
Beyond the X Factor: Relevance of Sex Hormones in NAFLD Pathophysiology


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. The Sex-Specific Regulation of the Healthy Liver Metabolism
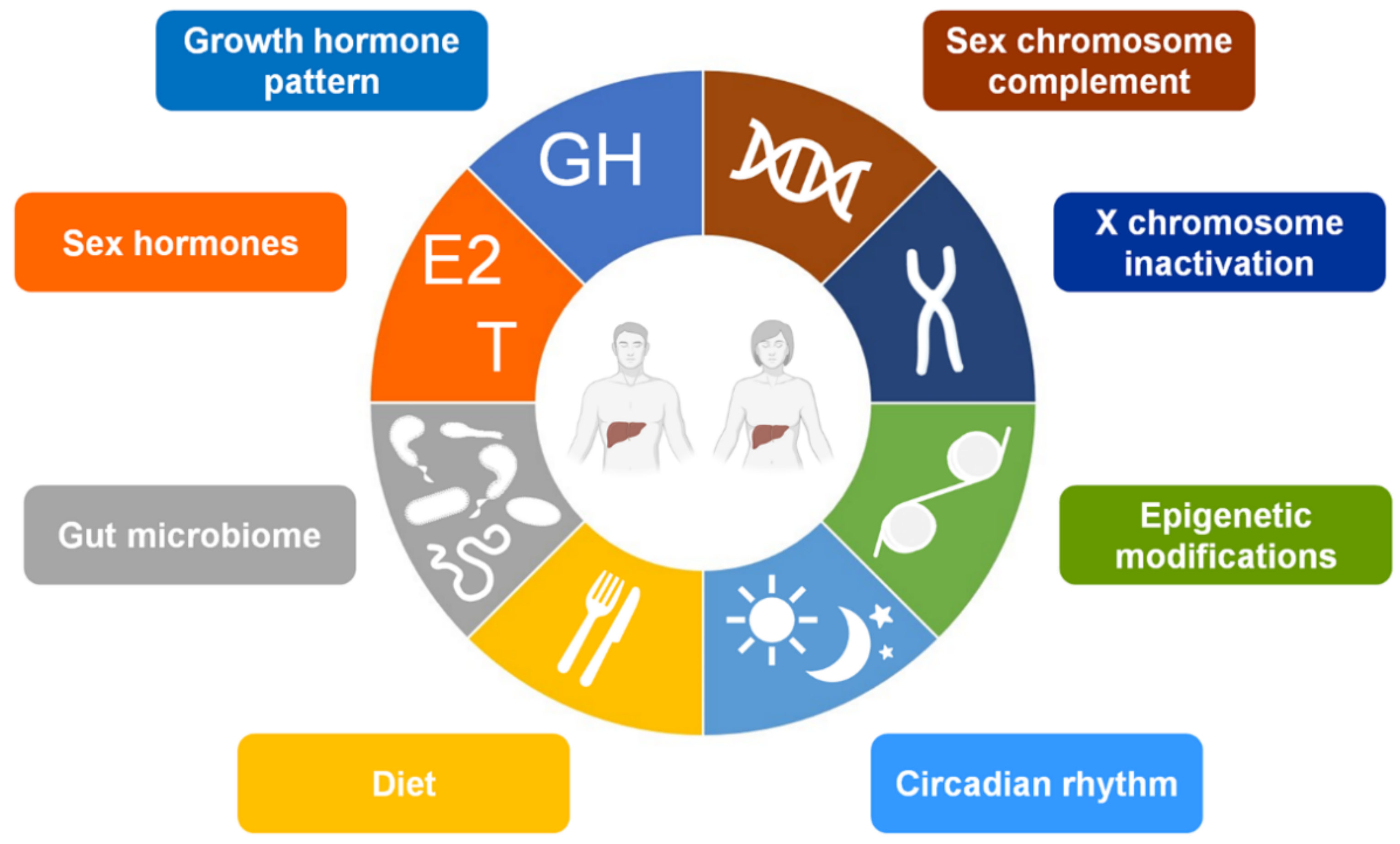
3. The Sex-Factors Involved in the Hepatic Sexual Dimorphism
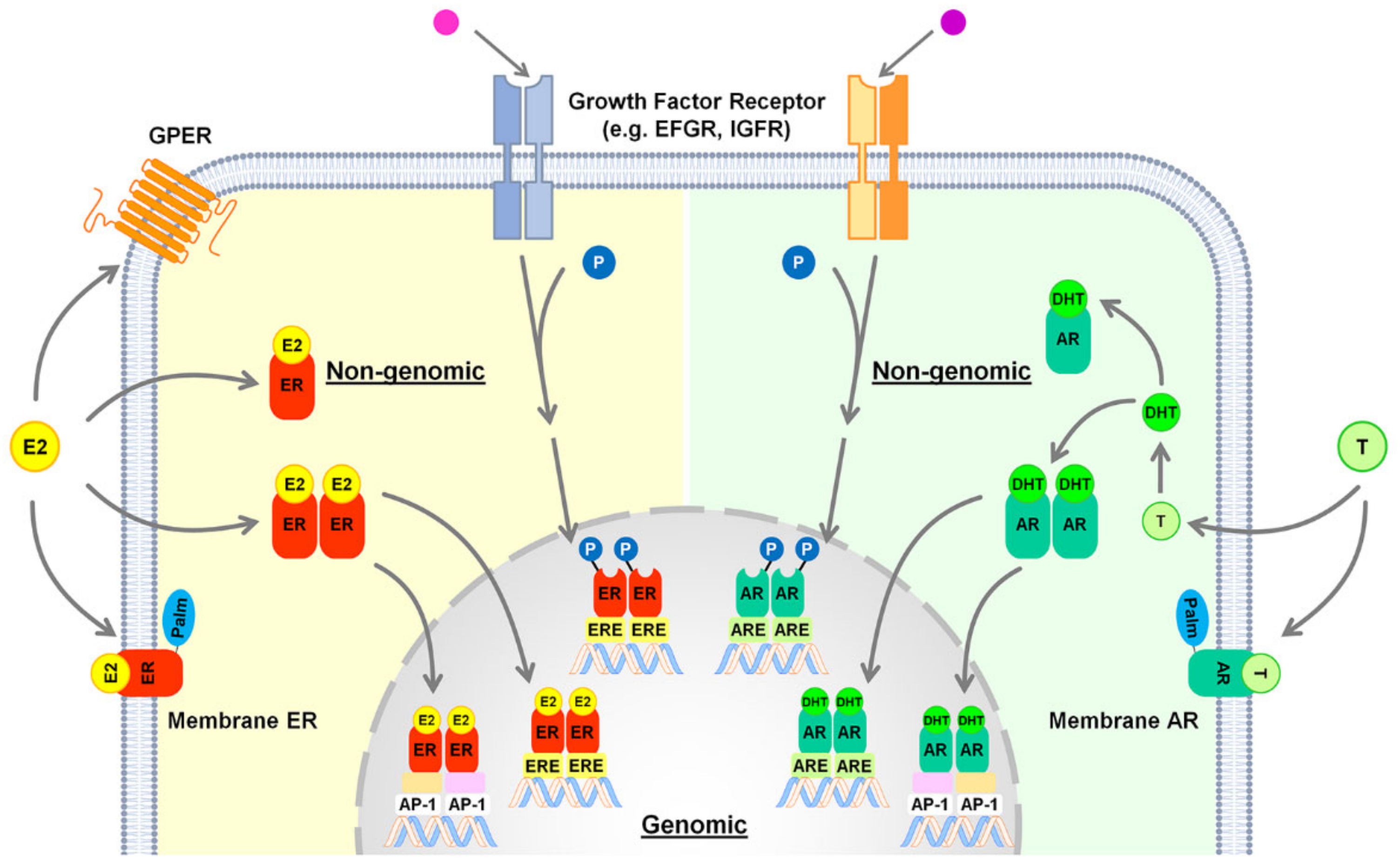
4. Estrogen Signaling in the Healthy Liver
5. Androgen Signaling in the Healthy Liver
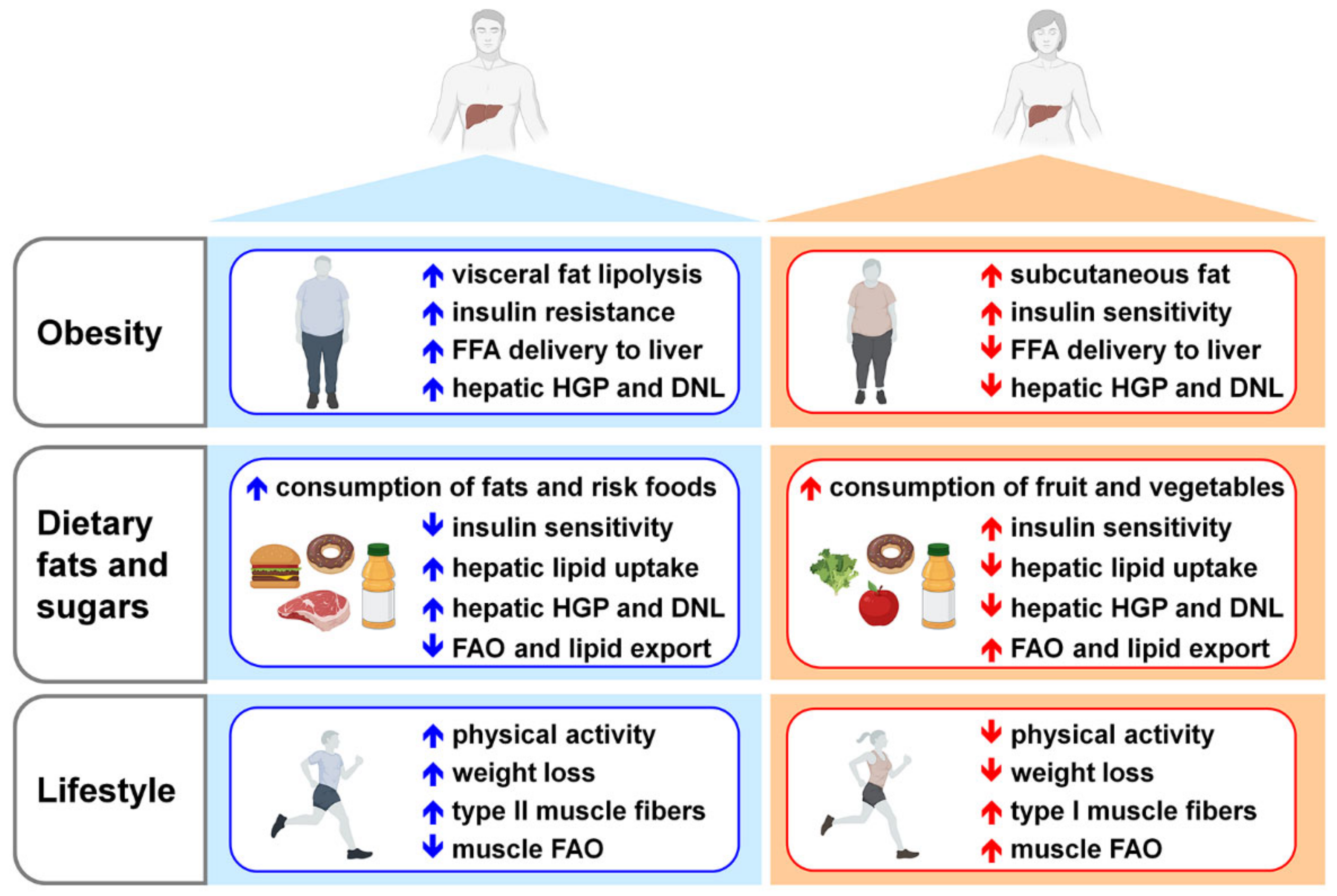
6. NAFLD, a Sex-Based Liver Disease
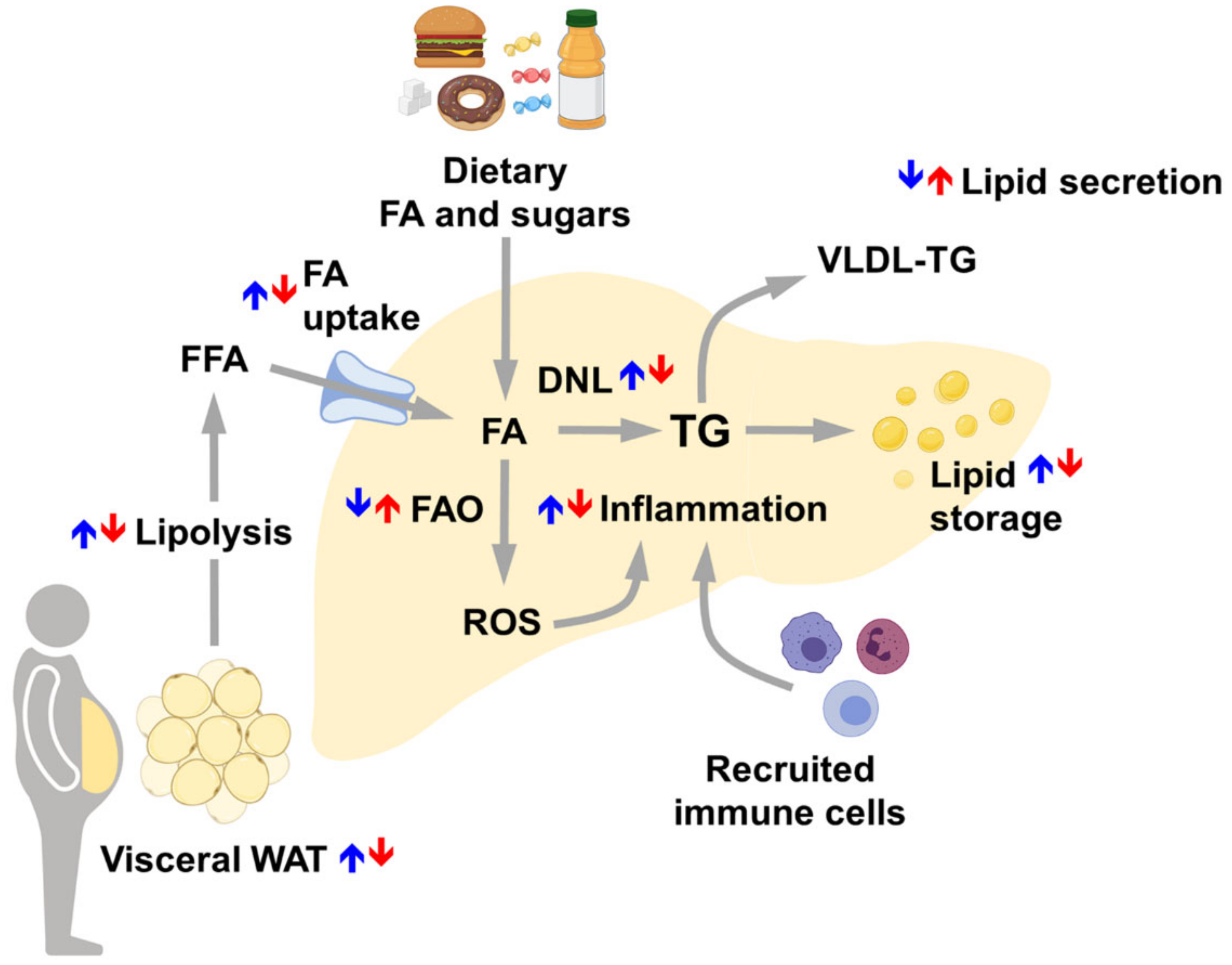
7. Risk Factors Triggering NAFLD Development and Progression
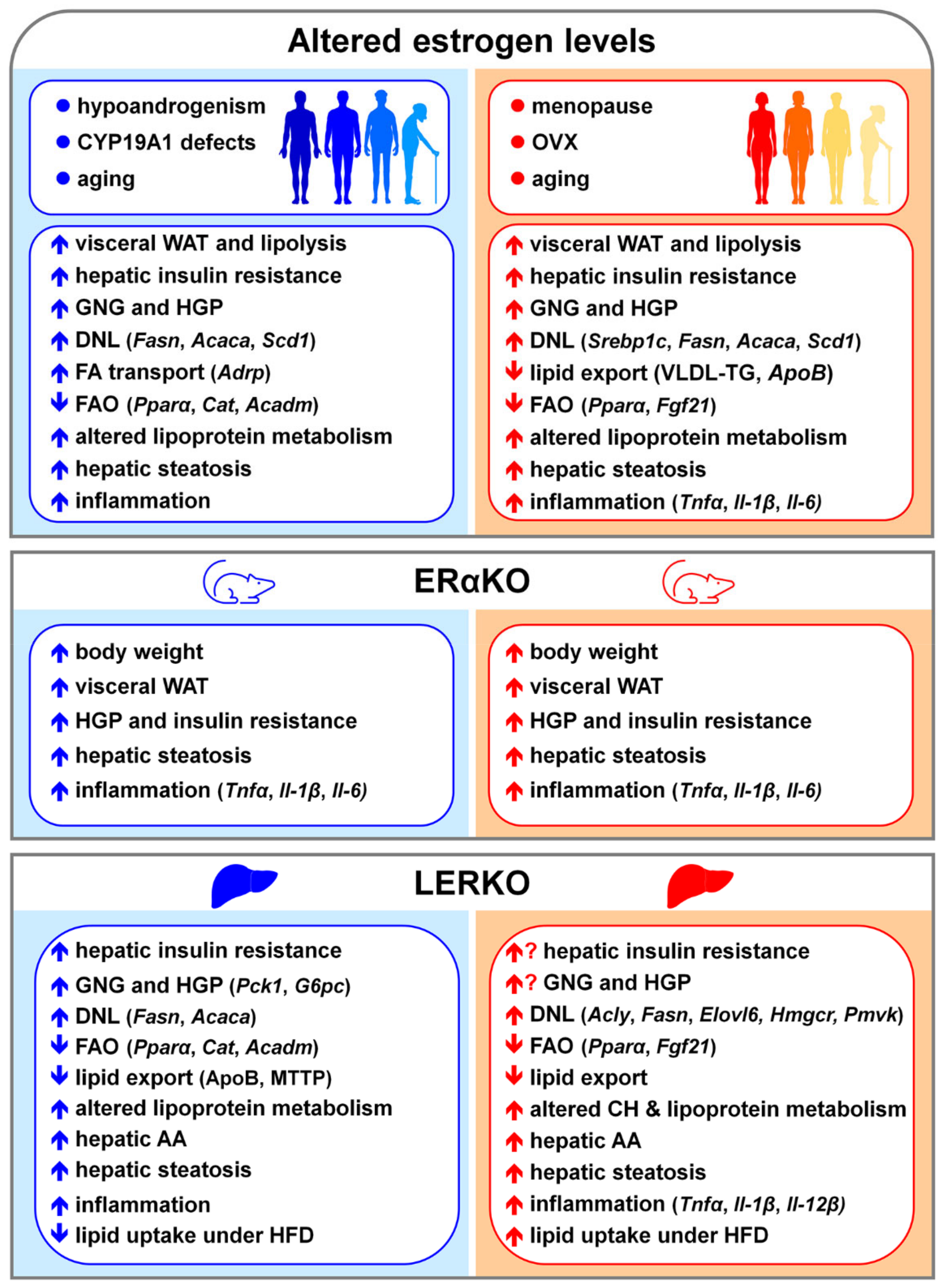
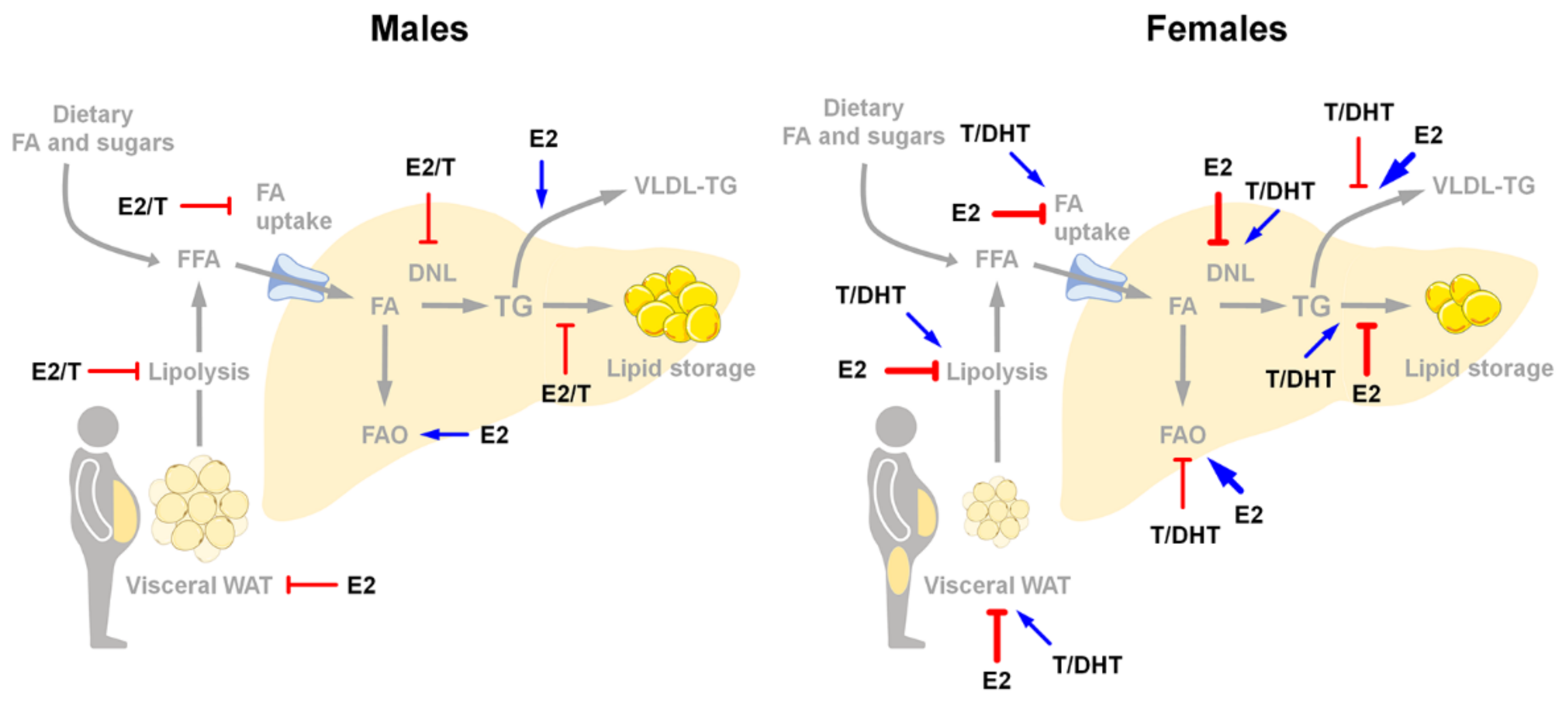
8. NAFLD and Estrogen Signaling
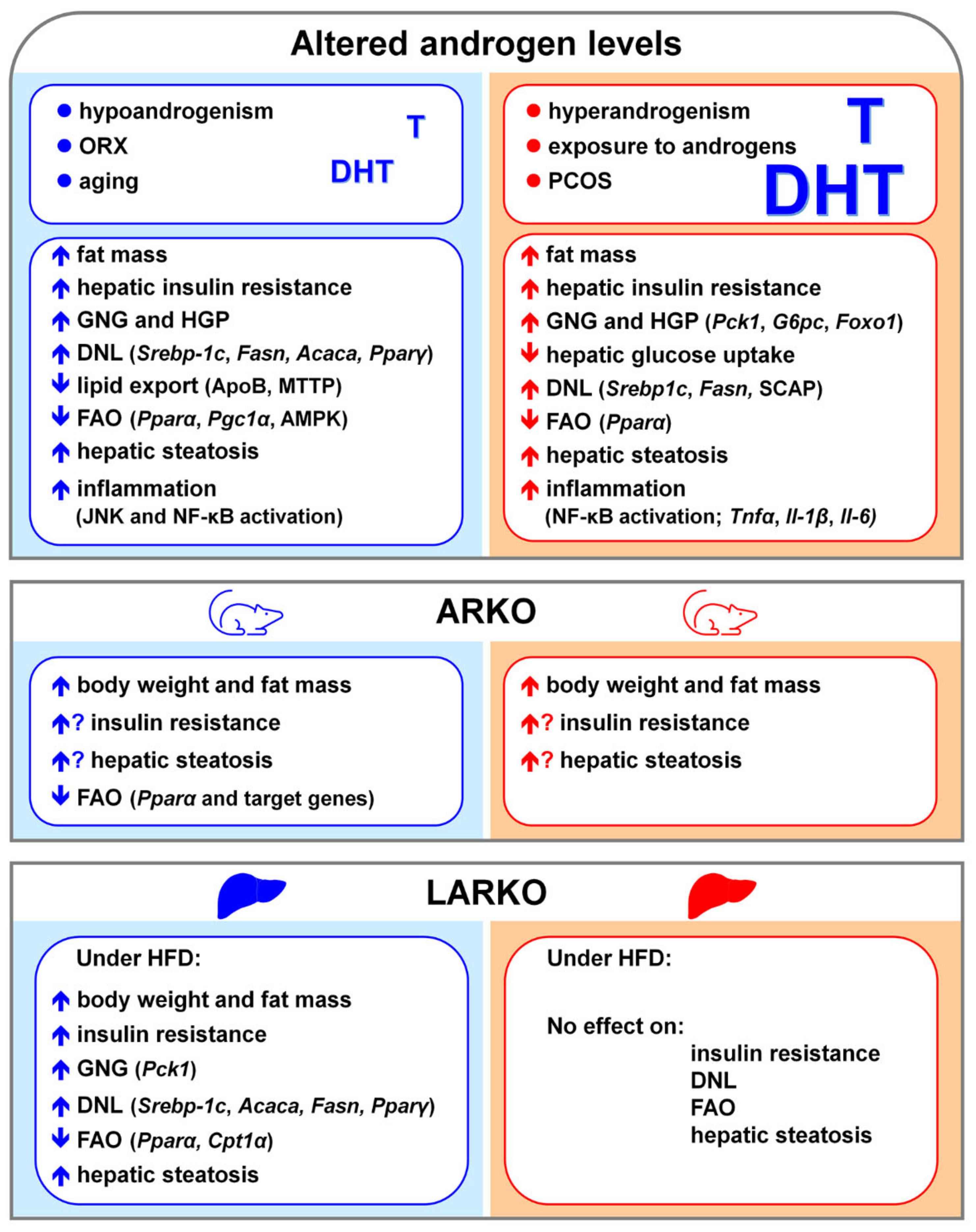
9. NAFLD and Androgen Signaling
10. Conclusions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Trefts, E.; Gannon, M.; Wasserman, D.H. The Liver. Curr. Biol. 2017, 27, R1147–R1151. [Google Scholar] [CrossRef]
- Yang, X. Tissue-Specific Expression and Regulation of Sexually Dimorphic Genes in Mice. Genome Res. 2006, 16, 995–1004. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Della Torre, S. Non-Alcoholic Fatty Liver Disease as a Canonical Example of Metabolic Inflammatory-Based Liver Disease Showing a Sex-Specific Prevalence: Relevance of Estrogen Signaling. Front. Endocrinol. 2020, 11, 572490. [Google Scholar] [CrossRef]
- Lonardo, A.; Suzuki, A. Sexual Dimorphism of NAFLD in Adults. Focus on Clinical Aspects and Implications for Practice and Translational Research. J. Clin. Med. 2020, 9, 1278. [Google Scholar] [CrossRef] [PubMed]
- Lonardo, A.; Nascimbeni, F.; Ballestri, S.; Fairweather, D.; Win, S.; Than, T.A.; Abdelmalek, M.F.; Suzuki, A. Sex Differences in Nonalcoholic Fatty Liver Disease: State of the Art and Identification of Research Gaps. Hepatology 2019, 70, 1457–1469. [Google Scholar] [CrossRef]
- Maggi, A.; Della Torre, S. Sex, Metabolism and Health. Mol. Metab. 2018, 15, 3–7. [Google Scholar] [CrossRef]
- Smith, R.L.; Soeters, M.R.; Wüst, R.C.I.; Houtkooper, R.H. Metabolic Flexibility as an Adaptation to Energy Resources and Requirements in Health and Disease. Endocr. Rev. 2018, 39, 489–517. [Google Scholar] [CrossRef] [Green Version]
- Han, H.-S.; Kang, G.; Kim, J.S.; Choi, B.H.; Koo, S.-H. Regulation of Glucose Metabolism from a Liver-Centric Perspective. Exp. Mol. Med. 2016, 48, e218. [Google Scholar] [CrossRef] [Green Version]
- Bazhan, N.; Jakovleva, T.; Feofanova, N.; Denisova, E.; Dubinina, A.; Sitnikova, N.; Makarova, E. Sex Differences in Liver, Adipose Tissue, and Muscle Transcriptional Response to Fasting and Refeeding in Mice. Cells 2019, 8, 1529. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lorbek, G.; Lewinska, M.; Rozman, D. Cytochrome P450s in the Synthesis of Cholesterol and Bile Acids--from Mouse Models to Human Diseases. FEBS J. 2012, 279, 1516–1533. [Google Scholar] [CrossRef] [PubMed]
- Phelps, T.; Snyder, E.; Rodriguez, E.; Child, H.; Harvey, P. The Influence of Biological Sex and Sex Hormones on Bile Acid Synthesis and Cholesterol Homeostasis. Biol. Sex Differ. 2019, 10, 52. [Google Scholar] [CrossRef]
- Della Torre, S.; Mitro, N.; Fontana, R.; Gomaraschi, M.; Favari, E.; Recordati, C.; Lolli, F.; Quagliarini, F.; Meda, C.; Ohlsson, C.; et al. An Essential Role for Liver ERα in Coupling Hepatic Metabolism to the Reproductive Cycle. Cell Rep. 2016, 15, 360–371. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Galan, X.; Llobera, M.; Ramírez, I. Lipoprotein Lipase and Hepatic Lipase in Wistar and Sprague-Dawley Rat Tissues. Differences in the Effects of Gender and Fasting. Lipids 1994, 29, 333–336. [Google Scholar] [CrossRef] [PubMed]
- Palmisano, B.T.; Zhu, L.; Eckel, R.H.; Stafford, J.M. Sex Differences in Lipid and Lipoprotein Metabolism. Mol. Metab. 2018, 15, 45–55. [Google Scholar] [CrossRef]
- Sorrentino, D.; Zhou, S.L.; Kokkotou, E.; Berk, P.D. Sex Differences in Hepatic Fatty Acid Uptake Reflect a Greater Affinity of the Transport System in Females. Am. J. Physiol. 1992, 263, G380–G385. [Google Scholar] [CrossRef]
- Ståhlberg, N.; Rico-Bautista, E.; Fisher, R.M.; Wu, X.; Cheung, L.; Flores-Morales, A.; Tybring, G.; Norstedt, G.; Tollet-Egnell, P. Female-Predominant Expression of Fatty Acid Translocase/CD36 in Rat and Human Liver. Endocrinology 2004, 145, 1972–1979. [Google Scholar] [CrossRef] [Green Version]
- Meda, C.; Barone, M.; Mitro, N.; Lolli, F.; Pedretti, S.; Caruso, D.; Maggi, A.; Della Torre, S. Hepatic ERα Accounts for Sex Differences in the Ability to Cope with an Excess of Dietary Lipids. Mol. Metab. 2020, 32, 97–108. [Google Scholar] [CrossRef]
- Matthan, N.R.; Jalbert, S.M.; Barrett, P.H.R.; Dolnikowski, G.G.; Schaefer, E.J.; Lichtenstein, A.H. Gender-Specific Differences in the Kinetics of Nonfasting TRL, IDL, and LDL Apolipoprotein B-100 in Men and Premenopausal Women. Arterioscler. Thromb. Vasc. Biol. 2008, 28, 1838–1843. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, X.; Magkos, F.; Mittendorfer, B. Sex differences in Lipid and Lipoprotein Metabolism: It’s Not Just about Sex Hormones. J. Clin. Endocrinol. Metab. 2011, 96, 885–893. [Google Scholar] [CrossRef] [Green Version]
- Goossens, G.H.; Jocken, J.W.E.; Blaak, E.E. Sexual Dimorphism in Cardiometabolic Health: The Role of Adipose Tissue, Muscle and Liver. Nat. Rev. Endocrinol. 2021, 17, 47–66. [Google Scholar] [CrossRef]
- Magkos, F.; Patterson, B.W.; Mohammed, B.S.; Klein, S.; Mittendorfer, B. Women Produce Fewer but Triglyceride-Richer Very Low-Density Lipoproteins than Men. J. Clin. Endocrinol. Metab. 2007, 92, 1311–1318. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Karpe, F.; Bickerton, A.S.; Hodson, L.; Fielding, B.A.; Tan, G.D.; Frayn, K.N. Removal of Triacylglycerols from Chylomicrons and VLDL by Capillary Beds: The Basis of Lipoprotein Remnant Formation. Biochem. Soc. Trans. 2007, 35, 472–476. [Google Scholar] [CrossRef] [Green Version]
- Magkos, F.; Wang, X.; Mittendorfer, B. Metabolic Actions of Insulin in Men and Women. Nutrition 2010, 26, 686–693. [Google Scholar] [CrossRef] [Green Version]
- Gustavsson, C.; Yassin, K.; Wahlström, E.; Cheung, L.; Lindberg, J.; Brismar, K.; Östenson, C.-G.; Norstedt, G.; Tollet-Egnell, P. Sex-Different Hepaticglycogen Content and Glucose Output in Rats. BMC Biochem. 2010, 11, 38. [Google Scholar] [CrossRef] [Green Version]
- Mauvais-Jarvis, F. Gender Differences in Glucose Homeostasis and Diabetes. Physiol. Behav. 2018, 187, 20–23. [Google Scholar] [CrossRef] [PubMed]
- Tramunt, B.; Smati, S.; Grandgeorge, N.; Lenfant, F.; Arnal, J.-F.; Montagner, A.; Gourdy, P. Sex Differences in Metabolic Regulation and Diabetes Susceptibility. Diabetologia 2020, 63, 453–461. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pramfalk, C.; Pavlides, M.; Banerjee, R.; McNeil, C.A.; Neubauer, S.; Karpe, F.; Hodson, L. Sex-Specific Differences in Hepatic Fat Oxidation and Synthesis May Explain the Higher Propensity for NAFLD in Men. J. Clin. Endocrinol. Metab. 2015, 100, 4425–4433. [Google Scholar] [CrossRef] [Green Version]
- Luo, J.; Yang, H.; Song, B.-L. Mechanisms and Regulation of Cholesterol Homeostasis. Nat. Rev. Mol. Cell Biol. 2020, 21, 225–245. [Google Scholar] [CrossRef]
- Freedman, D.S.; Otvos, J.D.; Jeyarajah, E.J.; Shalaurova, I.; Cupples, L.A.; Parise, H.; D’Agostino, R.B.; Wilson, P.W.F.; Schaefer, E.J. Sex and Age Differences in Lipoprotein Subclasses Measured by Nuclear Magnetic Resonance Spectroscopy: The Framingham Study. Clin. Chem. 2004, 50, 1189–1200. [Google Scholar] [CrossRef]
- Johnson, J.L.; Slentz, C.A.; Duscha, B.D.; Samsa, G.P.; McCartney, J.S.; Houmard, J.A.; Kraus, W.E. Gender and Racial Differences in Lipoprotein Subclass Distributions: The STRRIDE Study. Atherosclerosis 2004, 176, 371–377. [Google Scholar] [CrossRef]
- Velez-Carrasco, W.; Lichtenstein, A.H.; Li, Z.; Dolnikowski, G.G.; Lamon-Fava, S.; Welty, F.K.; Schaefer, E.J. Apolipoprotein A-I and A-II Kinetic Parameters as Assessed by Endogenous Labeling with [(2)H(3)]Leucine in Middle-Aged and Elderly Men and Women. Arterioscler. Thromb. Vasc. Biol. 2000, 20, 801–806. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hewitt, K.N.; Boon, W.C.; Murata, Y.; Jones, M.E.E.; Simpson, E.R. The Aromatase Knockout Mouse Presents with a Sexually Dimorphic Disruption to Cholesterol Homeostasis. Endocrinology 2003, 144, 3895–3903. [Google Scholar] [CrossRef] [Green Version]
- Fisher, F.M.; Maratos-Flier, E. Understanding the Physiology of FGF21. Annu. Rev. Physiol. 2016, 78, 223–241. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gabay, C.; Kushner, I. Acute-Phase Proteins and Other Systemic Responses to Inflammation. N. Engl. J. Med. 1999, 340, 448–454. [Google Scholar] [CrossRef] [PubMed]
- Jensen-Cody, S.O.; Potthoff, M.J. Hepatokines and Metabolism: Deciphering Communication from the Liver. Mol. Metab. 2021, 44, 101138. [Google Scholar] [CrossRef]
- Ohlsson, C.; Mohan, S.; Sjögren, K.; Tivesten, Å.; Isgaard, J.; Isaksson, O.; Jansson, J.-O.; Svensson, J. The Role of Liver-Derived Insulin-Like Growth Factor-I. Endocr. Rev. 2009, 30, 494–535. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thalacker-Mercer, A.E.; Johnson, C.A.; Yarasheski, K.E.; Carnell, N.S.; Campbell, W.W. Nutrient Ingestion, Protein Intake, and Sex, but Not Age, Affect the Albumin Synthesis Rate in Humans. J. Nutr. 2007, 137, 1734–1740. [Google Scholar] [CrossRef] [Green Version]
- Jaruvongvanich, V.; Sanguankeo, A.; Riangwiwat, T.; Upala, S. Testosterone, Sex Hormone-Binding Globulin and Nonalcoholic Fatty Liver Disease: A Systematic Review and Meta-Analysis. Ann. Hepatol. 2017, 16, 382–394. [Google Scholar] [CrossRef] [PubMed]
- Della Torre, S.; Mitro, N.; Meda, C.; Lolli, F.; Pedretti, S.; Barcella, M.; Ottobrini, L.; Metzger, D.; Caruso, D.; Maggi, A. Short-Term Fasting Reveals Amino Acid Metabolism as a Major Sex-Discriminating Factor in the Liver. Cell Metab. 2018, 28, 256–267.e5. [Google Scholar] [CrossRef] [PubMed]
- Heymann, F.; Tacke, F. Immunology in the Liver–from Homeostasis to Disease. Nat. Rev. Gastroenterol. Hepatol. 2016, 13, 88–110. [Google Scholar] [CrossRef]
- Kubes, P.; Jenne, C. Immune Responses in the Liver. Annu. Rev. Immunol. 2018, 36, 247–277. [Google Scholar] [CrossRef]
- Buzzetti, E.; Parikh, P.M.; Gerussi, A.; Tsochatzis, E. Gender Differences in Liver Disease and the Drug-Dose Gender Gap. Pharmacol. Res. 2017, 120, 97–108. [Google Scholar] [CrossRef]
- Klein, S.L.; Flanagan, K.L. Sex Differences in Immune Responses. Nat. Rev. Immunol. 2016, 16, 626–638. [Google Scholar] [CrossRef] [PubMed]
- Sutti, S.; Tacke, F. Liver Inflammation and Regeneration in Drug-Induced Liver Injury: Sex Matters! Clin. Sci. 2018, 132, 609–613. [Google Scholar] [CrossRef]
- Fuscoe, J.C.; Vijay, V.; Hanig, J.P.; Han, T.; Ren, L.; Greenhaw, J.J.; Beger, R.D.; Pence, L.M.; Shi, Q. Hepatic Transcript Profiles of Cytochrome P450 Genes Predict Sex Differences in Drug Metabolism. Drug. Metab. Dispos. 2020, 48, 447–458. [Google Scholar] [CrossRef] [PubMed]
- Klaassen, C.D.; Liu, L.; Dunn, R.T. Regulation of Sulfotransferase MRNA Expression in Male and Female Rats of Various Ages. Chem. Biol. Interact. 1998, 109, 299–313. [Google Scholar] [CrossRef]
- Waxman, D.J.; Holloway, M.G. Sex Differences in the Expression of Hepatic Drug Metabolizing Enzymes. Mol. Pharmacol. 2009, 76, 215–228. [Google Scholar] [CrossRef] [Green Version]
- Lichanska, A.M.; Waters, M.J. How Growth Hormone Controls Growth, Obesity and Sexual Dimorphism. Trends Genet. 2008, 24, 41–47. [Google Scholar] [CrossRef] [PubMed]
- Waxman, D.J.; O’Connor, C. Growth Hormone Regulation of Sex-Dependent Liver Gene Expression. Mol. Endocrinol. 2006, 20, 2613–2629. [Google Scholar] [CrossRef]
- Liu, Z.; Cordoba-Chacon, J.; Kineman, R.D.; Cronstein, B.N.; Muzumdar, R.; Gong, Z.; Werner, H.; Yakar, S. Growth Hormone Control of Hepatic Lipid Metabolism. Diabetes 2016, 65, 3598–3609. [Google Scholar] [CrossRef] [Green Version]
- Roy, A.K.; Chatterjee, B. Sexual Dimorphism in the Liver. Annu. Rev. Physiol. 1983, 45, 37–50. [Google Scholar] [CrossRef]
- Clodfelter, K.H.; Holloway, M.G.; Hodor, P.; Park, S.-H.; Ray, W.J.; Waxman, D.J. Sex-Dependent Liver Gene Expression Is Extensive and Largely Dependent upon Signal Transducer and Activator of Transcription 5b (STAT5b): STAT5b-Dependent Activation of Male Genes and Repression of Female Genes Revealed by Microarray Analysis. Mol. Endocrinol. 2006, 20, 1333–1351. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Y.; Laz, E.V.; Waxman, D.J. Dynamic, Sex-Differential STAT5 and BCL6 Binding to Sex-Biased, Growth Hormone-Regulated Genes in Adult Mouse Liver. Mol. Cell. Biol. 2012, 32, 880–896. [Google Scholar] [CrossRef] [Green Version]
- Holloway, M.G.; Miles, G.D.; Dombkowski, A.A.; Waxman, D.J. Liver-Specific Hepatocyte Nuclear Factor-4α Deficiency: Greater Impact on Gene Expression in Male than in Female Mouse Liver. Mol. Endocrinol. 2008, 22, 1274–1286. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wiwi, C.A.; Gupte, M.; Waxman, D.J. Sexually Dimorphic P450 Gene Expression in Liver-Specific Hepatocyte Nuclear Factor 4α-Deficient Mice. Mol. Endocrinol. 2004, 18, 1975–1987. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Conforto, T.L.; Steinhardt, G.F.; Waxman, D.J. Cross Talk between GH-Regulated Transcription Factors HNF6 and CUX2 in Adult Mouse Liver. Mol. Endocrinol. 2015, 29, 1286–1302. [Google Scholar] [CrossRef] [PubMed]
- Holloway, M.G.; Laz, E.V.; Waxman, D.J. Codependence of Growth Hormone-Responsive, Sexually Dimorphic Hepatic Gene Expression on Signal Transducer and Activator of Transcription 5b and Hepatic Nuclear Factor 4α. Mol. Endocrinol. 2006, 20, 647–660. [Google Scholar] [CrossRef] [Green Version]
- Park, S.-H.; Wiwi, C.A.; Waxman, D.J. Signalling Cross-Talk between Hepatocyte Nuclear Factor 4α and Growth-Hormone-Activated STAT5b. Biochem. J. 2006, 397, 159–168. [Google Scholar] [CrossRef] [Green Version]
- Lau-Corona, D.; Suvorov, A.; Waxman, D.J. Feminization of Male Mouse Liver by Persistent Growth Hormone Stimulation: Activation of Sex-Biased Transcriptional Networks and Dynamic Changes in Chromatin States. Mol. Cell. Biol. 2017, 37, e00301-17. [Google Scholar] [CrossRef] [Green Version]
- Ling, G.; Sugathan, A.; Mazor, T.; Fraenkel, E.; Waxman, D.J. Unbiased, Genome-Wide In Vivo Mapping of Transcriptional Regulatory Elements Reveals Sex Differences in Chromatin Structure Associated with Sex-Specific Liver Gene Expression. Mol. Cell. Biol. 2010, 30, 5531–5544. [Google Scholar] [CrossRef] [Green Version]
- Ramirez, M.C.; Zubeldía-Brenner, L.; Wargon, V.; Ornstein, A.M.; Becu-Villalobos, D. Expression and Methylation Status of Female-Predominant GH-Dependent Liver Genes Are Modified by Neonatal Androgenization in Female Mice. Mol. Cell. Endocrinol. 2014, 382, 825–834. [Google Scholar] [CrossRef]
