1. Introduction
Progenitor cells are present in living tissues and are involved in inflammatory reactions and tissue homeostasis [
1]. In muscle tissue, satellite cells, known as muscle stem cells, reside beneath the sarcolemma and basement membrane [
2]. Muscle contains not only myofibroblasts but also mesenchymal cells that differentiate into fat [
3]. Human-derived tissue progenitor cells are being studied worldwide to treat diseases and injuries that cannot be alleviated with current treatments. Tissue-resident progenitor cells are also present in livestock tissues, and meat is being cultivated outside the body as a new field of sustainable development [
4].
In recent years, depending on the demand for meat, cultured meat has attracted attention as a means to acquire animal proteins [
5,
6,
7]. Via this method, cells can be obtained by culturing and amplifying proliferative cells from bovine tissue (two-dimensional cell culture). Moreover, Ding et al. verified the expression of Pax7, an undifferentiated marker of muscle satellite cells, in cultured CD29+ CD56+ cells that had been sorted from bovine muscle tissue by fluorescence-activated cell sorting (FACS) [
8]. Furthermore, it was revealed that the undifferentiated state of bovine satellite cells can be maintained by suppressing proliferation and differentiation with a p38 inhibitor.
Previously, we developed a technique to isolate proliferative tissue stem cells that exist in mouse and human tissues [
9,
10]. Platelet-derived growth factor receptor a (Pdgfra)-positive cells are fibroblast-like stem cells present in the bone marrow and muscle tissue and have been identified as the origin of adipose cells [
