
The Nerve Growth Factor Metabolic Pathway Dysregulation as Cause of Alzheimer’s Cholinergic Atrophy

{kind=link}
{kind=link}
Abstract
1. Introduction
2. NGF as the First Nervous System Trophic Factor
3. Role of NGF in the Maintenance of the Cholinergic Phenotype of Basal Forebrain Neurons and Synapses in the Adult CNS
4. Does a Metabolic NGF Dysregulation Explain the Cholinergic Atrophy in the Alzheimer’s Pathology?
4.1. The BFCN Compromise in AD Pathology
4.2. The NGF Metabolic Cascade
4.3. NGF Dysmetabolism in Down Syndrome
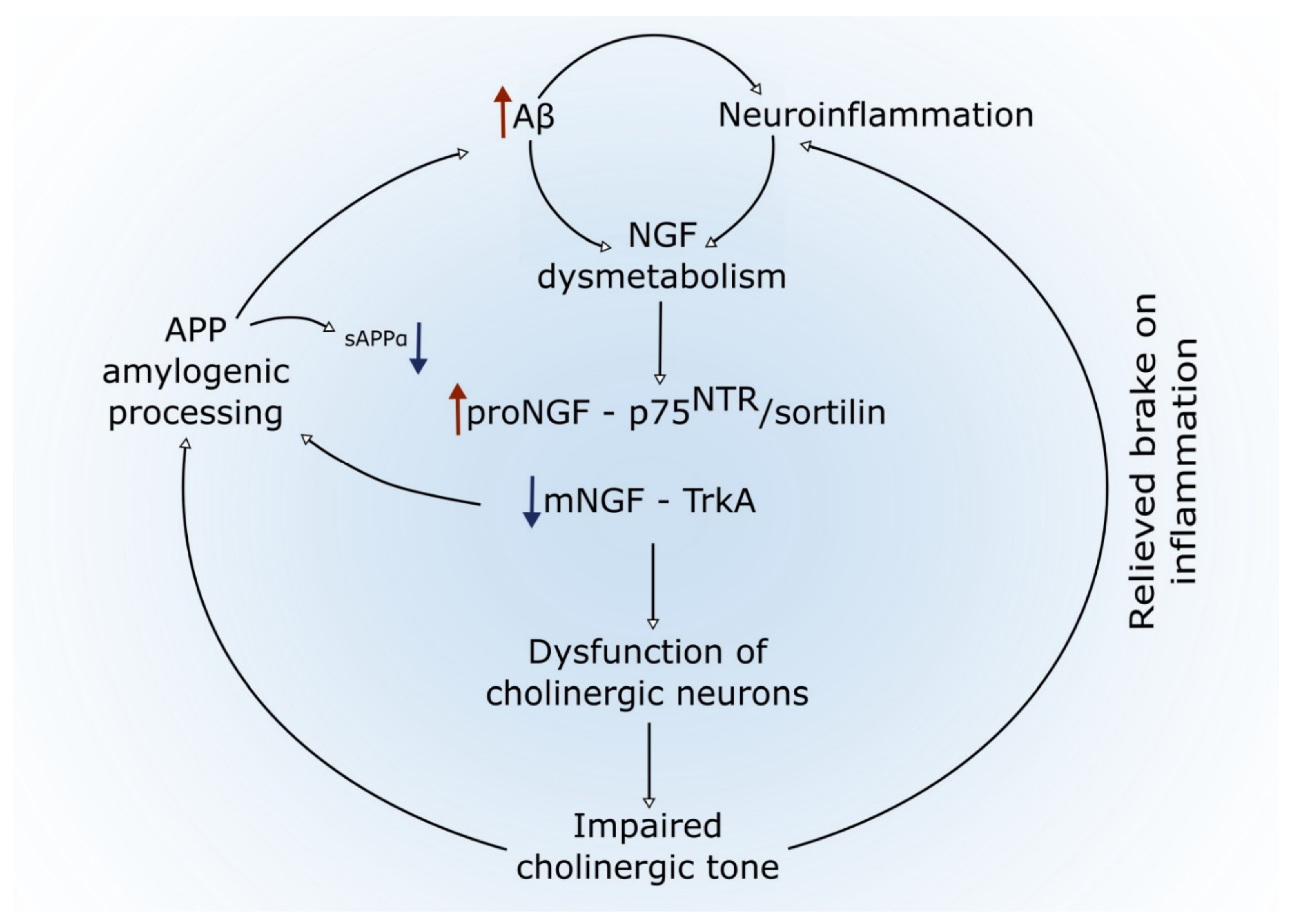
4.4. A Link between Amyloid Pathology, Inflammation, NGF Dysregulation, and Basal Forebrain Cholinergic Atrophy
5. Will the NGF Metabolic Dysregulation Assist the Identification of Preclinical AD Stages?
6. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Hamburger, V. The effects of wing bud extirpation on the development of the central nervous system in chick embryos. J. Exp. Zool. 1934, 68, 449–494. [Google Scholar] [CrossRef]
- Levi, L.-M.a.R.G. Les consequences de la destruction d’un territoire d’innervation peripheique sur le developments des centres nerveux correspondents dans l’embryon de poulet. Arch. Biol. 1942, 53, 537–545. [Google Scholar]
- Bueker, E.D. Implantation of tumors in the hind limb field of the embryonic chick and the developmental response of the lumbosacral nervous system. Anat. Rec. 1948, 102, 369–389. [Google Scholar] [CrossRef] [PubMed]
- Levi-Montalcini, R.; Hamburger, V. Selective growth stimulating effects of mouse sarcoma on the sensory and sympathetic nervous system of the chick embryo. J. Exp. Zool. 1951, 116, 321–361. [Google Scholar] [CrossRef]
- Levi-Montalcini, R.; Meyer, H.; Hamburger, V. In vitro experiments on the effects of mouse sarcomas 180 and 37 on the spinal and sympathetic ganglia of the chick embryo. Cancer Res. 1954, 14, 49–57. [Google Scholar] [PubMed]
- Levi-Montalcini, R.; Hamburger, V. A diffusible agent of mouse sarcoma, producing hyperplasia of sympathetic ganglia and hyperneurotization of viscera in the chick embryo. J. Exp. Zool. 1953, 123, 233–287. [Google Scholar] [CrossRef]
- Cohen, S.; Levi-Montalcini, R.; Hamburger, V. A nerve growth-stimulating factor isolated from sarcom as 37 and 180. Proc. Natl. Acad. Sci. USA 1954, 40, 1014. [Google Scholar] [CrossRef]
- Cohen, S.; Levi-Montalcini, R. A nerve growth-stimulating factor isolated from snake venom. Proc. Natl. Acad. Sci. USA 1956, 42, 571. [Google Scholar] [CrossRef]
