
Uncovering the Metabolic and Stress Responses of Human Embryonic Stem Cells to FTH1 Gene Silencing

 , , , ,
, , , , 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Cell Culture
2.2. Lentiviral Production and Generation of Stable Knock-Down ESCs
2.3. RNA Isolation and Quantitative Real-Time PCR
2.4. Immunofluorescence
2.5. Western Blotting
2.6. Measurement of Intracellular Iron, Total ROS and Mitochondrial Superoxide Species
2.7. Analysis of Mitochondrial Membrane Potential (ΔΨ)
2.8. Embryoid Bodies (EBs) Formation Assay
2.9. Alkaline Phosphatase Assay
2.10. Metabolic and Bioenergetic Function Profile
2.11. Apoptosis Analysis
2.12. Statistics
3. Results
3.1. Effects of FTH1-Silencing on ESCs Pluripotency
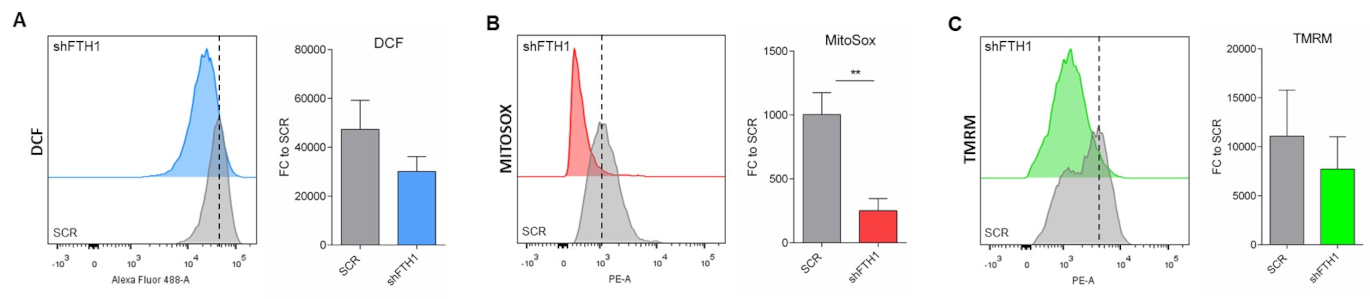
3.2. ROS Levels Are Reduced upon FTH1-Gene Silencing in hESCs
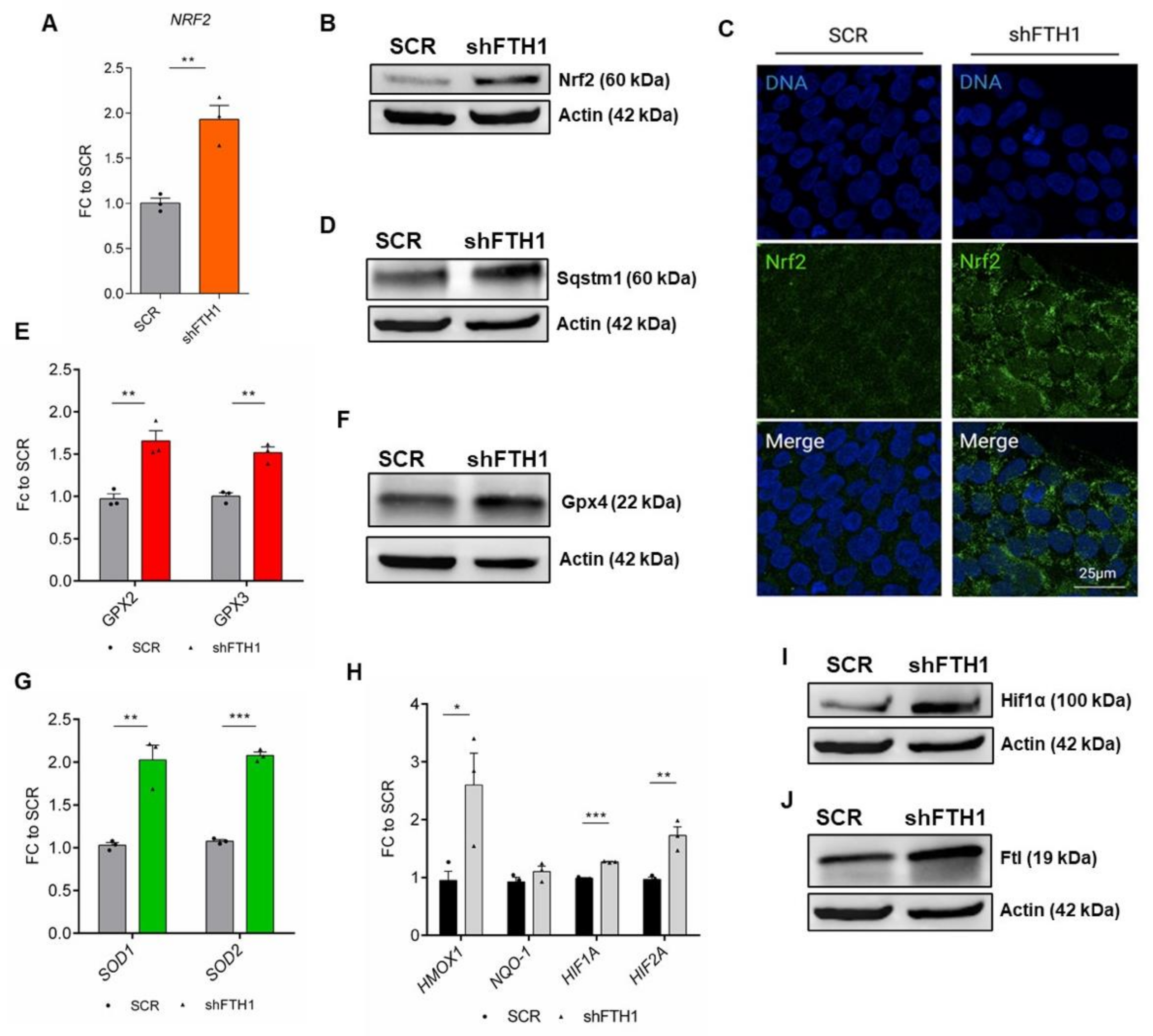
3.3. Nrf2 Signaling Pathway Drives the Antioxidant Response in hESCs
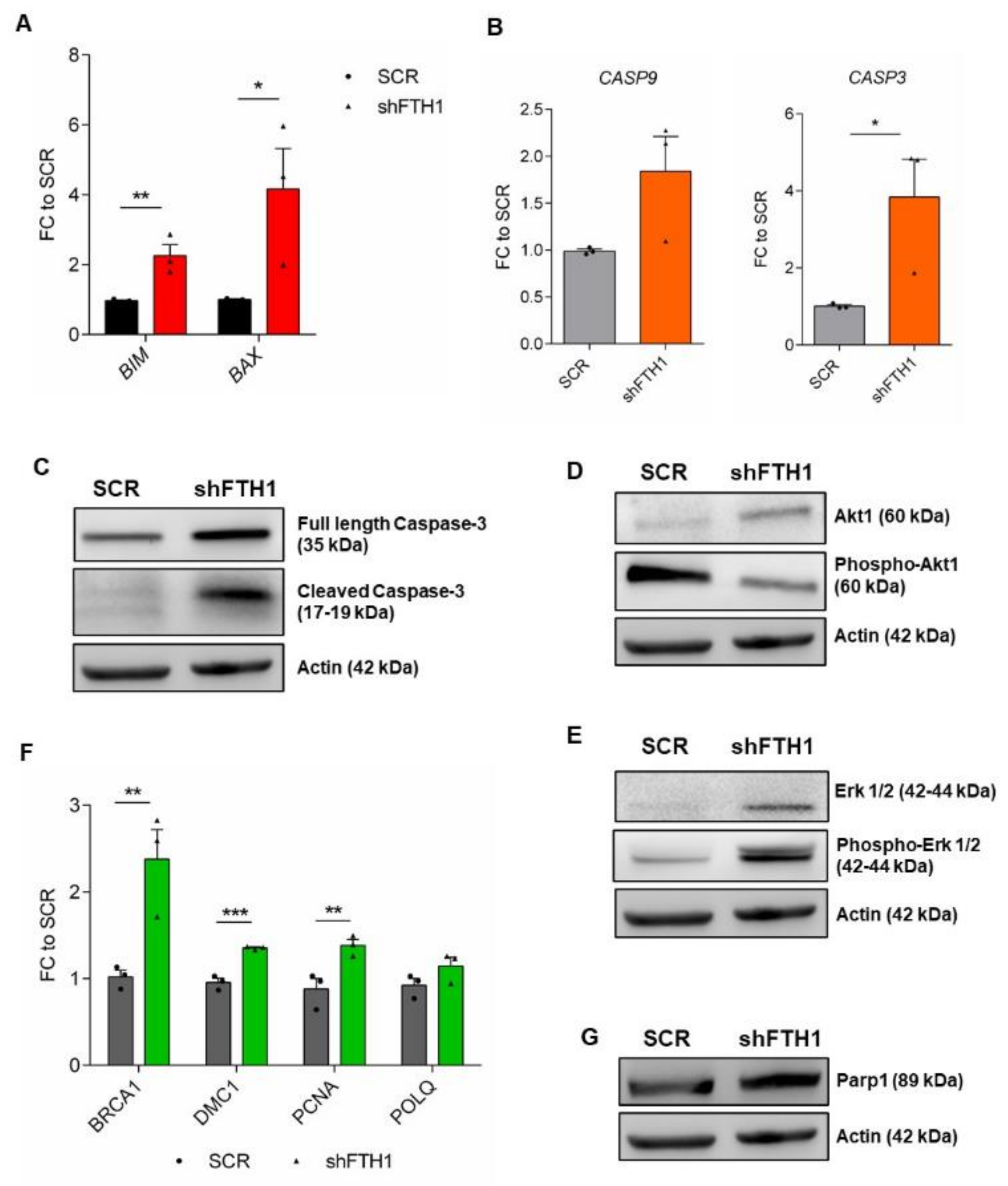
3.4. Effects of FTH1 Knock-Down on Apoptosis
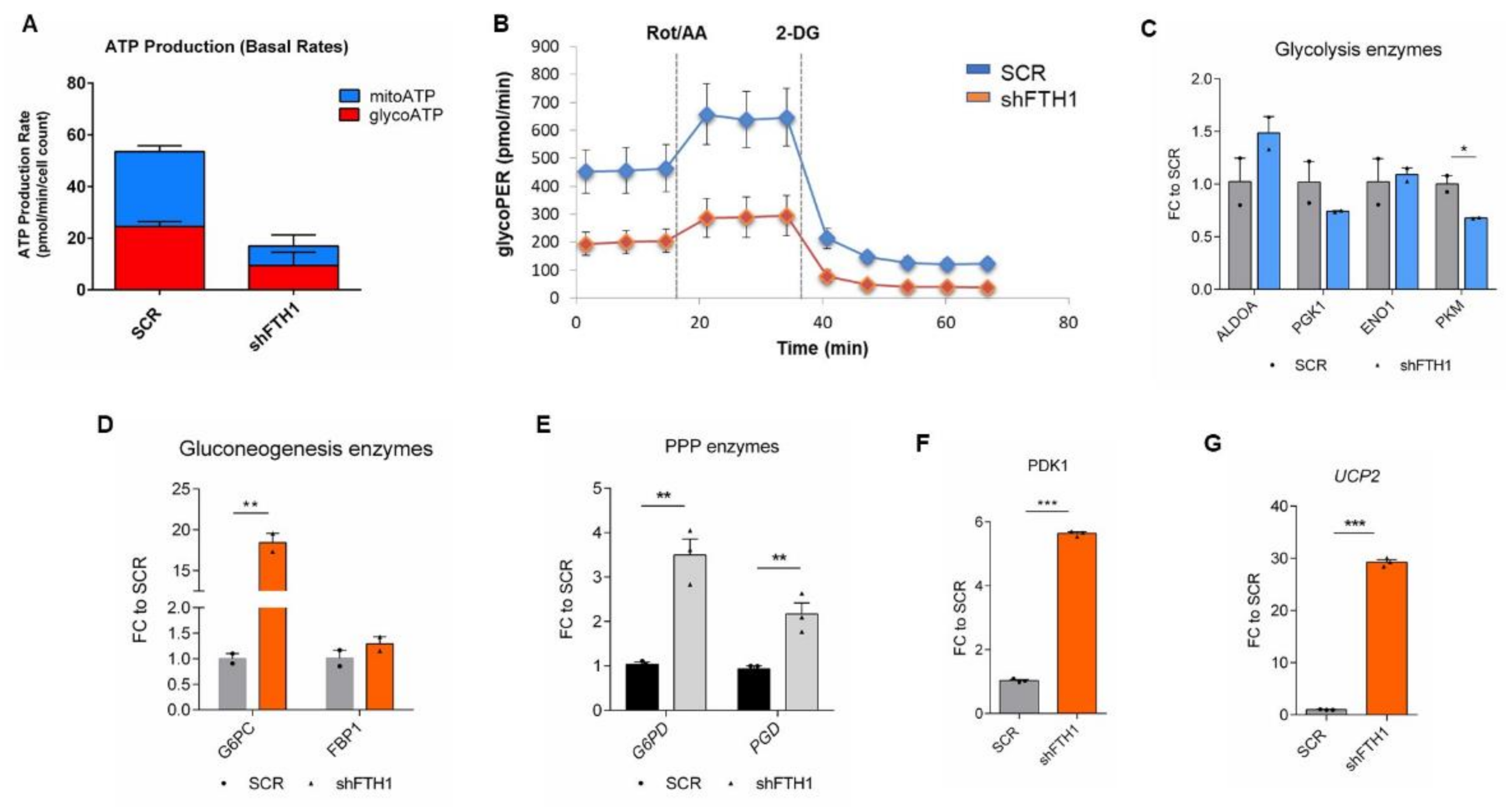
3.5. Effects of FTH1 Repression on Cellular Metabolism
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Forrester, S.J.; Kikuchi, D.S.; Hernandes, M.S.; Xu, Q.; Griendling, K.K. Reactive Oxygen Species in Metabolic and Inflammatory Signaling. Circ. Res. 2018, 122, 877–902. [Google Scholar] [CrossRef]
- Martindale, J.L.; Holbrook, N.J. Cellular Response to Oxidative Stress: Signaling for Suicide and Survival*. J. Cell. Physiol. 2002, 192, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Holmström, K.M.; Finkel, T. Cellular Mechanisms and Physiological Consequences of Redox-Dependent Signalling. Nat. Rev. Mol. Cell Biol. 2014, 15, 411–421. [Google Scholar] [CrossRef] [PubMed]
- Zhong, W.; Qian, K.; Xiong, J.; Ma, K.; Wang, A.; Zou, Y. Curcumin Alleviates Lipopolysaccharide Induced Sepsis and Liver Failure by Suppression of Oxidative Stress-Related Inflammation via PI3K/AKT and NF-ΚB Related Signaling. Biomed. Pharmacother. 2016, 83, 302–313. [Google Scholar] [CrossRef] [PubMed]
- Guo, Y.-L.; Chakraborty, S.; Rajan, S.S.; Wang, R.; Huang, F. Effects of Oxidative Stress on Mouse Embryonic Stem Cell Proliferation, Apoptosis, Senescence, and Self-Renewal. Stem Cells Dev. 2010, 19, 1321–1331. [Google Scholar] [CrossRef]
- Forsyth, N.R.; Musio, A.; Vezzoni, P.; Simpson, A.H.R.W.; Noble, B.S.; McWhir, J. Physiologic Oxygen Enhances Human Embryonic Stem Cell Clonal Recovery and Reduces Chromosomal Abnormalities. Cloning Stem Cells 2006, 8, 16–23. [Google Scholar] [CrossRef]
- Ji, A.-R.; Ku, S.-Y.; Cho, M.S.; Kim, Y.Y.; Kim, Y.J.; Oh, S.K.; Kim, S.H.; Moon, S.Y.; Choi, Y.M. Reactive Oxygen Species Enhance Differentiation of Human Embryonic Stem Cells into Mesendodermal Lineage. Exp. Mol. Med. 2010, 42, 175. [Google Scholar] [CrossRef] [Green Version]
- Saito, S.; Lin, Y.-C.; Tsai, M.-H.; Lin, C.-S.; Murayama, Y.; Sato, R.; Yokoyama, K.K. Emerging Roles of Hypoxia-Inducible Factors and Reactive Oxygen Species in Cancer and Pluripotent Stem Cells. Kaohsiung J. Med. Sci. 2015, 31, 279–286. [Google Scholar] [CrossRef] [Green Version]
