
Combined Use of Whole Exome Sequencing and CRISPR/Cas9 to Study the Etiology of Non-Obstructive Azoospermia: Demonstration of the Dispensable Role of the Testis-Specific Genes C1orf185 and CCT6B

 , , ,
, , , 
Abstract
:1. Introduction
2. Material and Methods
2.1. Patients and Biological Samples
2.2. WES and Variant Filtering
2.3. Sanger Verification of the Variant
2.4. CRISPR/Cas9—Mediated Mice Genome Edition
2.5. Mice Genotyping Strategy
2.6. Phenotypic Analysis of Mutant Mice
2.7. Statistical Analyses
3. Results
3.1. Medical Assessment of Two Infertile Men Displaying Idiopathic Non-Obstructive Azoospermia
3.2. WES and Variant Filtering
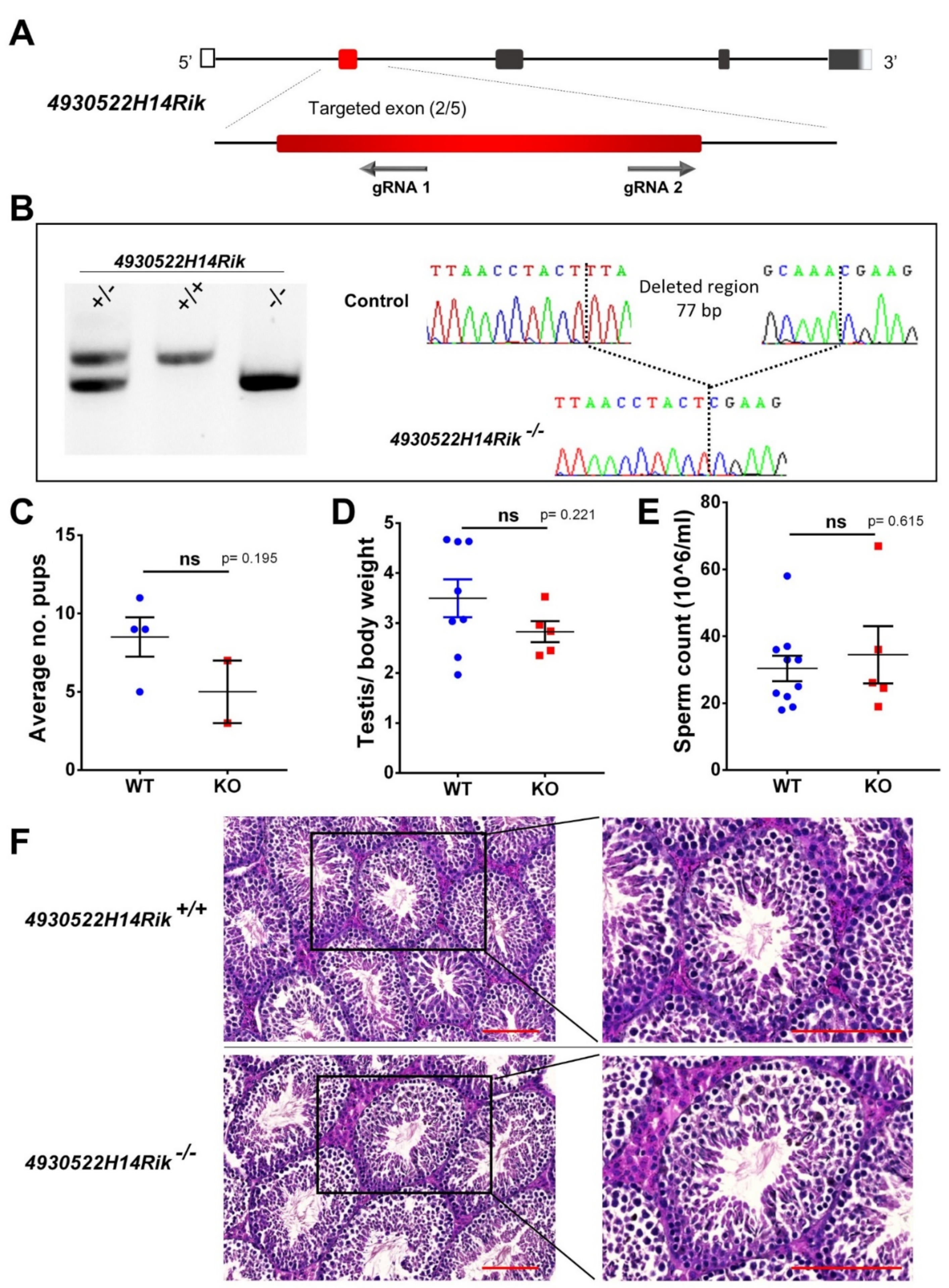
3.3. Generation of KO Mice by CRISPR/Cas9 System
3.4. Phenotypic Analysis of Adult KO Male Mice
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Inhorn, M.C.; Patrizio, P. Infertility around the Globe: New thinking on gender, reproductive technologies and global movements in the 21st Century. Hum. Reprod. Update 2015, 21, 411–426. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Boivin, J.; Bunting, L.; Collins, J.A.; Nygren, K.G. International estimates of infertility prevalence and treatment-seeking: Potential need and demand for infertility medical care. Hum. Reprod. Oxf. Engl. 2007, 22, 1506–1512. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pathak, U.I.; Gabrielsen, J.S.; Lipshultz, L.I. Cutting-edge evaluation of male infertility. Urol. Clin. North Am. 2020, 47, 129–138. [Google Scholar] [CrossRef] [PubMed]
- Fainberg, J.; Kashanian, J.A. Recent advances in understanding and managing male infertility. F1000Research 2019, 8. F1000 Faculty Rev-670. [Google Scholar] [CrossRef]
- Jan, S.Z.; Vormer, T.L.; Jongejan, A.; Röling, M.; Silber, S.J.; de Rooij, D.G.; Hamer, G.; Repping, S.; van Pelt, A.M.M. Unraveling transcriptome dynamics in human spermatogenesis. Dev. Camb. Engl. 2017, 144, 3659–3673. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Krausz, C.; Riera-Escamilla, A. Genetics of male infertility. Nat. Rev. Urol. 2018, 15, 369–384. [Google Scholar] [CrossRef]
- Cioppi, F.; Rosta, V.; Krausz, C. Genetics of azoospermia. Int. J. Mol. Sci. 2021, 22, 3264. [Google Scholar] [CrossRef]
- Boeri, L.; Ventimiglia, E.; Cazzaniga, W.; Pederzoli, F.; Fallara, G.; Pozzi, E.; Belladelli, F.; Baudo, A.; Frego, N.; Capogrosso, P.; et al. Risk of health status worsening in primary infertile men: A prospective 10-year follow-up study. Andrology 2021. [Google Scholar] [CrossRef]