- Sugathan, A.; Waxman, D.J. Genome-Wide Analysis of Chromatin States Reveals Distinct Mechanisms of Sex-Dependent Gene Regulation in Male and Female Mouse Liver. Mol. Cell. Biol. 2013, 33, 3594–3610. [Google Scholar] [CrossRef] [Green Version]
- Conforto, T.L.; Waxman, D.J. Sex-Specific Mouse Liver Gene Expression: Genome-Wide Analysis of Developmental Changes from Pre-Pubertal Period to Young Adulthood. Biol. Sex Differ. 2012, 3, 9. [Google Scholar] [CrossRef] [Green Version]
- Lowe, R.; Gemma, C.; Rakyan, V.K.; Holland, M.L. Sexually Dimorphic Gene Expression Emerges with Embryonic Genome Activation and Is Dynamic throughout Development. BMC Genom. 2015, 16, 295. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wauthier, V.; Sugathan, A.; Meyer, R.D.; Dombkowski, A.A.; Waxman, D.J. Intrinsic Sex Differences in the Early Growth Hormone Responsiveness of Sex-Specific Genes in Mouse Liver. Mol. Endocrinol. 2010, 24, 667–678. [Google Scholar] [CrossRef] [Green Version]
- Laz, E.V.; Holloway, M.G.; Chen, C.-S.; Waxman, D.J. Characterization of Three Growth Hormone-Responsive Transcription Factors Preferentially Expressed in Adult Female Liver. Endocrinology 2007, 148, 3327–3337. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bur, I.M.; Cohen-Solal, A.M.; Carmignac, D.; Abecassis, P.-Y.; Chauvet, N.; Martin, A.O.; van der Horst, G.T.J.; Robinson, I.C.A.F.; Maurel, P.; Mollard, P.; et al. The Circadian Clock Components CRY1 and CRY2 Are Necessary to Sustain Sex Dimorphism in Mouse Liver Metabolism. J. Biol. Chem. 2009, 284, 9066–9073. [Google Scholar] [CrossRef] [Green Version]
- Lomas-Soria, C.; Reyes-Castro, L.A.; Rodríguez-González, G.L.; Ibáñez, C.A.; Bautista, C.J.; Cox, L.A.; Nathanielsz, P.W.; Zambrano, E. Maternal Obesity Has Sex-Dependent Effects on Insulin, Glucose and Lipid Metabolism and the Liver Transcriptome in Young Adult Rat Offspring: Maternal Obesity Programs Liver Transcriptome Changes in Rat Offspring. J. Physiol. 2018, 596, 4611–4628. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lu, Y.-F.; Jin, T.; Xu, Y.; Zhang, D.; Wu, Q.; Zhang, Y.-K.J.; Liu, J. Sex Differences in the Circadian Variation of Cytochrome P450 Genes and Corresponding Nuclear Receptors in Mouse Liver. Chronobiol. Int. 2013, 30, 1135–1143. [Google Scholar] [CrossRef]
- Nohara, K.; Baba, T.; Murai, H.; Kobayashi, Y.; Suzuki, T.; Tateishi, Y.; Matsumoto, M.; Nishimura, N.; Sano, T. Global DNA Methylation in the Mouse Liver Is Affected by Methyl Deficiency and Arsenic in a Sex-Dependent Manner. Arch. Toxicol. 2011, 85, 653–661. [Google Scholar] [CrossRef]
- Stanimirovic, J.; Obradovic, M.; Jovanovic, A.; Sudar-Milovanovic, E.; Zafirovic, S.; Pitt, S.J.; Stewart, A.J.; Isenovic, E.R. A High Fat Diet Induces Sex-Specific Differences in Hepatic Lipid Metabolism and Nitrite/Nitrate in Rats. Nitric Oxide 2016, 54, 51–59. [Google Scholar] [CrossRef] [Green Version]
- Takasugi, M.; Hayakawa, K.; Arai, D.; Shiota, K. Age- and Sex-Dependent DNA Hypomethylation Controlled by Growth Hormone in Mouse Liver. Mech. Ageing Dev. 2013, 134, 331–337. [Google Scholar] [CrossRef]
- Wankhade, U.D.; Zhong, Y.; Kang, P.; Alfaro, M.; Chintapalli, S.V.; Piccolo, B.D.; Mercer, K.E.; Andres, A.; Thakali, K.M.; Shankar, K. Maternal High-Fat Diet Programs Offspring Liver Steatosis in a Sexually Dimorphic Manner in Association with Changes in Gut Microbial Ecology in Mice. Sci. Rep. 2018, 8, 16502. [Google Scholar] [CrossRef] [PubMed]
- Weger, B.D.; Gobet, C.; Yeung, J.; Martin, E.; Jimenez, S.; Betrisey, B.; Foata, F.; Berger, B.; Balvay, A.; Foussier, A.; et al. The Mouse Microbiome Is Required for Sex-Specific Diurnal Rhythms of Gene Expression and Metabolism. Cell Metab. 2019, 29, 362–382.e8. [Google Scholar] [CrossRef] [Green Version]
- Della Torre, S.; Maggi, A. Sex Differences: A Resultant of an Evolutionary Pressure? Cell Metab. 2017, 25, 499–505. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zheng, D.; Wang, X.; Antonson, P.; Gustafsson, J.-Å.; Li, Z. Genomics of Sex Hormone Receptor Signaling in Hepatic Sexual Dimorphism. Mol. Cell. Endocrinol. 2018, 471, 33–41. [Google Scholar] [CrossRef]
- van Nas, A.; GuhaThakurta, D.; Wang, S.S.; Yehya, N.; Horvath, S.; Zhang, B.; Ingram-Drake, L.; Chaudhuri, G.; Schadt, E.E.; Drake, T.A.; et al. Elucidating the Role of Gonadal Hormones in Sexually Dimorphic Gene Coexpression Networks. Endocrinology 2009, 150, 1235–1249. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhuang, Q.K.-W.; Galvez, J.H.; Xiao, Q.; AlOgayil, N.; Hyacinthe, J.; Taketo, T.; Bourque, G.; Naumova, A.K. Sex Chromosomes and Sex Phenotype Contribute to Biased DNA Methylation in Mouse Liver. Cells 2020, 9, 1436. [Google Scholar] [CrossRef]
- Della Torre, S.; Benedusi, V.; Fontana, R.; Maggi, A. Energy Metabolism and Fertility—A Balance Preserved for Female Health. Nat. Rev. Endocrinol. 2014, 10, 13–23. [Google Scholar] [CrossRef] [PubMed]
- Qiu, S.; Vazquez, J.T.; Boulger, E.; Liu, H.; Xue, P.; Hussain, M.A.; Wolfe, A. Hepatic Estrogen Receptor α Is Critical for Regulation of Gluconeogenesis and Lipid Metabolism in Males. Sci. Rep. 2017, 7, 1661. [Google Scholar] [CrossRef] [Green Version]
- Villa, A.; Della Torre, S.; Stell, A.; Cook, J.; Brown, M.; Maggi, A. Tetradian Oscillation of Estrogen Receptor Is Necessary to Prevent Liver Lipid Deposition. Proc. Natl. Acad. Sci. USA 2012, 109, 11806–11811. [Google Scholar] [CrossRef] [Green Version]
- Della Torre, S.; Lolli, F.; Ciana, P.; Maggi, A. Sexual Dimorphism and Estrogen Action in Mouse Liver. In Sex and Gender Factors Affecting Metabolic Homeostasis, Diabetes and Obesity; Mauvais-Jarvis, F., Ed.; Advances in Experimental Medicine and Biology; Springer International Publishing: Cham, Switzerland, 2017; Volume 1043, pp. 141–151. ISBN 978-3-319-70177-6. [Google Scholar]
- Palmisano, B.T.; Zhu, L.; Stafford, J.M. Role of Estrogens in the Regulation of Liver Lipid Metabolism. In Sex and Gender Factors Affecting Metabolic Homeostasis, Diabetes and Obesity; Mauvais-Jarvis, F., Ed.; Advances in Experimental Medicine and Biology; Springer International Publishing: Cham, Switzerland, 2017; Volume 1043, pp. 227–256. ISBN 978-3-319-70177-6. [Google Scholar]
- Zhu, L.; Shi, J.; Luu, T.N.; Neuman, J.C.; Trefts, E.; Yu, S.; Palmisano, B.T.; Wasserman, D.H.; Linton, M.F.; Stafford, J.M. Hepatocyte Estrogen Receptor Alpha Mediates Estrogen Action to Promote Reverse Cholesterol Transport during Western-Type Diet Feeding. Mol. Metab. 2018, 8, 106–116. [Google Scholar] [CrossRef] [PubMed]
- Addison, M.L.; Rissman, E.F. Sexual Dimorphism of Growth Hormone in the Hypothalamus: Regulation by Estradiol. Endocrinology 2012, 153, 1898–1907. [Google Scholar] [CrossRef] [PubMed]
- Fernández-Pérez, L.; Guerra, B.; Díaz-Chico, J.C.; Flores-Morales, A. Estrogens Regulate the Hepatic Effects of Growth Hormone, a Hormonal Interplay with Multiple Fates. Front. Endocrinol. 2013, 4, 66. [Google Scholar] [CrossRef] [Green Version]
- Della Torre, S.; Rando, G.; Meda, C.; Stell, A.; Chambon, P.; Krust, A.; Ibarra, C.; Magni, P.; Ciana, P.; Maggi, A. Amino Acid-Dependent Activation of Liver Estrogen Receptor Alpha Integrates Metabolic and Reproductive Functions via IGF-1. Cell Metab. 2011, 13, 205–214. [Google Scholar] [CrossRef] [Green Version]
- Fontana, R.; Torre, S. The Deep Correlation between Energy Metabolism and Reproduction: A View on the Effects of Nutrition for Women Fertility. Nutrients 2016, 8, 87. [Google Scholar] [CrossRef]
- Arnold, A.P.; Gorski, R.A. Gonadal Steroid Induction of Structural Sex differences in the Central Nervous System. Annu. Rev. Neurosci. 1984, 7, 413–442. [Google Scholar] [CrossRef]
- Gorski, R.A.; Wagner, J.W. Gonadal activity and sexual differentiation of the hypothalamus. Endocrinology 1965, 76, 226–239. [Google Scholar] [CrossRef]
- Meinhardt, U.J.; Ho, K.K.Y. Modulation of Growth Hormone Action by Sex Steroids. Clin. Endocrinol. 2006, 65, 413–422. [Google Scholar] [CrossRef]
- Biddie, S.C. Chromatin Architecture and the Regulation of Nuclear Receptor Inducible Transcription: Nuclear Receptors and the Epigenetic Regulation of Chromatin. J. Neuroendocrinol. 2011, 23, 94–106. [Google Scholar] [CrossRef] [PubMed]
- He, H.H.; Meyer, C.A.; Chen, M.W.; Jordan, V.C.; Brown, M.; Liu, X.S. Differential DNase I Hypersensitivity Reveals Factor-Dependent Chromatin Dynamics. Genome Res. 2012, 22, 1015–1025. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Della Torre, S.; Benedusi, V.; Pepe, G.; Meda, C.; Rizzi, N.; Uhlenhaut, N.H.; Maggi, A. Dietary Essential Amino Acids Restore Liver Metabolism in Ovariectomized Mice via Hepatic Estrogen Receptor α. Nat. Commun. 2021, in press. [Google Scholar]
- Ackerman, G.E.; Carr, B.R. Estrogens. Rev. Endocr. Metab. Disord. 2002, 3, 225–230. [Google Scholar] [CrossRef]
- Gruber, C.J.; Tschugguel, W.; Schneeberger, C.; Huber, J.C. Production and Actions of Estrogens. N. Engl. J. Med. 2002, 346, 340–352. [Google Scholar] [CrossRef]
- Frederiksen, H.; Johannsen, T.H.; Andersen, S.E.; Albrethsen, J.; Landersoe, S.K.; Petersen, J.H.; Andersen, A.N.; Vestergaard, E.T.; Schorring, M.E.; Linneberg, A.; et al. Sex-Specific Estrogen Levels and Reference Intervals from Infancy to Late Adulthood Determined by LC-MS/MS. J. Clin. Endocrinol. Metab. 2020, 105, 754–768. [Google Scholar] [CrossRef] [PubMed]
- Holinka, C.F.; Diczfalusy, E.; Coelingh Bennink, H.J.T. Estetrol: A Unique Steroid in Human Pregnancy. J. Steroid. Biochem. Mol. Biol. 2008, 110, 138–143. [Google Scholar] [CrossRef]
- Kobayashi, H. Estrogen Synthesis in Gastric Parietal Cells and Secretion into Portal Vein. Anat. Sci. Int. 2020, 95, 22–30. [Google Scholar] [CrossRef] [PubMed]
- Kobayashi, H.; Yoshida, S.; Sun, Y.-J.; Shirasawa, N.; Naito, A. Postnatal Development of Gastric Aromatase and Portal Venous Estradiol-17β Levels in Male Rats. J. Endocrinol. 2013, 218, 117–124. [Google Scholar] [CrossRef]
- Hetemäki, N.; Savolainen-Peltonen, H.; Tikkanen, M.J.; Wang, F.; Paatela, H.; Hämäläinen, E.; Turpeinen, U.; Haanpää, M.; Vihma, V.; Mikkola, T.S. Estrogen Metabolism in Abdominal Subcutaneous and Visceral Adipose Tissue in Postmenopausal Women. J. Clin. Endocrinol. Metab. 2017, 102, 4588–4595. [Google Scholar] [CrossRef]
- Misso, M.L.; Jang, C.; Adams, J.; Tran, J.; Murata, Y.; Bell, R.; Boon, W.C.; Simpson, E.R.; Davis, S.R. Adipose Aromatase Gene Expression Is Greater in Older Women and Is Unaffected by Postmenopausal Estrogen Therapy. Menopause 2005, 12, 210–215. [Google Scholar] [CrossRef]
- Dahlman-Wright, K.; Cavailles, V.; Fuqua, S.A.; Jordan, V.C.; Katzenellenbogen, J.A.; Korach, K.S.; Maggi, A.; Muramatsu, M.; Parker, M.G.; Gustafsson, J.-A. International Union of Pharmacology. LXIV. Estrogen Receptors. Pharmacol. Rev. 2006, 58, 773–781. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Levin, E.R. Plasma Membrane Estrogen Receptors. Trends Endocrinol. Metab. 2009, 20, 477–482. [Google Scholar] [CrossRef] [Green Version]
- Sharma, G.; Prossnitz, E.R. G-Protein-Coupled Estrogen Receptor (GPER) and Sex-Specific Metabolic Homeostasis. In Sex and Gender Factors Affecting Metabolic Homeostasis, Diabetes and Obesity; Mauvais-Jarvis, F., Ed.; Advances in Experimental Medicine and Biology; Springer International Publishing: Cham, Switzerland, 2017; Volume 1043, pp. 427–453. ISBN 978-3-319-70177-6. [Google Scholar]
- Della Torre, S.; Biserni, A.; Rando, G.; Monteleone, G.; Ciana, P.; Komm, B.; Maggi, A. The Conundrum of Estrogen Receptor Oscillatory Activity in the Search for an Appropriate Hormone Replacement Therapy. Endocrinology 2011, 152, 2256–2265. [Google Scholar] [CrossRef]
- Jakacka, M.; Ito, M.; Weiss, J.; Chien, P.Y.; Gehm, B.D.; Jameson, J.L. Estrogen Receptor Binding to DNA Is Not Required for Its Activity through the Nonclassical AP1 Pathway. J. Biol. Chem. 2001, 276, 13615–13621. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Levin, E.R. Integration of the Extranuclear and Nuclear Actions of Estrogen. Mol. Endocrinol. 2005, 19, 1951–1959. [Google Scholar] [CrossRef] [Green Version]
- Acconcia, F.; Ascenzi, P.; Bocedi, A.; Spisni, E.; Tomasi, V.; Trentalance, A.; Visca, P.; Marino, M. Palmitoylation-Dependent Estrogen Receptor Alpha Membrane Localization: Regulation by 17beta-Estradiol. Mol. Biol. Cell 2005, 16, 231–237. [Google Scholar] [CrossRef]
- Almazroo, O.A.; Miah, M.K.; Venkataramanan, R. Drug Metabolism in the Liver. Clin. Liver Dis. 2017, 21, 1–20. [Google Scholar] [CrossRef] [PubMed]
- Fabris, G.; Dumortier, O.; Pisani, D.F.; Gautier, N.; Van Obberghen, E. Amino Acid-Induced Regulation of Hepatocyte Growth: Possible Role of Drosha. Cell Death Dis. 2019, 10, 566. [Google Scholar] [CrossRef]
- Jenne, C.N.; Kubes, P. Immune Surveillance by the Liver. Nat. Immunol. 2013, 14, 996–1006. [Google Scholar] [CrossRef]
- Nagarajan, S.R.; Paul-Heng, M.; Krycer, J.R.; Fazakerley, D.J.; Sharland, A.F.; Hoy, A.J. Lipid and Glucose Metabolism in Hepatocyte Cell Lines and Primary Mouse Hepatocytes: A Comprehensive Resource for in Vitro Studies of Hepatic Metabolism. Am. J. Physiol. Endocrinol. Metab. 2019, 316, E578–E589. [Google Scholar] [CrossRef] [PubMed]
- Vickers, A.E.M.; Lucier, G.W. Estrogen Receptor Levels and Occupancy in Hepatic Sinusoidal Endothelial and Kupffer Cells Are Enhanced by Initiation with Diethylnitrosamine and Promotion with 17α-Ethinylestradiol in Rats. Carcinogenesis 1996, 17, 1235–1242. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kazankov, K.; Jørgensen, S.M.D.; Thomsen, K.L.; Møller, H.J.; Vilstrup, H.; George, J.; Schuppan, D.; Grønbæk, H. The Role of Macrophages in Nonalcoholic Fatty Liver Disease and Nonalcoholic Steatohepatitis. Nat. Rev. Gastroenterol. Hepatol. 2019, 16, 145–159. [Google Scholar] [CrossRef] [PubMed]
- Krenkel, O.; Tacke, F. Liver Macrophages in Tissue Homeostasis and Disease. Nat. Rev. Immunol. 2017, 17, 306–321. [Google Scholar] [CrossRef]
- Dixon, L.J.; Barnes, M.; Tang, H.; Pritchard, M.T.; Nagy, L.E. Kupffer Cells in the Liver. In Comprehensive Physiology; Terjung, R., Ed.; John Wiley & Sons, Inc.: Hoboken, NJ, USA, 2013; p. c120026. ISBN 978-0-470-65071-4. [Google Scholar]
- Zhou, Y.; Shimizu, I.; Lu, G.; Itonaga, M.; Okamura, Y.; Shono, M.; Honda, H.; Inoue, S.; Muramatsu, M.; Ito, S. Hepatic Stellate Cells Contain the Functional Estrogen Receptor β but Not the Estrogen Receptor α in Male and Female Rats. Biochem. Biophys. Res. Commun. 2001, 286, 1059–1065. [Google Scholar] [CrossRef] [PubMed]
- Zhang, B.; Zhang, C.-G.; Ji, L.-H.; Zhao, G.; Wu, Z.-Y. Estrogen Receptor β Selective Agonist Ameliorates Liver Cirrhosis in Rats by Inhibiting the Activation and Proliferation of Hepatic Stellate Cells: ERβ Agonist and Liver Cirrhosis. J. Gastroenterol. Hepatol. 2018, 33, 747–755. [Google Scholar] [CrossRef]
- Cortes, E.; Lachowski, D.; Rice, A.; Thorpe, S.D.; Robinson, B.; Yeldag, G.; Lee, D.A.; Ghemtio, L.; Rombouts, K.; del Río Hernández, A.E. Tamoxifen Mechanically Deactivates Hepatic Stellate Cells via the G Protein-Coupled Estrogen Receptor. Oncogene 2019, 38, 2910–2922. [Google Scholar] [CrossRef] [Green Version]