- Trachootham, D.; Lu, W.; Ogasawara, M.A.; Valle, N.R.-D.; Huang, P. Redox Regulation of Cell Survival. Antioxid. Redox Signal. 2008, 10, 1343–1374. [Google Scholar] [CrossRef] [Green Version]
- Armstrong, L.; Tilgner, K.; Saretzki, G.; Atkinson, S.P.; Stojkovic, M.; Moreno, R.; Przyborski, S.; Lako, M. Human Induced Pluripotent Stem Cell Lines Show Stress Defense Mechanisms and Mitochondrial Regulation Similar to Those of Human Embryonic Stem Cells. Stem Cells 2010, 28, 661–673. [Google Scholar] [CrossRef] [PubMed]
- Murry, C.E.; Keller, G. Differentiation of Embryonic Stem Cells to Clinically Relevant Populations: Lessons from Embryonic Development. Cell 2008, 132, 661–680. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ogasawara, M.A.; Zhang, H. Redox Regulation and Its Emerging Roles in Stem Cells and Stem-Like Cancer Cells. Antioxid. Redox Signal. 2009, 11, 1107–1122. [Google Scholar] [CrossRef] [PubMed]
- Saretzki, G.; Walter, T.; Atkinson, S.; Passos, J.F.; Bareth, B.; Keith, W.N.; Stewart, R.; Hoare, S.; Stojkovic, M.; Armstrong, L.; et al. Downregulation of Multiple Stress Defense Mechanisms During Differentiation of Human Embryonic Stem Cells. Stem Cells 2008, 26, 455–464. [Google Scholar] [CrossRef] [PubMed]
- Lin, C.-Y.; Peng, C.-Y.; Huang, T.-T.; Wu, M.-L.; Lai, Y.-L.; Peng, D.H.; Chen, P.-F.; Chen, H.-F.; Yen, B.L.; Wu, K.K.; et al. Exacerbation of Oxidative Stress-Induced Cell Death and Differentiation in Induced Pluripotent Stem Cells Lacking Heme Oxygenase-1. Stem Cells Dev. 2012, 21, 1675–1687. [Google Scholar] [CrossRef] [Green Version]
- Sanzo, M.D.; Aversa, I.; Santamaria, G.; Gagliardi, M.; Panebianco, M.; Biamonte, F.; Zolea, F.; Faniello, M.C.; Cuda, G.; Costanzo, F. FTH1P3, a Novel H-Ferritin Pseudogene Transcriptionally Active, Is Ubiquitously Expressed and Regulated during Cell Differentiation. PLoS ONE 2016, 11, e0151359. [Google Scholar] [CrossRef]
- Solari, C.; Petrone, M.V.; Vazquez Echegaray, C.; Cosentino, M.S.; Waisman, A.; Francia, M.; Barañao, L.; Miriuka, S.; Guberman, A. Superoxide Dismutase 1 Expression Is Modulated by the Core Pluripotency Transcription Factors Oct4, Sox2 and Nanog in Embryonic Stem Cells. Mech. Dev. 2018, 154, 116–121. [Google Scholar] [CrossRef]
- Folmes, C.D.L.; Dzeja, P.P.; Nelson, T.J.; Terzic, A. Metabolic Plasticity in Stem Cell Homeostasis and Differentiation. Cell. Stem Cell 2012, 11, 596–606. [Google Scholar] [CrossRef] [Green Version]
- Prigione, A.; Fauler, B.; Lurz, R.; Lehrach, H.; Adjaye, J. The Senescence-Related Mitochondrial/Oxidative Stress Pathway Is Repressed in Human Induced Pluripotent Stem Cells. Stem Cells 2010, 28, 721–733. [Google Scholar] [CrossRef]
- Zhang, J.; Khvorostov, I.; Hong, J.S.; Oktay, Y.; Vergnes, L.; Nuebel, E.; Wahjudi, P.N.; Setoguchi, K.; Wang, G.; Do, A.; et al. UCP2 Regulates Energy Metabolism and Differentiation Potential of Human Pluripotent Stem Cells. EMBO J. 2011, 30, 4860–4873. [Google Scholar] [CrossRef] [Green Version]