- Petersen, B.-S.; Fredrich, B.; Hoeppner, M.P.; Ellinghaus, D.; Franke, A. Opportunities and challenges of whole-genome and -exome sequencing. BMC Genet. 2017, 18, 14. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, H.; Durbin, R. Fast and accurate long-read alignment with burrows-wheeler transform. Bioinformatics 2010, 26, 589–595. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, S.; Zhou, Y.; Chen, Y.; Gu, J. Fastp: An ultra-fast all-in-one FASTQ preprocessor. Bioinformatics 2018, 34, i884–i890. [Google Scholar] [CrossRef]
- Li, H. Aligning sequence reads, clone sequences and assembly contigs with BWA-MEM. arXiv 2013, arXiv:1303.3997. [Google Scholar]
- Faust, G.G.; Hall, I.M. SAMBLASTER: Fast duplicate marking and structural variant read extraction. Bioinformatics 2014, 30, 2503–2505. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, H.; Handsaker, B.; Wysoker, A.; Fennell, T.; Ruan, J.; Homer, N.; Marth, G.; Abecasis, G.; Durbin, R.; 1000 Genome Project Data Processing Subgroup. The sequence alignment/map format and SAMtools. Bioinformatics 2009, 25, 2078–2079. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, S.; Scheffler, K.; Halpern, A.L.; Bekritsky, M.A.; Noh, E.; Källberg, M.; Chen, X.; Kim, Y.; Beyter, D.; Krusche, P.; et al. Strelka2: Fast and accurate calling of germline and somatic variants. Nat. Methods 2018, 15, 591–594. [Google Scholar] [CrossRef]
- McLaren, W.; Gil, L.; Hunt, S.E.; Riat, H.S.; Ritchie, G.R.S.; Thormann, A.; Flicek, P.; Cunningham, F. The Ensembl variant effect predictor. Genome Biol. 2016, 17, 122. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bouker, A.; Halouani, L.; Kharouf, M.; Latrous, H.; Makni, M.; Marrakchi, O.; Zouari, R.; Fourati, S. Step-by-step loupes-MTESE in non-obstructive azoospermic men, a retrospective study. Basic Clin. Androl. 2019, 29, 11. [Google Scholar] [CrossRef] [Green Version]
- Beurois, J.; Cazin, C.; Kherraf, Z.-E.; Martinez, G.; Celse, T.; Touré, A.; Arnoult, C.; Ray, P.F.; Coutton, C. Genetics of teratozoospermia: Back to the head. Best Pract. Res. Clin. Endocrinol. Metab. 2020, 34, 101473. [Google Scholar] [CrossRef] [PubMed]
- Mitchell, M.J.; Metzler-Guillemain, C.; Toure, A.; Coutton, C.; Arnoult, C.; Ray, P.F. Single gene defects leading to sperm quantitative anomalies. Clin. Genet. 2017, 91, 208–216. [Google Scholar] [CrossRef] [Green Version]