3,
11]. However, various kinds of cells reside in tissues, and it is known that their gene expression and cellular properties are heterogeneous [
12]. Therefore, the identification and isolation of the progenitor cells that make up tissues are in progress in livestock, but there are problems that must be solved in order to produce meat in this way. First, an efficient method for amplifying muscle cells has not yet been established because muscle cells alone have limited proliferative capacity and require other factors to be present in meat. Second, there are many technical barriers to producing three-dimensional structures with a similar organization to livestock meat.
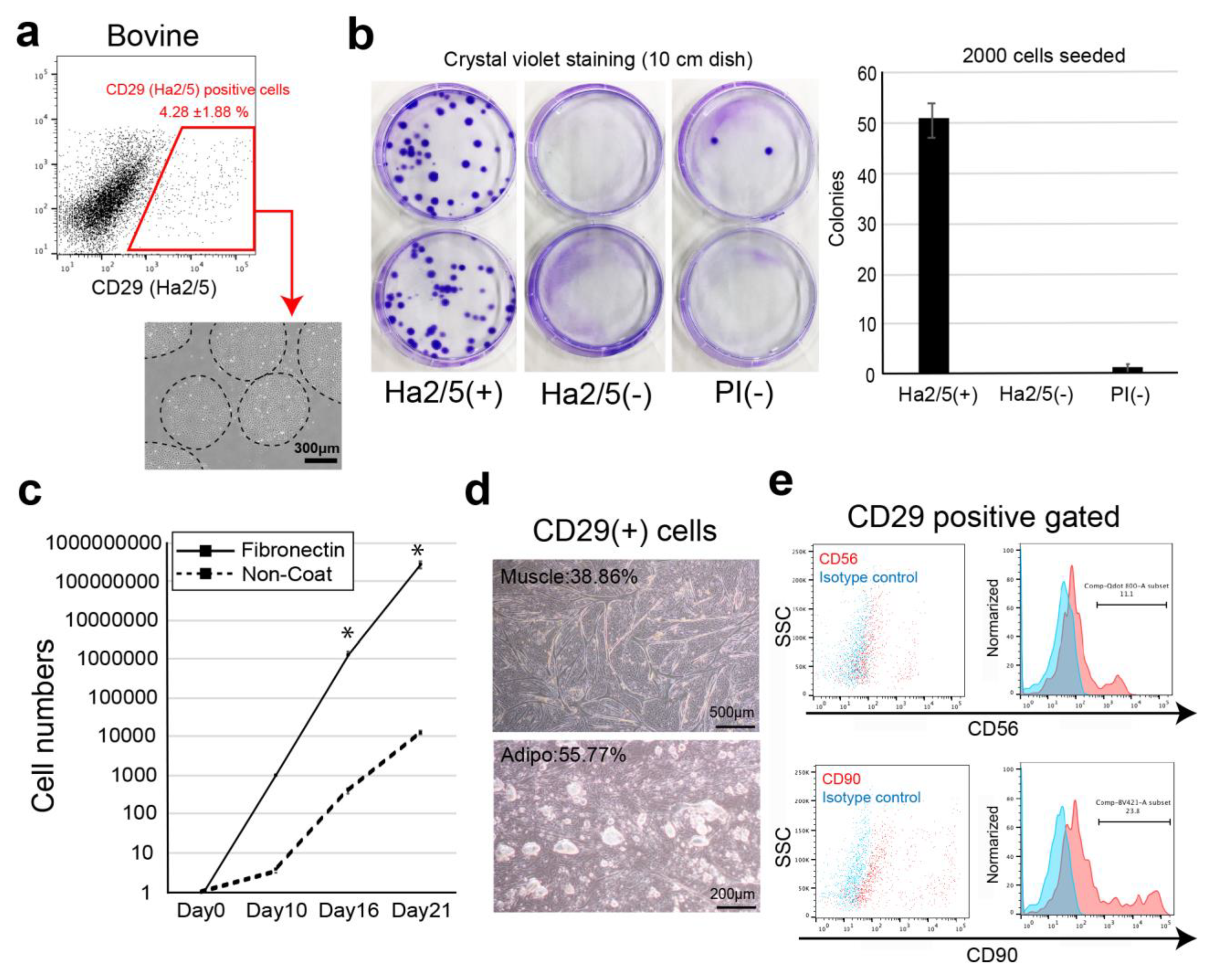
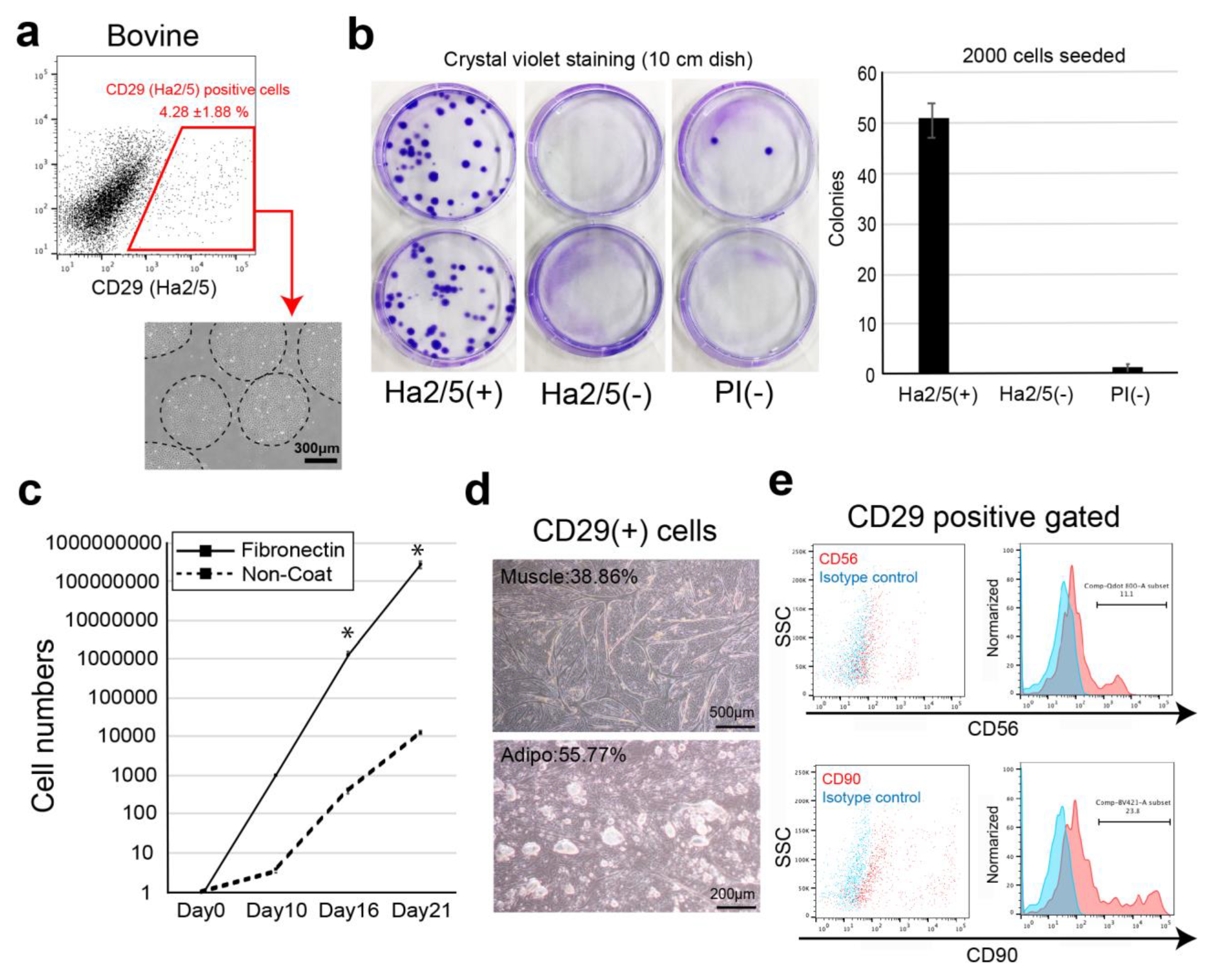
In this study, we screened cell-surface antibodies (anti-human, anti-mouse, anti-rat, and anti-sheep) using a flow cytometer and assessed their capacity to generate cell colonies. The bovine cell population recognized by CD29 antigen (clone: Ha2/5) was highly proliferative with the capacity to differentiate into muscle and adipose lineages. We also established a culture condition to improve the proliferative capacity of cells using fibronectin. Therefore, we isolated Ha2/5+ cells from bovine skeletal muscle, which have a self-aggregating capacity, to determine whether they are a suitable cell source for the construction of meat-like cell mass “meat buds”. The characteristics of meat buds were examined by analyzing the constituent cells that make up meat to gain insights into the production of meat alternatives in vitro.