- Banales, J.M.; Huebert, R.C.; Karlsen, T.; Strazzabosco, M.; LaRusso, N.F.; Gores, G.J. Cholangiocyte Pathobiology. Nat. Rev. Gastroenterol. Hepatol. 2019, 16, 269–281. [Google Scholar] [CrossRef]
- Raven, A.; Lu, W.-Y.; Man, T.Y.; Ferreira-Gonzalez, S.; O’Duibhir, E.; Dwyer, B.J.; Thomson, J.P.; Meehan, R.R.; Bogorad, R.; Koteliansky, V.; et al. Cholangiocytes Act as Facultative Liver Stem Cells during Impaired Hepatocyte Regeneration. Nature 2017, 547, 350–354. [Google Scholar] [CrossRef]
- Alvaro, D. Estrogens and the Pathophysiology of the Biliary Tree. World J. Gastroenterol. 2006, 12, 3537. [Google Scholar] [CrossRef]
- Alvaro, D.; Alpini, G.; Onori, P.; Perego, L.; Baroni, G.S.; Franchitto, A.; Baiocchi, L.; Glaser, S.S.; Le Sage, G.; Folli, F.; et al. Estrogens Stimulate Proliferation of Intrahepatic Biliary Epithelium in Rats. Gastroenterology 2000, 119, 1681–1691. [Google Scholar] [CrossRef]
- Bryzgalova, G.; Gao, H.; Ahren, B.; Zierath, J.R.; Galuska, D.; Steiler, T.L.; Dahlman-Wright, K.; Nilsson, S.; Gustafsson, J.-Å.; Efendic, S.; et al. Evidence That Oestrogen Receptor-α Plays an Important Role in the Regulation of Glucose Homeostasis in Mice: Insulin Sensitivity in the Liver. Diabetologia 2006, 49, 588–597. [Google Scholar] [CrossRef] [Green Version]
- Khristi, V.; Ratri, A.; Ghosh, S.; Borosha, S.; Dai, E.; Chakravarthi, V.P.; Rumi, M.A.K.; Wolfe, M.W. Liver Transcriptome Data of Esr1 Knockout Male Rats Reveals Altered Expression of Genes Involved in Carbohydrate and Lipid Metabolism. Data Brief 2019, 22, 771–780. [Google Scholar] [CrossRef] [PubMed]
- Mauvais-Jarvis, F.; Clegg, D.J.; Hevener, A.L. The Role of Estrogens in Control of Energy Balance and Glucose Homeostasis. Endocr. Rev. 2013, 34, 309–338. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Manson, J.E.; Chlebowski, R.T.; Stefanick, M.L.; Aragaki, A.K.; Rossouw, J.E.; Prentice, R.L.; Anderson, G.; Howard, B.V.; Thomson, C.A.; LaCroix, A.Z.; et al. Menopausal Hormone Therapy and Health Outcomes during the Intervention and Extended Poststopping Phases of the Women’s Health Initiative Randomized Trials. JAMA 2013, 310, 1353–1368. [Google Scholar] [CrossRef]
- Mauvais-Jarvis, F.; Manson, J.E.; Stevenson, J.C.; Fonseca, V.A. Menopausal Hormone Therapy and Type 2 Diabetes Prevention: Evidence, Mechanisms, and Clinical Implications. Endocr. Rev. 2017, 38, 173–188. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Van Pelt, R.E.; Gozansky, W.S.; Schwartz, R.S.; Kohrt, W.M. Intravenous Estrogens Increase Insulin Clearance and Action in Postmenopausal Women. Am. J. Physiol. Endocrinol. Metab. 2003, 285, E311–E317. [Google Scholar] [CrossRef] [Green Version]
- D’Eon, T.M.; Souza, S.C.; Aronovitz, M.; Obin, M.S.; Fried, S.K.; Greenberg, A.S. Estrogen Regulation of Adiposity and Fuel Partitioning: Evidence of Genomic and Non-Genomic Regulation of Lipogenic and Oxidative Pathways. J. Biol. Chem. 2005, 280, 35983–35991. [Google Scholar] [CrossRef] [Green Version]
- Jones, M.E.E.; Thorburn, A.W.; Britt, K.L.; Hewitt, K.N.; Wreford, N.G.; Proietto, J.; Oz, O.K.; Leury, B.J.; Robertson, K.M.; Yao, S.; et al. Aromatase-Deficient (ArKO) Mice Have a Phenotype of Increased Adiposity. Proc. Natl. Acad. Sci. USA 2000, 97, 12735–12740. [Google Scholar] [CrossRef] [Green Version]
- Tomaz, L.M.; Barbosa, M.R.; Farahnak, Z.; Lagoeiro, C.G.; Magosso, N.S.S.; Lavoie, J.-M.; Perez, S.E.A. GLUT2 Proteins and PPARγ Transcripts Levels Are Increased in Liver of Ovariectomized Rats: Reversal Effects of Resistance Training. J. Exerc. Nutr. Biochem. 2016, 20, 51–57. [Google Scholar] [CrossRef]
- Ahmed-Sowur, H.; Bailey, C.J. Role of Ovarian Hormones in the Long-Term Control of Glucose Homeostasis Glycogen Formation and Gluconeogenesis. Ann. Nutr. Metab. 1981, 25, 208–212. [Google Scholar] [CrossRef]
- Yan, H.; Yang, W.; Zhou, F.; Li, X.; Pan, Q.; Shen, Z.; Han, G.; Newell-Fugate, A.; Tian, Y.; Majeti, R.; et al. Estrogen Improves Insulin Sensitivity and Suppresses Gluconeogenesis via the Transcription Factor Foxo1. Diabetes 2019, 68, 291–304. [Google Scholar] [CrossRef] [Green Version]
- Heine, P.A.; Taylor, J.A.; Iwamoto, G.A.; Lubahn, D.B.; Cooke, P.S. Increased Adipose Tissue in Male and Female Estrogen Receptor-Alpha Knockout Mice. Proc. Natl. Acad. Sci. USA 2000, 97, 12729–12734. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Allard, C.; Morford, J.J.; Xu, B.; Salwen, B.; Xu, W.; Desmoulins, L.; Zsombok, A.; Kim, J.K.; Levin, E.R.; Mauvais-Jarvis, F. Loss of Nuclear and Membrane Estrogen Receptor-α Differentially Impairs Insulin Secretion and Action in Male and Female Mice. Diabetes 2019, 68, 490–501. [Google Scholar] [CrossRef] [Green Version]
- Zhu, L.; Martinez, M.N.; Emfinger, C.H.; Palmisano, B.T.; Stafford, J.M. Estrogen Signaling Prevents Diet-Induced Hepatic Insulin Resistance in Male Mice with Obesity. Am. J. Physiol. Endocrinol. Metab. 2014, 306, E1188–E1197. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Matic, M.; Bryzgalova, G.; Gao, H.; Antonson, P.; Humire, P.; Omoto, Y.; Portwood, N.; Pramfalk, C.; Efendic, S.; Berggren, P.-O.; et al. Estrogen Signalling and the Metabolic Syndrome: Targeting the Hepatic Estrogen Receptor Alpha Action. PLoS ONE 2013, 8, e57458. [Google Scholar] [CrossRef] [Green Version]
- Grossmann, M.; Wierman, M.E.; Angus, P.; Handelsman, D.J. Reproductive Endocrinology of Nonalcoholic Fatty Liver Disease. Endocr. Rev. 2019, 40, 417–446. [Google Scholar] [CrossRef] [PubMed]
- Hewitt, K.N.; Pratis, K.; Jones, M.E.E.; Simpson, E.R. Estrogen Replacement Reverses the Hepatic Steatosis Phenotype in the Male Aromatase Knockout Mouse. Endocrinology 2004, 145, 1842–1848. [Google Scholar] [CrossRef] [Green Version]
- Macut, D.; Tziomalos, K.; Božić-Antić, I.; Bjekić-Macut, J.; Katsikis, I.; Papadakis, E.; Andrić, Z.; Panidis, D. Non-Alcoholic Fatty Liver Disease Is Associated with Insulin Resistance and Lipid Accumulation Product in Women with Polycystic Ovary Syndrome. Hum. Reprod. 2016, 31, 1347–1353. [Google Scholar] [CrossRef]
- Zhu, L.; Brown, W.C.; Cai, Q.; Krust, A.; Chambon, P.; McGuinness, O.P.; Stafford, J.M. Estrogen Treatment After Ovariectomy Protects Against Fatty Liver and May Improve Pathway-Selective Insulin Resistance. Diabetes 2013, 62, 424–434. [Google Scholar] [CrossRef] [Green Version]
- Paquette, A.; Wang, D.; Jankowski, M.; Gutkowska, J.; Lavoie, J.-M. Effects of Ovariectomy on PPARα, SREBP-1c, and SCD-1 Gene Expression in the Rat Liver. Menopause 2008, 15, 1169–1175. [Google Scholar] [CrossRef]
- Paquette, A.; Chapados, N.; Bergeron, R.; Lavoie, J.-M. Fatty Acid Oxidation Is Decreased in the Liver of Ovariectomized Rats. Horm. Metab. Res. 2009, 41, 511–515. [Google Scholar] [CrossRef]
- Chen, Z.; Wang, O.; Nie, M.; Elison, K.; Zhou, D.; Li, M.; Jiang, Y.; Xia, W.; Meng, X.; Chen, S.; et al. Aromatase Deficiency in a Chinese Adult Man Caused by Novel Compound Heterozygous CYP19A1 Mutations: Effects of Estrogen Replacement Therapy on the Bone, Lipid, Liver and Glucose Metabolism. Mol. Cell. Endocrinol. 2015, 399, 32–42. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Van Sinderen, M.L.; Steinberg, G.R.; Jørgensen, S.B.; To, S.Q.; Knower, K.C.; Clyne, C.D.; Honeyman, J.; Chow, J.D.; Herridge, K.A.; Jones, M.E.E.; et al. Hepatic Glucose Intolerance Precedes Hepatic Steatosis in the Male Aromatase Knockout (ArKO) Mouse. PLoS ONE 2014, 9, e87230. [Google Scholar] [CrossRef] [Green Version]
- Amano, A.; Kondo, Y.; Noda, Y.; Ohta, M.; Kawanishi, N.; Machida, S.; Mitsuhashi, K.; Senmaru, T.; Fukui, M.; Takaoka, O.; et al. Abnormal Lipid/Lipoprotein Metabolism and High Plasma Testosterone Levels in Male but Not Female Aromatase-Knockout Mice. Arch. Biochem. Biophys. 2017, 622, 47–58. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Lu, Y.; Wang, E.; Zhang, Z.; Xiong, X.; Zhang, H.; Lu, J.; Zheng, S.; Yang, J.; Xia, X.; et al. Hepatic Estrogen Receptor α Improves Hepatosteatosis through Upregulation of Small Heterodimer Partner. J. Hepatol. 2015, 63, 183–190. [Google Scholar] [CrossRef]
- Zhang, Z.-C.; Liu, Y.; Xiao, L.-L.; Li, S.-F.; Jiang, J.-H.; Zhao, Y.; Qian, S.-W.; Tang, Q.-Q.; Li, X. Upregulation of MiR-125b by Estrogen Protects against Non-Alcoholic Fatty Liver in Female Mice. J. Hepatol. 2015, 63, 1466–1475. [Google Scholar] [CrossRef]
- Gerdts, E.; Regitz-Zagrosek, V. Sex Differences in Cardiometabolic Disorders. Nat. Med. 2019, 25, 1657–1666. [Google Scholar] [CrossRef]
- Man, J.J.; Beckman, J.A.; Jaffe, I.Z. Sex as a Biological Variable in Atherosclerosis. Circ. Res. 2020, 126, 1297–1319. [Google Scholar] [CrossRef]
- Regitz-Zagrosek, V.; Kararigas, G. Mechanistic Pathways of Sex differences in Cardiovascular Disease. Physiol. Rev. 2017, 97, 1–37. [Google Scholar] [CrossRef] [Green Version]
- Lake, A.D.; Novak, P.; Shipkova, P.; Aranibar, N.; Robertson, D.G.; Reily, M.D.; Lehman-McKeeman, L.D.; Vaillancourt, R.R.; Cherrington, N.J. Branched Chain Amino Acid Metabolism Profiles in Progressive Human Nonalcoholic Fatty Liver Disease. Amino Acids 2015, 47, 603–615. [Google Scholar] [CrossRef] [PubMed]
- Obayashi, M.; Shimomura, Y.; Nakai, N.; Jeoung, N.H.; Nagasaki, M.; Murakami, T.; Sato, Y.; Harris, R.A. Estrogen Controls Branched-Chain Amino Acid Catabolism in Female Rats. Nutr. J. 2004, 134, 2628–2633. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Leung, K.-C.; Johannsson, G.; Leong, G.M.; Ho, K.K.Y. Estrogen Regulation of Growth Hormone Action. Endocr. Rev. 2004, 25, 693–721. [Google Scholar] [CrossRef]
- Schiffer, L.; Arlt, W.; Storbeck, K.-H. Intracrine Androgen Biosynthesis, Metabolism and Action Revisited. Mol. Cell. Endocrinol. 2018, 465, 4–26. [Google Scholar] [CrossRef]
- Courant, F.; Aksglaede, L.; Antignac, J.-P.; Monteau, F.; Sorensen, K.; Andersson, A.-M.; Skakkebaek, N.E.; Juul, A.; Bizec, B.L. Assessment of Circulating Sex Steroid Levels in Prepubertal and Pubertal Boys and Girls by a Novel Ultrasensitive Gas Chromatography-Tandem Mass Spectrometry Method. J. Clin. Endocrinol. Metab. 2010, 95, 82–92. [Google Scholar] [CrossRef] [Green Version]
- Travison, T.G.; Vesper, H.W.; Orwoll, E.; Wu, F.; Kaufman, J.M.; Wang, Y.; Lapauw, B.; Fiers, T.; Matsumoto, A.M.; Bhasin, S. Harmonized Reference Ranges for Circulating Testosterone Levels in Men of Four Cohort Studies in the United States and Europe. J. Clin. Endocrinol. Metab. 2017, 102, 1161–1173. [Google Scholar] [CrossRef] [PubMed]
- Davison, S.L.; Bell, R.; Donath, S.; Montalto, J.G.; Davis, S.R. Androgen Levels in Adult Females: Changes with Age, Menopause, and Oophorectomy. J. Clin. Endocrinol. Metab. 2005, 90, 3847–3853. [Google Scholar] [CrossRef]
- Kostakis, E.K.; Gkioni, L.N.; Macut, D.; Mastorakos, G. Androgens in Menopausal Women: Not Only Polycystic Ovary Syndrome. In Hyperandrogenism in Women; Pasquali, R., Pignatelli, D., Eds.; Frontiers of Hormone Research; Karger: Basel, Germany, 2019; Volume 53, pp. 135–161. ISBN 978-3-318-06470-4. [Google Scholar]
- Matsumoto, T.; Sakari, M.; Okada, M.; Yokoyama, A.; Takahashi, S.; Kouzmenko, A.; Kato, S. The Androgen Receptor in Health and Disease. Annu. Rev. Physiol. 2013, 75, 201–224. [Google Scholar] [CrossRef]
- Vlahopoulos, S.; Zimmer, W.E.; Jenster, G.; Belaguli, N.S.; Balk, S.P.; Brinkmann, A.O.; Lanz, R.B.; Zoumpourlis, V.C.; Schwartz, R.J. Recruitment of the Androgen Receptor via Serum Response Factor Facilitates Expression of a Myogenic Gene. J. Biol. Chem. 2005, 280, 7786–7792. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Heinlein, C.A.; Chang, C. The Roles of Androgen Receptors and Androgen-Binding Proteins in Nongenomic Androgen Actions. Mol. Endocrinol. 2002, 16, 2181–2187. [Google Scholar] [CrossRef]
- Treviño, L.S.; Gorelick, D.A. The Interface of Nuclear and Membrane Steroid Signaling. Endocrinology 2021, 162, bqab107. [Google Scholar] [CrossRef] [PubMed]
- Thomas, P. Membrane Androgen Receptors Unrelated to Nuclear Steroid Receptors. Endocrinology 2019, 160, 772–781. [Google Scholar] [CrossRef]
- Takeda, H.; Chodak, G.; Mutchnik, S.; Nakamoto, T.; Chang, C. Immunohistochemical Localization of Androgen Receptors with Mono- and Polyclonal Antibodies to Androgen Receptor. J. Endocrinol. 1990, 126, 17–25. [Google Scholar] [CrossRef]
- Navarro, G.; Allard, C.; Xu, W.; Mauvais-Jarvis, F. The Role of Androgens in Metabolism, Obesity, and Diabetes in Males and Females: Androgens and Diabetes. Obesity 2015, 23, 713–719. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Azziz, R.; Carmina, E.; Chen, Z.; Dunaif, A.; Laven, J.S.E.; Legro, R.S.; Lizneva, D.; Natterson-Horowtiz, B.; Teede, H.J.; Yildiz, B.O. Polycystic Ovary Syndrome. Nat. Rev. Dis. Primers 2016, 2, 16057. [Google Scholar] [CrossRef] [PubMed]
- Grossmann, M. Low Testosterone in Men with Type 2 Diabetes: Significance and Treatment. J. Clin. Endocrinol. Metab. 2011, 96, 2341–2353. [Google Scholar] [CrossRef]
- Movérare-Skrtic, S.; Venken, K.; Andersson, N.; Lindberg, M.K.; Svensson, J.; Swanson, C.; Vanderschueren, D.; Oscarsson, J.; Gustafsson, J.-Å.; Ohlsson, C. Dihydrotestosterone Treatment Results in Obesity and Altered Lipid Metabolism in Orchidectomized Mice. Obesity 2006, 14, 662–672. [Google Scholar] [CrossRef]
- Parthasarathy, C.; Renuka, V.N.; Balasubramanian, K. Sex Steroids Enhance Insulin Receptors and Glucose Oxidation in Chang Liver Cells. Clin. Chim. Acta 2009, 399, 49–53. [Google Scholar] [CrossRef]
- Kelly, D.M.; Jones, T.H. Testosterone: A Metabolic Hormone in Health and Disease. J. Endocrinol. 2013, 217, R25–R45. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Muthusamy, T.; Murugesan, P.; Balasubramanian, K. Sex Steroids Deficiency Impairs Glucose Transporter 4 Expression and Its Translocation through Defective Akt Phosphorylation in Target Tissues of Adult Male Rat. Metabolism 2009, 58, 1581–1592. [Google Scholar] [CrossRef]
- Pal, M.; Gupta, S. Testosterone Supplementation Improves Glucose Homeostasis despite Increasing Hepatic Insulin Resistance in Male Mouse Model of Type 2 Diabetes Mellitus. Nutr. Diabetes 2016, 6, e236. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kur, P.; Kolasa-Wołosiuk, A.; Grabowska, M.; Kram, A.; Tarnowski, M.; Baranowska-Bosiacka, I.; Rzeszotek, S.; Piasecka, M.; Wiszniewska, B. The Postnatal Offspring of Finasteride-Treated Male Rats Shows Hyperglycaemia, Elevated Hepatic Glycogen Storage and Altered GLUT2, IR, and AR Expression in the Liver. Int. J. Mol. Sci. 2021, 22, 1242. [Google Scholar] [CrossRef]
- Lin, H.-Y.; Yu, I.-C.; Wang, R.-S.; Chen, Y.-T.; Liu, N.-C.; Altuwaijri, S.; Hsu, C.-L.; Ma, W.-L.; Jokinen, J.; Sparks, J.D.; et al. Increased Hepatic Steatosis and Insulin Resistance in Mice Lacking Hepatic Androgen Receptor. Hepatology 2008, 47, 1924–1935. [Google Scholar] [CrossRef] [PubMed]