- Cohen, S. Purification of a nerve-growth promoting protein from the mouse salivary gland and its neuro-cytotoxic antiserum. Proc. Natl. Acad. Sci. USA 1960, 46, 302. [Google Scholar] [CrossRef]
- Angeletti, R.H.; Bradshaw, R.A. Nerve growth factor from mouse submaxillary gland: Amino acid sequence. Proc. Natl. Acad. Sci. USA 1971, 68, 2417–2420. [Google Scholar] [CrossRef]
- McDonald, N.Q.; Lapatto, R.; Rust, J.M.; Gunning, J.; Wlodawer, A.; Blundell, T.L. New protein fold revealed by a 2.3-Å resolution crystal structure of nerve growth factor. Nature 1991, 354, 411–414. [Google Scholar] [CrossRef]
- Levi-Montalcini, R.; Booker, B. Excessive growth of the sympathetic ganglia evoked by a protein isolated from mouse salivary glands. Proc. Natl. Acad. Sci. USA 1960, 46, 373. [Google Scholar] [CrossRef] [PubMed]
- Levi-Montalcini, R.; Booker, B. Destruction of the sympathetic ganglia in mammals by an antiserum to a nerve-growth protein. Proc. Natl. Acad. Sci. USA 1960, 46, 384–391. [Google Scholar] [CrossRef] [PubMed]
- Levi-Montalcini, R.; Angeletti, P. Noradrenaline and monoaminoxidase content in immunosympathectomized animals. Int. J. Neuropharmacol. 1962, 1, 161–164. [Google Scholar] [CrossRef]
- Sabatini, M.; De Iraldi, A.P.; De Robertis, E. Early effects of antiserum against the nerve growth factor on fine structure of sympathetic neurons. Exp. Neurol. 1965, 12, 370–383. [Google Scholar] [CrossRef]
- Iversen, L.L.; Glowinski, J.; Axelrod, J. Reduced uptake of tritiated noradrenaline in tissues of immunosympathectomized animals. Nature 1965, 206, 1222–1223. [Google Scholar] [CrossRef]
- Levi-Montalcini, R.; Angeletti, P.U.H. Immunosympathectomy. Pharmacol. Rev. 1966, 18, 619–628. [Google Scholar] [PubMed]
- Allen, S.J.; Dawbarn, D. Clinical relevance of the neurotrophins and their receptors. Clin. Sci. 2006, 110, 175–191. [Google Scholar] [CrossRef]
- Teng, K.; Hempstead, B. Neurotrophins and their receptors: Signaling trios in complex biological systems. Cell. Mol. Life Sci. 2004, 61, 35–48. [Google Scholar] [CrossRef]
- Chao, M.V. Neurotrophins and their receptors: A convergence point for many signalling pathways. Nat. Rev. Neurosci. 2003, 4, 299–309. [Google Scholar] [CrossRef]
- Ibáñez, C.F. Structure–function relationships in the neurotrophin family. J. Neurobiol. 1994, 25, 1349–1361. [Google Scholar] [CrossRef]
- Ceni, C.; Unsain, N.; Zeinieh, M.P.; Barker, P.A. Neurotrophins in the regulation of cellular survival and death. Neurotrophic Factors 2014, 220, 193–221. [Google Scholar]