2. Materials and Methods
2.1. Livestock Tissues
The bovine muscle and adipose tissues in this study were derived from two-year-old Japanese black beef (n = 8). These meats were obtained from Tokyo Meat Center (Yamanashi, Japan). The pig (n = 3) and sheep (n = 3) tissues were purchased from Tokyo Meat Center and Charomen Sheep Pasture (Hokkaido, Japan).
2.2. Cell Preparation
The cells were isolated from tissue samples as previously described [
9,
10]. Briefly, the tissues (bovine, sheep, and pig) were mechanically dissected and digested with 2 mg/mL collagenase (#032-22364, FUJIFILM Wako Pure Chemical, Osaka, Japan), 10 mM HEPES (#15630080, Life Technologies, Carlsbad, CA, USA), and 1% penicillin/streptomycin (#15140-122, gibco, New York, NY, USA) prepared in Dulbecco’s modified Eagle medium (DMEM) with GlutaMax (#10569-100, gibco, Bleiswijk, The Netherlands). After shaking for 1 h at 37 °C, the tissue fragments were removed. The supernatant was centrifuged at 800×
g for 10 min at 20 °C and then collected as mononuclear cell pellets. The cells were washed twice in HBSS and were filtered through a 100-μm cell strainer (#352360, Corning, Durham, NC, USA). The red blood cells in the suspension were lysed by adding 1 mL of ice-cold sterile H
2O for 6 s. Immediately after lysis, 1 mL of 2 × phosphate-buffered saline (PBS) with 4% fetal bovine serum (FBS) was added to quench the reaction. The cells were then filtered through a 70-μm cell strainer and washed twice. The cell pellet was reconstituted with HBSS (2% fetal bovine serum, FBS) or frozen in CELLBANKER 1 plus (NIPPON ZENYAKU KOGYO CO., LTD., Fukushima, Japan).
2.3. Flow Cytometry and Cell Isolation
The cells were resuspended in HBSS and stained with the following antibodies for cell sorting and analysis: allophycocyanin (APC)-conjugated anti-CD29 (anti-rat, clone: Ha2/5, anti-human, clone: TS2/16 and MAR4), anti-CD44 (anti-mouse, clone: IM7), and anti-CD344/Frizzled-4 (anti-human, clone: CH3A4A7) (all antibodies were from BD Biosciences, Franklin Lakes, NJ, USA). For cell surface marker analysis, we screened 246 cell-surface antibodies shown in
Supplementary Materials Table S1. After antibody incubation on ice, the cells were washed and reconstituted in HBSS. Propidium iodide fluorescence was used to gate out the dead cells. Flow cytometry and cell sorting were performed using a FACS Aria II cell sorter (BD Biosciences).
2.4. Cell Culture
The isolated cells were plated on plastic dishes or glass chamber slides coated with fibronectin (#F0635, Sigma-Aldrich MO, USA) in DMEM with GlutaMax (#10569-100, gibco, Bleiswijk, Netherlands) containing 20% FBS (#10270106, gibco, Paisely, UK), 100 units/mL penicillin, 100 μg/mL streptomycin (#15140-122, gibco, New York, NY, USA), and 5 ng/mL basic fibroblast growth factor (#RCHEOT003, ReproCell, Beltsville, MD, USA) in 5% CO2 at 37 °C. After cell expansion, the cells were passaged to maintain a density of <80% confluence (passage 2–5 cells used in the experiments). The colony-forming unit fibroblast assay was performed by culturing 2000 sorted cells on a fibronectin-coated 100 mm dish for 14 days in DMEM-GlutaMax containing 20% FBS (#10270106, gibco, Paisely, UK), 1% penicillin/streptomycin, and 5 ng/mL basic fibroblast growth factor (#RCHEOT003, ReproCell, Beltsville, MD, USA). The medium was changed twice weekly.