- Golden, S.H.; Dobs, A.S.; Vaidya, D.; Szklo, M.; Gapstur, S.; Kopp, P.; Liu, K.; Ouyang, P. Endogenous Sex Hormones and Glucose Tolerance Status in Postmenopausal Women. J. Clin. Endocrinol. Metab. 2007, 92, 1289–1295. [Google Scholar] [CrossRef] [Green Version]
- Larsson, H.; Ahren, B. Androgen Activity as a Risk Factor for Impaired Glucose Tolerance in Postmenopausal Women. Diabetes Care 1996, 19, 1399–1403. [Google Scholar] [CrossRef]
- Andrisse, S.; Childress, S.; Ma, Y.; Billings, K.; Chen, Y.; Xue, P.; Stewart, A.; Sonko, M.L.; Wolfe, A.; Wu, S. Low-Dose Dihydrotestosterone Drives Metabolic Dysfunction via Cytosolic and Nuclear Hepatic Androgen Receptor Mechanisms. Endocrinology 2017, 158, 531–544. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dubois, V.; Laurent, M.R.; Jardi, F.; Antonio, L.; Lemaire, K.; Goyvaerts, L.; Deldicque, L.; Carmeliet, G.; Decallonne, B.; Vanderschueren, D.; et al. Androgen Deficiency Exacerbates High-Fat Diet-Induced Metabolic Alterations in Male Mice. Endocrinology 2016, 157, 648–665. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mårin, P.; Holmäng, S.; Gustafsson, C.; Jönsson, L.; Kvist, H.; Elander, A.; Eldh, J.; Sjöström, L.; Holm, G.; Björntorp, P. Androgen Treatment of Abdominally Obese Men. Obes. Res. 1993, 1, 245–251. [Google Scholar] [CrossRef] [PubMed]
- Rochira, V.; Madeo, B.; Zirilli, L.; Caffagni, G.; Maffei, L.; Carani, C. Oestradiol Replacement Treatment and Glucose Homeostasis in Two Men with Congenital Aromatase Deficiency: Evidence for a Role of Oestradiol and Sex Steroids Imbalance on Insulin Sensitivity in Men. Diabet. Med. 2007, 24, 1491–1495. [Google Scholar] [CrossRef] [PubMed]
- Van Sinderen, M.; Steinberg, G.; Jorgensen, S.B.; Honeyman, J.; Chow, J.D.Y.; Simpson, E.R.; Jones, M.E.E.; Boon, W.C. Sexual Dimorphism in the Glucose Homeostasis Phenotype of the Aromatase Knockout (ArKO) Mice. J. Steroid Biochem. Mol. Biol. 2017, 170, 39–48. [Google Scholar] [CrossRef] [PubMed]
- Birzniece, V. Hepatic Actions of Androgens in the Regulation of Metabolism. Curr. Opin. Endocrinol. Diabetes Obes. 2018, 25, 201–208. [Google Scholar] [CrossRef] [PubMed]
- Abruzzese, G.A.; Heber, M.F.; Ferreira, S.R.; Velez, L.M.; Reynoso, R.; Pignataro, O.P.; Motta, A.B. Prenatal Hyperandrogenism Induces Alterations That Affect Liver Lipid Metabolism. J. Endocrinol. 2016, 230, 67–79. [Google Scholar] [CrossRef]
- Dowman, J.K.; Hopkins, L.J.; Reynolds, G.M.; Armstrong, M.J.; Nasiri, M.; Nikolaou, N.; van Houten, E.L.A.F.; Visser, J.A.; Morgan, S.A.; Lavery, G.G.; et al. Loss of 5α-Reductase Type 1 Accelerates the Development of Hepatic Steatosis but Protects Against Hepatocellular Carcinoma in Male Mice. Endocrinology 2013, 154, 4536–4547. [Google Scholar] [CrossRef] [PubMed]
- Kelly, D.M.; Nettleship, J.E.; Akhtar, S.; Muraleedharan, V.; Sellers, D.J.; Brooke, J.C.; McLaren, D.S.; Channer, K.S.; Jones, T.H. Testosterone Suppresses the Expression of Regulatory Enzymes of Fatty Acid Synthesis and Protects against Hepatic Steatosis in Cholesterol-Fed Androgen Deficient Mice. Life Sci. 2014, 109, 95–103. [Google Scholar] [CrossRef] [PubMed]
- Seidu, T.; McWhorter, P.; Myer, J.; Alamgir, R.; Eregha, N.; Bogle, D.; Lofton, T.; Ecelbarger, C.; Andrisse, S. DHT Causes Liver Steatosis via Transcriptional Regulation of SCAP in Normal Weight Female Mice. J. Endocrinol. 2021, 250, 49–65. [Google Scholar] [CrossRef]
- Xia, F.; Xu, X.; Zhai, H.; Meng, Y.; Zhang, H.; Du, S.; Xu, H.; Wu, H.; Lu, Y. Castration-Induced Testosterone Deficiency Increases Fasting Glucose Associated with Hepatic and Extra-Hepatic Insulin Resistance in Adult Male Rats. Reprod. Biol. Endocrinol. 2013, 11, 106. [Google Scholar] [CrossRef] [Green Version]
- Cui, P.; Hu, W.; Ma, T.; Hu, M.; Tong, X.; Zhang, F.; Shi, J.; Xu, X.; Li, X.; Shao, L.R.; et al. Long-Term Androgen Excess Induces Insulin Resistance and Non-Alcoholic Fatty Liver Disease in PCOS-like Rats. J. Steroid Biochem. Mol. Biol. 2021, 208, 105829. [Google Scholar] [CrossRef]
- Livingstone, D.E.W.; Barat, P.; Di Rollo, E.M.; Rees, G.A.; Weldin, B.A.; Rog-Zielinska, E.A.; MacFarlane, D.P.; Walker, B.R.; Andrew, R. 5α-Reductase Type 1 Deficiency or Inhibition Predisposes to Insulin Resistance, Hepatic Steatosis, and Liver Fibrosis in Rodents. Diabetes 2015, 64, 447–458. [Google Scholar] [CrossRef] [Green Version]
- Kelly, D.M.; Akhtar, S.; Sellers, D.J.; Muraleedharan, V.; Channer, K.S.; Jones, T.H. Testosterone Differentially Regulates Targets of Lipid and Glucose Metabolism in Liver, Muscle and Adipose Tissues of the Testicular Feminised Mouse. Endocrine 2016, 54, 504–515. [Google Scholar] [CrossRef] [Green Version]
- Krycer, J.R.; Brown, A.J. Cross-Talk between the Androgen Receptor and the Liver X Receptor. J. Biol. Chem. 2011, 286, 20637–20647. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Birzniece, V.; Meinhardt, U.J.; Umpleby, M.A.; Handelsman, D.J.; Ho, K.K.Y. Interaction between Testosterone and Growth Hormone on Whole-Body Protein Anabolism Occurs in the Liver. J. Clin. Endocrinol. Metab. 2011, 96, 1060–1067. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lam, T.; Poljak, A.; McLean, M.; Bahl, N.; Ho, K.K.Y.; Birzniece, V. Testosterone Prevents Protein Loss via the Hepatic Urea Cycle in Human. Eur. J. Endocrinol. 2017, 176, 489–496. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Birzniece, V.; Umpleby, M.A.; Poljak, A.; Handelsman, D.J.; Ho, K.K.Y. Oral Low-Dose Testosterone Administration Induces Whole-Body Protein Anabolism in Postmenopausal Women: A Novel Liver-Targeted Therapy. Eur. J. Endocrinol. 2013, 169, 321–327. [Google Scholar] [CrossRef] [Green Version]
- Basualto-Alarcón, C.; Varela, D.; Duran, J.; Maass, R.; Estrada, M. Sarcopenia and Androgens: A Link between Pathology and Treatment. Front. Endocrinol. 2014, 5, 217. [Google Scholar] [CrossRef] [Green Version]
- Anderson, L.J.; Liu, H.; Garcia, J.M. Sex Differences in Muscle Wasting. In Sex and Gender Factors Affecting Metabolic Homeostasis, Diabetes and Obesity; Mauvais-Jarvis, F., Ed.; Advances in Experimental Medicine and Biology; Springer International Publishing: Cham, Switzerland, 2017; Volume 1043, pp. 153–197. ISBN 978-3-319-70177-6. [Google Scholar]
- Anzai, Á.; Marcondes, R.R.; Gonçalves, T.H.; Carvalho, K.C.; Simões, M.J.; Garcia, N.; Soares, J.M.; Padmanabhan, V.; Baracat, E.C.; da Silva, I.D.C.G.; et al. Impaired Branched-Chain Amino Acid Metabolism May Underlie the Nonalcoholic Fatty Liver Disease-like Pathology of Neonatal Testosterone-Treated Female Rats. Sci. Rep. 2017, 7, 13167. [Google Scholar] [CrossRef] [Green Version]
- Rizvi, S.; Weinbauer, G.; Arslan, M.; Partsch, C.; Nieschlag, E. Testosterone Modulates Growth Hormone Secretion at the Hypothalamic but Not at the Hypophyseal Level in the Adult Male Rhesus Monkey. J. Endocrinol. 2000, 165, 337–344. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Roelfsema, F.; Yang, R.J.; Takahashi, P.Y.; Erickson, D.; Bowers, C.Y.; Veldhuis, J.D. Aromatized Estrogens Amplify Nocturnal Growth Hormone Secretion in Testosterone-Replaced Older Hypogonadal Men. J. Clin. Endocrinol. Metab. 2018, 103, 4419–4427. [Google Scholar] [CrossRef] [Green Version]
- Saggese, G.; Cesaretti, G.; Franchi, G.; Startari, L. Testosterone-Induced Increase of Insulin-like Growth Factor I Levels Depends upon Normal Levels of Growth Hormone. Eur. J. Endocrinol. 1996, 135, 211–215. [Google Scholar] [CrossRef]
- Weissberger, A.J.; Ho, K.K. Activation of the Somatotropic Axis by Testosterone in Adult Males: Evidence for the Role of Aromatization. J. Clin. Endocrinol. Metab. 1993, 76, 1407–1412. [Google Scholar] [CrossRef]
- Veldhuis, J.D.; Metzger, D.L.; Martha, P.M.; Mauras, N.; Kerrigan, J.R.; Keenan, B.; Rogol, A.D.; Pincus, S.M. Estrogen and Testosterone, but Not a Nonaromatizable Androgen, Direct Network Integration of the Hypothalamo-Somatotrope (Growth Hormone)-Insulin-like Growth Factor I Axis in the Human: Evidence from Pubertal Pathophysiology and Sex-Steroid Hormone Replacement. J. Clin. Endocrinol. Metab. 1997, 82, 3414–3420. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rochira, V.; Zirilli, L.; Maffei, L.; Premrou, V.; Aranda, C.; Baldi, M.; Ghigo, E.; Aimaretti, G.; Carani, C.; Lanfranco, F. Tall Stature without Growth Hormone: Four Male Patients with Aromatase Deficiency. J. Clin. Endocrinol. Metab. 2010, 95, 1626–1633. [Google Scholar] [CrossRef] [Green Version]
- Gibney, J.; Wolthers, T.; Johannsson, G.; Umpleby, A.M.; Ho, K.K.Y. Growth Hormone and Testosterone Interact Positively to Enhance Protein and Energy Metabolism in Hypopituitary Men. Am. J. Physiol. Endocrinol. Metab. 2005, 289, E266–E271. [Google Scholar] [CrossRef] [Green Version]
- Mauras, N.; Rini, A.; Welch, S.; Sager, B.; Murphy, S.P. Synergistic Effects of Testosterone and Growth Hormone on Protein Metabolism and Body Composition in Prepubertal Boys. Metabolism 2003, 52, 964–969. [Google Scholar] [CrossRef]
- Yu, Y.M.; Domené, H.M.; Sztein, J.; Counts, D.R.; Cassorla, F. Developmental Changes and Differential Regulation by Testosterone and Estradiol of Growth Hormone Receptor Expression in the Rabbit. Eur. J. Endocrinol. 1996, 135, 583–590. [Google Scholar] [CrossRef] [PubMed]
- Ramirez, M.C.; Luque, G.M.; Ornstein, A.M.; Becu-Villalobos, D. Differential Neonatal Testosterone Imprinting of GH-Dependent Liver Proteins and Genes in Female Mice. J. Endocrinol. 2010, 207, 301–308. [Google Scholar] [CrossRef] [Green Version]
- Ellefson, W.M.; Lakner, A.M.; Hamilton, A.; McKillop, I.H.; Bonkovsky, H.L.; Steuerwald, N.M.; Huet, Y.M.; Schrum, L.W. Neonatal Androgenization Exacerbates Alcohol-Induced Liver Injury in Adult Rats, an Effect Abrogated by Estrogen. PLoS ONE 2011, 6, e29463. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Buzzetti, E.; Pinzani, M.; Tsochatzis, E.A. The Multiple-Hit Pathogenesis of Non-Alcoholic Fatty Liver Disease (NAFLD). Metabolism 2016, 65, 1038–1048. [Google Scholar] [CrossRef]
- Ferguson, D.; Finck, B.N. Emerging Therapeutic Approaches for the Treatment of NAFLD and Type 2 Diabetes Mellitus. Nat. Rev. Endocrinol. 2021, 17, 484–495. [Google Scholar] [CrossRef] [PubMed]
- Friedman, S.L.; Neuschwander-Tetri, B.A.; Rinella, M.; Sanyal, A.J. Mechanisms of NAFLD Development and Therapeutic Strategies. Nat. Med. 2018, 24, 908–922. [Google Scholar] [CrossRef]
- Adams, L.A.; Anstee, Q.M.; Tilg, H.; Targher, G. Non-Alcoholic Fatty Liver Disease and its Relationship with Cardiovascular Disease and Other Extrahepatic Diseases. Gut 2017, 66, 1138–1153. [Google Scholar] [CrossRef] [Green Version]
- Estes, C.; Razavi, H.; Loomba, R.; Younossi, Z.; Sanyal, A.J. Modeling the Epidemic of Nonalcoholic Fatty Liver Disease Demonstrates an Exponential Increase in Burden of Disease: Estes et Al. Hepatology 2018, 67, 123–133. [Google Scholar] [CrossRef]
- Mikolasevic, I.; Milic, S.; Turk Wensveen, T.; Grgic, I.; Jakopcic, I.; Stimac, D.; Wensveen, F.; Orlic, L. Nonalcoholic Fatty Liver Disease—A Multisystem Disease? World J. Gastroenterol. 2016, 22, 9488. [Google Scholar] [CrossRef]
- Younossi, Z.; Anstee, Q.M.; Marietti, M.; Hardy, T.; Henry, L.; Eslam, M.; George, J.; Bugianesi, E. Global Burden of NAFLD and NASH: Trends, Predictions, Risk Factors and Prevention. Nat. Rev. Gastroenterol. Hepatol. 2018, 15, 11–20. [Google Scholar] [CrossRef]
- Ipsen, D.H.; Lykkesfeldt, J.; Tveden-Nyborg, P. Molecular Mechanisms of Hepatic Lipid Accumulation in Non-Alcoholic Fatty Liver Disease. Cell. Mol. Life Sci. 2018, 75, 3313–3327. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kawano, Y.; Cohen, D.E. Mechanisms of Hepatic Triglyceride Accumulation in Non-Alcoholic Fatty Liver Disease. J. Gastroenterol. 2013, 48, 434–441. [Google Scholar] [CrossRef] [Green Version]
- Lambert, J.E.; Ramos–Roman, M.A.; Browning, J.D.; Parks, E.J. Increased De Novo Lipogenesis is a Distinct Characteristic of Individuals with Nonalcoholic Fatty Liver Disease. Gastroenterology 2014, 146, 726–735. [Google Scholar] [CrossRef]
- Engin, A. Non-Alcoholic Fatty Liver Disease. In Obesity and Lipotoxicity; Engin, A.B., Engin, A., Eds.; Advances in Experimental Medicine and Biology; Springer International Publishing: Cham, Switzerland, 2017; Volume 960, pp. 443–467. ISBN 978-3-319-48380-1. [Google Scholar]
- Lebeaupin, C.; Vallée, D.; Hazari, Y.; Hetz, C.; Chevet, E.; Bailly-Maitre, B. Endoplasmic Reticulum Stress Signalling and the Pathogenesis of Non-Alcoholic Fatty Liver Disease. J. Hepatol. 2018, 69, 927–947. [Google Scholar] [CrossRef] [PubMed]
- Satapati, S.; Kucejova, B.; Duarte, J.A.G.; Fletcher, J.A.; Reynolds, L.; Sunny, N.E.; He, T.; Nair, L.A.; Livingston, K.A.; Livingston, K.; et al. Mitochondrial Metabolism Mediates Oxidative Stress and Inflammation in Fatty Liver. J. Clin. Investig. 2015, 125, 4447–4462. [Google Scholar] [CrossRef] [Green Version]
- Sunny, N.E.; Bril, F.; Cusi, K. Mitochondrial Adaptation in Nonalcoholic Fatty Liver Disease: Novel Mechanisms and Treatment Strategies. Trends Endocrinol. Metab. 2017, 28, 250–260. [Google Scholar] [CrossRef] [PubMed]
- Day, C.P. From Fat to Inflammation. Gastroenterology 2006, 130, 207–210. [Google Scholar] [CrossRef]
- Hotamisligil, G.S. Inflammation and Metabolic Disorders. Nature 2006, 444, 860–867. [Google Scholar] [CrossRef]
- Luedde, T.; Schwabe, R.F. NF-ΚB in the Liver—Linking Injury, Fibrosis and Hepatocellular Carcinoma. Nat. Rev. Gastroenterol. Hepatol. 2011, 8, 108–118. [Google Scholar] [CrossRef] [Green Version]
- Papa, S.; Bubici, C.; Zazzeroni, F.; Franzoso, G. Mechanisms of Liver Disease: Cross-Talk between the NF-ΚB and JNK Pathways. Biol. Chem. 2009, 390, 965–976. [Google Scholar] [CrossRef] [Green Version]
- Sharma, M.; Mitnala, S.; Vishnubhotla, R.K.; Mukherjee, R.; Reddy, D.N.; Rao, P.N. The Riddle of Nonalcoholic Fatty Liver Disease: Progression From Nonalcoholic Fatty Liver to Nonalcoholic Steatohepatitis. J. Clin. Exp. Hepatol. 2015, 5, 147–158. [Google Scholar] [CrossRef] [Green Version]
- Chen, Z.; Yu, R.; Xiong, Y.; Du, F.; Zhu, S. A Vicious Circle between Insulin Resistance and Inflammation in Nonalcoholic Fatty Liver Disease. Lipids Health Dis. 2017, 16, 203. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kanda, T.; Matsuoka, S.; Yamazaki, M.; Shibata, T.; Nirei, K.; Takahashi, H.; Kaneko, T.; Fujisawa, M.; Higuchi, T.; Nakamura, H.; et al. Apoptosis and Non-Alcoholic Fatty Liver Diseases. World J. Gastroenterol. 2018, 24, 2661–2672. [Google Scholar] [CrossRef] [PubMed]
- Schuster, S.; Cabrera, D.; Arrese, M.; Feldstein, A.E. Triggering and Resolution of Inflammation in NASH. Nat. Rev. Gastroenterol. Hepatol. 2018, 15, 349–364. [Google Scholar] [CrossRef]
- Solinas, G.; Becattini, B. JNK at the Crossroad of Obesity, Insulin Resistance, and Cell Stress Response. Mol. Metab. 2017, 6, 174–184. [Google Scholar] [CrossRef]
- Yan, H.; Gao, Y.; Zhang, Y. Inhibition of JNK Suppresses Autophagy and Attenuates Insulin Resistance in a Rat Model of Nonalcoholic Fatty Liver Disease. Mol. Med. Rep. 2017, 15, 180–186. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stanger, B.Z. Cellular Homeostasis and Repair in the Mammalian Liver. Annu. Rev. Physiol. 2015, 77, 179–200. [Google Scholar] [CrossRef] [Green Version]
