1. Introduction
Clonal, maternal inheritance of mitochondria and mitochondrial DNA is observed in humans and prevalent in most animals [
1]. Sperm mitophagy, the elimination of sperm-borne mitochondria after fertilization, assures normal preimplantation development and might prevent adverse effects on fitness and fertility arising from heteroplasmy, the coexistence of two distinct mitochondrial genomes in one organism [
2,
3,
4,
5]. Early studies of mammalian sperm mitophagy reported that the sperm mitochondrial sheath becomes ubiquitinated during spermatogenesis and surrounded by the oocyte-derived lysosome-like structures after fertilization, thus implicating both the ubiquitin-proteasome system (UPS) and the autophagic pathway in the elimination of paternal mitochondria [
6,
7]. The porcine zygote is a particularly useful model system as sperm mitophagy is observed prior to the first embryo cleavage, earlier than in other relevant mammalian models. Cell-free systems are yet to be applied to this area of research, relying on monitoring mitochondria from one spermatozoon per zygote. While frog egg extracts are commonly used in cell biology, they are not suitable for the study of mammalian sperm mitophagy, which appears to be species-specific and not observed in interspecific crosses. Our recent observations in zygotes of the domestic pig and rhesus monkey indicate that post-fertilization sperm mitophagy depends on both autophagy driven by ubiquitin-binding autophagy receptor, sequestosome 1 (SQSTM1). Presentation of ubiquitinated sperm mitochondrial membrane proteins to the 26S proteasome is mediated by the ubiquitinated protein dislocase, valosin-containing protein (VCP) [
8]. SQSTM1 was found to exclusively associate with boar and rhesus monkey sperm mitochondria after fertilization. While VCP was detectable in the sperm midpiece, both before and after in vitro fertilization, transient pharmacological inhibition of VCP function delayed the progress of sperm mitophagy in porcine zygotes. A combined treatment interfering concomitantly with SQSTM1 and VCP in the zygotes delayed sperm mitophagy up to the two–four-cell stages of porcine embryonic development, at which time the control zygotes no longer contained detectable sperm mitochondria. These findings raised the question of whether the post-fertilization sperm mitophagy could be reconstituted in a cell-free system, an in vitro research tool that could simplify the identification of additional mitophagy regulators.
Cell-free systems are widely used to study biochemical and molecular events, which reduce the complexity of interactions seen in whole cells [
9]. Cell-free systems derived commonly from
Xenopus laevis, or rarely from mammalian oocytes have been used to reconstitute early events of zygotic development [
9,
10,
11]. The large size of
Xenopus oocytes assures large quantities of cell extract suitable for analyzing molecular and subcellular events occurring at low detection levels in a zygote fertilized by a single spermatozoon. For example, the assembly and incorporation of the nuclear pore complexes (NPC) into the nuclear envelope formed
de novo around the demembranated bull sperm nuclei was reconstituted in
Xenopus oocyte extracts [
11]. Under cell-free conditions, treatment with microtubule inhibitors reduced the assembly of NPCs around the sperm nuclei as well as sperm head expansion that signals the onset of paternal pronuclear development. The morphological events observed under cell-free conditions thus replicated NPC assembly observed during natural fertilization. The
Xenopus oocyte extracts have also been used to induce nuclear reprogramming of differentiated mammalian somatic cells. Expression of pluripotent marker genes was confirmed in the differentiated mammalian cells treated with
Xenopus oocyte extracts [
10]. In addition, cell-free extracts derived from mammalian oocytes were used to induce nuclear reprogramming in somatic cells [
9,
10]. A semi-cell-free system composed of live, capacitated boar spermatozoa and solubilized porcine zona pellucida (ZP) proteins were used to study the role of sperm-borne proteasomes in the digestion of ZP proteins, mimicking the events of sperm-ZP penetration during natural fertilization [
12]. Together, such studies document the feasibility of reconstituting early developmental events in a cell-free system consisting of permeabilized mammalian spermatozoa co-incubated with intra- or inter-specific oocyte extracts.
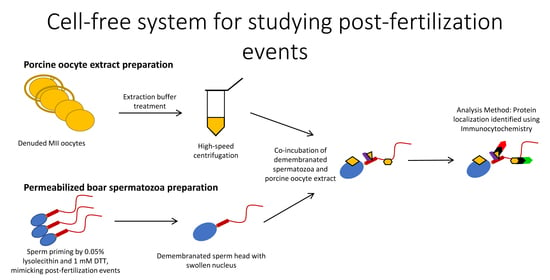
Porcine oocytes are obtainable in large numbers and extract volumes, sufficient to treat thousands of spermatozoa in one trial. Thus, cell-free systems lend themselves to large-scale studies of post-fertilization sperm mitophagy, which is limited to one spermatozoon/sperm mitochondrial sheath per zygote in a conventional monospermic IVF system. Our cell-free system is able to mimic the proteomic interactions taking place between the fertilizing spermatozoa and the ooplasm during natural fertilization. The priming process of removing the plasma membrane and reducing disulfide bonds mimics spermatozoa processing which takes place prior to sperm-ooplasm contact. At that point, the demembranated and disulfide-reduced spermatozoa would enter the ooplasm, and proteomic interactions such as pronuclear development and sperm mitochondrial sheath degradation would begin. Our unique and novel cell-free system was designed with capturing these protein-protein interactions in mind. Here, we established a cell-free system consisting of permeabilized boar and bull spermatozoa co-incubated with cell extracts from porcine metaphase II (MII) oocytes and used it to examine the binding of oocyte-derived mitophagy factors, SQSTM1 and VCP, to sperm mitochondria. Furthermore, our cell-free system was used to study the localization of candidate sperm-borne mitophagy determinants, parkin co-regulated gene (PACRG), and spermatogenesis associated 18 (SPATA18) in spermatozoa before and after coincubation with the oocyte extract.