2.5. Generation of Bovine Meat Buds
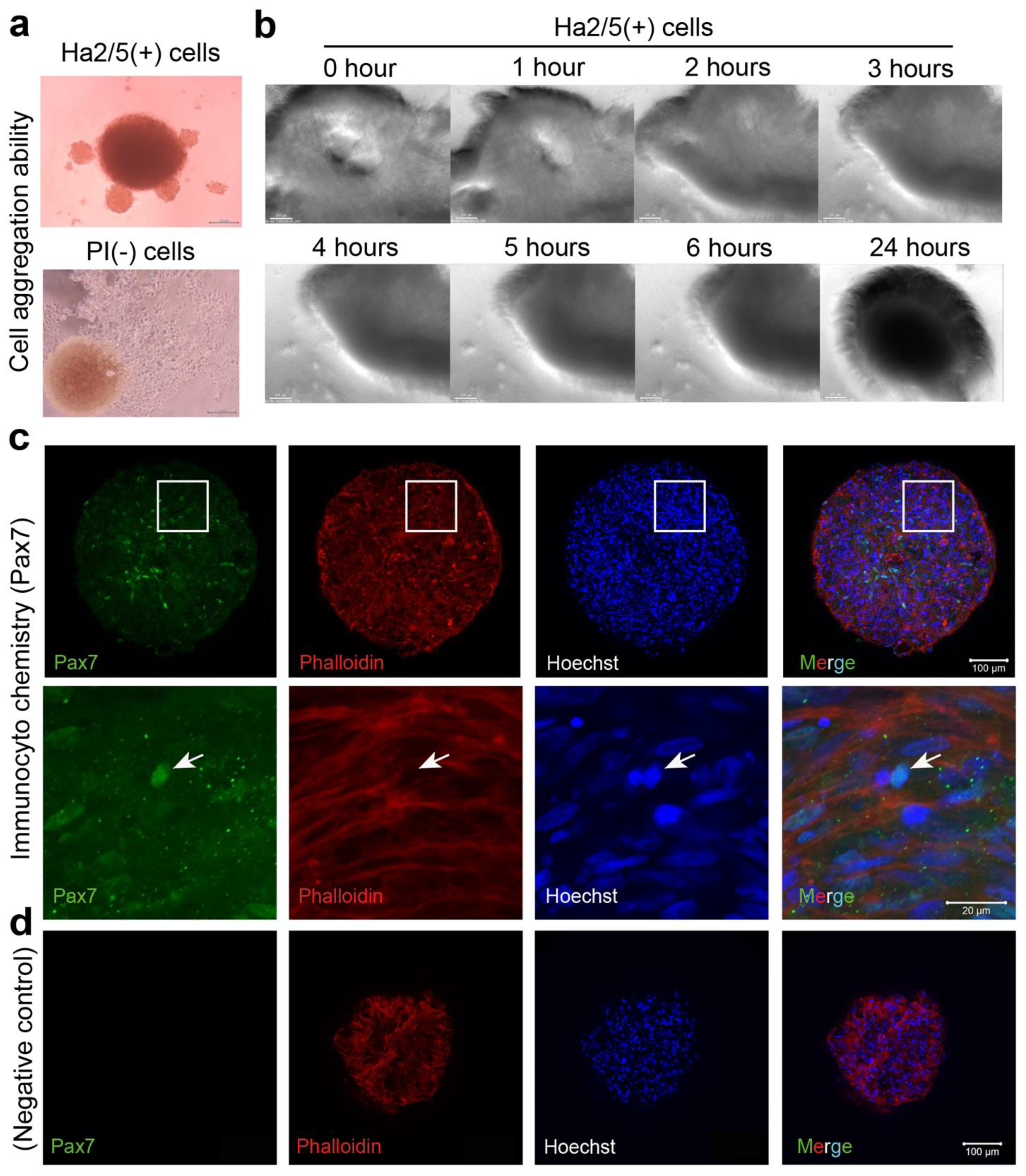
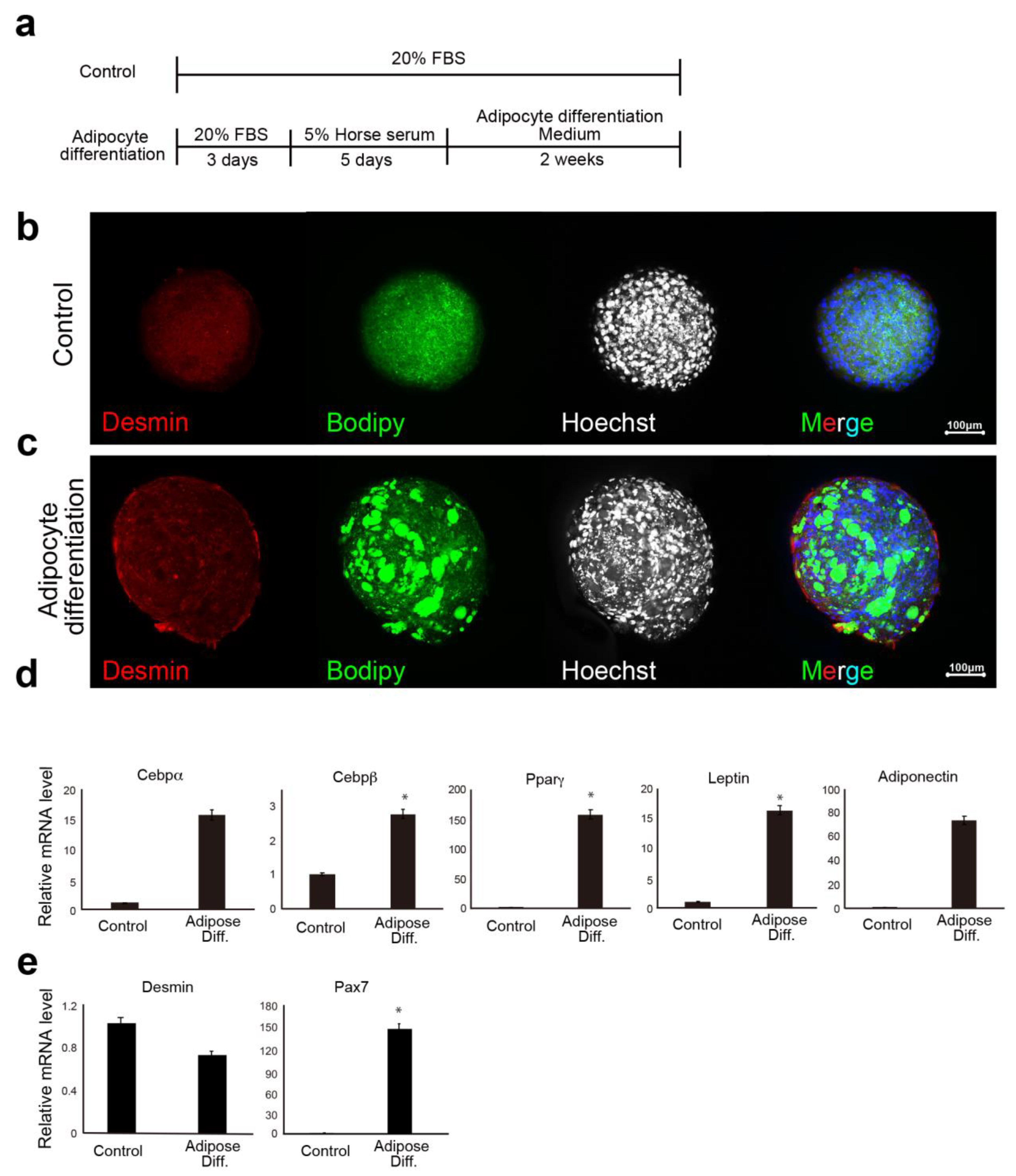
Cultured CD29+ cells (1 × 105) were seeded in a 96-well U-bottom plate (#4870-800SP, IWAKI, Chiyoda City, Japan) in DMEM with GlutaMax (Life Technologies, Carlsbad, CA, USA) containing 20% FBS (#10270106, gibco, Paisely, UK), 100 units/mL penicillin, 100 μg/mL streptomycin (#15140-122, gibco, New York, NY, USA), and 5 ng/mL basic fibroblast growth factor (ReproCell, Beltsville, MD, USA). The plate was then centrifuged at 400× g for 3 min (4 degrees Celsius). The cells were cultured in a state where the cells aggregated