- Touré, A.; Martinez, G.; Kherraf, Z.-E.; Cazin, C.; Beurois, J.; Arnoult, C.; Ray, P.F.; Coutton, C. The genetic architecture of morphological abnormalities of the sperm tail. Hum. Genet. 2021, 140, 21–42. [Google Scholar] [CrossRef]
- Cerván-Martín, M.; Castilla, J.A.; Palomino-Morales, R.J.; Carmona, F.D. Genetic landscape of nonobstructive azoospermia and new perspectives for the clinic. J. Clin. Med. 2020, 9, 300. [Google Scholar] [CrossRef] [Green Version]
- Fakhro, K.A.; Elbardisi, H.; Arafa, M.; Robay, A.; Rodriguez-Flores, J.L.; Al-Shakaki, A.; Syed, N.; Mezey, J.G.; Abi Khalil, C.; Malek, J.A.; et al. Point-of-care whole-exome sequencing of idiopathic male infertility. Genet. Med. Off. J. Am. Coll. Med. Genet. 2018, 20, 1365–1373. [Google Scholar] [CrossRef] [PubMed]
- Chen, S.; Wang, G.; Zheng, X.; Ge, S.; Dai, Y.; Ping, P.; Chen, X.; Liu, G.; Zhang, J.; Yang, Y.; et al. Whole-exome sequencing of a large chinese azoospermia and severe oligospermia cohort identifies novel infertility causative variants and genes. Hum. Mol. Genet. 2020, 29, 2451–2459. [Google Scholar] [CrossRef] [PubMed]
- Krausz, C.; Riera-Escamilla, A.; Moreno-Mendoza, D.; Holleman, K.; Cioppi, F.; Algaba, F.; Pybus, M.; Friedrich, C.; Wyrwoll, M.J.; Casamonti, E.; et al. Genetic dissection of spermatogenic arrest through exome analysis: Clinical implications for the management of azoospermic men. Genet. Med. Off. J. Am. Coll. Med. Genet. 2020, 22, 1956–1966. [Google Scholar] [CrossRef]
- Oud, M.S.; Volozonoka, L.; Smits, R.M.; Vissers, L.E.L.M.; Ramos, L.; Veltman, J.A. A systematic review and standardized clinical validity assessment of male infertility genes. Hum. Reprod. Oxf. Engl. 2019, 34, 932–941. [Google Scholar] [CrossRef] [Green Version]
- Lu, Y.; Oura, S.; Matsumura, T.; Oji, A.; Sakurai, N.; Fujihara, Y.; Shimada, K.; Miyata, H.; Tobita, T.; Noda, T.; et al. CRISPR/Cas9-mediated genome editing reveals 30 testis-enriched genes dispensable for male fertility in mice. Biol. Reprod. 2019, 101, 501–511. [Google Scholar] [CrossRef]
- Schultz, N.; Hamra, F.K.; Garbers, D.L. A multitude of genes expressed solely in meiotic or postmeiotic spermatogenic cells offers a myriad of contraceptive targets. Proc. Natl. Acad. Sci. USA 2003, 100, 12201–12206. [Google Scholar] [CrossRef] [Green Version]
- Coutton, C.; Vargas, A.S.; Amiri-Yekta, A.; Kherraf, Z.-E.; Mustapha, S.F.; Tanno, P.; Wambergue-Legrand, C.; Karaouzène, T.; Martinez, G.; Crouzy, S.; et al. Mutations in CFAP43 and CFAP44 cause male infertility and flagellum defects in trypanosoma and human. Nat. Commun. 2018, 9, 686. [Google Scholar] [CrossRef]