2. Materials and Methods
2.1. Semen Preparation
Boars were raised at the University of Missouri swine farm. Fresh boar semen was collected in one regular collection per week, transferred into 15 mL centrifuge tubes, and centrifuged at 800× g for 10 min to separate spermatozoa from seminal plasma. Sperm concentration was assessed by using a light microscope and a hemocytometer (ThermoFisher Scientific, Houston, TX, USA). Only semen collections with acceptable sperm motility were used. Spermatozoa were diluted with BTS extender (IMV Technologies, Maple Grove, MN, USA) to a final concentration of 1 × 108 spermatozoa/mL and stored in a styrofoam box at room temperature for up to 5 days.
Cryopreserved bull semen was thawed at 37 °C for 40 s and washed by centrifugation in HEPES-buffered Tyrode lactate medium containing 0.01% (
w/
v) polyvinyl alcohol (TL-HEPES-PVA, pH = 7.4) [
13]. The frozen-thawed bull spermatozoa were used without dilution.
2.2. Sperm Priming for Cell-Free System
Boar or bull spermatozoa were washed with phosphate-buffered saline (PBS, 137 mM NaCl, 2.7 mM KCl, 10 mM, 11 Na
2HPO
4, 1.8 mM KH
2HPO
4, pH = 7.2) containing 0.1% PVA (PBS-PVA) two times by centrifugation at 800×
g for 5 min. To stain sperm mitochondria, the spermatozoa were labeled with a fixable, vital, mitochondrion selective probe MitoTracker
® Red CMXRos (Invitrogen-Molecular Probes, Eugene, OR, USA) for 10 min at 37 °C. At the previously tested concentration of 400 nM, the probe specifically stains bull sperm mitochondria, while in the boar, the probe is also taken up by the sperm head structures [
8]. However, this is not detrimental to the colocalization of autophagy proteins with sperm mitochondria in zygotes or the cell-free system described here.
Sperm heads and tails were separated to examine sperm morphological changes after oocyte extract co-incubation. Spermatozoa pre-labeled with MitoTracker were sonicated with a digital sonifier (Branson) at 30% intensity for 1 min in RIPA buffer (200 mM Tris∙HCI, pH = 8; 150 mM NaCI; 1% (
v/
v) Triton X-100; 0.5% (
w/
v) sodium deoxycholate; 0.1% (
w/
v) SDS; and 1 mM PMSF) [
14].
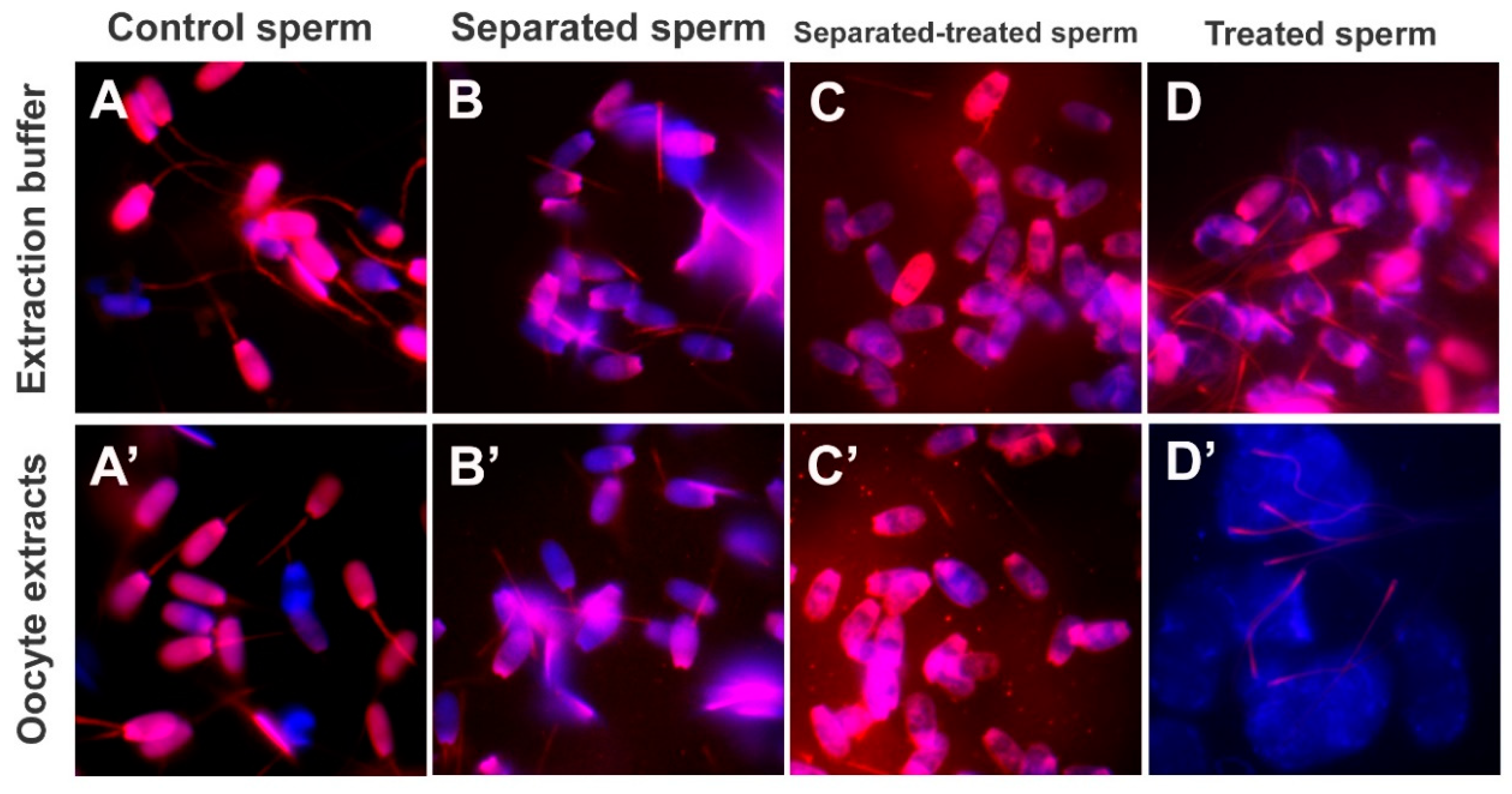
To prime sperm mitochondrial sheaths for cell-free studies, spermatozoa pre-labeled with MitoTracker were demembranated/permeabilized with 0.05% (
w/
v) lysolecithin (L-α-lysophosphatidylcholine, Sigma, St. Louis, MO, USA) in KMT (20 mM KCl, 5 mM MgCl
2, 50 mM TRIS∙HCl, pH = 7.0) for 10 min at 37 °C, and washed twice with the KMT for 5 min by centrifugation, to terminate the reaction. The spermatozoa were subsequently incubated with 0.0, 0.1, 1.0, and 10.0 mM dithiothreitol (DTT; disulfide bonds/S-S reduction agent; Sigma, St. Louis, MO, USA) diluted in KMT, pH = 8.2 for 20 min at 37 °C and washed twice with KMT for 5 min by centrifugation, to terminate the reaction. Such sequential lysolecithin-DTT treatment was applied to mimic the demembranation and S-S reduction in the sperm mitochondrial sheath following natural fertilization [
15].
2.3. In Vitro Maturation of Porcine Oocytes