at the bottom of the U-bottom wells. A meat bud was formed within 24 h and used in the experiment after culturing for three days. In the case of long-term culture such as differentiation experiments, the medium was changed every three to four days.
2.6. Cell Differentiation
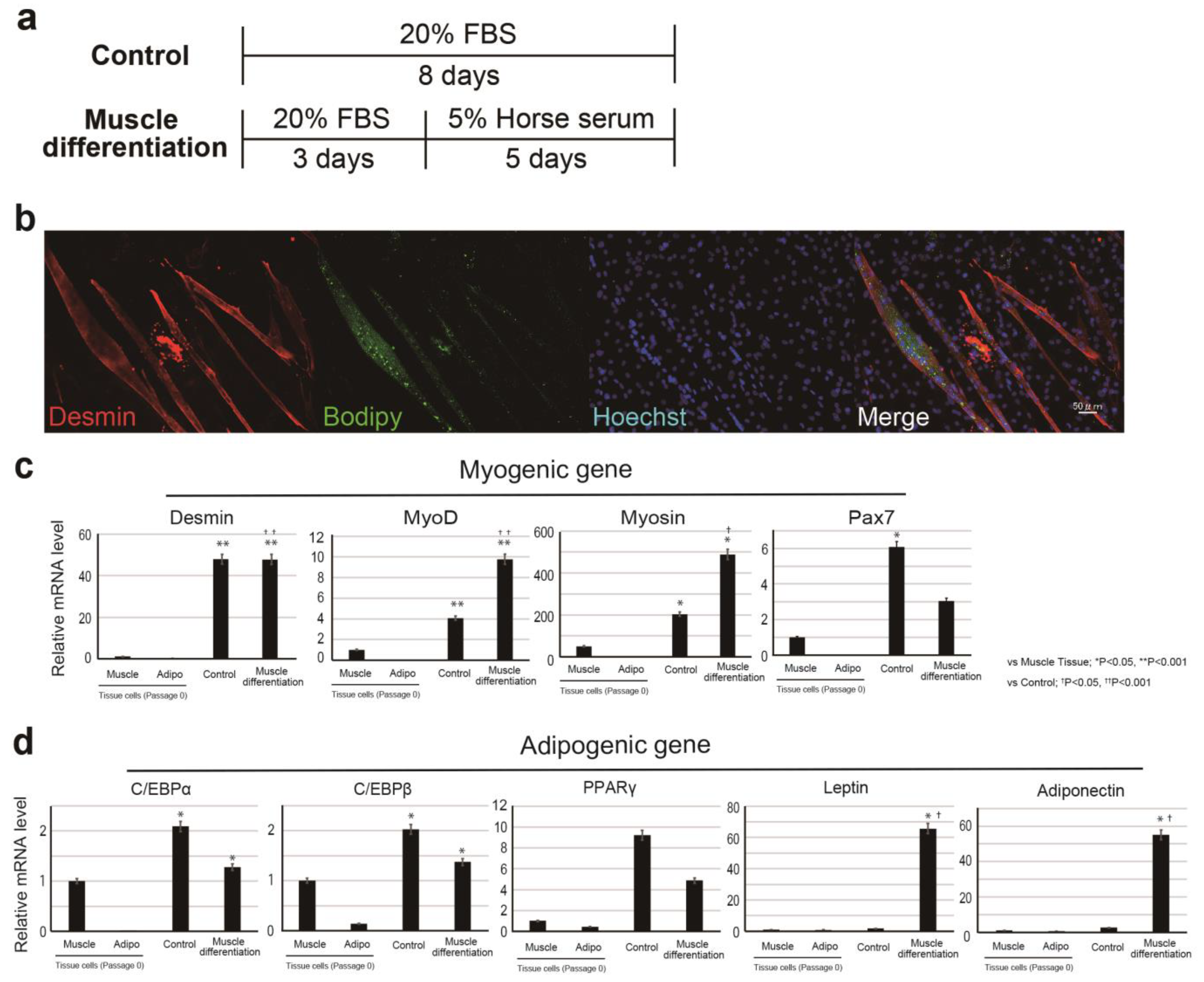
In the muscle-differentiation assay, CD29+ cells (or meat buds) were cultured in a differentiation medium consisting of DMEM with GlutaMax (Life Technologies) supplemented with 5% horse serum (#16050122, Gibco, USA), 100 units/mL penicillin, and 100 μg/mL streptomycin (#15140-122, gibco, New York, NY, USA) for five days.