- Fausto, N.; Campbell, J.S.; Riehle, K.J. Liver Regeneration. Hepatology 2006, 43, S45–S53. [Google Scholar] [CrossRef] [PubMed]
- Kumar, S.; Duan, Q.; Wu, R.; Harris, E.N.; Su, Q. Pathophysiological Communication between Hepatocytes and Non-Parenchymal Cells in Liver Injury from NAFLD to Liver Fibrosis. Adv. Drug Deliv. Rev. 2021, 176, 113869. [Google Scholar] [CrossRef] [PubMed]
- Koyama, Y.; Brenner, D.A. Liver Inflammation and Fibrosis. J. Clin. Investig. 2017, 127, 55–64. [Google Scholar] [CrossRef]
- Tsuchida, T.; Friedman, S.L. Mechanisms of Hepatic Stellate Cell Activation. Nat. Rev. Gastroenterol. Hepatol. 2017, 14, 397–411. [Google Scholar] [CrossRef] [PubMed]
- Zhang, C.-Y.; Yuan, W.-G.; He, P.; Lei, J.-H.; Wang, C.-X. Liver Fibrosis and Hepatic Stellate Cells: Etiology, Pathological Hallmarks and Therapeutic Targets. World J. Gastroenterol. 2016, 22, 10512. [Google Scholar] [CrossRef]
- Anstee, Q.M.; Reeves, H.L.; Kotsiliti, E.; Govaere, O.; Heikenwalder, M. From NASH to HCC: Current Concepts and Future Challenges. Nat. Rev. Gastroenterol. Hepatol. 2019, 16, 411–428. [Google Scholar] [CrossRef] [PubMed]
- Wree, A.; Broderick, L.; Canbay, A.; Hoffman, H.M.; Feldstein, A.E. From NAFLD to NASH to Cirrhosis—New Insights into Disease Mechanisms. Nat. Rev. Gastroenterol. Hepatol. 2013, 10, 627–636. [Google Scholar] [CrossRef]
- Mantovani, A.; Zaza, G.; Byrne, C.D.; Lonardo, A.; Zoppini, G.; Bonora, E.; Targher, G. Nonalcoholic Fatty Liver Disease Increases Risk of Incident Chronic Kidney Disease: A Systematic Review and Meta-Analysis. Metabolism 2018, 79, 64–76. [Google Scholar] [CrossRef] [PubMed]
- Motamed, N.; Rabiee, B.; Poustchi, H.; Dehestani, B.; Hemasi, G.R.; Khonsari, M.R.; Maadi, M.; Saeedian, F.S.; Zamani, F. Non-Alcoholic Fatty Liver Disease (NAFLD) and 10-Year Risk of Cardiovascular Diseases. Clin. Res. Hepatol. Gastroenterol. 2017, 41, 31–38. [Google Scholar] [CrossRef]
- Principi, M.; Iannone, A.; Losurdo, G.; Mangia, M.; Shahini, E.; Albano, F.; Rizzi, S.F.; La Fortezza, R.F.; Lovero, R.; Contaldo, A.; et al. Nonalcoholic Fatty Liver Disease in Inflammatory Bowel Disease: Prevalence and Risk Factors. Inflamm. Bowel Dis. 2018, 24, 1589–1596. [Google Scholar] [CrossRef] [PubMed]
- Stahl, E.P.; Dhindsa, D.S.; Lee, S.K.; Sandesara, P.B.; Chalasani, N.P.; Sperling, L.S. Nonalcoholic Fatty Liver Disease and the Heart. J. Am. Coll. Cardiol. 2019, 73, 948–963. [Google Scholar] [CrossRef]
- Stols-Gonçalves, D.; Hovingh, G.K.; Nieuwdorp, M.; Holleboom, A.G. NAFLD and Atherosclerosis: Two Sides of the Same Dysmetabolic Coin? Trends Endocrinol. Metab. 2019, 30, 891–902. [Google Scholar] [CrossRef]
- Targher, G.; Byrne, C.D.; Tilg, H. NAFLD and Increased Risk of Cardiovascular Disease: Clinical Associations, Pathophysiological Mechanisms and Pharmacological Implications. Gut 2020, 69, 1691–1705. [Google Scholar] [CrossRef] [PubMed]
- Upala, S.; Jaruvongvanich, V.; Wijarnpreecha, K.; Sanguankeo, A. Nonalcoholic Fatty Liver Disease and Osteoporosis: A Systematic Review and Meta-Analysis. J. Bone Miner. Metab. 2017, 35, 685–693. [Google Scholar] [CrossRef]
- Ballestri, S.; Nascimbeni, F.; Baldelli, E.; Marrazzo, A.; Romagnoli, D.; Lonardo, A. NAFLD as a Sexual Dimorphic Disease: Role of Gender and Reproductive Status in the Development and Progression of Nonalcoholic Fatty Liver Disease and Inherent Cardiovascular Risk. Adv. Ther. 2017, 34, 1291–1326. [Google Scholar] [CrossRef]
- Gong, Z.; Tas, E.; Yakar, S.; Muzumdar, R. Hepatic Lipid Metabolism and Non-Alcoholic Fatty Liver Disease in Aging. Mol. Cell. Endocrinol. 2017, 455, 115–130. [Google Scholar] [CrossRef] [PubMed]
- Papatheodoridi, A.-M.; Chrysavgis, L.; Koutsilieris, M.; Chatzigeorgiou, A. The Role of Senescence in the Development of Nonalcoholic Fatty Liver Disease and Progression to Nonalcoholic Steatohepatitis. Hepatology 2020, 71, 363–374. [Google Scholar] [CrossRef]
- Stahl, E.C.; Haschak, M.J.; Popovic, B.; Brown, B.N. Macrophages in the Aging Liver and Age-Related Liver Disease. Front. Immunol. 2018, 9, 2795. [Google Scholar] [CrossRef] [Green Version]
- Gutierrez-Grobe, Y.; Ponciano-Rodríguez, G.; Ramos, M.H.; Uribe, M.; Méndez-Sánchez, N. Prevalence of Non Alcoholic Fatty Liver Disease in Premenopausal, Posmenopausal and Polycystic Ovary Syndrome Women. The Role of Estrogens. Ann. Hepatol. 2010, 9, 402–409. [Google Scholar] [CrossRef]
- Matsuo, K.; Gualtieri, M.R.; Cahoon, S.S.; Jung, C.E.; Paulson, R.J.; Shoupe, D.; Muderspach, L.I.; Wakatsuki, A.; Wright, J.D.; Roman, L.D. Surgical Menopause and Increased Risk of Nonalcoholic Fatty Liver Disease in Endometrial Cancer. Menopause 2016, 23, 189–196. [Google Scholar] [CrossRef] [PubMed]
- Clark, J.M.; Brancati, F.L.; Diehl, A.M. Nonalcoholic Fatty Liver Disease. Gastroenterology 2002, 122, 1649–1657. [Google Scholar] [CrossRef] [Green Version]
- Anderson, E.L.; Howe, L.D.; Jones, H.E.; Higgins, J.P.T.; Lawlor, D.A.; Fraser, A. The Prevalence of Non-Alcoholic Fatty Liver Disease in Children and Adolescents: A Systematic Review and Meta-Analysis. PLoS ONE 2015, 10, e0140908. [Google Scholar] [CrossRef] [Green Version]
- Lu, J.; Zhang, J.; Du, R.; Wang, T.; Xu, M.; Xu, Y.; Wang, W.; Bi, Y.; Li, D.; Chen, Y.; et al. Age at Menarche Is Associated with the Prevalence of Non-Alcoholic Fatty Liver Disease Later in Life. J. Diabetes 2017, 9, 53–60. [Google Scholar] [CrossRef]
- Mueller, N.T.; Pereira, M.A.; Demerath, E.W.; Dreyfus, J.G.; MacLehose, R.F.; Carr, J.J.; Terry, J.G.; Jacobs, D.R. Earlier Menarche Is Associated with Fatty Liver and Abdominal Ectopic Fat in Midlife, Independent of Young Adult BMI: The CARDIA Study: Menarcheal Timing, Abdominal Fat, and NAFLD. Obesity 2015, 23, 468–474. [Google Scholar] [CrossRef]
- Suzuki, A.; Abdelmalek, M.F.; Schwimmer, J.B.; Lavine, J.E.; Scheimann, A.O.; Unalp–Arida, A.; Yates, K.P.; Sanyal, A.J.; Guy, C.D.; Diehl, A.M. Association Between Puberty and Features of Nonalcoholic Fatty Liver Disease. Clin. Gastroenterol. Hepatol. 2012, 10, 786–794. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yi, K.H.; Hwang, J.S.; Lim, S.W.; Lee, J.A.; Kim, D.H.; Lim, J.S. Early Menarche is Associated with Non-Alcoholic Fatty Liver Disease in Adulthood. Pediatr. Int. 2017, 59, 1270–1275. [Google Scholar] [CrossRef]
- Eslam, M.; Valenti, L.; Romeo, S. Genetics and Epigenetics of NAFLD and NASH: Clinical Impact. J. Hepatol. 2018, 68, 268–279. [Google Scholar] [CrossRef]
- Kovalic, A.J.; Banerjee, P.; Tran, Q.T.; Singal, A.K.; Satapathy, S.K. Genetic and Epigenetic Culprits in the Pathogenesis of Nonalcoholic Fatty Liver Disease. J. Clin. Exp. Hepatol. 2018, 8, 390–402. [Google Scholar] [CrossRef]
- Fazel, Y.; Koenig, A.B.; Sayiner, M.; Goodman, Z.D.; Younossi, Z.M. Epidemiology and Natural History of Non-Alcoholic Fatty Liver Disease. Metabolism 2016, 65, 1017–1025. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Donnelly, K.L.; Smith, C.I.; Schwarzenberg, S.J.; Jessurun, J.; Boldt, M.D.; Parks, E.J. Sources of Fatty Acids Stored in Liver and Secreted via Lipoproteins in Patients with Nonalcoholic Fatty Liver Disease. J. Clin. Investig. 2005, 115, 1343–1351. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fabbrini, E.; Magkos, F.; Mohammed, B.S.; Pietka, T.; Abumrad, N.A.; Patterson, B.W.; Okunade, A.; Klein, S. Intrahepatic Fat, Not Visceral Fat, Is Linked with Metabolic Complications of Obesity. Proc. Natl. Acad. Sci. USA 2009, 106, 15430–15435. [Google Scholar] [CrossRef] [Green Version]
- Wilson, C.G.; Tran, J.L.; Erion, D.M.; Vera, N.B.; Febbraio, M.; Weiss, E.J. Hepatocyte-Specific Disruption of CD36 Attenuates Fatty Liver and Improves Insulin Sensitivity in HFD-Fed Mice. Endocrinology 2016, 157, 570–585. [Google Scholar] [CrossRef] [Green Version]
- Petersen, K.F.; Dufour, S.; Savage, D.B.; Bilz, S.; Solomon, G.; Yonemitsu, S.; Cline, G.W.; Befroy, D.; Zemany, L.; Kahn, B.B.; et al. The Role of Skeletal Muscle Insulin Resistance in the Pathogenesis of the Metabolic Syndrome. Proc. Natl. Acad. Sci. USA 2007, 104, 12587–12594. [Google Scholar] [CrossRef] [Green Version]
- Farrell, G.C.; Haczeyni, F.; Chitturi, S. Pathogenesis of NASH: How Metabolic Complications of Overnutrition Favour Lipotoxicity and Pro-Inflammatory Fatty Liver Disease. In Obesity, Fatty Liver and Liver Cancer; Yu, J., Ed.; Advances in Experimental Medicine and Biology; Springer: Singapore, 2018; Volume 1061, pp. 19–44. ISBN 978-981-10-8683-0. [Google Scholar]
- Mota, M.; Banini, B.A.; Cazanave, S.C.; Sanyal, A.J. Molecular Mechanisms of Lipotoxicity and Glucotoxicity in Nonalcoholic Fatty Liver Disease. Metabolism 2016, 65, 1049–1061. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Henao-Mejia, J.; Elinav, E.; Jin, C.; Hao, L.; Mehal, W.Z.; Strowig, T.; Thaiss, C.A.; Kau, A.L.; Eisenbarth, S.C.; Jurczak, M.J.; et al. Inflammasome-Mediated Dysbiosis Regulates Progression of NAFLD and Obesity. Nature 2012, 482, 179–185. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jornayvaz, F.R.; Samuel, V.T.; Shulman, G.I. The Role of Muscle Insulin Resistance in the Pathogenesis of Atherogenic Dyslipidemia and Nonalcoholic Fatty Liver Disease Associated with the Metabolic Syndrome. Annu. Rev. Nutr. 2010, 30, 273–290. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nachit, M.; Leclercq, I.A. Emerging Awareness on the Importance of Skeletal Muscle in Liver Diseases: Time to Dig Deeper into Mechanisms! Clin. Sci. 2019, 133, 465–481. [Google Scholar] [CrossRef]
- Polyzos, S.A.; Kountouras, J.; Mantzoros, C.S. Leptin in Nonalcoholic Fatty Liver Disease: A Narrative Review. Metabolism 2015, 64, 60–78. [Google Scholar] [CrossRef] [PubMed]
- Safari, Z.; Gérard, P. The Links between the Gut Microbiome and Non-Alcoholic Fatty Liver Disease (NAFLD). Cell. Mol. Life Sci. 2019, 76, 1541–1558. [Google Scholar] [CrossRef] [PubMed]
- Kautzky-Willer, A.; Handisurya, A. Metabolic Diseases and Associated Complications: Sex and Gender Matter! Eur. J. Clin. Investig. 2009, 39, 631–648. [Google Scholar] [CrossRef] [PubMed]
- Ye, Q.; Zou, B.; Yeo, Y.H.; Li, J.; Huang, D.Q.; Wu, Y.; Yang, H.; Liu, C.; Kam, L.Y.; Tan, X.X.E.; et al. Global Prevalence, Incidence, and Outcomes of Non-Obese or Lean Non-Alcoholic Fatty Liver Disease: A Systematic Review and Meta-Analysis. Lancet Gastroenterol. Hepatol. 2020, 5, 739–752. [Google Scholar] [CrossRef]
- Petersen, M.C.; Vatner, D.F.; Shulman, G.I. Regulation of Hepatic Glucose Metabolism in Health and Disease. Nat. Rev. Endocrinol. 2017, 13, 572–587. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Samuel, V.T.; Shulman, G.I. The Pathogenesis of Insulin Resistance: Integrating Signaling Pathways and Substrate Flux. J. Clin. Investig. 2016, 126, 12–22. [Google Scholar] [CrossRef] [Green Version]
- Watt, M.J.; Miotto, P.M.; De Nardo, W.; Montgomery, M.K. The Liver as an Endocrine Organ—Linking NAFLD and Insulin Resistance. Endocr. Rev. 2019, 40, 1367–1393. [Google Scholar] [CrossRef]
- Santoleri, D.; Titchenell, P.M. Resolving the Paradox of Hepatic Insulin Resistance. Cell. Mol. Gastroenterol. Hepatol. 2019, 7, 447–456. [Google Scholar] [CrossRef] [Green Version]
- Sunny, N.E.; Parks, E.J.; Browning, J.D.; Burgess, S.C. Excessive Hepatic Mitochondrial TCA Cycle and Gluconeogenesis in Humans with Nonalcoholic Fatty Liver Disease. Cell Metab. 2011, 14, 804–810. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De Bandt, J.-P.; Jegatheesan, P.; Tennoune-El-Hafaia, N. Muscle Loss in Chronic Liver Diseases: The Example of Nonalcoholic Liver Disease. Nutrients 2018, 10, 1195. [Google Scholar] [CrossRef] [Green Version]
- Seko, Y.; Sumida, Y.; Tanaka, S.; Mori, K.; Taketani, H.; Ishiba, H.; Hara, T.; Okajima, A.; Umemura, A.; Nishikawa, T.; et al. Insulin Resistance Increases the Risk of Incident Type 2 Diabetes Mellitus in Patients with Non-Alcoholic Fatty Liver Disease: HOMA-IR Predicts Development T2DM in NAFLD. Hepatol. Res. 2018, 48, E42–E51. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tilg, H.; Moschen, A.R.; Roden, M. NAFLD and Diabetes Mellitus. Nat. Rev. Gastroenterol. Hepatol. 2017, 14, 32–42. [Google Scholar] [CrossRef]
- Couchepin, C.; Le, K.-A.; Bortolotti, M.; da Encarnacao, J.A.; Oboni, J.-B.; Tran, C.; Schneiter, P.; Tappy, L. Markedly Blunted Metabolic Effects of Fructose in Healthy Young Female Subjects Compared With Male Subjects. Diabetes Care 2008, 31, 1254–1256. [Google Scholar] [CrossRef] [Green Version]
- Ter Horst, K.W.; Gilijamse, P.W.; de Weijer, B.A.; Kilicarslan, M.; Ackermans, M.T.; Nederveen, A.J.; Nieuwdorp, M.; Romijn, J.A.; Serlie, M.J. Sexual Dimorphism in Hepatic, Adipose Tissue, and Peripheral Tissue Insulin Sensitivity in Obese Humans. Front. Endocrinol. 2015, 6, 182. [Google Scholar] [CrossRef]
- Perdomo, C.M.; Frühbeck, G.; Escalada, J. Impact of Nutritional Changes on Nonalcoholic Fatty Liver Disease. Nutrients 2019, 11, 677. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Eslamparast, T.; Tandon, P.; Raman, M. Dietary Composition Independent of Weight Loss in the Management of Non-Alcoholic Fatty Liver Disease. Nutrients 2017, 9, 800. [Google Scholar] [CrossRef] [Green Version]
- Ter Horst, K.W.; Serlie, M.J. Fructose Consumption, Lipogenesis, and Non-Alcoholic Fatty Liver Disease. Nutrients 2017, 9, 981. [Google Scholar] [CrossRef] [Green Version]
- Lim, J.S.; Mietus-Snyder, M.; Valente, A.; Schwarz, J.-M.; Lustig, R.H. The Role of Fructose in the Pathogenesis of NAFLD and the Metabolic Syndrome. Nat. Rev. Gastroenterol. Hepatol. 2010, 7, 251–264. [Google Scholar] [CrossRef]
- Yki-Järvinen, H.; Luukkonen, P.K.; Hodson, L.; Moore, J.B. Dietary Carbohydrates and Fats in Nonalcoholic Fatty Liver Disease. Nat. Rev. Gastroenterol. Hepatol. 2021. [Google Scholar] [CrossRef] [PubMed]
- Shiferaw, B.; Verrill, L.; Booth, H.; Zansky, S.M.; Norton, D.M.; Crim, S.; Henao, O.L. Sex-Based Differences in Food Consumption: Foodborne Diseases Active Surveillance Network (FoodNet) Population Survey, 2006–2007. Clin. Infect. Dis. 2012, 54, S453–S457. [Google Scholar] [CrossRef] [Green Version]
- Wardle, J.; Haase, A.M.; Steptoe, A.; Nillapun, M.; Jonwutiwes, K.; Bellisle, F. Gender Differences in Food Choice: The Contribution of Health Beliefs and Dieting. Ann. Behav. Med. 2004, 27, 107–116. [Google Scholar] [CrossRef] [PubMed]
- Wolongevicz, D.M.; Zhu, L.; Pencina, M.J.; Kimokoti, R.W.; Newby, P.K.; D’Agostino, R.B.; Millen, B.E. Diet Quality and Obesity in Women: The Framingham Nutrition Studies. Br. J. Nutr. 2010, 103, 1223–1229. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vitale, M.; Masulli, M.; Cocozza, S.; Anichini, R.; Babini, A.C.; Boemi, M.; Bonora, E.; Buzzetti, R.; Carpinteri, R.; Caselli, C.; et al. Sex Differences in Food Choices, Adherence to Dietary Recommendations and Plasma Lipid Profile in Type 2 Diabetes—The TOSCA.IT Study. Nutr. Metab. Cardiovasc. Dis. 2016, 26, 879–885. [Google Scholar] [CrossRef]