Porcine ovaries were obtained from a local slaughterhouse. Cumulus-oocyte complexes (COCs) were aspirated from ovarian follicles sized 3–6 mm, divided into groups of 100–150 in 4-well culture dishes (Nunc, Roskilde, Denmark), and matured in TCM-199 medium (Mediatech, Inc., Manassas, VA, USA) supplemented with 0.1% PVA, 3.05 mM D-glucose, 0.91 mM sodium pyruvate, 0.57 mM cysteine, 0.5 μg/mL FSH, 0.5 μg/mL LH, 10 ng/mL EGF, 10% porcine follicular fluid, 75 μg/mL penicillin G and 50 μg/mL streptomycin. The COCs were incubated at 38.5 °C, with 5% CO2 in the air. After 22 h of culture, the COCs were transferred into TCM199 medium without FSH and LH and then cultured for an additional 22 h at 38.5 °C and 5% CO2 in the air.
2.4. In Vitro Fertilization (IVF) and In Vitro Culture (IVC) of Pig Oocytes/Zygotes
Cumulus cells from matured COCs were removed with 0.1% hyaluronidase in TL-HEPES-PVA medium and washed three times with TL-HEPES-PVA medium. The oocytes were washed one more time with Tris-buffered medium (mTBM) containing 0.3% BSA (A7888, Sigma). Between 25–30 oocytes/drop were placed into 100 µL drops of the mTBM covered with mineral oil in a 35 mm polystyrene culture dish, then incubated until spermatozoa were prepared for fertilization. Liquid semen preserved in BTS extender solution was washed with PBS containing 0.1% PVA (PBS-PVA) two times by centrifugation at 800× g for 5 min. To stain mitochondria in the sperm tail, the boar spermatozoa were incubated with vital, fixable, mitochondrion-specific probe MitoTracker® Red CMXRos (Molecular Probes, Inc., Eugene, OR) for 10 min at 38.5 °C. The spermatozoa pre-labeled with MitoTracker were resuspended in mTBM, and added to the 100 µL drops of mTBM for a final concentration of 2.5 to 5 × 104 spermatozoa/mL. Matured oocytes were incubated with spermatozoa for 6 h at 38.5 °C, 5% CO2 in the air, then transferred to 500 µL drops of MU3 medium containing 0.3% BSA (A6003; Sigma) for additional culture.
2.5. Preparation of Porcine Oocyte Extracts
Cumulus cells from matured COCs were denuded with 0.1% hyaluronidase in TL-HEPES-PVA, and zonae pellucidae (ZP) were removed by protease (0.1%, w/v) (Pronase, Sigma) in TL-HEPES-PVA. The ZP-free, mature MII oocytes were transferred into an extraction buffer (50 mM KCl, 5 mM MgCl2, 5 mM ethylene glycol-bis(β-aminoethyl ether)-N,N,N′,N′-tetraacetic acid (EGTA), 2 mM β-mercaptoethanol, 0.1 mM PMSF, protease inhibitor cocktail (cat# 78410, ThermoFisher Scientific, Houston, TX, USA), 50 mM HEPES, pH = 7.6) containing an energy-regenerating system (2 mM ATP, 20 mM phosphocreatine, 20 U/mL creatine kinase, and 2 mM GTP), and submerged three times into liquid nitrogen for 5 min each. Next, the frozen-thawed oocytes were crushed by high-speed centrifugation at 13,000 rpm for 20 min at 4 °C in a Sorvall Biofuge Fresco (Kendro Laboratory Products). The supernatants were harvested, transferred into a 1.5 mL tube, and stored in a deep freezer (−80 °C). Unless otherwise noted, all chemicals used in this study were purchased from Sigma Chemical Co. (St. Louis, MO, USA).