- De Angelis, M.T.; Santamaria, G.; Parrotta, E.I.; Scalise, S.; Lo Conte, M.; Gasparini, S.; Ferlazzo, E.; Aguglia, U.; Ciampi, C.; Sgura, A.; et al. Establishment and Characterization of Induced Pluripotent Stem Cells (IPSCs) from Central Nervous System Lupus Erythematosus. J. Cell. Mol. Med. 2019, 23, 7382–7394. [Google Scholar] [CrossRef]
- Galaris, D.; Barbouti, A.; Pantopoulos, K. Iron Homeostasis and Oxidative Stress: An Intimate Relationship. Biochim. Biophys. Acta BBA-Mol. Cell Res. 2019, 1866, 118535. [Google Scholar] [CrossRef]
- Han, Z.; Yu, Y.; Xu, J.; Bao, Z.; Xu, Z.; Hu, J.; Yu, M.; Bamba, D.; Ma, W.; Ding, F.; et al. Iron Homeostasis Determines Fate of Human Pluripotent Stem Cells Via Glycerophospholipids-Epigenetic Circuit. Stem Cells 2019, 37, 489–503. [Google Scholar] [CrossRef]
- Han, Z.; Xu, Z.; Chen, L.; Ye, D.; Yu, Y.; Zhang, Y.; Cao, Y.; Djibril, B.; Guo, X.; Gao, X.; et al. Iron Overload Inhibits Self-renewal of Human Pluripotent Stem Cells via DNA Damage and Generation of Reactive Oxygen Species. FEBS Open Bio 2020, 10, 726–733. [Google Scholar] [CrossRef] [Green Version]
- Kondoh, H.; Lleonart, M.E.; Nakashima, Y.; Yokode, M.; Tanaka, M.; Bernard, D.; Gil, J.; Beach, D. A High Glycolytic Flux Supports the Proliferative Potential of Murine Embryonic Stem Cells. Antioxid. Redox Signal. 2007, 9, 293–299. [Google Scholar] [CrossRef] [PubMed]
- Liu, K.; Cao, J.; Shi, X.; Wang, L.; Zhao, T. Cellular Metabolism and Homeostasis in Pluripotency Regulation. Protein Cell 2020, 11, 630–640. [Google Scholar] [CrossRef]
- Zhang, J.; Nuebel, E.; Daley, G.Q.; Koehler, C.M.; Teitell, M.A. Metabolic Regulation in Pluripotent Stem Cells during Reprogramming and Self-Renewal. Cell Stem Cell 2012, 11, 589–595. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Caiazza, C.; D’Agostino, M.; Passaro, F.; Faicchia, D.; Mallardo, M.; Paladino, S.; Pierantoni, G.M.; Tramontano, D. Effects of Long-Term Citrate Treatment in the PC3 Prostate Cancer Cell Line. Int. J. Mol. Sci. 2019, 20, 2613. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Heiden, M.G.V.; Locasale, J.W.; Swanson, K.D.; Sharfi, H.; Heffron, G.J.; Amador-Noguez, D.; Christofk, H.R.; Wagner, G.; Rabinowitz, J.D.; Asara, J.M.; et al. Evidence for an Alternative Glycolytic Pathway in Rapidly Proliferating Cells. Science 2010, 329, 1492–1499. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lisowski, P.; Kannan, P.; Mlody, B.; Prigione, A. Mitochondria and the Dynamic Control of Stem Cell Homeostasis. EMBO Rep. 2018, 19, e45432. [Google Scholar] [CrossRef]
- Ralser, M.; Wamelink, M.M.; Kowald, A.; Gerisch, B.; Heeren, G.; Struys, E.A.; Klipp, E.; Jakobs, C.; Breitenbach, M.; Lehrach, H.; et al. Dynamic Rerouting of the Carbohydrate Flux Is Key to Counteracting Oxidative Stress. J. Biol. 2007, 6, 10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, H.; Badur, M.G.; Divakaruni, A.S.; Parker, S.J.; Jäger, C.; Hiller, K.; Murphy, A.N.; Metallo, C.M. Distinct Metabolic States Can Support Self-Renewal and Lipogenesis in Human Pluripotent Stem Cells under Different Culture Conditions. Cell Rep. 2016, 16, 1536–1547. [Google Scholar] [CrossRef] [Green Version]