- Zámbó, V.; Simon-Szabó, L.; Szelényi, P.; Kereszturi, É.; Bánhegyi, G.; Csala, M. Lipotoxicity in the Liver. World J. Hepatol. 2013, 5, 550. [Google Scholar] [CrossRef] [Green Version]
- Jump, D.B. Fatty Acid Regulation of Hepatic Lipid Metabolism. Curr. Opin. Clin. Nutr. Metab. Care 2011, 14, 115–120. [Google Scholar] [CrossRef] [Green Version]
- Leamy, A.K.; Egnatchik, R.A.; Young, J.D. Molecular Mechanisms and the Role of Saturated Fatty Acids in the Progression of Non-Alcoholic Fatty Liver Disease. Prog. Lipid Res. 2013, 52, 165–174. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dhibi, M.; Brahmi, F.; Mnari, A.; Houas, Z.; Chargui, I.; Bchir, L.; Gazzah, N.; Alsaif, M.A.; Hammami, M. The Intake of High Fat Diet with Different Trans Fatty Acid Levels Differentially Induces Oxidative Stress and Non Alcoholic Fatty Liver Disease (NAFLD) in Rats. Nutr. Metab. 2011, 8, 65. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- da Silva-Santi, L.; Antunes, M.; Caparroz-Assef, S.; Carbonera, F.; Masi, L.; Curi, R.; Visentainer, J.; Bazotte, R. Liver Fatty Acid Composition and Inflammation in Mice Fed with High-Carbohydrate Diet or High-Fat Diet. Nutrients 2016, 8, 682. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yki-Järvinen, H. Nutritional Modulation of Non-Alcoholic Fatty Liver Disease and Insulin Resistance. Nutrients 2015, 7, 9127–9138. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ranković, S.; Popović, T.; Martačić, J.D.; Petrović, S.; Tomić, M.; Ignjatović, Đ.; Tovilović-Kovačević, G.; Glibetić, M. Liver Phospholipids Fatty Acids Composition in Response to Different Types of Diets in Rats of Both Sexes. Lipids Health Dis. 2017, 16, 94. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Basaranoglu, M. Fructose as a Key Player in the Development of Fatty Liver Disease. World J. Gastroenterol. 2013, 19, 1166. [Google Scholar] [CrossRef] [PubMed]
- Jegatheesan, P.; De Bandt, J. Fructose and NAFLD: The Multifaceted Aspects of Fructose Metabolism. Nutrients 2017, 9, 230. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jensen, T.; Abdelmalek, M.F.; Sullivan, S.; Nadeau, K.J.; Green, M.; Roncal, C.; Nakagawa, T.; Kuwabara, M.; Sato, Y.; Kang, D.-H.; et al. Fructose and Sugar: A Major Mediator of Non-Alcoholic Fatty Liver Disease. J. Hepatol. 2018, 68, 1063–1075. [Google Scholar] [CrossRef] [Green Version]
- Alwahsh, S.M.; Gebhardt, R. Dietary Fructose as a Risk Factor for Non-Alcoholic Fatty Liver Disease (NAFLD). Arch. Toxicol. 2017, 91, 1545–1563. [Google Scholar] [CrossRef]
- Softic, S.; Stanhope, K.L.; Boucher, J.; Divanovic, S.; Lanaspa, M.A.; Johnson, R.J.; Kahn, C.R. Fructose and Hepatic Insulin Resistance. Crit. Rev. Clin. Lab. Sci. 2020, 57, 308–322. [Google Scholar] [CrossRef] [PubMed]
- Tappy, L.; Lê, K.-A. Metabolic Effects of Fructose and the Worldwide Increase in Obesity. Physiol. Rev. 2010, 90, 23–46. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wei, Y.; Wang, D.; Pagliassotti, M.J. Fructose Selectively Modulates C-Jun N-Terminal Kinase Activity and Insulin Signaling in Rat Primary Hepatocytes. J. Nutr. 2005, 135, 1642–1646. [Google Scholar] [CrossRef] [Green Version]
- Lambertz, J.; Weiskirchen, S.; Landert, S.; Weiskirchen, R. Fructose: A Dietary Sugar in Crosstalk with Microbiota Contributing to the Development and Progression of Non-Alcoholic Liver Disease. Front. Immunol. 2017, 8, 1159. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Comitato, R.; Saba, A.; Turrini, A.; Arganini, C.; Virgili, F. Sex Hormones and Macronutrient Metabolism. Crit. Rev. Food Sci. Nutr. 2015, 55, 227–241. [Google Scholar] [CrossRef] [Green Version]
- Hyer, M.M.; Dyer, S.K.; Kloster, A.; Adrees, A.; Taetzsch, T.; Feaster, J.; Valdez, G.; Neigh, G.N. Sex Modifies the Consequences of Extended Fructose Consumption on Liver Health, Motor Function, and Physiological Damage in Rats. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2019, 317, R903–R911. [Google Scholar] [CrossRef]
- Low, W.; Cornfield, T.; Charlton, C.; Tomlinson, J.; Hodson, L. Sex Differences in Hepatic De Novo Lipogenesis with Acute Fructose Feeding. Nutrients 2018, 10, 1263. [Google Scholar] [CrossRef] [Green Version]
- Morrell, A.; Tripet, B.P.; Eilers, B.J.; Tegman, M.; Thompson, D.; Copié, V.; Burkhead, J.L. Copper Modulates Sex-Specific Fructose Hepatoxicity in Nonalcoholic Fatty Liver Disease (NALFD) Wistar Rat Models. J. Nutr. Biochem. 2020, 78, 108316. [Google Scholar] [CrossRef]
- De Chiara, F.; Ureta Checcllo, C.; Ramón Azcón, J. High Protein Diet and Metabolic Plasticity in Non-Alcoholic Fatty Liver Disease: Myths and Truths. Nutrients 2019, 11, 2985. [Google Scholar] [CrossRef] [Green Version]
- Harris, R.A.; Joshi, M.; Jeoung, N.H.; Obayashi, M. Overview of the Molecular and Biochemical Basis of Branched-Chain Amino Acid Catabolism. J. Nutr. 2005, 135, 1527S–1530S. [Google Scholar] [CrossRef] [Green Version]
- Honda, T.; Ishigami, M.; Luo, F.; Lingyun, M.; Ishizu, Y.; Kuzuya, T.; Hayashi, K.; Nakano, I.; Ishikawa, T.; Feng, G.-G.; et al. Branched-Chain Amino Acids Alleviate Hepatic Steatosis and Liver Injury in Choline-Deficient High-Fat Diet Induced NASH Mice. Metabolism 2017, 69, 177–187. [Google Scholar] [CrossRef] [PubMed]
- Takegoshi, K.; Honda, M.; Okada, H.; Takabatake, R.; Matsuzawa-Nagata, N.; Campbell, J.S.; Nishikawa, M.; Shimakami, T.; Shirasaki, T.; Sakai, Y.; et al. Branched-Chain Amino Acids Prevent Hepatic Fibrosis and Development of Hepatocellular Carcinoma in a Non-Alcoholic Steatohepatitis Mouse Model. Oncotarget 2017, 8, 18191–18205. [Google Scholar] [CrossRef]
- Gaggini, M.; Carli, F.; Rosso, C.; Buzzigoli, E.; Marietti, M.; Della Latta, V.; Ciociaro, D.; Abate, M.L.; Gambino, R.; Cassader, M.; et al. Altered Amino Acid Concentrations in NAFLD: Impact of Obesity and Insulin Resistance: Gaggini et Al. Hepatology 2018, 67, 145–158. [Google Scholar] [CrossRef] [Green Version]
- Grzych, G.; Vonghia, L.; Bout, M.-A.; Weyler, J.; Verrijken, A.; Dirinck, E.; Joncquel, M.; Van Gaal, L.; Paumelle, R.; Francque, S.; et al. Plasma BCAA Changes in Patients with NAFLD Are Sex Dependent. J. Clin. Endocrinol. Metab. 2020, 105, dgaa175. [Google Scholar] [CrossRef] [PubMed]
- Kim, D.; Vazquez-Montesino, L.M.; Li, A.A.; Cholankeril, G.; Ahmed, A. Inadequate Physical Activity and Sedentary Behavior Are Independent Predictors of Nonalcoholic Fatty Liver Disease. Hepatology 2020, 72, 1556–1568. [Google Scholar] [CrossRef]
- Rodriguez, B.; Torres, D.M.; Harrison, S.A. Physical Activity: An Essential Component of Lifestyle Modification in NAFLD. Nat. Rev. Gastroenterol. Hepatol. 2012, 9, 726–731. [Google Scholar] [CrossRef]
- Romero-Gómez, M.; Zelber-Sagi, S.; Trenell, M. Treatment of NAFLD with Diet, Physical Activity and Exercise. J. Hepatol. 2017, 67, 829–846. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pereira, R.M.; da Cruz Rodrigues, K.C.; Anaruma, C.P.; Sant’Ana, M.R.; de Campos, T.D.P.; Gaspar, R.S.; Canciglieri, R.D.S.; de Melo, D.G.; Mekary, R.A.; da Silva, A.S.R.; et al. Short-Term Strength Training Reduces Gluconeogenesis and NAFLD in Obese Mice. J. Endocrinol. 2019, 241, 59–70. [Google Scholar] [CrossRef] [Green Version]
- Sung, K.-C.; Ryu, S.; Lee, J.-Y.; Kim, J.-Y.; Wild, S.H.; Byrne, C.D. Effect of Exercise on the Development of New Fatty Liver and the Resolution of Existing Fatty Liver. J. Hepatol. 2016, 65, 791–797. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Van der Windt, D.J.; Sud, V.; Zhang, H.; Tsung, A.; Huang, H. The Effects of Physical Exercise on Fatty Liver Disease. Gene Expr. 2018, 18, 89–101. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Caudwell, P.; Gibbons, C.; Finlayson, G.; Näslund, E.; Blundell, J. Exercise and Weight Loss: No Sex Differences in Body Weight Response to Exercise. Exerc. Sport Sci. Rev. 2014, 42, 92–101. [Google Scholar] [CrossRef]
- Christensen, P.; Meinert Larsen, T.; Westerterp-Plantenga, M.; Macdonald, I.; Martinez, J.A.; Handjiev, S.; Poppitt, S.; Hansen, S.; Ritz, C.; Astrup, A.; et al. Men and Women Respond Differently to Rapid Weight Loss: Metabolic Outcomes of a Multi-Centre Intervention Study after a Low-Energy Diet in 2500 Overweight, Individuals with Pre-Diabetes (PREVIEW). Diabetes Obes. Metab. 2018, 20, 2840–2851. [Google Scholar] [CrossRef] [Green Version]
- Doucet, E.; St-Pierre, S.; Alméras, N.; Imbeault, P.; Mauriège, P.; Pascot, A.; Després, J.-P.; Tremblay, A. Reduction of Visceral Adipose Tissue during Weight Loss. Eur. J. Clin. Nutr. 2002, 56, 297–304. [Google Scholar] [CrossRef]
- Hagobian, T.A.; Evero, N. Exercise and Weight Loss: What Is the Evidence of Sex Differences? Curr. Obes. Rep. 2013, 2, 86–92. [Google Scholar] [CrossRef] [Green Version]
- Lovejoy, J.C.; Sainsbury, A. Stock Conference 2008 Working Group Sex Differences in Obesity and the Regulation of Energy Homeostasis. Obes. Rev. 2009, 10, 154–167. [Google Scholar] [CrossRef] [PubMed]
- White, U.A.; Tchoukalova, Y.D. Sex Dimorphism and Depot Differences in Adipose Tissue Function. Biochim. Biophys. Acta 2014, 1842, 377–392. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vilar-Gomez, E.; Martinez-Perez, Y.; Calzadilla-Bertot, L.; Torres-Gonzalez, A.; Gra-Oramas, B.; Gonzalez-Fabian, L.; Friedman, S.L.; Diago, M.; Romero-Gomez, M. Weight Loss Through Lifestyle Modification Significantly Reduces Features of Nonalcoholic Steatohepatitis. Gastroenterology 2015, 149, 367–378. [Google Scholar] [CrossRef]
- McGee, S.L.; Hargreaves, M. Exercise Adaptations: Molecular Mechanisms and Potential Targets for Therapeutic Benefit. Nat. Rev. Endocrinol. 2020, 16, 495–505. [Google Scholar] [CrossRef]
- Melo, L.; Tilmant, K.; Hagar, A.; Klaunig, J.E. Effect of Endurance Exercise Training on Liver Gene Expression in Male and Female Mice. Appl. Physiol. Nutr. Metab. 2021, 46, 356–367. [Google Scholar] [CrossRef]
- Severinsen, M.C.K.; Pedersen, B.K. Muscle–Organ Crosstalk: The Emerging Roles of Myokines. Endocr. Rev. 2020, 41, 594–609. [Google Scholar] [CrossRef]
- Camera, D.M.; Anderson, M.J.; Hawley, J.A.; Carey, A.L. Short-Term Endurance Training Does Not Alter the Oxidative Capacity of Human Subcutaneous Adipose Tissue. Eur. J. Appl. Physiol. 2010, 109, 307–316. [Google Scholar] [CrossRef]
- Stanford, K.I.; Goodyear, L.J. Exercise Regulation of Adipose Tissue. Adipocyte 2016, 5, 153–162. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tsiloulis, T.; Carey, A.L.; Bayliss, J.; Canny, B.; Meex, R.C.R.; Watt, M.J. No Evidence of White Adipocyte Browning after Endurance Exercise Training in Obese Men. Int. J. Obes. 2018, 42, 721–727. [Google Scholar] [CrossRef] [PubMed]
- Beaudry, K.M.; Devries, M.C. Sex-Based Differences in Hepatic and Skeletal Muscle Triglyceride Storage and Metabolism 1. Appl. Physiol. Nutr. Metab. 2019, 44, 805–813. [Google Scholar] [CrossRef]
- Burra, P.; Bizzaro, D.; Gonta, A.; Shalaby, S.; Gambato, M.; Morelli, M.C.; Trapani, S.; Floreani, A.; Marra, F.; Brunetto, M.R.; et al. Clinical Impact of Sexual Dimorphism in Non-alcoholic Fatty Liver Disease (NAFLD) and Non-alcoholic Steatohepatitis (NASH). Liver Int. 2021, 41, 1713–1733. [Google Scholar] [CrossRef]
- Henderson, G.C. Sexual Dimorphism in the Effects of Exercise on Metabolism of Lipids to Support Resting Metabolism. Front. Endocrinol. 2014, 5, 162. [Google Scholar] [CrossRef] [Green Version]
- Haizlip, K.M.; Harrison, B.C.; Leinwand, L.A. Sex-Based Differences in Skeletal Muscle Kinetics and Fiber-Type Composition. Physiology 2015, 30, 30–39. [Google Scholar] [CrossRef]
- Lundsgaard, A.-M.; Kiens, B. Gender Differences in Skeletal Muscle Substrate Metabolism-Molecular Mechanisms and Insulin Sensitivity. Front. Endocrinol. 2014, 5, 195. [Google Scholar] [CrossRef] [Green Version]
- McCoin, C.S.; Von Schulze, A.; Allen, J.; Fuller, K.N.Z.; Xia, Q.; Koestler, D.C.; Houchen, C.J.; Maurer, A.; Dorn, G.W.; Shankar, K.; et al. Sex Modulates Hepatic Mitochondrial Adaptations to High-Fat Diet and Physical Activity. Am. J. Physiol. Endocrinol. Metab. 2019, 317, E298–E311. [Google Scholar] [CrossRef] [PubMed]
- Brady, C.W. Liver Disease in Menopause. World J. Gastroenterol. 2015, 21, 7613. [Google Scholar] [CrossRef]
- DiStefano, J.K. NAFLD and NASH in Postmenopausal Women: Implications for Diagnosis and Treatment. Endocrinology 2020, 161, bqaa134. [Google Scholar] [CrossRef]
- Barsalani, R.; Chapados, N.; Lavoie, J.-M. Hepatic VLDL-TG Production and MTP Gene Expression Are Decreased in Ovariectomized Rats: Effects of Exercise Training. Horm. Metab. Res. 2010, 42, 860–867. [Google Scholar] [CrossRef] [PubMed]
- Palmisano, B.T.; Le, T.D.; Zhu, L.; Lee, Y.K.; Stafford, J.M. Cholesteryl Ester Transfer Protein Alters Liver and Plasma Triglyceride Metabolism through Two Liver Networks in Female Mice. J. Lipid Res. 2016, 57, 1541–1551. [Google Scholar] [CrossRef] [Green Version]
- Kim, J.H.; Meyers, M.S.; Khuder, S.S.; Abdallah, S.L.; Muturi, H.T.; Russo, L.; Tate, C.R.; Hevener, A.L.; Najjar, S.M.; Leloup, C.; et al. Tissue-Selective Estrogen Complexes with Bazedoxifene Prevent Metabolic Dysfunction in Female Mice. Mol. Metab. 2014, 3, 177–190. [Google Scholar] [CrossRef] [PubMed]
- Smith, G.I.; Reeds, D.N.; Okunade, A.L.; Patterson, B.W.; Mittendorfer, B. Systemic Delivery of Estradiol, but Not Testosterone or Progesterone, Alters Very Low Density Lipoprotein-Triglyceride Kinetics in Postmenopausal Women. J. Clin. Endocrinol. Metab. 2014, 99, E1306–E1310. [Google Scholar] [CrossRef] [Green Version]
- Zegura, B.; Guzic-Salobir, B.; Sebestjen, M.; Keber, I. The Effect of Various Menopausal Hormone Therapies on Markers of Inflammation, Coagulation, Fibrinolysis, Lipids, and Lipoproteins in Healthy Postmenopausal Women. Menopause 2006, 13, 643–650. [Google Scholar] [CrossRef]
- Moro, L.; Arbini, A.A.; Hsieh, J.-T.; Ford, J.; Simpson, E.R.; Hajibeigi, A.; Öz, O.K. Aromatase Deficiency Inhibits the Permeability Transition in Mouse Liver Mitochondria. Endocrinology 2010, 151, 1643–1652. [Google Scholar] [CrossRef] [Green Version]
- Nemoto, Y.; Toda, K.; Ono, M.; Fujikawa-Adachi, K.; Saibara, T.; Onishi, S.; Enzan, H.; Okada, T.; Shizuta, Y. Altered Expression of Fatty Acid–Metabolizing Enzymes in Aromatase-Deficient Mice. J. Clin. Investig. 2000, 105, 1819–1825. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yoshikawa, T. Aromatase-Deficient (ArKO) Mice Are Retrieved from Severe Hepatic Steatosis by Peroxisome Proliferator Administration. Hepatol. Res. 2002, 22, 278–287. [Google Scholar] [CrossRef]
- Djouadi, F.; Weinheimer, C.J.; Saffitz, J.E.; Pitchford, C.; Bastin, J.; Gonzalez, F.J.; Kelly, D.P. A Gender-Related Defect in Lipid Metabolism and Glucose Homeostasis in Peroxisome Proliferator- Activated Receptor Alpha- Deficient Mice. J. Clin. Investig. 1998, 102, 1083–1091. [Google Scholar] [CrossRef]