2.6. Co-Incubation of Permeabilized Mammalian Spermatozoa with Porcine Oocyte Extracts
The permeabilized boar or bull spermatozoa were added to porcine oocyte extracts at a concentration of 1 × 104 spermatozoa/10 μL of an extract derived from 2000 porcine oocytes/batch (200 μL) and co-incubated for 4–24 h in a humid atmosphere at 38.5 °C.
Mouse monoclonal anti-SQSTM1 antibody (cat. #ab56416; Abcam, Cambridge, MA, USA), previously shown to interfere with sperm mitophagy when injected in porcine zygotes [
8], was added to the co-culture to examine the consequences of the inhibition of ooplasmic SQSTM1 binding to sperm mitochondria in the cell-free system. The permeabilized boar spermatozoa were co-incubated with porcine oocyte extracts at the same concentration as above for 4 h in a humid atmosphere at 38.5 °C, with or without the addition of 2 μg of anti-SQSTM1 antibody or control non-immune rabbit serum.
At the end of co-incubation, the spermatozoa exposed to oocyte extracts and the control spermatozoa were sedimented onto poly-L-lysine-coated 18 mm coverslips in a drop of KMT, pH = 7.0 at 38.5 °C on a slide warmer. The sperm-coated coverslips were fixed in 2% formaldehyde in PBS-NaN3 for 40 min at room temperature and used for immunocytochemistry immediately.
2.7. Immunocytochemistry to Visualize the Mitophagy Factors in The Cell-Free System
Spermatozoa affixed onto poly-L-lysine coated coverslips were permeabilized in PBS-NaN
3 with 0.1% (
v/
v) Triton-X-100 (PBS-TX) for 40 min and blocked in PBS-TX containing 5% (
v/
v) normal goat serum for 25 min at the ambient temperature. The sperm-coated coverslips were immunolabeled with mouse monoclonal anti-SQSTM1 (cat. #ab56416; Abcam, Cambridge, MA, USA), rabbit polyclonal anti-VCP (cat. #sc-20799; Santa Cruz Biotechnology, Dallas, TX, USA), rabbit polyclonal anti-PACRG (Cat. #ab4090; Abcam), or rabbit monoclonal anti-SPATA18 (Cat. #ab180154; Abcam) antibodies overnight at 4 °C, followed by goat-anti-mouse (GAM)-IgG-FITC (1:200 dilution) or goat-anti-rabbit (GAR)-IgG-FITC (1:200) to demonstrate the localization of SQSTM1, VCP, PACRG, and SPATA18 in the cell-free system. Studies of PACRG were replicated with comparable results by using custom-produced monoclonal antibodies described previously [
16]. Sperm DNA was counterstained with 2.5 μg/mL DAPI (Invitrogen-Molecular Probes, Eugene, OR, USA) added into the secondary antibody solution. Coverslips were mounted on microscopy slides in the VectaShield mounting medium (Vector Laboratories, Burlingame, CA, USA) and examined under a Nikon Eclipse 800 epifluorescence microscope (Nikon Instruments Inc., Melville, NY, USA) with a Retiga QI-R6 camera (Teledyne QImaging, Surrey, BC, Canada) operated by MetaMorph 7.10.2.240 software (Molecular Devices, San Jose, CA, USA).
2.8. Immunocytochemistry to Visualize the Mitophagy Factors in Oocytes and Zygotes
Oocytes and embryos were fixed in 2% formaldehyde for 40 min at room temperature. In some cases, the zona pellucida was removed using protease in TL-HEPES-PVA prior to fixation. Formaldehyde was then removed via washing in PBS. Oocytes/embryos were then permeabilized in PBS with 0.1% Triton-X-100 (PBS-TX) at room temperature for 40 min, then blocked in PBS-TX containing 5% normal goat serum (NGS) for 25 min. Oocytes/embryos were incubated with primary antibodies diluted in PBS containing 1% NGS and 0.1% Triton-X-100 overnight at 4 °C. The samples were incubated with a secondary antibody solution including 2.5 µg/mL DNA stain DAPI (Molecular Probes, Eugene, OR, USA), and goat-anti-rabbit (GAR)-IgG-FITC (1:100 dilution), GAR-IgG-TRITC (1:100), goat-anti-mouse (GAM)-IgG-FITC (1:100), or GAM-IgG-TRITC (1:100) for 40 min at room temperature. The oocytes were mounted on microscopy slides and photographed as described above for sperm samples.
2.9. SQSTM1 Fluorescence Intensity Measurement and Statistical Analysis
Images for pixel intensity measurements were acquired with identical magnification, exposure time, and acquisition settings for all treatment groups. The fluorescence intensity of SQSTM1 was analyzed by NIS-Elements software (v4, Nikon Inc., Melville, NY, USA). Values were represented as the mean of SQSTM1 fluorescence intensity ± SEM. Fluorescence intensities were evaluated by analysis of variance (ANOVA) using the SAS package (v9.3). Duncan’s multiple range test was used to compare significance between individual measurements when F-value was significant (p < 0.05).