- Lees, J.G.; Rathjen, J.; Sheedy, J.R.; Gardner, D.K.; Harvey, A.J. Distinct Profiles of Human Embryonic Stem Cell Metabolism and Mitochondria Identified by Oxygen. Reproduction 2015, 150, 367–382. [Google Scholar] [CrossRef] [Green Version]
- Varum, S.; Rodrigues, A.S.; Moura, M.B.; Momcilovic, O.; Easley, C.A.; Ramalho-Santos, J.; Van Houten, B.; Schatten, G. Energy Metabolism in Human Pluripotent Stem Cells and Their Differentiated Counterparts. PLoS ONE 2011, 6, e20914. [Google Scholar] [CrossRef] [Green Version]
- Tian, Y.; Lu, J.; Hao, X.; Li, H.; Zhang, G.; Liu, X.; Li, X.; Zhao, C.; Kuang, W.; Chen, D.; et al. FTH1 Inhibits Ferroptosis Through Ferritinophagy in the 6-OHDA Model of Parkinson’s Disease. Neurotherapeutics 2020, 17, 1796–1812. [Google Scholar] [CrossRef]
- Kerins, M.J.; Ooi, A. The Roles of NRF2 in Modulating Cellular Iron Homeostasis. Antioxid. Redox Signal. 2018, 29, 1756–1773. [Google Scholar] [CrossRef] [Green Version]
- Salatino, A.; Aversa, I.; Battaglia, A.M.; Sacco, A.; Di Vito, A.; Santamaria, G.; Chirillo, R.; Veltri, P.; Tradigo, G.; Di Cello, A.; et al. H-Ferritin Affects Cisplatin-Induced Cytotoxicity in Ovarian Cancer Cells through the Modulation of ROS. Oxid. Med. Cell. Longev. 2019, 2019, e3461251. [Google Scholar] [CrossRef] [PubMed]
- McMahon, M.; Itoh, K.; Yamamoto, M.; Hayes, J.D. Keap1-Dependent Proteasomal Degradation of Transcription Factor Nrf2 Contributes to the Negative Regulation of Antioxidant Response Element-Driven Gene Expression. J. Biol. Chem. 2003, 278, 21592–21600. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kobayashi, A.; Kang, M.-I.; Okawa, H.; Ohtsuji, M.; Zenke, Y.; Chiba, T.; Igarashi, K.; Yamamoto, M. Oxidative Stress Sensor Keap1 Functions as an Adaptor for Cul3-Based E3 Ligase To Regulate Proteasomal Degradation of Nrf2. Mol. Cell. Biol. 2004, 24, 7130–7139. [Google Scholar] [CrossRef] [Green Version]
- Kansanen, E.; Kuosmanen, S.M.; Leinonen, H.; Levonen, A.-L. The Keap1-Nrf2 Pathway: Mechanisms of Activation and Dysregulation in Cancer. Redox Biol. 2013, 1, 45–49. [Google Scholar] [CrossRef] [Green Version]
- Hawkins, K.E.; Joy, S.; Delhove, J.M.K.M.; Kotiadis, V.N.; Fernandez, E.; Fitzpatrick, L.M.; Whiteford, J.R.; King, P.J.; Bolanos, J.P.; Duchen, M.R.; et al. NRF2 Orchestrates the Metabolic Shift during Induced Pluripotent Stem Cell Reprogramming. Cell Rep. 2016, 14, 1883–1891. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jang, J.; Wang, Y.; Kim, H.-S.; Lalli, M.A.; Kosik, K.S. Nrf2, a Regulator of the Proteasome, Controls Self-Renewal and Pluripotency in Human Embryonic Stem Cells. Stem Cells 2014, 32, 2616–2625. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Katsuragi, Y.; Ichimura, Y.; Komatsu, M. Regulation of the Keap1–Nrf2 Pathway by P62/SQSTM1. Curr. Opin. Toxicol. 2016, 1, 54–61. [Google Scholar] [CrossRef] [Green Version]
- Jain, A.; Lamark, T.; Sjøttem, E.; Larsen, K.B.; Awuh, J.A.; Øvervatn, A.; McMahon, M.; Hayes, J.D.; Johansen, T. P62/SQSTM1 Is a Target Gene for Transcription Factor NRF2 and Creates a Positive Feedback Loop by Inducing Antioxidant Response Element-Driven Gene Transcription. J. Biol. Chem. 2010, 285, 22576–22591. [Google Scholar] [CrossRef] [Green Version]