In the lipid accumulation assay, CD29+ cells (or meat buds) were cultured in adipogenic differentiation medium (Lonza, Basel, Switzerland) with 5% horse serum (#16050122, Gibco, New York, NY, USA) and 500 μM oleic acid (#O0180, Tokyo Chemical Industry Co., Ltd., Tokyo, Japan) for two weeks. The adipogenic differentiation medium consisted of adipose induction medium (containing indomethachin, 3-isobutyl-1-methylxanthine (IBMX), insulin, dexamethasone, and L-glutamine: Cat# PT-4136, Lonza, Basel, Switzerland) and maintenance medium (containing insulin and l-glutamine: Cat# PT-4122, Lonza Basel, Switzerland). Two different media were used alternately (every three to four days). Oil Red O staining was performed with cells that had been washed twice with 250 µL of PBS. After 15 min at 25 °C, the Oil Red O solution (#4049-1, Muto Pure Chemicals, Tokyo, Japan) was removed and the cells were washed three times with water. The stained cells were visualized and scanned using a BZX-710 microscope (Keyence, Osaka, Japan) or an LSM 700 confocal microscope (Zeiss, Oberkochen, Germany).
2.7. Immunofluorescent Staining
CD29+ cells in the glass chamber were fixed in 4% paraformaldehyde for 10 min. After washing with PBS, the cells were permeabilized with 0.2% Triton-X/PBS for 5 min and blocked with Blocking One solution (#03953-95, Nacalai Tesque, Kyoto, Japan) for 1 h. The following primary antibodies were used: anti-desmin antibody (1:100 dilution, #D1033; Sigma-Aldrich, St Louis, MO, USA), anti-myosin antibody (1:100 dilution, # M1570; Sigma-Aldrich, MO, USA), anti-Pax7 antibody (1:50 dilution, #sc-81648; Santa Cruz, CA, USA), anti-phalloidin antibody (1:100 dilution, #R415; Invitrogen, Carlsbad, CA, USA), and anti-CD29 (Ha2/5) antibody (1:500, #555004; BD Biosciences, Franklin Lakes, NJ, USA). BODIPY® 493/503 was used for lipid staining (Thermo Fisher Scientific, Waltham, MA, USA). The primary antibodies were incubated with 2% BSA/PBS at 4 °C overnight. The cells were washed with PBS and stained with APC-labeled anti-goat mouse IgG (1:1000, Alexa Fluor 594; Abcam, Cambridge, UK) and Hoechst 33342 (#PN226, Dojindo, Kumamoto, Japan). The cells were then washed and mounted using Fluoroshield™ mounting medium (TA-030-FM, Thermo Scientific, Waltham, MA, USA. The stained cells were visualized and scanned using a BZX-710 microscope (Keyence, Osaka, Japan).
For whole mount immunofluorescent staining, CD29+ meat buds were harvested by pipetting and fixed in 4% paraformaldehyde for 24 h. For whole-mount staining, the fixed CD29+ meat buds were washed with PBS, permeabilized with 0.2% Triton-X/PBS for 5 min and blocked with Blocking One solution for 1 h. Primary antibodies (the same as those for normal Immunofluorescent staining) were used with 2% BSA/PBS and 0.1% Triton-X at 4 °C overnight. CD29+ meat buds were then washed with PBS and stained with APC-labeled anti-goat mouse IgG1 (1:1000, #A21125, Alexa Fluor 594, Life Technologies, Austin, TX, USA). CD29+ meat buds were washed and mounted using Fluoroshield mounting medium (TA-030-FM, Thermo Scientific, Waltham, MA, USA). The stained CD29+ meat buds were visualized and scanned using an LSM 700 confocal microscope (Zeiss, Oberkochen, Germany).
2.8. RNA Isolation and Quantitative Real-Time PCR
The total RNA was extracted from cells using the RNeasy Plus mini kit according to the manufacturer’s instructions (#74136, Qiagen, Hilden, Germany), and cDNA was synthesized with 100 ng RNA, oligo (dt) primers, and PrimeScript II Reverse Transcriptase (#6210A, Takara, Shiga, Japan). The 200-fold diluted cDNA samples were used for quantitative real-time PCR (95 °C followed by 40 cycles for 15 s at 95 °C and 60 s at 60 °C) in the presence of 10 μM specific forward and reverse primers and Power Track SYBR Green Master Mix (#A46109, Thermo Fisher Scientific, Waltham, MA, USA). Amplification reactions were performed using a Quant Studio 5 Real-Time PCR system (Thermo Fisher Scientific, Waltham, MA, USA). Gene expression was normalized to that of β-actin expression. Each sample was analyzed in duplicate. As a negative control, water was used as the template for each reaction. The primer sequences used for RT-qPCR are described in
Table 1.