2.10. SDS-PAGE and Western Blotting
Oocytes were suspended in 1× LDS loading buffer (106 mM Tris∙HCl, 141 mM Tris base, 2% (w/v) LDS, 10% (v/v) Glycerol, 0.75% (w/v) Coomasie Blue G250, 0.025% (w/v) Phenol Red, pH = 8.5) and sperm protein extracts were mixed with 4× LDS loading buffer in ratio 3:1, both supplemented with 2.5% β-mercaptoethanol. Spermatozoa were incubated at room temperature in 1× LDS loading buffer for 1 h on a rocking platform, spun, and the extract was supplemented with 2.5% β-mercaptoethanol. MII-oocytes and cultured embryos were incubated with protease to remove ZP and mixed with 1× LDS loading buffer supplemented with 2.5% β-mercaptoethanol. Prior to PAGE, all extracts were incubated at 70 °C for 10 min. Equal numbers of oocytes 50 or 100 per lane, as specified in images, spermatozoa (50 to 100 million per lane, as specified in images), and protein extract (the equivalent of 20 µg per lane) were loaded in each lane on a NuPAGE 4–12% Bis-Tris gel (Invitrogen, Waltham, MA, USA). Electrophoresis was carried out in Bis-Tris system using MOPS SDS running buffer (50 mM MOPS, 50 mM Tris base, 0.1% (w/v) SDS, 1 mM EDTA, pH = 7.7) with the cathode buffer supplemented with 5 mM sodium bisulfite. The molecular masses of separated proteins were estimated using Novex® Sharp Pre-stained Protein Standard (cat # LC5800, Invitrogen, Carlsbad, CA, USA) run in parallel. PAGE was carried out for 5 min at 80 V to let the samples delve into the gel and then for another 60–70 min at 160 V. The power was limited to 20 W. After PAGE, proteins were electrotransferred to polyvinylidene fluoride (PVDF) membranes (Millipore) using Owl wet transfer system (Fischer Scientific, Waltham, MA, USA) at 300 mA for 90 min for immunodetection, using Bis-Tris-Bicine transfer buffer (25 mM Bis-Tris base, 25 mM Bicine, 1 mM EDTA, pH = 7.2) supplemented with 10% (v/v) methanol, and 2.5 mM sodium bisulfite. The membranes with the transferred proteins were blocked with 10% (w/v) non-fat milk in TBS with 0.05% (v/v) Tween 20 (TBST; Sigma-Aldrich) for 1 h and incubated with primary antibodies described in the Immunocytochemistry section, overnight at 4 °C and subsequently incubated with the HRP-conjugated goat anti-mouse IgG (GAM-IgG-HRP), or goat anti-rabbit (GAR-IgG-HRP) as secondary antibodies for 40 min at room temperature. The membranes were reacted with chemiluminescent substrate (Millipore Corporation, Billerica, MA, USA), detected using ChemiDoc Touch Imaging System (Bio-Rad, Hercules, CA, USA) to record the protein bands and analyzed by Image Lab Software (ver. 5.2.1, Bio-Rad, Hercules, CA, USA). The membranes were stained with CBB R-250 after chemiluminescence detection for protein load control.
4. Discussion
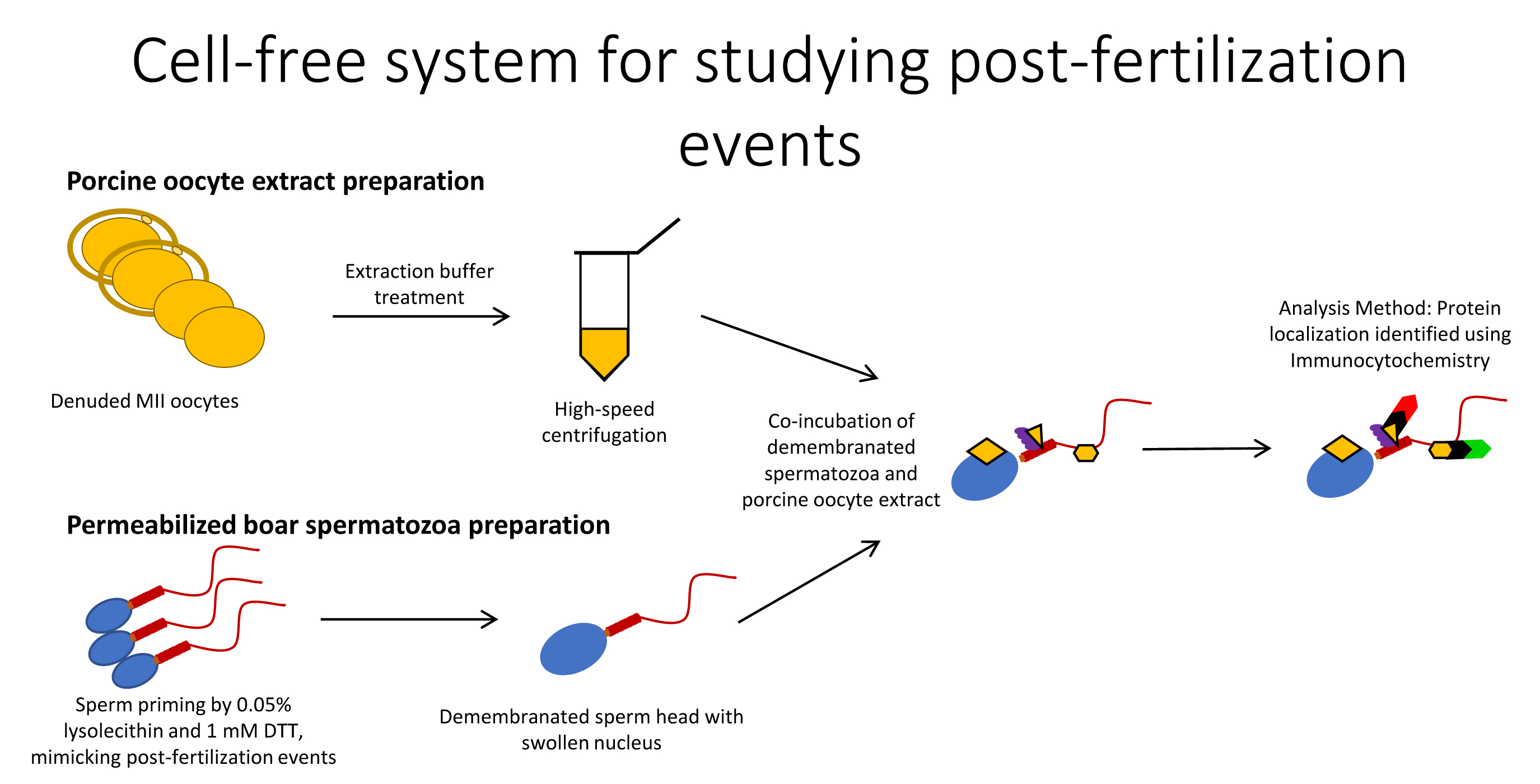
Using the cell-free system consisting of permeabilized, disulfide-reduced boar spermatozoa co-incubated with porcine oocyte extracts, we reconstituted the post-fertilization recognition of sperm mitochondria by autophagy/mitophagy factors SQSTM1 and VCP, as observed previously in mammalian (SQSTM1 and VCP [
8]) and non-mammalian (SQSTM1 only) zygotes and embryos [
17,
18]. Cell-free extracts were derived from mature porcine MII oocytes. A volume of 10 μL extract derived from 2000 porcine oocytes/batch (200 μL) was sufficient to reconstitute the initial step of sperm mitophagy in 1 × 10
4 boar spermatozoa primed by the demembranation and disulfide reduction, demonstrating that the oocyte extract can be used to study the mechanisms promoting clonal, maternal inheritance of mitochondria and mtDNA [
8].
Boar sperm priming with lysolecithin and DTT, which mimics sperm plasma membrane removal during sperm oolemma fusion and subsequent disulfide reduction by ooplasmic factors, was sufficient to expose the sperm mitochondrial sheaths to the binding of autophagy factors from the oocyte extract. Sperm mitochondrial membrane proteins are hardened by disulfide cross-linking [
15,
19,
20,
21]. By using disulfide bond-reducing agents such as DTT and β-mercaptoethanol, sperm mitochondria can be completely dissociated from the sperm tail structure [
22]. The stabilization of sperm accessory structures by disulfide bridges may be a unique feature of mammalian spermatozoa [