- Jin, X.; He, X.; Cao, X.; Xu, P.; Xing, Y.; Sui, S.; Wang, L.; Meng, J.; Lu, W.; Cui, R.; et al. Iron Overload Impairs Normal Hematopoietic Stem and Progenitor Cells through Reactive Oxygen Species and Shortens Survival in Myelodysplastic Syndrome Mice. Haematologica 2018, 103, 1627–1634. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dixon, S.J.; Stockwell, B.R. The Role of Iron and Reactive Oxygen Species in Cell Death. Nat. Chem. Biol. 2014, 10, 9–17. [Google Scholar] [CrossRef] [PubMed]
- Stoyanovsky, D.A.; Billiar, T.R. Cellular non-heme iron modulates apoptosis and caspase 3 activity. In Radicals for Life; Elsevier: Amsterdam, The Netherlands, 2007; pp. 253–268. ISBN 978-0-444-52236-8. [Google Scholar]
- Fang, S.; Yu, X.; Ding, H.; Han, J.; Feng, J. Effects of Intracellular Iron Overload on Cell Death and Identification of Potent Cell Death Inhibitors. Biochem. Biophys. Res. Commun. 2018, 503, 297–303. [Google Scholar] [CrossRef]
- Zhuang, S.; Yan, Y.; Daubert, R.A.; Han, J.; Schnellmann, R.G. ERK Promotes Hydrogen Peroxide-Induced Apoptosis through Caspase-3 Activation and Inhibition of Akt in Renal Epithelial Cells. Am. J. Physiol.-Ren. Physiol. 2007, 292, F440–F447. [Google Scholar] [CrossRef] [Green Version]
- Kennedy, S.G.; Kandel, E.S.; Cross, T.K.; Hay, N. Akt/Protein Kinase B Inhibits Cell Death by Preventing the Release of Cytochrome c from Mitochondria. Mol. Cell. Biol. 1999, 19, 5800–5810. [Google Scholar] [CrossRef] [Green Version]
- Cal, M.; Matyjaszczyk, I.; Litwin, I.; Augustyniak, D.; Ogórek, R.; Ko, Y.; Ułaszewski, S. The Anticancer Drug 3-Bromopyruvate Induces DNA Damage Potentially Through Reactive Oxygen Species in Yeast and in Human Cancer Cells. Cells 2020, 9, 1161. [Google Scholar] [CrossRef]
- Rowe, L.A.; Degtyareva, N.; Doetsch, P.W. DNA Damage-Induced Reactive Oxygen Species (ROS) Stress Response in Saccharomyces Cerevisiae. Free Radic. Biol. Med. 2008, 45, 1167–1177. [Google Scholar] [CrossRef] [Green Version]
- Storr, H.L.; Kind, B.; Parfitt, D.A.; Chapple, J.P.; Lorenz, M.; Koehler, K.; Huebner, A.; Clark, A.J.L. Deficiency of Ferritin Heavy-Chain Nuclear Import in Triple A Syndrome Implies Nuclear Oxidative Damage as the Primary Disease Mechanism. Mol. Endocrinol. 2009, 23, 2086–2094. [Google Scholar] [CrossRef] [Green Version]
- Yu, T.-W.; Anderson, D. Reactive Oxygen Species-Induced DNA Damage and Its Modification: A Chemical Investigation. Mutat. Res. Mol. Mech. Mutagen. 1997, 379, 201–210. [Google Scholar] [CrossRef]
- Zheng, J. Energy Metabolism of Cancer: Glycolysis versus Oxidative Phosphorylation (Review). Oncol. Lett. 2012, 4, 1151–1157. [Google Scholar] [CrossRef] [Green Version]
- Wanet, A.; Arnould, T.; Najimi, M.; Renard, P. Connecting Mitochondria, Metabolism, and Stem Cell Fate. Stem Cells Dev. 2015, 24, 1957–1971. [Google Scholar] [CrossRef] [Green Version]
