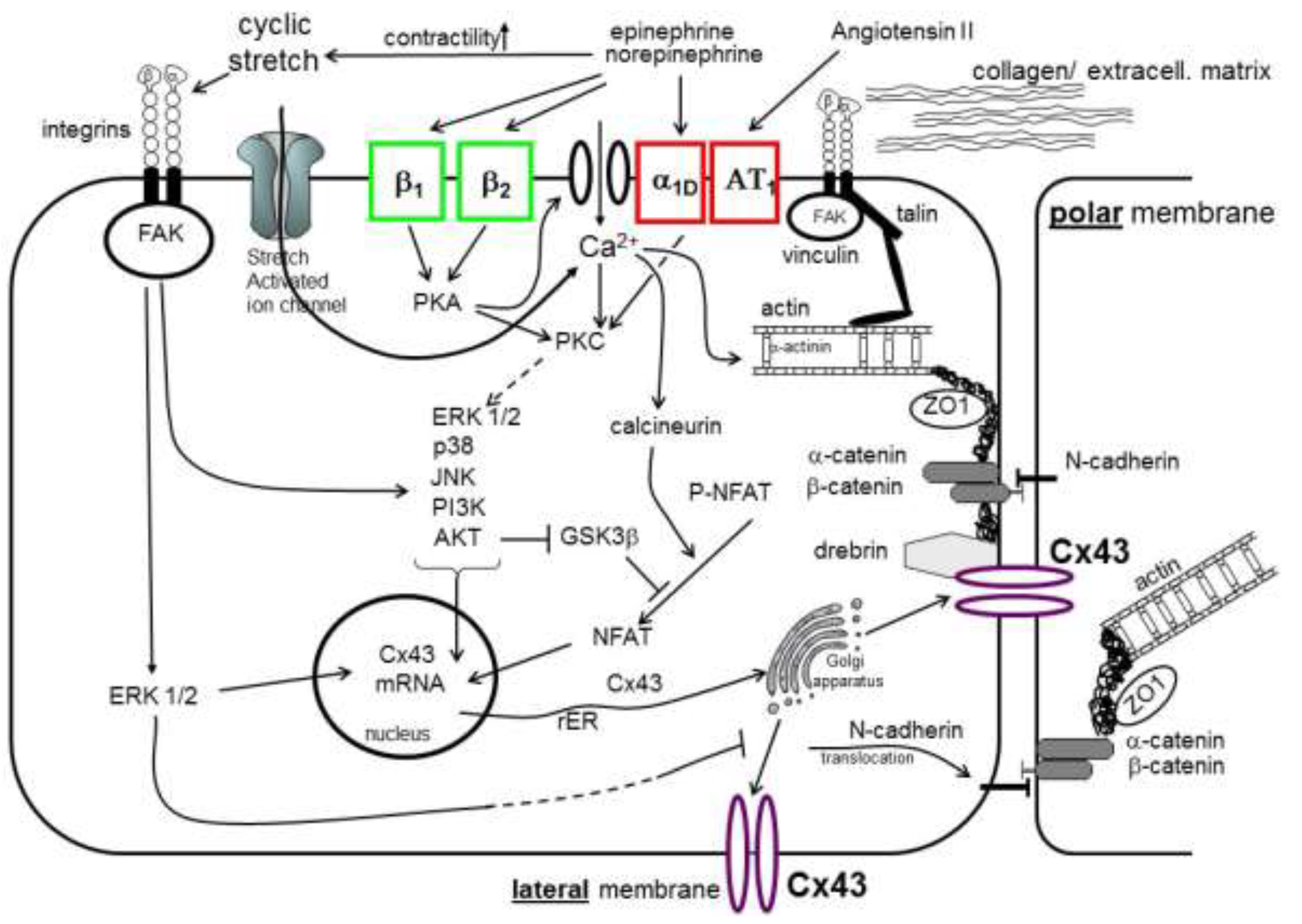{kind=link}
| Stimulus | Opening | Closure | Comment |
|---|---|---|---|
| H+; CO2 | C | takes 7–10 min. | |
| Na+, Ca2+ | C | e.g., in hypoxia | |
| acylcarnitine, lysophosphoglycerides | C | in ischemia/ reperfusion injury | |
| ATP loss | C | in ischema | |
| arachidonic acid, oleic acid, palmitoleic acid, hepantol, octanol, narcotics (halothane) | C | via incorporation? or via altered phosphorylation? | |
| Unspecific PKC stimulation (e.g., phorbol esters) | C | depends on PKC isoform present in the targeted cell | |
| cAMP and direct or indirect stimulators of adenylylcyclase (isoprenalin, forskolin) | O | cAMP activates PKA, in Cx40 or Cx45 coupled cells | |
| PKA | O | in Cx40 or Cx45 coupled cells | |
| antiarrhythmic peptides, AAP10, rotigaptide, danegaptide | O | via PKCα |
| Stimulus | Enhanced Number | Enhanced Degradation | Comment |
|---|---|---|---|
| phenylephrine (α-adrenergic stimulator) | Cx43 | via PKC, MAPKs (cardiomyocytes) | |
| isoprenaline (β-adrenergic stimulator) | Cx43 | via PKA, MAPKs (cardiomyocytes) | |
| angiotensin-II | Cx43 | via AT1 receptors (cardiomyocytes) | |
| Endothelin | Cx43 | via ETA-receptors (cardiomyocytes) | |
| thyroid hormone | Cx40, Cx43 | mechanism unclear | |
| VEGF | Cx43 | via TGFβ | |
| bFGF | Cx43 | via PKCε | |
| EGF | Cx43 | enhanced internalization | |
| Nicotine | Cx37, Cx43 | via nACh-R (endothelium) |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Dhein, S.; Salameh, A. Remodeling of Cardiac Gap Junctional Cell–Cell Coupling. Cells 2021, 10, 2422. https://doi.org/10.3390/cells10092422
Dhein S, Salameh A. Remodeling of Cardiac Gap Junctional Cell–Cell Coupling. Cells. 2021; 10(9):2422. https://doi.org/10.3390/cells10092422
Chicago/Turabian StyleDhein, Stefan, and Aida Salameh. 2021. "Remodeling of Cardiac Gap Junctional Cell–Cell Coupling" Cells 10, no. 9: 2422. https://doi.org/10.3390/cells10092422
APA StyleDhein, S., & Salameh, A. (2021). Remodeling of Cardiac Gap Junctional Cell–Cell Coupling. Cells, 10(9), 2422. https://doi.org/10.3390/cells10092422