23]. Invertebrates and lower vertebrates, including sea urchin and
Xenopus laevis spermatozoa, lack the disulfide stabilization of sperm accessory structures, showing no structural changes after treatment with disulfide bond reducing agents [
15,
24]. Experiments aimed at optimizing sperm priming prior to the extract co-incubation indicate that disulfide reduction is necessary for establishing a porcine oocyte cell-free system.
Prominent among mammalian sperm structures stabilized with disulfide bridges is the sperm nucleus in which protamines become cross-linked during epididymal sperm maturation [
15,
25]. Protamines are arginine-rich DNA binding proteins that supplant nuclear histones during spermatid differentiation to mediate the hypercondensation and packaging of sperm DNA into a compact sperm nucleus [
26]. Sperm nucleus protamination is reversed at fertilization during the disassembly of sperm accessory structures at the onset of embryonic development. During natural fertilization, the reduction in disulfide bonds in the cysteine residues of protamines facilitates sperm nuclear decondensation, male pronuclear development, and replacement of protamines with oocyte-derived histones [
27]. This protamination reversal is mediated by oocyte-produced glutathione (the tripeptide γ-glutamyl-cysteinyl-glycine; GSH) [
28] and may facilitate proteasomal degradation of sperm nuclear protamines, as proteasomal inhibitors hinder paternal pronuclear development [
29]. The above findings support our observation in which sperm head expansion was induced by an oocyte extract following sperm priming by demembranation and disulfide bond reduction, thus mimicking the early stages of paternal pronucleus formation in the zygote. This sperm priming process certainly may influence the localization of some spermatozoa proteins. However, this shift in localization should be mimicking physiological events because both plasma membrane removal and disulfide bond reduction are notable in vivo fertilization events.
Ubiquitin-binding autophagy receptor SQSTM1 links ubiquitinated proteins to the autophagic pathway [
30]. The SQSTM1 binds to polyubiquitinated proteins through its C-terminal polyubiquitin-binding UBA domain [
31]. Mammalian sperm mitochondria carry (poly)ubiquitinated proteins of spermatogenic origin, while additional mitochondrial substrates may become ubiquitinated after sperm incorporation in the ooplasm at fertilization [
6,
7,
32]. Specifically, sperm mitochondrial membrane protein prohibitin (PHB) and mitochondrial transcription factor A (TFAM) are ubiquitinated in bull and boar spermatozoa, respectively [
32,
33]. Our recent study identified three SQSTM1/UBA domain co-purifying boar sperm mitochondrial proteins, including mitochondrial trifunctional enzyme subunit alpha/HADHA, mitochondrial aconitase ACO2, and mitochondrial ATP synthase H
+ transporting F1 complex β-subunit/ATPF1B [
8]. These and other mitochondrial proteins are plausible binding partners/substrates of SQSTM1 during post-fertilization sperm mitophagy. Our previous finding that SQSTM1 derived from the oocyte exclusively associated with sperm mitochondria in porcine and rhesus monkey zygotes [
8] was replicated in a cell-free system in the present study. In addition, the microinjection of autophagy-targeting antibodies specific to SQSTM1 and GABARAP interfered with mammalian sperm mitophagy [
8], and the addition of anti-SQSMT1 antibody to sperm-oocyte extract mixtures consistently prevented the oocyte-cytosolic SQSTM1 from binding to sperm mitochondria in the present study. Parallel to sperm mitochondrion changes, the sperm heads in the present study became enlarged during the prolonged incubation in oocyte extract, mimicking the initial steps of paternal pronucleus development. The addition of anti-SQSTM1 antibody to the sperm-oocyte extract mixture prevented the binding of ooplasmic SQSTM1 to sperm mitochondria, again mimicking the events of early zygotic development. In addition, SQSTM1 derived from porcine oocyte extracts was detected in bull sperm mitochondria, showing interspecific recognition of ooplasmic SQSTM1.