- Heiss, E.H.; Schachner, D.; Zimmermann, K.; Dirsch, V.M. Glucose Availability Is a Decisive Factor for Nrf2-Mediated Gene Expression. Redox Biol. 2013, 1, 359–365. [Google Scholar] [CrossRef] [Green Version]
- Mitsuishi, Y.; Taguchi, K.; Kawatani, Y.; Shibata, T.; Nukiwa, T.; Aburatani, H.; Yamamoto, M.; Motohashi, H. Nrf2 Redirects Glucose and Glutamine into Anabolic Pathways in Metabolic Reprogramming. Cancer Cell 2012, 22, 66–79. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, J.; Tchernyshyov, I.; Semenza, G.L.; Dang, C.V. HIF-1-Mediated Expression of Pyruvate Dehydrogenase Kinase: A Metabolic Switch Required for Cellular Adaptation to Hypoxia. Cell Metab. 2006, 3, 177–185. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Iurlaro, R.; León-Annicchiarico, C.L.; Muñoz-Pinedo, C. Chapter Three-Regulation of Cancer Metabolism by Oncogenes and Tumor Suppressors. In Methods in Enzymology; Galluzzi, L., Kroemer, G., Eds.; Conceptual Background and Bioenergetic/Mitochondrial Aspects of Oncometabolism; Academic Press: Cambridge, MA, USA, 2014; Volume 542, pp. 59–80. [Google Scholar]
- Pecqueur, C.; Alves-Guerra, M.-C.; Gelly, C.; Lévi-Meyrueis, C.; Couplan, E.; Collins, S.; Ricquier, D.; Bouillaud, F.; Miroux, B. Uncoupling Protein 2, in Vivo Distribution, Induction upon Oxidative Stress, and Evidence for Translational Regulation. J. Biol. Chem. 2001, 276, 8705–8712. [Google Scholar] [CrossRef] [Green Version]
- Parrotta, E.I.; Scalise, S.; Taverna, D.; De Angelis, M.T.; Sarro, G.; Gaspari, M.; Santamaria, G.; Cuda, G. Comprehensive Proteogenomic Analysis of Human Embryonic and Induced Pluripotent Stem Cells. J. Cell. Mol. Med. 2019, 23, 5440–5453. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Avior, Y.; Sagi, I.; Benvenisty, N. Pluripotent Stem Cells in Disease Modelling and Drug Discovery. Nat. Rev. Mol. Cell Biol. 2016, 17, 170–182. [Google Scholar] [CrossRef] [PubMed]
- Fiedler, L.R.; Chapman, K.; Xie, M.; Maifoshie, E.; Jenkins, M.; Golforoush, P.A.; Bellahcene, M.; Noseda, M.; Faust, D.; Jarvis, A.; et al. MAP4K4 Inhibition Promotes Survival of Human Stem Cell-Derived Cardiomyocytes and Reduces Infarct Size In Vivo. Cell. Stem Cell 2019, 24, 579–591.e12. [Google Scholar] [CrossRef] [Green Version]
- Wainger, B.J.; Kiskinis, E.; Mellin, C.; Wiskow, O.; Han, S.S.W.; Sandoe, J.; Perez, N.P.; Williams, L.A.; Lee, S.; Boulting, G.; et al. Intrinsic Membrane Hyperexcitability of Amyotrophic Lateral Sclerosis Patient-Derived Motor Neurons. Cell Rep. 2014, 7, 1–11. [Google Scholar] [CrossRef] [Green Version]
- Zhu, Z.; Huangfu, D. Human Pluripotent Stem Cells: An Emerging Model in Developmental Biology. Development 2013, 140, 705–717. [Google Scholar] [CrossRef] [Green Version]
- Lucchino, V.; Scaramuzzino, L.; Scalise, S.; Grillone, K.; Lo Conte, M.; Esposito, C.; Aguglia, U.; Ferlazzo, E.; Perrotti, N.; Malatesta, P.; et al. Generation of Human Induced Pluripotent Stem Cell Lines (UNIMGi003-A and UNIMGi004-A) from Two Italian Siblings Affected by Unverricht-Lundborg Disease. Stem Cell Res. 2021, 53, 102329. [Google Scholar] [CrossRef]