- Kherraf, Z.-E.; Christou-Kent, M.; Karaouzene, T.; Amiri-Yekta, A.; Martinez, G.; Vargas, A.S.; Lambert, E.; Borel, C.; Dorphin, B.; Aknin-Seifer, I.; et al. SPINK2 deficiency causes infertility by inducing sperm defects in heterozygotes and azoospermia in homozygotes. EMBO Mol. Med. 2017, 9, 1132–1149. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pierre, V.; Martinez, G.; Coutton, C.; Delaroche, J.; Yassine, S.; Novella, C.; Pernet-Gallay, K.; Hennebicq, S.; Ray, P.F.; Arnoult, C. Absence of Dpy19l2, a new inner nuclear membrane protein, causes globozoospermia in mice by preventing the anchoring of the acrosome to the nucleus. Dev. Camb. Engl. 2012, 139, 2955–2965. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kherraf, Z.-E.; Conne, B.; Amiri-Yekta, A.; Kent, M.C.; Coutton, C.; Escoffier, J.; Nef, S.; Arnoult, C.; Ray, P.F. Creation of knock out and knock in mice by CRISPR/Cas9 to validate candidate genes for human male infertility, interest, difficulties and feasibility. Mol. Cell. Endocrinol. 2018, 468, 70–80. [Google Scholar] [CrossRef] [PubMed]
- Yang, P.; Tang, W.; Li, H.; Hua, R.; Yuan, Y.; Zhang, Y.; Zhu, Y.; Cui, Y.; Sha, J. T-Complex protein 1 subunit zeta-2 (CCT6B) deficiency induces murine teratospermia. PeerJ 2021, 9, e11545. [Google Scholar] [CrossRef]
- Miyata, H.; Castaneda, J.M.; Fujihara, Y.; Yu, Z.; Archambeault, D.R.; Isotani, A.; Kiyozumi, D.; Kriseman, M.L.; Mashiko, D.; Matsumura, T.; et al. Genome engineering uncovers 54 evolutionarily conserved and testis-enriched genes that are not required for male fertility in mice. Proc. Natl. Acad. Sci. USA 2016, 113, 7704–7710. [Google Scholar] [CrossRef] [Green Version]
- Oyama, Y.; Miyata, H.; Shimada, K.; Fujihara, Y.; Tokuhiro, K.; Garcia, T.X.; Matzuk, M.M.; Ikawa, M. CRISPR/Cas9-mediated genome editing reveals 12 testis-enriched genes dispensable for male fertility in mice. Asian J. Androl. 2021. [Google Scholar] [CrossRef]
- Park, S.; Shimada, K.; Fujihara, Y.; Xu, Z.; Shimada, K.; Larasati, T.; Pratiwi, P.; Matzuk, R.M.; Devlin, D.J.; Yu, Z.; et al. CRISPR/Cas9-mediated genome-edited mice reveal 10 testis-enriched genes are dispensable for male fecundity. Biol. Reprod. 2020, 103, 195–204. [Google Scholar] [CrossRef] [PubMed]
- Sun, J.; Lu, Y.; Nozawa, K.; Xu, Z.; Morohoshi, A.; Castaneda, J.M.; Noda, T.; Miyata, H.; Abbasi, F.; Shawki, H.H.; et al. CRISPR/Cas9-based genome editing in mice uncovers 13 testis- or epididymis-enriched genes individually dispensable for male reproduction. Biol. Reprod. 2020, 103, 183–194. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