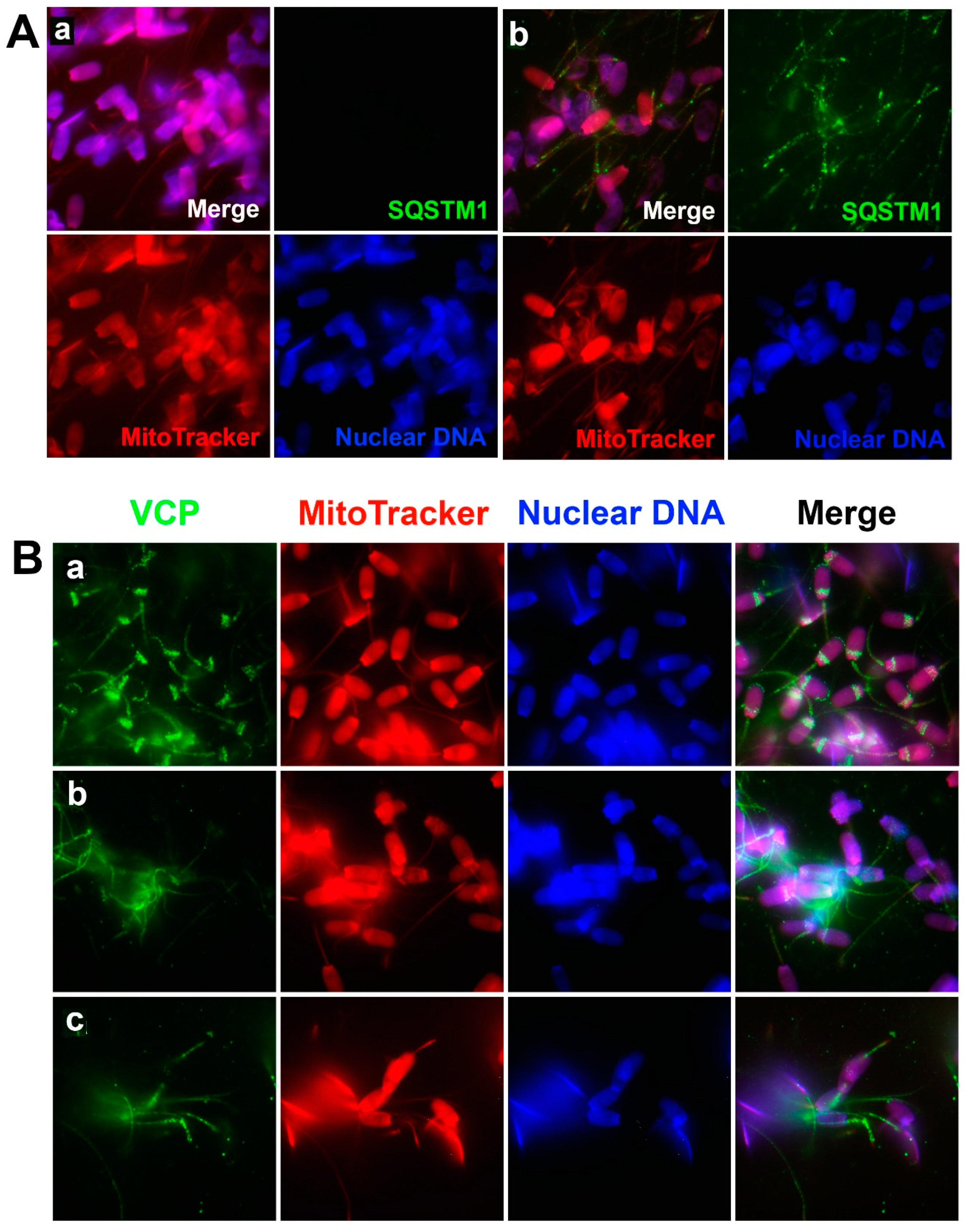
The VCP protein belongs to the AAA+ (ATPase associated with diverse cellular activities) protein family that participates in various cellular events, including cell division, protein degradation by UPS, mitochondria-associated protein degradation, and autophagosome maturation [
34,
35]. Acting as a protein dislocase, VCP extracts ubiquitinated mitochondrial membrane proteins and presents them to the 26S proteasome for degradation during organelle renewal mitophagy in somatic cells [
35]. Association of the VCP with sperm mitochondria exposed to oocyte extracts agrees with previous studies showing that VCP protein was detectable in the mitochondrial sheaths of boar spermatozoa and remained associated with sperm mitochondria after fertilization [
8].