2.9. Analysis of Meat Buds Composition
The samples (muscle tissue, adipose tissue, normal bud, muscle bud, and adipose bud) were harvested and stored at −80 °C. To assess the composition of samples, we used attenuated total reflection-Fourier transform infrared (ATR-FTIR) spectroscopy (IRSptit-T, Shimadzu, Kyoto, Japan). The FT-IR spectra were collected at room temperature using a diamond ATR unit (QATR-S; Shimadzu, crystal diameter of 1.8 mm) in the middle IR region (4000–400 cm−1) with a spectral resolution of 4 cm−1. To eliminate the influence of moisture on the spectrum as much as possible, the sample was left to stand for several minutes after installation, and the measurement was carried out after drying.
2.10. Embedding Meat Buds in Collagen Gel
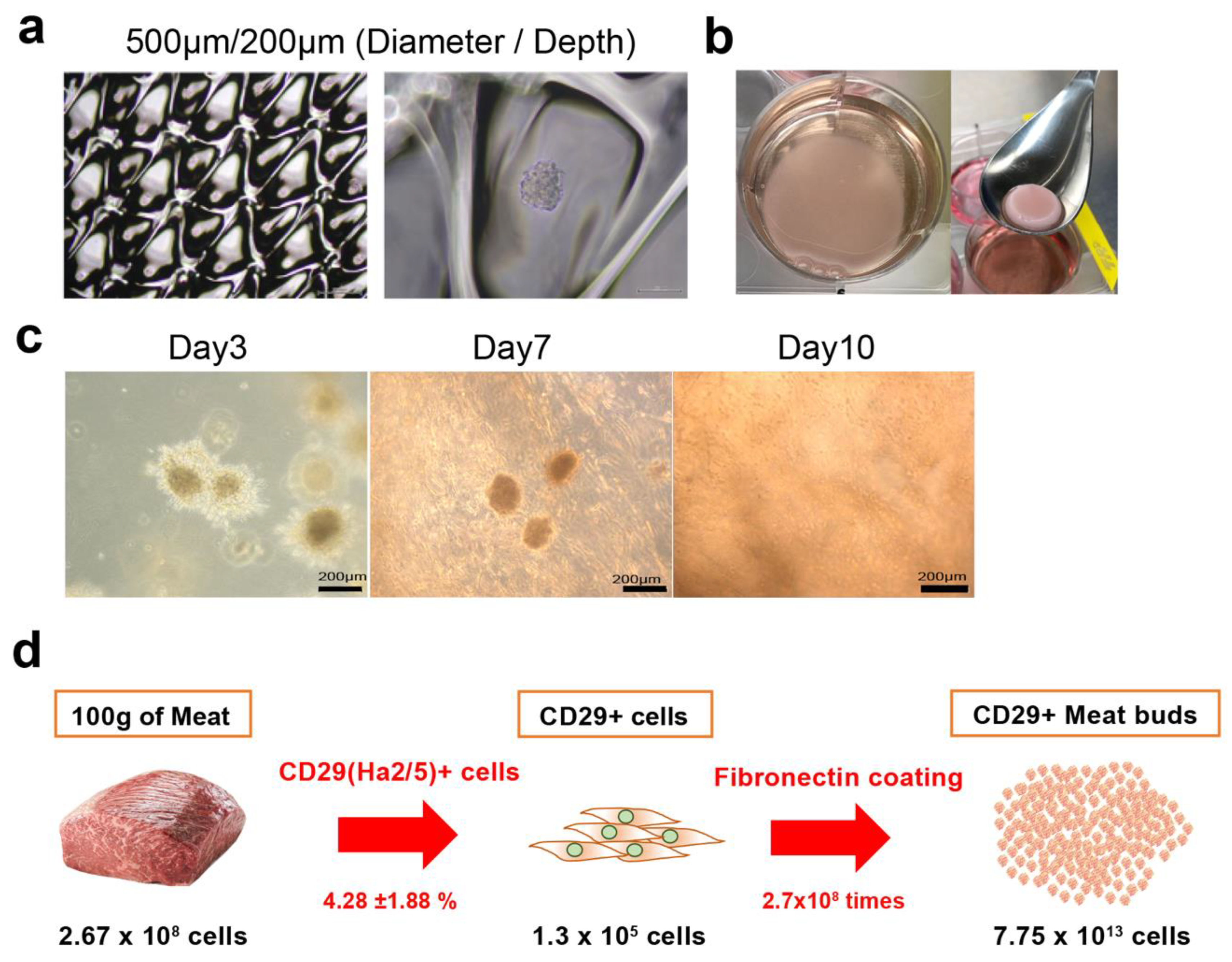
Cultured bovine Ha2/5+ cells were seeded and cultured in EZ sphere dishes of diameter ~500 µm and depth ~200 µm (#4020-900SP, IWAKI, Tokyo, Japan). A meat bud was used in the experiment after culturing for three days. For embedding meat buds in collagen gel, 1% BSA/PBS (5 mL) was placed into a six-well plate and incubated for blocking (30 min). The pH of collagen (Collagen I, Rat Tail, A10483, Gibco, NC, USA) was adjusted to pH 7.0 on ice. CD29-positive meat buds and collagen were mixed on ice at a ratio of 1:1 and seeded in a six-well plate. To investigate cell apoptosis, meat buds were retrieved from collagen gel with collagenase and analyzed by flow cytometer using Active Caspase-3 Apoptosis Kit (#550480, BD Pharmingen, San Diego, CA, USA).