- Cano, R.; Pérez, J.L.; Dávila, L.A.; Ortega, Á.; Gómez, Y.; Valero-Cedeño, N.J.; Parra, H.; Manzano, A.; Véliz Castro, T.I.; Albornoz, M.P.D.; et al. Role of Endocrine-Disrupting Chemicals in the Pathogenesis of Non-Alcoholic Fatty Liver Disease: A Comprehensive Review. Int. J. Mol. Sci. 2021, 22, 4807. [Google Scholar] [CrossRef] [PubMed]
- Foulds, C.E.; Treviño, L.S.; York, B.; Walker, C.L. Endocrine-Disrupting Chemicals and Fatty Liver Disease. Nat. Rev. Endocrinol. 2017, 13, 445–457. [Google Scholar] [CrossRef] [Green Version]
- Stratakis, N.; Conti, D.V.; Jin, R.; Margetaki, K.; Valvi, D.; Siskos, A.P.; Maitre, L.; Garcia, E.; Varo, N.; Zhao, Y.; et al. Prenatal Exposure to Perfluoroalkyl Substances Associated With Increased Susceptibility to Liver Injury in Children. Hepatology 2020, 72, 1758–1770. [Google Scholar] [CrossRef]
- Treviño, L.S.; Katz, T.A. Endocrine Disruptors and Developmental Origins of Nonalcoholic Fatty Liver Disease. Endocrinology 2018, 159, 20–31. [Google Scholar] [CrossRef]
- Ministrini, S.; Montecucco, F.; Sahebkar, A.; Carbone, F. Macrophages in the Pathophysiology of NAFLD: The Role of Sex Differences. Eur. J. Clin. Investig. 2020, 50, e13236. [Google Scholar] [CrossRef] [Green Version]
- Naugler, W.E.; Sakurai, T.; Kim, S.; Maeda, S.; Kim, K.; Elsharkawy, A.M.; Karin, M. Gender Disparity in Liver Cancer Due to Sex Differences in MyD88-Dependent IL-6 Production. Science 2007, 317, 121–124. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Evans, M.J.; Eckert, A.; Lai, K.; Adelman, S.J.; Harnish, D.C. Reciprocal Antagonism between Estrogen Receptor and NF-ΚB Activity In Vivo. Circ. Res. 2001, 89, 823–830. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Galmés-Pascual, B.M.; Martínez-Cignoni, M.R.; Morán-Costoya, A.; Bauza-Thorbrügge, M.; Sbert-Roig, M.; Valle, A.; Proenza, A.M.; Lladó, I.; Gianotti, M. 17β-Estradiol Ameliorates Lipotoxicity-Induced Hepatic Mitochondrial Oxidative Stress and Insulin Resistance. Free Radic. Biol. Med. 2020, 150, 148–160. [Google Scholar] [CrossRef]
- Kalaitzidis, D.; Gilmore, T.D. Transcription Factor Cross-Talk: The Estrogen Receptor and NF-ΚB. Trends Endocrinol. Metab. 2005, 16, 46–52. [Google Scholar] [CrossRef] [PubMed]
- Win, S.; Min, R.W.M.; Chen, C.Q.; Zhang, J.; Chen, Y.; Li, M.; Suzuki, A.; Abdelmalek, M.F.; Wang, Y.; Aghajan, M.; et al. Expression of Mitochondrial Membrane–Linked SAB Determines Severity of Sex-Dependent Acute Liver Injury. J. Clin. Investig. 2019, 129, 5278–5293. [Google Scholar] [CrossRef]
- Benedusi, V.; Martini, E.; Kallikourdis, M.; Villa, A.; Meda, C.; Maggi, A. Ovariectomy Shortens the Life Span of Female Mice. Oncotarget 2015, 6, 10801–10811. [Google Scholar] [CrossRef] [Green Version]
- Kireev, R.A.; Tresguerres, A.C.F.; Garcia, C.; Borras, C.; Ariznavarreta, C.; Vara, E.; Vina, J.; Tresguerres, J.A.F. Hormonal Regulation of Pro-Inflammatory and Lipid Peroxidation Processes in Liver of Old Ovariectomized Female Rats. Biogerontology 2010, 11, 229–243. [Google Scholar] [CrossRef] [PubMed]
- Rogers, N.H.; Perfield, J.W.; Strissel, K.J.; Obin, M.S.; Greenberg, A.S. Reduced Energy Expenditure and Increased Inflammation Are Early Events in the Development of Ovariectomy-Induced Obesity. Endocrinology 2009, 150, 2161–2168. [Google Scholar] [CrossRef] [PubMed]
- Kamada, Y.; Kiso, S.; Yoshida, Y.; Chatani, N.; Kizu, T.; Hamano, M.; Tsubakio, M.; Takemura, T.; Ezaki, H.; Hayashi, N.; et al. Estrogen Deficiency Worsens Steatohepatitis in Mice Fed High-Fat and High-Cholesterol Diet. Am. J. Physiol. Gastrointest. Liver Physiol. 2011, 301, G1031–G1043. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Klair, J.S.; Yang, J.D.; Abdelmalek, M.F.; Guy, C.D.; Gill, R.M.; Yates, K.; Unalp-Arida, A.; Lavine, J.E.; Clark, J.M.; Diehl, A.M.; et al. A Longer Duration of Estrogen Deficiency Increases Fibrosis Risk among Postmenopausal Women with Nonalcoholic Fatty Liver Disease: Steatohepatitis/Metabolic Liver Disease. Hepatology 2016, 64, 85–91. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, C.; Xu, M.M.; Wang, K.; Adler, A.J.; Vella, A.T.; Zhou, B. Macrophage Polarization and Meta-Inflammation. Transl. Res. 2018, 191, 29–44. [Google Scholar] [CrossRef] [PubMed]
- Sica, A.; Invernizzi, P.; Mantovani, A. Macrophage Plasticity and Polarization in Liver Homeostasis and Pathology: Sica et Al. Hepatology 2014, 59, 2034–2042. [Google Scholar] [CrossRef]
- Villa, A.; Rizzi, N.; Vegeto, E.; Ciana, P.; Maggi, A. Estrogen Accelerates the Resolution of Inflammation in Macrophagic Cells. Sci. Rep. 2015, 5, 15224. [Google Scholar] [CrossRef]
- Verrijken, A.; Francque, S.; Gaal, L.V. The Role of Visceral Adipose Tissue in the Pathogenesis of Non-Alcoholic Fatty Liver Disease. Eur. Endocrinol. 2010, 7, 96. [Google Scholar] [CrossRef] [Green Version]
- Meyer, M.R.; Clegg, D.J.; Prossnitz, E.R.; Barton, M. Obesity, Insulin Resistance and Diabetes: Sex Differences and Role of Oestrogen Receptors: Obesity, Insulin Resistance and Diabetes: Role of Oestrogen. Acta Physiol. 2011, 203, 259–269. [Google Scholar] [CrossRef]
- Azzu, V.; Vacca, M.; Virtue, S.; Allison, M.; Vidal-Puig, A. Adipose Tissue-Liver Cross Talk in the Control of Whole-Body Metabolism: Implications in Nonalcoholic Fatty Liver Disease. Gastroenterology 2020, 158, 1899–1912. [Google Scholar] [CrossRef] [PubMed]
- Gancheva, S.; Jelenik, T.; Álvarez-Hernández, E.; Roden, M. Interorgan Metabolic Crosstalk in Human Insulin Resistance. Physiol. Rev. 2018, 98, 1371–1415. [Google Scholar] [CrossRef] [Green Version]
- Kurt, Z.; Barrere-Cain, R.; LaGuardia, J.; Mehrabian, M.; Pan, C.; Hui, S.T.; Norheim, F.; Zhou, Z.; Hasin, Y.; Lusis, A.J.; et al. Tissue-Specific Pathways and Networks Underlying Sexual Dimorphism in Non-Alcoholic Fatty Liver Disease. Biol. Sex Differ. 2018, 9, 46. [Google Scholar] [CrossRef]
- Varghese, M.; Griffin, C.; McKernan, K.; Eter, L.; Lanzetta, N.; Agarwal, D.; Abrishami, S.; Singer, K. Sex Differences in Inflammatory Responses to Adipose Tissue Lipolysis in Diet-Induced Obesity. Endocrinology 2019, 160, 293–312. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Besse-Patin, A.; Léveillé, M.; Oropeza, D.; Nguyen, B.N.; Prat, A.; Estall, J.L. Estrogen Signals Through Peroxisome Proliferator-Activated Receptor-γ Coactivator 1α to Reduce Oxidative Damage Associated With Diet-Induced Fatty Liver Disease. Gastroenterology 2017, 152, 243–256. [Google Scholar] [CrossRef]
- Strakovsky, R.S.; Wang, H.; Engeseth, N.J.; Flaws, J.A.; Helferich, W.G.; Pan, Y.-X.; Lezmi, S. Developmental Bisphenol A (BPA) Exposure Leads to Sex-Specific Modification of Hepatic Gene Expression and Epigenome at Birth That May Exacerbate High-Fat Diet-Induced Hepatic Steatosis. Toxicol. Appl. Pharmacol. 2015, 284, 101–112. [Google Scholar] [CrossRef] [Green Version]
- Wei, J.; Sun, X.; Chen, Y.; Li, Y.; Song, L.; Zhou, Z.; Xu, B.; Lin, Y.; Xu, S. Perinatal Exposure to Bisphenol A Exacerbates Nonalcoholic Steatohepatitis-like Phenotype in Male Rat Offspring Fed on a High-Fat Diet. J. Endocrinol. 2014, 222, 313–325. [Google Scholar] [CrossRef] [Green Version]
- Figueiredo, L.S.; Oliveira, K.M.; Freitas, I.N.; Silva, J.A.; Silva, J.N.; Favero-Santos, B.C.; Bonfleur, M.L.; Carneiro, E.M.; Ribeiro, R.A. Bisphenol-A Exposure Worsens Hepatic Steatosis in Ovariectomized Mice Fed on a High-Fat Diet: Role of Endoplasmic Reticulum Stress and Fibrogenic Pathways. Life Sci. 2020, 256, 118012. [Google Scholar] [CrossRef]
- Ohashi, T.; Kato, M.; Yamasaki, A.; Kuwano, A.; Suzuki, H.; Kohjima, M.; Ogawa, Y. Effects of High Fructose Intake on Liver Injury Progression in High Fat Diet Induced Fatty Liver Disease in Ovariectomized Female Mice. Food Chem. Toxicol. 2018, 118, 190–197. [Google Scholar] [CrossRef] [PubMed]
- Buniam, J.; Chukijrungroat, N.; Khamphaya, T.; Weerachayaphorn, J.; Saengsirisuwan, V. Estrogen and Voluntary Exercise Attenuate Cardiometabolic Syndrome and Hepatic Steatosis in Ovariectomized Rats Fed a High-Fat High-Fructose Diet. Am. J. Physiol. Endocrinol. Metab. 2019, 316, E908–E921. [Google Scholar] [CrossRef] [PubMed]
- Barros, R.P.A.; Gustafsson, J.-Å. Estrogen Receptors and the Metabolic Network. Cell Metab. 2011, 14, 289–299. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Evans, M.J.; Lai, K.; Shaw, L.J.; Harnish, D.C.; Chadwick, C.C. Estrogen Receptor α Inhibits IL-1β Induction of Gene Expression in the Mouse Liver. Endocrinology 2002, 143, 2559–2570. [Google Scholar] [CrossRef]
- Ribas, V.; Nguyen, M.T.A.; Henstridge, D.C.; Nguyen, A.-K.; Beaven, S.W.; Watt, M.J.; Hevener, A.L. Impaired Oxidative Metabolism and Inflammation Are Associated with Insulin Resistance in ERα-Deficient Mice. Am. J. Physiol. Endocrinol. Metab. 2010, 298, E304–E319. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chambliss, K.L.; Barrera, J.; Umetani, M.; Umetani, J.; Kim, S.H.; Madak-Erdogan, Z.; Huang, L.; Katzenellenbogen, B.S.; Katzenellenbogen, J.A.; Mineo, C.; et al. Nonnuclear Estrogen Receptor Activation Improves Hepatic Steatosis in Female Mice. Endocrinology 2016, 157, 3731–3741. [Google Scholar] [CrossRef]
- Pedram, A.; Razandi, M.; O’Mahony, F.; Harvey, H.; Harvey, B.J.; Levin, E.R. Estrogen Reduces Lipid Content in the Liver Exclusively from Membrane Receptor Signaling. Sci. Signal. 2013, 6, ra36. [Google Scholar] [CrossRef] [PubMed]
- Guillaume, M.; Riant, E.; Fabre, A.; Raymond-Letron, I.; Buscato, M.; Davezac, M.; Tramunt, B.; Montagner, A.; Smati, S.; Zahreddine, R.; et al. Selective Liver Estrogen Receptor α Modulation Prevents Steatosis, Diabetes, and Obesity Through the Anorectic Growth Differentiation Factor 15 Hepatokine in Mice. Hepatol. Commun. 2019, 3, 908–924. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Khristi, V.; Ratri, A.; Ghosh, S.; Pathak, D.; Borosha, S.; Dai, E.; Roy, R.; Chakravarthi, V.P.; Wolfe, M.W.; Karim Rumi, M.A. Disruption of ESR1 Alters the Expression of Genes Regulating Hepatic Lipid and Carbohydrate Metabolism in Male Rats. Mol. Cell. Endocrinol. 2019, 490, 47–56. [Google Scholar] [CrossRef] [PubMed]
- Winn, N.C.; Jurrissen, T.J.; Grunewald, Z.I.; Cunningham, R.P.; Woodford, M.L.; Kanaley, J.A.; Lubahn, D.B.; Manrique-Acevedo, C.; Rector, R.S.; Vieira-Potter, V.J.; et al. Estrogen Receptor-α Signaling Maintains Immunometabolic Function in Males and Is Obligatory for Exercise-Induced Amelioration of Nonalcoholic Fatty Liver. Am. J. Physiol. Endocrinol. Metab. 2019, 316, E156–E167. [Google Scholar] [CrossRef]
- Sun, B.; Karin, M. NF-ΚB Signaling, Liver Disease and Hepatoprotective Agents. Oncogene 2008, 27, 6228–6244. [Google Scholar] [CrossRef] [Green Version]
- Erkan, G.; Yilmaz, G.; Konca Degertekin, C.; Akyol, G.; Ozenirler, S. Presence and Extent of Estrogen Receptor-Alpha Expression in Patients with Simple Steatosis and NASH. Pathol. Res. Pract. 2013, 209, 429–432. [Google Scholar] [CrossRef]
- Bizzaro, D.; Crescenzi, M.; Di Liddo, R.; Arcidiacono, D.; Cappon, A.; Bertalot, T.; Amodio, V.; Tasso, A.; Stefani, A.; Bertazzo, V.; et al. Sex-Dependent Differences in Inflammatory Responses during Liver Regeneration in a Murine Model of Acute Liver Injury. Clin. Sci. 2018, 132, 255–272. [Google Scholar] [CrossRef] [PubMed]
- Kao, T.-L.; Kuan, Y.-P.; Cheng, W.-C.; Chang, W.-C.; Jeng, L.-B.; Yeh, S.; Ma, W.-L. Estrogen Receptors Orchestrate Cell Growth and Differentiation to Facilitate Liver Regeneration. Theranostics 2018, 8, 2672–2682. [Google Scholar] [CrossRef] [PubMed]
- Tsugawa, Y.; Natori, M.; Handa, H.; Imai, T. Estradiol Accelerates Liver Regeneration through Estrogen Receptor α. Clin. Exp. Gastroenterol. 2019, 12, 331–336. [Google Scholar] [CrossRef] [Green Version]
- Uebi, T.; Umeda, M.; Imai, T. Estrogen Induces Estrogen Receptor Alpha Expression and Hepatocyte Proliferation in the Livers of Male Mice. Genes Cells 2015, 20, 217–223. [Google Scholar] [CrossRef]
- Srisowanna, N.; Choijookhuu, N.; Yano, K.; Batmunkh, B.; Ikenoue, M.; Nhat Huynh Mai, N.; Yamaguchi, Y.; Hishikawa, Y. The Effect of Estrogen on Hepatic Fat Accumulation during Early Phase of Liver Regeneration after Partial Hepatectomy in Rats. Acta Histochem. Cytochem. 2019, 52, 67–75. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lavoie, J.-M.; Pighon, A. NAFLD, Estrogens, and Physical Exercise: The Animal Model. J. Nutr. Metab. 2012, 2012, 914938. [Google Scholar] [CrossRef] [Green Version]
- Venetsanaki, V.; Polyzos, S.A. Menopause and Non-Alcoholic Fatty Liver Disease: A Review Focusing on Therapeutic Perspectives. Curr. Vasc. Pharmacol. 2019, 17, 546–555. [Google Scholar] [CrossRef]
- Witayavanitkul, N.; Werawatganon, D.; Chayanupatkul, M.; Klaikeaw, N.; Sanguanrungsirikul, S.; Siriviriyakul, P. Genistein and Exercise Modulated Lipid Peroxidation and Improved Steatohepatitis in Ovariectomized Rats. BMC Complement. Med. Ther. 2020, 20, 162. [Google Scholar] [CrossRef]
- Fuller, K.N.Z.; McCoin, C.S.; Von Schulze, A.T.; Houchen, C.J.; Choi, M.A.; Thyfault, J.P. Estradiol Treatment or Modest Exercise Improves Hepatic Health and Mitochondrial Outcomes in Female Mice Following Ovariectomy. Am. J. Physiol. Endocrinol. Metab. 2021, 320, E1020–E1031. [Google Scholar] [CrossRef]
- Pighon, A.; Barsalani, R.; Yasari, S.; Prud’homme, D.; Lavoie, J.-M. Does Exercise Training Prior to Ovariectomy Protect against Liver and Adipocyte Fat Accumulation in Rats? Climacteric 2010, 13, 238–248. [Google Scholar] [CrossRef]
- Meoli, L.; Isensee, J.; Zazzu, V.; Nabzdyk, C.S.; Soewarto, D.; Witt, H.; Foryst-Ludwig, A.; Kintscher, U.; Noppinger, P.R. Sex- and Age-Dependent Effects of Gpr30 Genetic Deletion on the Metabolic and Cardiovascular Profiles of Diet-Induced Obese Mice. Gene 2014, 540, 210–216. [Google Scholar] [CrossRef] [PubMed]
- Wei, T.; Chen, W.; Wen, L.; Zhang, J.; Zhang, Q.; Yang, J.; Liu, H.; Chen, B.W.; Zhou, Y.; Feng, X.; et al. G Protein-Coupled Estrogen Receptor Deficiency Accelerates Liver Tumorigenesis by Enhancing Inflammation and Fibrosis. Cancer Lett. 2016, 382, 195–202. [Google Scholar] [CrossRef]
- Fu, W.; Gao, X.-P.; Zhang, S.; Dai, Y.-P.; Zou, W.-J.; Yue, L.-M. 17β-Estradiol Inhibits PCSK9-Mediated LDLR Degradation Through GPER/PLC Activation in HepG2 Cells. Front. Endocrinol. 2020, 10, 930. [Google Scholar] [CrossRef] [Green Version]
- Hussain, Y.; Ding, Q.; Connelly, P.W.; Brunt, J.H.; Ban, M.R.; McIntyre, A.D.; Huff, M.W.; Gros, R.; Hegele, R.A.; Feldman, R.D. G-Protein Estrogen Receptor as a Regulator of Low-Density Lipoprotein Cholesterol Metabolism: Cellular and Population Genetic Studies. Arterioscler. Thromb. Vasc. Biol. 2015, 35, 213–221. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cai, J.; Wu, C.H.; Zhang, Y.; Wang, Y.Y.; Xu, W.D.; Lin, T.C.; Li, S.X.; Wang, L.H.; Zheng, J.; Sun, Y.; et al. High-Free Androgen Index Is Associated with Increased Risk of Non-Alcoholic Fatty Liver Disease in Women with Polycystic Ovary Syndrome, Independent of Obesity and Insulin Resistance. Int. J. Obes. 2017, 41, 1341–1347. [Google Scholar] [CrossRef]