In addition to SQSTM1 and VCP, we used our complementary cell-free and IVF systems to explore the post-fertilization fate of pro-mitophagic proteins, PACRG and SPATA18. Parkin co-regulated gene product, PACRG, is regulated by the UPS. The E3 type ubiquitin ligases which regulate it have yet to be identified [
36], though the components of the linear ubiquitin chain assembly complex (LUBAC), including ubiquitin ligases HOIP and HOIL-1L, are plausible candidates [
37]. The formation of ubiquitinated protein aggregates, the autophagy-targeted aggresome particles, is mediated by PACRG, particularly under conditions of repressed proteasomal proteolysis. These PACRG-containing aggresomes are then removed through macroautophagic pathways. In this sense, mitochondrial sheaths of mammalian spermatozoa are similar to aggresomes and are detectable using aggresome-detecting probes [
8,
38]. Thus, the whole sperm mitochondrial sheath may be recognized as a large aggresome by the oocyte autophagy machinery. PACRG was selected as a target protein in the present study because it shares a bidirectional promoter with the gene
parkin, which codes for the PRKN protein [
8]. The PRKN protein plays a role in a well-defined somatic cell mitophagic pathway, the PINK1-PRKN mitophagic axis [
39], which contributes to post-fertilization sperm mitophagy in the mouse zygotes, in synergy with the SQSTM1-regulated mitophagy branch [
40]. The identification of PACRG on the mitochondrial sheath prior to fertilization and throughout the tail after 4 and 24 h of oocyte extract exposure could build upon this theory. PACRG may serve as a sperm-borne substrate that signals for aggresome-clearing autophagy of the mitochondrial sheath. Further supporting this theory, it has been shown that PACRG levels are directly correlated with the activity of certain autophagic pathway branches. In particular, PACRG and SQSTM1 (alias P62/p62) directly interact following proteasomal inhibition. However, an increase in PACRG alone did not result in aggresome formation; the inhibition of the proteasome was necessary, indicating that PACRG seems to function in a compensatory role [
41]. Perhaps this PACRG pathway is also used during early fertilization to degrade the sperm tail and the mitochondrial sheath through autophagy. PACRG has also been shown to be crucial for spermiogenesis, and its carryover in the sperm mitochondria may reflect such function. Male PACRG knock-out mice are unable to assemble sperm flagellum and undergo spermiogenesis arrest [
42]. PACRG mediates both the formation of the sperm tail [
42] and somatic cell aggresomes [
43] via interactions with the microtubule network. Mutations in the
PACRG gene have been implicated in the etiology of human asthenozoospermia [
44]. The progressive change in localization of sperm PACRG after the oocyte extract treatment in this study (
Figure 3), which resulted in its disappearance at the one-cell stage after fertilization, may help elucidate its function within the context of post-fertilization sperm mitophagy. The localization of PACRG to the mitochondrial sheath of freshly ejaculated spermatozoa appears to be a remanent of its role in sperm tail formation (
Figure 3B), with perhaps a dual function related to post-fertilization sperm mitophagy. It was observed after 4 and 24 h of the cell-free system exposure that the presumed oocyte-derived PACRG molecules appeared to localize throughout the sperm tail principal piece tail and the post-acrosomal region of the sperm head (
Figure 3D,E). In somatic cells, PACRG plays a role in aggresome formation around mitochondria and then plays a role in signaling for degradation. The sperm tail localization pattern of PACRG after 4 and 24 h of cell-free system exposure may be evidence of PACRG acting in conjunction with UPS and signal to recruit SQSTM1, which could explain the absence of PACRG from sperm mitochondria in a one-cell embryo where SQSTM1 has clearly relocated to the sperm mitochondrial sheath (
Figure 3F). Comparatively lesser intensity of SQSTM1 and incomplete removal of PACRG is observed after sperm exposure to oocyte extracts in the cell-free system (present study), which may also lack some of the organelle membranes and lysosomes necessary for the completion of sperm mitophagy. Our future experiments will thus include varied proportions of oocyte cytosol and membrane fractions used in the cell-free system.
Mitochondria eating protein SPATA18 serves for mitochondrial quality control in various cell types. It mediates the degradation of mitochondria using vacuole-like structures, which engulf and degrade unhealthy mitochondria [
45]. The SPATA18 protein can facilitate mitophagy because of the recognition of DNA damage through the p53-SPATA18 axis. This axis serves as a mitochondrial homeostasis maintaining mechanism rather than a direct DNA damage response. SPATA18 induces the formation of intramitochondrial lysosome-like organelle after mitochondrial damage, which seem to act independently of conventional autophagy [
46]. An increase in mitochondrial ATP output under stress leads to the mitochondria producing more reactive oxygen species (ROS), which, in turn, increases mitochondrial stress. This is where SPATA18 comes into play as a mitochondrial quality control measure. Likewise, sperm mitochondria face stress and possible mtDNA damage by ROS as the spermatozoa journey across the male and female reproductive tracts to reach the site of fertilization in the oviduct [
45]. Perhaps SPATA18 works along the mitochondrial quality control axis on the sperm mitochondria upon entry into the oocyte cytoplasm, assuring that potentially damaged paternal mtDNA is not passed onto the next generation. Paternal mtDNA transmission to the offspring has been observed in various interspecific hybrids throughout early embryonic development, whereas sperm mtDNA was eliminated in intraspecific hybrids [
6,
47,
48,
49]. Therefore, mitophagy machinery may be insufficient to eliminate sperm mitochondria in interspecific crosses, although our observation showed bull sperm mitochondria were recognized by the mitophagy factor SQSTM1 in the interspecific cell-free system. Such a system could be used in the near future to identify factors responsible for the species specificity of mammalian sperm mitophagy.


 ,
, 

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}