- Orban, M.; Goedel, A.; Haas, J.; Sandrock-Lang, K.; Gärtner, F.; Jung, C.B.; Zieger, B.; Parrotta, E.; Kurnik, K.; Sinnecker, D.; et al. Functional Comparison of Induced Pluripotent Stem Cell- and Blood-Derived GPIIbIIIa Deficient Platelets. PLoS ONE 2015, 10, e0115978. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Parrotta, E.I.; Lucchino, V.; Scaramuzzino, L.; Scalise, S.; Cuda, G. Modeling Cardiac Disease Mechanisms Using Induced Pluripotent Stem Cell-Derived Cardiomyocytes: Progress, Promises and Challenges. Int. J. Mol. Sci. 2020, 21, 4354. [Google Scholar] [CrossRef] [PubMed]
- Parrotta, E.I.; Procopio, A.; Scalise, S.; Esposito, C.; Nicoletta, G.; Santamaria, G.; De Angelis, M.T.; Dorn, T.; Moretti, A.; Laugwitz, K.-L.; et al. Deciphering the Role of Wnt and Rho Signaling Pathway in IPSC-Derived ARVC Cardiomyocytes by In Silico Mathematical Modeling. Int. J. Mol. Sci. 2021, 22, 2004. [Google Scholar] [CrossRef] [PubMed]
- Scalise, S.; Scaramuzzino, L.; Lucchino, V.; Esposito, C.; Malatesta, P.; Grillone, K.; Perrotti, N.; Cuda, G.; Parrotta, E.I. Generation of IPSC Lines from Two Patients Affected by Febrile Seizure Due to Inherited Missense Mutation in SCN1A Gene. Stem Cell Res. 2020, 49, 102083. [Google Scholar] [CrossRef] [PubMed]
- Yamanaka, S. Pluripotent Stem Cell-Based Cell Therapy-Promise and Challenges. Cell. Stem Cell 2020, 27, 523–531. [Google Scholar] [CrossRef] [PubMed]
- Passaro, F.; Testa, G. Implications of Cellular Aging in Cardiac Reprogramming. Front. Cardiovasc. Med. 2018, 18, 43. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Parrotta, E.I.; Scalise, S.; Scaramuzzino, L.; Cuda, G. Stem Cells: The Game Changers of Human Cardiac Disease Modelling and Regenerative Medicine. Int. J. Mol. Sci. 2019, 20, 5760. [Google Scholar] [CrossRef] [Green Version]
- Harrison, P.M.; Banyard, S.H.; Hoare, R.J.; Russell, S.M.; Treffry, A. The Structure and Function of Ferritin. Ciba Found. Symp. 1976, 20, 19–40. [Google Scholar] [CrossRef]
- Xu, B.; Chasteen, N.D. Iron Oxidation Chemistry in Ferritin. Increasing Fe/O2 Stoichiometry during Core Formation. J. Biol. Chem. 1991, 266, 19965–19970. [Google Scholar] [CrossRef]
- Cheng, H.-T.; Yen, C.-J.; Chang, C.-C.; Huang, K.-T.; Chen, K.-H.; Zhang, R.-Y.; Lee, P.-Y.; Miaw, S.-C.; Huang, J.-W.; Chiang, C.-K.; et al. Ferritin Heavy Chain Mediates the Protective Effect of Heme Oxygenase-1 against Oxidative Stress. Biochim. Biophys. Acta BBA-Gen. Subj. 2015, 1850, 2506–2517. [Google Scholar] [CrossRef] [PubMed]
- Dennery, P.A. Effects of Oxidative Stress on Embryonic Development. Birth Defects Res. Part C Embryo Today Rev. 2007, 81, 155–162. [Google Scholar] [CrossRef] [PubMed]
- Han, Y.; Ishibashi, S.; Iglesias-Gonzalez, J.; Chen, Y.; Love, N.R.; Amaya, E. Ca2+-Induced Mitochondrial ROS Regulate the Early Embryonic Cell Cycle. Cell Rep. 2018, 22, 218–231. [Google Scholar] [CrossRef] [Green Version]
- Rodesch, F.; Simon, P.; Donner, C.; Jauniaux, E. Oxygen Measurements in Endometrial and Trophoblastic Tissues during Early Pregnancy. Obstet. Gynecol. 1992, 80, 283–285. [Google Scholar]