- Kim, S.; Kwon, H.; Park, J.-H.; Cho, B.; Kim, D.; Oh, S.-W.; Lee, C.M.; Choi, H.-C. A Low Level of Serum Total Testosterone Is Independently Associated with Nonalcoholic Fatty Liver Disease. BMC Gastroenterol. 2012, 12, 69. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Magyar, Z.; Bekesi, G.; Racz, K.; Feher, J.; Schaff, Z.; Lengyel, G.; Blazovics, A.; Illyes, G.; Szombath, D.; Hrabak, A.; et al. Increased Total Scavenger Capacity and Decreased Liver Fat Content in Rats Fed Dehydroepiandrosterone and Its Sulphate on a High-Fat Diet. Gerontology 2011, 57, 343–349. [Google Scholar] [CrossRef]
- Sarkar, M.; Wellons, M.; Cedars, M.I.; VanWagner, L.; Gunderson, E.P.; Ajmera, V.; Torchen, L.; Siscovick, D.; Carr, J.J.; Terry, J.G.; et al. Testosterone Levels in Pre-Menopausal Women Are Associated With Nonalcoholic Fatty Liver Disease in Midlife. Am. J. Gastroenterol. 2017, 112, 755–762. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, H.; Liu, Y.; Wang, L.; Li, Z.; Zhang, H.; Wu, J.; Rahman, N.; Guo, Y.; Li, D.; Li, N.; et al. Differential Effects of Estrogen/Androgen on the Prevention of Nonalcoholic Fatty Liver Disease in the Male Rat. J. Lipid Res. 2013, 54, 345–357. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cerda, C.; Pérez-Ayuso, R.M.; Riquelme, A.; Soza, A.; Villaseca, P.; Sir-Petermann, T.; Espinoza, M.; Pizarro, M.; Solis, N.; Miquel, J.F.; et al. Nonalcoholic Fatty Liver Disease in Women with Polycystic Ovary Syndrome. J. Hepatol. 2007, 47, 412–417. [Google Scholar] [CrossRef]
- Paschou, S.A.; Polyzos, S.A.; Anagnostis, P.; Goulis, D.G.; Kanaka-Gantenbein, C.; Lambrinoudaki, I.; Georgopoulos, N.A.; Vryonidou, A. Nonalcoholic Fatty Liver Disease in Women with Polycystic Ovary Syndrome. Endocrine 2020, 67, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Nikolaenko, L.; Jia, Y.; Wang, C.; Diaz-Arjonilla, M.; Yee, J.K.; French, S.W.; Liu, P.Y.; Laurel, S.; Chong, C.; Lee, K.; et al. Testosterone Replacement Ameliorates Nonalcoholic Fatty Liver Disease in Castrated Male Rats. Endocrinology 2014, 155, 417–428. [Google Scholar] [CrossRef] [Green Version]
- Senmaru, T.; Fukui, M.; Okada, H.; Mineoka, Y.; Yamazaki, M.; Tsujikawa, M.; Hasegawa, G.; Kitawaki, J.; Obayashi, H.; Nakamura, N. Testosterone Deficiency Induces Markedly Decreased Serum Triglycerides, Increased Small Dense LDL, and Hepatic Steatosis Mediated by Dysregulation of Lipid Assembly and Secretion in Mice Fed a High-Fat Diet. Metabolism 2013, 62, 851–860. [Google Scholar] [CrossRef]
- Münzker, J.; Hofer, D.; Trummer, C.; Ulbing, M.; Harger, A.; Pieber, T.; Owen, L.; Keevil, B.; Brabant, G.; Lerchbaum, E.; et al. Testosterone to Dihydrotestosterone Ratio as a New Biomarker for an Adverse Metabolic Phenotype in the Polycystic Ovary Syndrome. J. Clin. Endocrinol. Metab. 2015, 100, 653–660. [Google Scholar] [CrossRef] [PubMed]
- Condorelli, R.A.; Calogero, A.E.; Di Mauro, M.; Mongioi’, L.M.; Cannarella, R.; Rosta, G.; La Vignera, S. Androgen Excess and Metabolic Disorders in Women with PCOS: Beyond the Body Mass Index. J. Endocrinol. Investig. 2018, 41, 383–388. [Google Scholar] [CrossRef]
- Petta, S.; Ciresi, A.; Bianco, J.; Geraci, V.; Boemi, R.; Galvano, L.; Magliozzo, F.; Merlino, G.; Craxì, A.; Giordano, C. Insulin Resistance and Hyperandrogenism Drive Steatosis and Fibrosis Risk in Young Females with PCOS. PLoS ONE 2017, 12, e0186136. [Google Scholar] [CrossRef]
- Caldwell, A.S.L.; Middleton, L.J.; Jimenez, M.; Desai, R.; McMahon, A.C.; Allan, C.M.; Handelsman, D.J.; Walters, K.A. Characterization of Reproductive, Metabolic, and Endocrine Features of Polycystic Ovary Syndrome in Female Hyperandrogenic Mouse Models. Endocrinology 2014, 155, 3146–3159. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gur, E.B.; Karadeniz, M.; Turan, G.A. Fetal Programming of Polycystic Ovary Syndrome. World J. Diabetes 2015, 6, 936–942. [Google Scholar] [CrossRef]
- Rocha, A.L.L.; Faria, L.C.; Guimarães, T.C.M.; Moreira, G.V.; Cândido, A.L.; Couto, C.A.; Reis, F.M. Non-Alcoholic Fatty Liver Disease in Women with Polycystic Ovary Syndrome: Systematic Review and Meta-Analysis. J. Endocrinol. Investig. 2017, 40, 1279–1288. [Google Scholar] [CrossRef]
- Salva-Pastor, N.; Chávez-Tapia, N.C.; Uribe, M.; Nuño-Lámbarri, N. Understanding the Association of Polycystic Ovary Syndrome and Non-Alcoholic Fatty Liver Disease. J. Steroid Biochem. Mol. Biol. 2019, 194, 105445. [Google Scholar] [CrossRef]
- Polyzos, S.A.; Kountouras, J.; Tsatsoulis, A.; Zafeiriadou, E.; Katsiki, E.; Patsiaoura, K.; Zavos, C.; Anastasiadou, V.V.; Slavakis, A. Sex Steroids and Sex Hormone-Binding Globulin in Postmenopausal Women with Nonalcoholic Fatty Liver Disease. Hormones 2013, 12, 405–416. [Google Scholar] [CrossRef] [PubMed]
- Qu, X.; Donnelly, R. Sex Hormone-Binding Globulin (SHBG) as an Early Biomarker and Therapeutic Target in Polycystic Ovary Syndrome. Int. J. Mol. Sci. 2020, 21, 8191. [Google Scholar] [CrossRef]
- Wang, X.; Xie, J.; Pang, J.; Zhang, H.; Chen, X.; Lin, J.; Li, Q.; Chen, Q.; Ma, J.; Xu, X.; et al. Serum SHBG Is Associated With the Development and Regression of Nonalcoholic Fatty Liver Disease: A Prospective Study. J. Clin. Endocrinol. Metab. 2020, 105, dgz244. [Google Scholar] [CrossRef] [PubMed]
- Sáez-López, C.; Salcedo-Allende, M.T.; Hernandez, C.; Simó-Servat, O.; Simó, R.; Selva, D.M. Sex Hormone-Binding Globulin Expression Correlates With Acetyl-Coenzyme A Carboxylase and Triglyceride Content in Human Liver. J. Clin. Endocrinol. Metab. 2019, 104, 1500–1507. [Google Scholar] [CrossRef] [PubMed]
- Le, T.N.; Nestler, J.E.; Strauss, J.F.; Wickham, E.P. Sex Hormone-Binding Globulin and Type 2 Diabetes Mellitus. Trends Endocrinol. Metab. 2012, 23, 32–40. [Google Scholar] [CrossRef] [Green Version]
- Winters, S.J.; Gogineni, J.; Karegar, M.; Scoggins, C.; Wunderlich, C.A.; Baumgartner, R.; Ghooray, D.T. Sex Hormone-Binding Globulin Gene Expression and Insulin Resistance. J. Clin. Endocrinol. Metab. 2014, 99, E2780–E2788. [Google Scholar] [CrossRef]
- Saez-Lopez, C.; Barbosa-Desongles, A.; Hernandez, C.; Dyer, R.A.; Innis, S.M.; Simó, R.; Selva, D.M. Sex Hormone-Binding Globulin Reduction in Metabolic Disorders May Play a Role in NAFLD Development. Endocrinology 2017, 158, 545–559. [Google Scholar] [CrossRef] [PubMed]
- Kornicka-Garbowska, K.; Bourebaba, L.; Röcken, M.; Marycz, K. Sex Hormone Binding Globulin (SHBG) Mitigates ER Stress in Hepatocytes In Vitro and Ex Vivo. Cells 2021, 10, 755. [Google Scholar] [CrossRef] [PubMed]
- Jia, Y.; Yee, J.K.; Wang, C.; Nikolaenko, L.; Diaz-Arjonilla, M.; Cohen, J.N.; French, S.W.; Liu, P.Y.; Lue, Y.; Lee, W.-N.P.; et al. Testosterone Protects High-Fat/Low-Carbohydrate Diet-Induced Nonalcoholic Fatty Liver Disease in Castrated Male Rats Mainly via Modulating Endoplasmic Reticulum Stress. Am. J. Physiol. Endocrinol. Metab. 2018, 314, E366–E376. [Google Scholar] [CrossRef] [PubMed]
- Schwinge, D.; Carambia, A.; Quaas, A.; Krech, T.; Wegscheid, C.; Tiegs, G.; Prinz, I.; Lohse, A.W.; Herkel, J.; Schramm, C. Testosterone Suppresses Hepatic Inflammation by the Downregulation of IL-17, CXCL-9, and CXCL-10 in a Mouse Model of Experimental Acute Cholangitis. J. Immunol. 2015, 194, 2522–2530. [Google Scholar] [CrossRef] [PubMed]
- Greco, D.; Kotronen, A.; Westerbacka, J.; Puig, O.; Arkkila, P.; Kiviluoto, T.; Laitinen, S.; Kolak, M.; Fisher, R.M.; Hamsten, A.; et al. Gene Expression in Human NAFLD. Am. J. Physiol. Gastrointest. Liver Physiol. 2008, 294, G1281–G1287. [Google Scholar] [CrossRef] [Green Version]
- Hogg, K.; Wood, C.; McNeilly, A.S.; Duncan, W.C. The in Utero Programming Effect of Increased Maternal Androgens and a Direct Fetal Intervention on Liver and Metabolic Function in Adult Sheep. PLoS ONE 2011, 6, e24877. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lin, H.-Y.; Xu, Q.; Yeh, S.; Wang, R.-S.; Sparks, J.D.; Chang, C. Insulin and Leptin Resistance with Hyperleptinemia in Mice Lacking Androgen Receptor. Diabetes 2005, 54, 1717–1725. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fan, W.; Yanase, T.; Nomura, M.; Okabe, T.; Goto, K.; Sato, T.; Kawano, H.; Kato, S.; Nawata, H. Androgen Receptor Null Male Mice Develop Late-Onset Obesity Caused by Decreased Energy Expenditure and Lipolytic Activity but Show Normal Insulin Sensitivity with High Adiponectin Secretion. Diabetes 2005, 54, 1000–1008. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rana, K.; Fam, B.C.; Clarke, M.V.; Pang, T.P.S.; Zajac, J.D.; MacLean, H.E. Increased Adiposity in DNA Binding-Dependent Androgen Receptor Knockout Male Mice Associated with Decreased Voluntary Activity and Not Insulin Resistance. Am. J. Physiol. Endocrinol. Metab. 2011, 301, E767–E778. [Google Scholar] [CrossRef] [PubMed]
- Derby, C.A.; Zilber, S.; Brambilla, D.; Morales, K.H.; McKinlay, J.B. Body Mass Index, Waist Circumference and Waist to Hip Ratio and Change in Sex Steroid Hormones: The Massachusetts Male Ageing Study. Clin. Endocrinol. 2006, 65, 125–131. [Google Scholar] [CrossRef] [PubMed]
- Giagulli, V.A.; Kaufman, J.M.; Vermeulen, A. Pathogenesis of the Decreased Androgen Levels in Obese Men. J. Clin. Endocrinol. Metab. 1994, 79, 997–1000. [Google Scholar] [CrossRef]
- Mohr, B.A.; Bhasin, S.; Link, C.L.; O’Donnell, A.B.; McKinlay, J.B. The Effect of Changes in Adiposity on Testosterone Levels in Older Men: Longitudinal Results from the Massachusetts Male Aging Study. Eur. J. Endocrinol. 2006, 155, 443–452. [Google Scholar] [CrossRef]
- Nielsen, T.L.; Hagen, C.; Wraae, K.; Brixen, K.; Petersen, P.H.; Haug, E.; Larsen, R.; Andersen, M. Visceral and Subcutaneous Adipose Tissue Assessed by Magnetic Resonance Imaging in Relation to Circulating Androgens, Sex Hormone-Binding Globulin, and Luteinizing Hormone in Young Men. J. Clin. Endocrinol. Metab. 2007, 92, 2696–2705. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Völzke, H.; Aumann, N.; Krebs, A.; Nauck, M.; Steveling, A.; Lerch, M.M.; Rosskopf, D.; Wallaschofski, H. Hepatic Steatosis Is Associated with Low Serum Testosterone and High Serum DHEAS Levels in Men. Int. J. Androl. 2010, 33, 45–53. [Google Scholar] [CrossRef]
- Pellitero, S.; Olaizola, I.; Alastrue, A.; Martínez, E.; Granada, M.L.; Balibrea, J.M.; Moreno, P.; Serra, A.; Navarro-Díaz, M.; Romero, R.; et al. Hypogonadotropic Hypogonadism in Morbidly Obese Males Is Reversed after Bariatric Surgery. Obes. Surg. 2012, 22, 1835–1842. [Google Scholar] [CrossRef] [PubMed]
- Zhu, C.; Zhang, Y.; Zhang, L.; Gao, J.; Mei, F.; Zhu, B.; Lu, L.; Zhou, D.; Qu, S. Changes in Sex Hormones After Laparoscopic Sleeve Gastrectomy in Chinese Obese Men: A 12-Month Follow-Up. Obes. Surg. 2019, 29, 869–877. [Google Scholar] [CrossRef] [PubMed]
- Bekaert, M.; Van Nieuwenhove, Y.; Calders, P.; Cuvelier, C.A.; Batens, A.-H.; Kaufman, J.-M.; Ouwens, D.M.; Ruige, J.B. Determinants of Testosterone Levels in Human Male Obesity. Endocrine 2015, 50, 202–211. [Google Scholar] [CrossRef] [Green Version]
- Michalakis, K.; Mintziori, G.; Kaprara, A.; Tarlatzis, B.C.; Goulis, D.G. The Complex Interaction between Obesity, Metabolic Syndrome and Reproductive Axis: A Narrative Review. Metabolism 2013, 62, 457–478. [Google Scholar] [CrossRef] [PubMed]
- Corbould, A.; Bhathal, P.S.; Dixon, J.B.; O’Brien, P.E. Interrelationships of Serum Androgens, Omental Adipose Tissue Metabolism, and Nonalcoholic Fatty Liver Disease in Obese Premenopausal Women. Metab. Syndr. Relat. Disord. 2014, 12, 311–319. [Google Scholar] [CrossRef]
- Kim, B.-J.; Ahn, S.H.; Lee, S.H.; Hong, S.; Hamrick, M.W.; Isales, C.M.; Koh, J.-M. Lower Hand Grip Strength in Older Adults with Non-Alcoholic Fatty Liver Disease: A Nationwide Population-Based Study. Aging 2019, 11, 4547–4560. [Google Scholar] [CrossRef]
- Wang, Y.-M.; Zhu, K.-F.; Zhou, W.-J.; Zhang, Q.; Deng, D.-F.; Yang, Y.-C.; Lu, W.-W.; Xu, J.; Yang, Y.-M. Sarcopenia Is Associated with the Presence of Nonalcoholic Fatty Liver Disease in Zhejiang Province, China: A Cross-Sectional Observational Study. BMC Geriatr. 2021, 21, 55. [Google Scholar] [CrossRef] [PubMed]
- Kazemi, M.; Jarrett, B.Y.; Parry, S.A.; Thalacker-Mercer, A.E.; Hoeger, K.M.; Spandorfer, S.D.; Lujan, M.E. Osteosarcopenia in Reproductive-Aged Women with Polycystic Ovary Syndrome: A Multicenter Case-Control Study. J. Clin. Endocrinol. Metab. 2020, 105, dgaa426. [Google Scholar] [CrossRef] [PubMed]
- Appiah, D.; Nwabuo, C.C.; Ebong, I.A.; Wellons, M.F.; Winters, S.J. Trends in Age at Natural Menopause and Reproductive Life Span Among US Women, 1959–2018. JAMA 2021, 325, 1328. [Google Scholar] [CrossRef]
- Shadyab, A.H.; Macera, C.A.; Shaffer, R.A.; Jain, S.; Gallo, L.C.; Gass, M.L.S.; Waring, M.E.; Stefanick, M.L.; LaCroix, A.Z. Ages at Menarche and Menopause and Reproductive Lifespan as Predictors of Exceptional Longevity in Women: The Women’s Health Initiative. Menopause 2017, 24, 35–44. [Google Scholar] [CrossRef] [Green Version]
- Chung, L.W.; Raymond, G.; Fox, S. Role of Neonatal Androgen in the Development of Hepatic Microsomal Drug-Metabolizing Enzymes. J. Pharmacol. Exp. Ther. 1975, 193, 621–630. [Google Scholar] [PubMed]
- Collins, J.M.; Wang, D. Co-Expression of Drug Metabolizing Cytochrome P450 Enzymes and Estrogen Receptor Alpha (ESR1) in Human Liver: Racial Differences and the Regulatory Role of ESR1. Drug. Metab. Pers. Ther. 2021, 36, 205–214. [Google Scholar] [CrossRef]
- Wang, D.; Lu, R.; Rempala, G.; Sadee, W. Ligand-Free Estrogen Receptor α (ESR1) as Master Regulator for the Expression of CYP3A4 and Other Cytochrome P450 Enzymes in the Human Liver. Mol. Pharmacol. 2019, 96, 430–440. [Google Scholar] [CrossRef] [PubMed]
- Huang, D.Q.; El-Serag, H.B.; Loomba, R. Global Epidemiology of NAFLD-Related HCC: Trends, Predictions, Risk Factors and Prevention. Nat. Rev. Gastroenterol. Hepatol. 2021, 18, 223–238. [Google Scholar] [CrossRef]
- Paik, J.M.; Golabi, P.; Younossi, Y.; Mishra, A.; Younossi, Z.M. Changes in the Global Burden of Chronic Liver Diseases From 2012 to 2017: The Growing Impact of NAFLD. Hepatology 2020, 72, 1605–1616. [Google Scholar] [CrossRef] [PubMed]
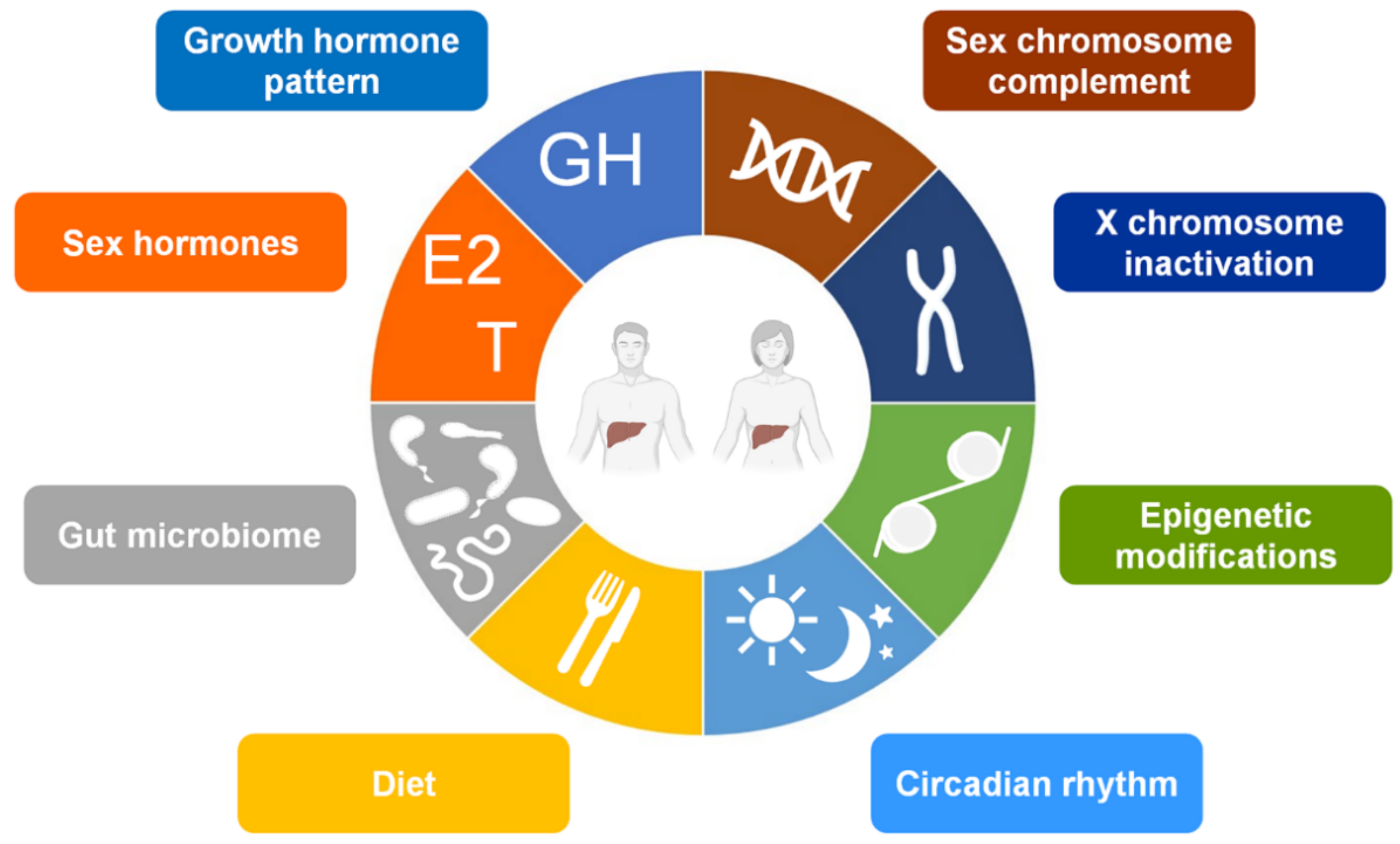
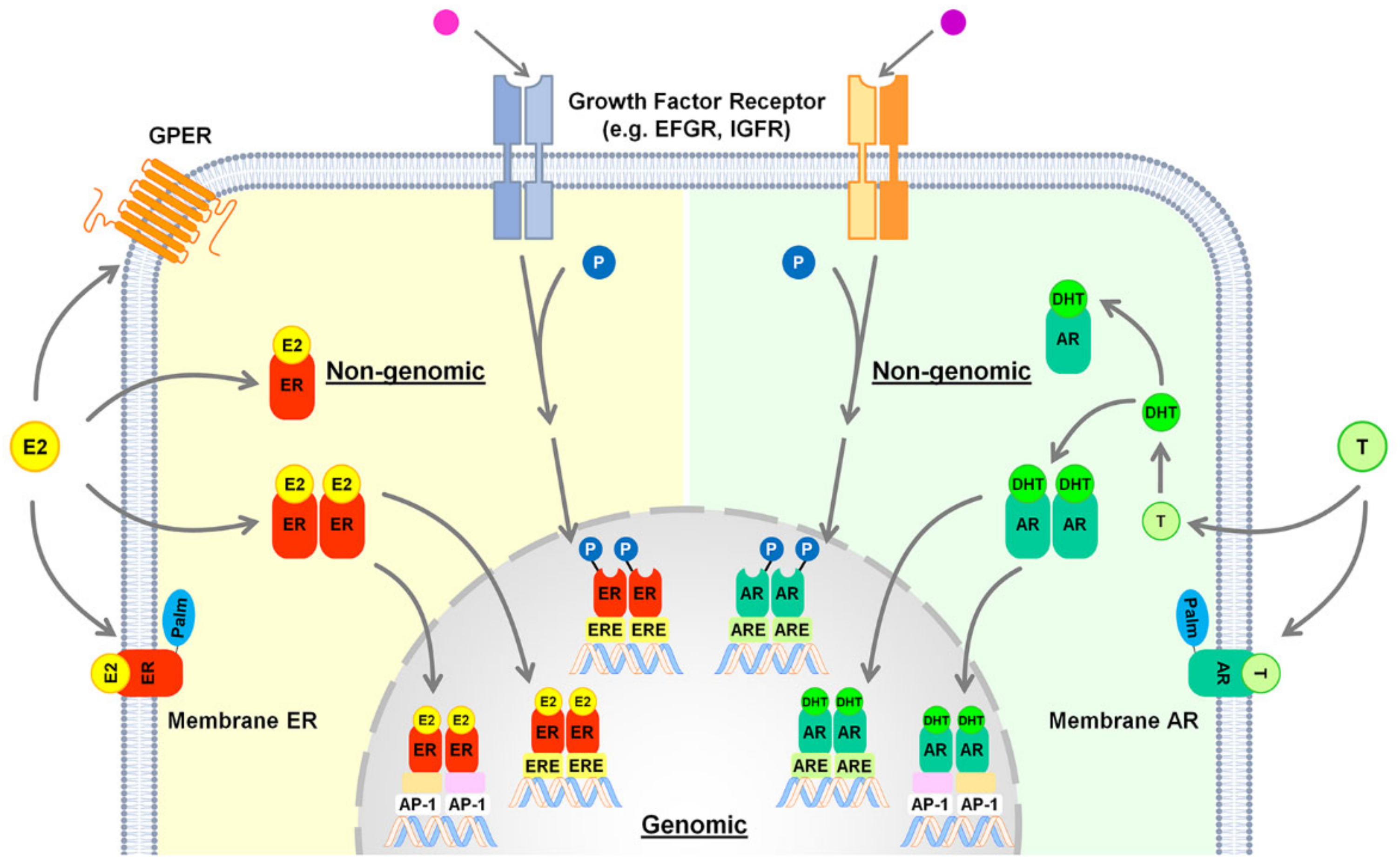

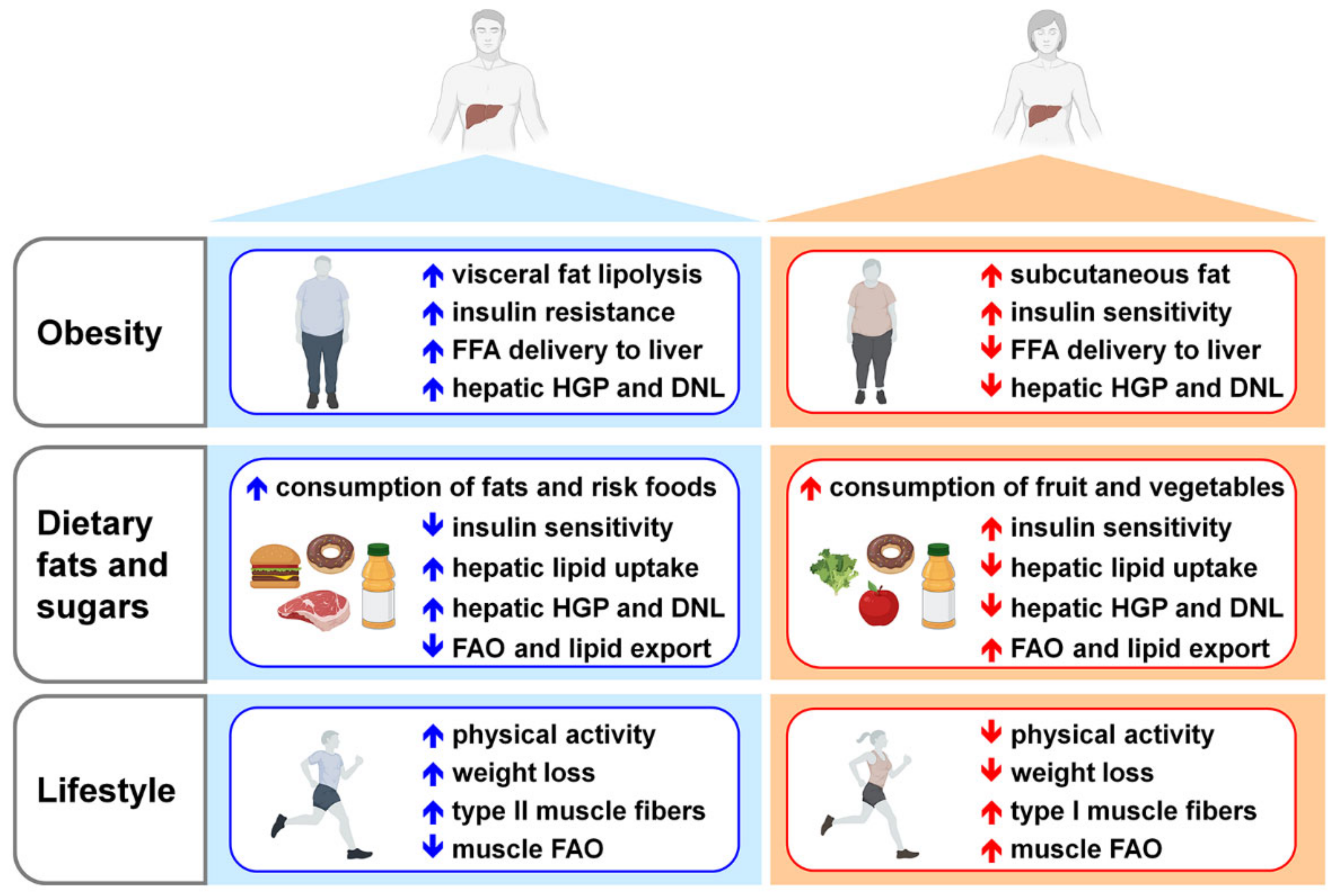

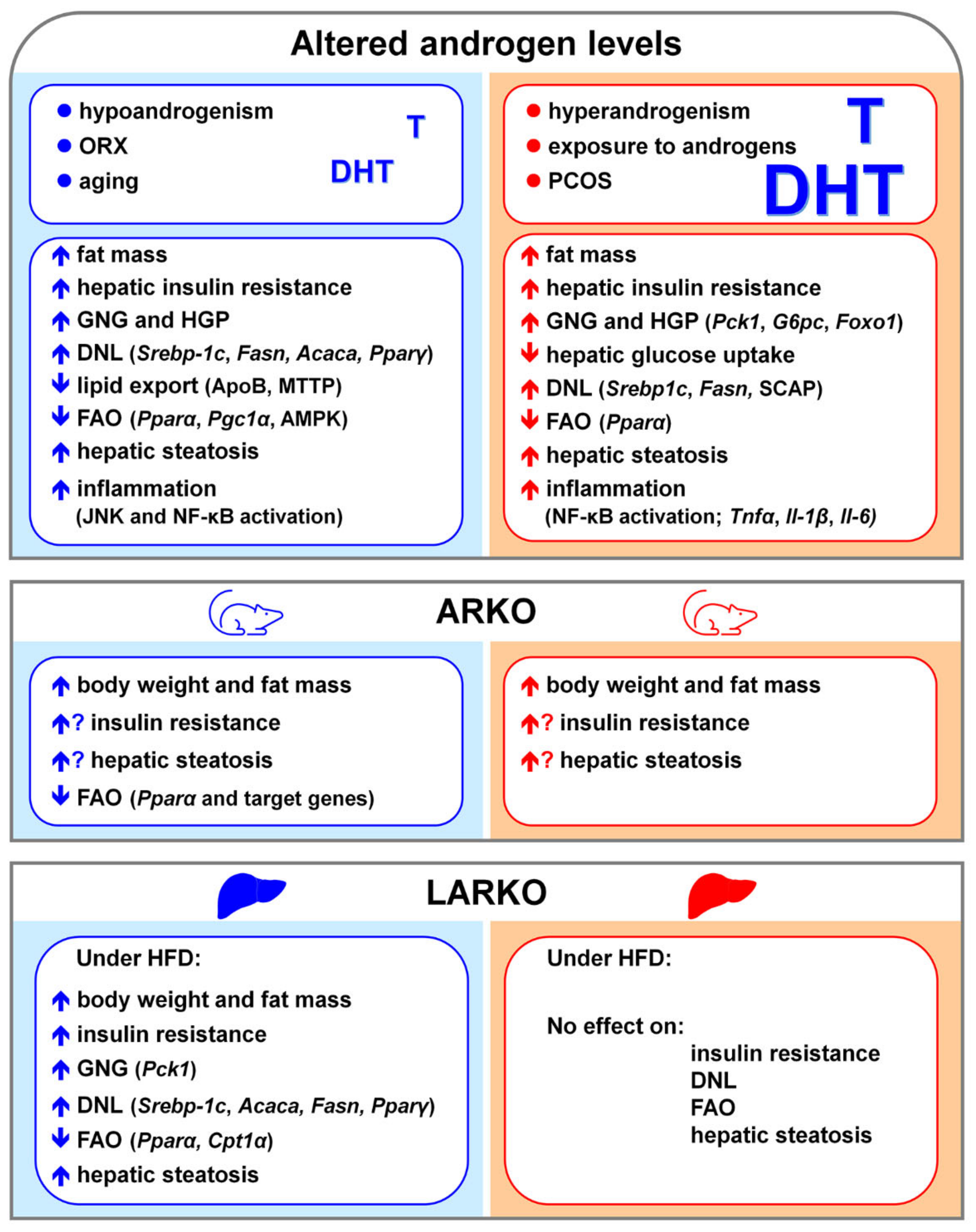

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Della Torre, S. Beyond the X Factor: Relevance of Sex Hormones in NAFLD Pathophysiology. Cells 2021, 10, 2502. https://doi.org/10.3390/cells10092502
Della Torre S. Beyond the X Factor: Relevance of Sex Hormones in NAFLD Pathophysiology. Cells. 2021; 10(9):2502. https://doi.org/10.3390/cells10092502
Chicago/Turabian StyleDella Torre, Sara. 2021. "Beyond the X Factor: Relevance of Sex Hormones in NAFLD Pathophysiology" Cells 10, no. 9: 2502. https://doi.org/10.3390/cells10092502
APA StyleDella Torre, S. (2021). Beyond the X Factor: Relevance of Sex Hormones in NAFLD Pathophysiology. Cells, 10(9), 2502. https://doi.org/10.3390/cells10092502