{kind=link}
| P0365 | P0280 | |
|---|---|---|
| Age (years) | 43 | 44 |
| Geographical origin | Tunisia | Tunisia |
| Consanguinity | Yes (1st degree) | Yes (1st degree) |
| Testosterone (ng/mL) (N: 2.5–10.6) | 5.52 | 3.45 |
| FSH (UI/l) (N: 1.5–12.4) | 5.73 | 37 |
| Karyotype | 46,XY | 46,XY |
| AZF microdeletions | Negative | Negative |
| Testicular volume (left/right, mL) (N: >15) | 10–15/10–15 | <5/<5 |
| Testicular histology | Severe hypospermatogenesis | Severe hypospermatogenesis associated with seminiferous tubules hyalinization |
| Sperm retrieval | Positive (rare spermatozoa) | Negative |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cazin, C.; Neirijnck, Y.; Loeuillet, C.; Wehrli, L.; Kühne, F.; Lordey, I.; Mustapha, S.F.B.; Bouker, A.; Zouari, R.; Thierry-Mieg, N.; et al. Combined Use of Whole Exome Sequencing and CRISPR/Cas9 to Study the Etiology of Non-Obstructive Azoospermia: Demonstration of the Dispensable Role of the Testis-Specific Genes C1orf185 and CCT6B. Cells 2022, 11, 118. https://doi.org/10.3390/cells11010118
Cazin C, Neirijnck Y, Loeuillet C, Wehrli L, Kühne F, Lordey I, Mustapha SFB, Bouker A, Zouari R, Thierry-Mieg N, et al. Combined Use of Whole Exome Sequencing and CRISPR/Cas9 to Study the Etiology of Non-Obstructive Azoospermia: Demonstration of the Dispensable Role of the Testis-Specific Genes C1orf185 and CCT6B. Cells. 2022; 11(1):118. https://doi.org/10.3390/cells11010118
Chicago/Turabian StyleCazin, Caroline, Yasmine Neirijnck, Corinne Loeuillet, Lydia Wehrli, Françoise Kühne, Isabelle Lordey, Selima Fourati Ben Mustapha, Amin Bouker, Raoudha Zouari, Nicolas Thierry-Mieg, and et al. 2022. "Combined Use of Whole Exome Sequencing and CRISPR/Cas9 to Study the Etiology of Non-Obstructive Azoospermia: Demonstration of the Dispensable Role of the Testis-Specific Genes C1orf185 and CCT6B" Cells 11, no. 1: 118. https://doi.org/10.3390/cells11010118
APA StyleCazin, C., Neirijnck, Y., Loeuillet, C., Wehrli, L., Kühne, F., Lordey, I., Mustapha, S. F. B., Bouker, A., Zouari, R., Thierry-Mieg, N., Nef, S., Arnoult, C., Ray, P. F., & Kherraf, Z.-E. (2022). Combined Use of Whole Exome Sequencing and CRISPR/Cas9 to Study the Etiology of Non-Obstructive Azoospermia: Demonstration of the Dispensable Role of the Testis-Specific Genes C1orf185 and CCT6B. Cells, 11(1), 118. https://doi.org/10.3390/cells11010118