- Ezashi, T.; Das, P.; Roberts, R.M. Low O2 Tensions and the Prevention of Differentiation of HES Cells. Proc. Natl. Acad. Sci. USA 2005, 102, 4783–4788. [Google Scholar] [CrossRef] [Green Version]
- Dai, X.; Yan, X.; Wintergerst, K.A.; Cai, L.; Keller, B.B.; Tan, Y. Nrf2: Redox and Metabolic Regulator of Stem Cell State and Function. Trends Mol. Med. 2020, 26, 185–200. [Google Scholar] [CrossRef] [Green Version]
- Pietsch, E.C.; Chan, J.Y.; Torti, F.M.; Torti, S.V. Nrf2 Mediates the Induction of Ferritin H in Response to Xenobiotics and Cancer Chemopreventive Dithiolethiones*. J. Biol. Chem. 2003, 278, 2361–2369. [Google Scholar] [CrossRef] [Green Version]
- Aung, W.; Hasegawa, S.; Furukawa, T.; Saga, T. Potential Role of Ferritin Heavy Chain in Oxidative Stress and Apoptosis in Human Mesothelial and Mesothelioma Cells: Implications for Asbestos-Induced Oncogenesis. Carcinogenesis 2007, 28, 2047–2052. [Google Scholar] [CrossRef] [PubMed]
- Burdon, T.; Smith, A.; Savatier, P. Signalling, Cell Cycle and Pluripotency in Embryonic Stem Cells. Trends Cell Biol. 2002, 12, 432–438. [Google Scholar] [CrossRef] [Green Version]
- Armstrong, L.; Hughes, O.; Yung, S.; Hyslop, L.; Stewart, R.; Wappler, I.; Peters, H.; Walter, T.; Stojkovic, P.; Evans, J.; et al. The Role of PI3K/AKT, MAPK/ERK and NFκβ Signalling in the Maintenance of Human Embryonic Stem Cell Pluripotency and Viability Highlighted by Transcriptional Profiling and Functional Analysis. Hum. Mol. Genet. 2006, 15, 1894–1913. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stincone, A.; Prigione, A.; Cramer, T.; Wamelink, M.M.C.; Campbell, K.; Cheung, E.; Olin-Sandoval, V.; Grüning, N.-M.; Krüger, A.; Alam, M.T.; et al. The Return of Metabolism: Biochemistry and Physiology of the Pentose Phosphate Pathway. Biol. Rev. 2015, 90, 927–963. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grant, C.M. Metabolic Reconfiguration Is a Regulated Response to Oxidative Stress. J. Biol. 2008, 7, 1. [Google Scholar] [CrossRef] [Green Version]
- Ralser, M.; Lehrach, H. Building a New Bridge between Metabolism, Free Radicals and Longevity. Aging 2009, 1, 836–838. [Google Scholar]

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Scaramuzzino, L.; Lucchino, V.; Scalise, S.; Lo Conte, M.; Zannino, C.; Sacco, A.; Biamonte, F.; Parrotta, E.I.; Costanzo, F.S.; Cuda, G. Uncovering the Metabolic and Stress Responses of Human Embryonic Stem Cells to FTH1 Gene Silencing. Cells 2021, 10, 2431. https://doi.org/10.3390/cells10092431
Scaramuzzino L, Lucchino V, Scalise S, Lo Conte M, Zannino C, Sacco A, Biamonte F, Parrotta EI, Costanzo FS, Cuda G. Uncovering the Metabolic and Stress Responses of Human Embryonic Stem Cells to FTH1 Gene Silencing. Cells. 2021; 10(9):2431. https://doi.org/10.3390/cells10092431
Chicago/Turabian StyleScaramuzzino, Luana, Valeria Lucchino, Stefania Scalise, Michela Lo Conte, Clara Zannino, Alessandro Sacco, Flavia Biamonte, Elvira Immacolata Parrotta, Francesco Saverio Costanzo, and Giovanni Cuda. 2021. "Uncovering the Metabolic and Stress Responses of Human Embryonic Stem Cells to FTH1 Gene Silencing" Cells 10, no. 9: 2431. https://doi.org/10.3390/cells10092431