2.11. Statistical Analysis
Quantitative data are presented as mean ± standard error of the mean (SEM) from at least three representative experiments. Statistical analyses were performed using SPSS v26.0 (IBM, Armonk, NY, USA). Statistical analyses on the gene expression were performed by one-way ANOVA with a Bonferroni post hoc analysis for comparison of three or more groups. For comparisons between groups, Student’s t-test was used. Results are expressed as mean ± SE. Results with * p < 0.05, ** p < 0.001 were considered significant.
4. Discussion
In this study, we isolated Ha2/5+ cells from bovine skeletal muscles to obtain cells with proliferative and self-renewal capacity. In addition, we demonstrated that Ha2/5+ cells possess muscle- and adipose-differentiation capacities. Furthermore, the meat buds created from Ha2/5+ cells could differentiate into muscle and adipose tissue when constructed three dimensionally. We found that approximately 10 trillion cells can be theoretically obtained from 100 g of bovine tissue by culturing and amplifying them in fibronectin-coated dishes.
Culturing muscle satellite cells in vitro for a long period has been challenging. Previous reports have shown that muscle satellite cells are activated during in vitro isolation, proceeding to differentiation in skeletal muscle [
15]. Our group has established a culture method to maintain muscle satellite cells of skeletal muscle stem cells in an undifferentiated state in vitro [
16]. In addition, a technique to self-assemble meat bud tissues in vitro using the liver is being developed [
17]. Based on these two fundamental techniques, we created hybrid meat buds using multiple cell populations as meat seeds and established an innovative meat-culture method. By culturing Ha2/5+ cells, Pax7+ cells were maintained for a long period (21 days) and they could proliferate on fibronectin. This suggests that the method is useful for the in vitro culture of muscle satellite cells. In previous studies, CD29 (integrin β1) and CD56 (NCAM) have been used as markers for muscle satellite cells. Choi et al. reported that sorting porcine muscle tissue with the CD29 marker increased the proportion of CD56+/CD29+ cells. These authors also reported that CD56 cells had insufficient muscle-differentiation capacity [
18]. Sun et al. performed FACS analysis of cell-surface antigens from pig muscle tissue and found that CD29 was expressed in both adipocytes and muscle cells [
19]. In the present study, Ha2/5-positive cells were capable of muscle differentiation and lipid accumulation. Ha2/5 is a cell population with special abilities in bovine tissue, and the identification of these cells and the establishment of a long-term culture method are major achievements in this study.
The previous report shows that adipogenesis of mesenchymal progenitor cells in muscle is strongly inhibited by the presence of satellite cell-derived myofibers [
3]. These results suggest that interaction between muscle cells and mesenchymal progenitors has a considerable impact on muscle homeostasis. The CD29 (Ha2/5)-positive cells are a heterogeneous population, and the satellite cells may be limited in number (
Figure 1d,e). On the contrary, fat gene expression was elevated when the cells were cultured in 5% horse serum (
Figure 2d). This phenomenon may be related to adipose progenitor cells contained in Ha2/5-positive or mature muscle cells. In fact, the muscle cells and adipocytes were observed by continuing the culture without inducing differentiation (
Figure 1d). Interestingly, the “dual induction” with adipose differentiation medium and 5% horse serum was confirmed to promote differentiation into fat and muscle (
Figure 4). These are unexpected results, which may be due to the properties of Ha2/5 cells. These results showed that the progenitor cell population contained in Ha2/5-positive cells expresses the muscle and adipose gene by culture stimulation.
Currently, our technique is at the laboratory level and is limited to a small scale. Thus, the technique needs to be developed (scaled up) for future mass cultures. If muscle satellite cells cannot be cultured alone, then a mass culture method for muscle satellite cells may be established by adding mesenchymal stem cells, which are known to support the survival and proliferation of muscle satellite cells. However, there is a limitation with regard to production costs. The use of bovine-induced pluripotent stem cells, which are stem cells capable of proliferating indefinitely, may be advantageous in terms of future costs, but the notion of edible induced pluripotent stem cells is not currently socially acceptable. As meat is not the only protein source today, we cannot evaluate whether this technique is superior to protein obtained from livestock only from the viewpoint of environmental effect and safety. For the liver tissue, a “mini-multi-organ” that can generate multiple cells (tissue) simultaneously has been successfully created, which was shown to have a high additional value that cannot be obtained with mixed constituent cells [
20]. If meat buds with freely adjustable cell components and nutrients can be created, then meat with a higher additional value to stimulate social demand could be developed.


 ,
, 





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}