- Honegger, P.; Lenoir, D. Nerve growth factor (NGF) stimulation of cholinergic telencephalic neurons in aggregating cell cultures. Dev. Brain Res. 1982, 3, 229–238. [Google Scholar] [CrossRef]
- Gnahn, H.; Hefti, F.; Heumann, R.; Schwab, M.; Thoenen, H. NGF-mediated increase of choline acetyltransferase (ChAT) in the neonatal rat forebrain: Evidence for a physiological role of NGF in the brain? Dev. Brain Res. 1983, 9, 45–52. [Google Scholar] [CrossRef]
- Hefti, F.; Hartikka, J.; Eckenstein, F.; Gnahn, H.; Heumann, R.; Schwab, M. Nerve growth factor increases choline acetyl-transferase but not survival or fiber outgrowth of cultured fetal septal cholinergic neurons. Neuroscience 1985, 14, 55–68. [Google Scholar] [CrossRef]
- Mobley, W.C.; Rutkowski, J.L.; Tennekoon, G.I.; Buchanan, K.; Johnston, M.V. Choline acetyltransferase activity in striatum of neonatal rats increased by nerve growth factor. Science 1985, 229, 284–287. [Google Scholar] [CrossRef] [PubMed]
- Martinez, H.J.; Dreyfus, C.F.; Jonakait, G.M.; Black, I.B. Nerve growth factor promotes cholinergic development in brain striatal cultures. Proc. Natl. Acad. Sci. USA 1985, 82, 7777–7781. [Google Scholar] [CrossRef]
- Mobley, W.C.; Rutkowski, J.L.; Tennekoon, G.I.; Gemski, J.; Buchanan, K.; Johnston, M.V. Nerve growth factor increases choline acetyltransferase activity in developing basal forebrain neurons. Mol. Brain Res. 1986, 1, 53–62. [Google Scholar] [CrossRef]
- Johnston, M.V.; Rutkowski, J.L.; Wainer, B.H.; Long, J.B.; Mobley, W.C. NGF effects on developing forebrain cholinergic neurons are regionally specific. Neurochem. Res. 1987, 12, 985–994. [Google Scholar] [CrossRef] [PubMed]
- Hatanaka, H.; Tsukui, H. Differential effects of nerve-growth factor and glioma-conditioned medium on neurons cultured from various regions of fetal rat central nervous system. Dev. Brain Res. 1986, 30, 47–56. [Google Scholar] [CrossRef]
- Honegger, P.; du Pasquier, P.; Tenot, M. Cholinergic neurons of fetal rat telencephalon in aggregating cell culture respond to NGF as well as to protein kinase C-activating tumor promoters. Dev. Brain Res. 1986, 29, 217–223. [Google Scholar] [CrossRef]
- Auburger, G.; Heumann, R.; Hellweg, R.; Korsching, S.; Thoenen, H. Developmental changes of nerve growth factor and its mRNA in the rat hippocampus: Comparison with choline acetyltransferase. Dev. Biol. 1987, 120, 322–328. [Google Scholar] [CrossRef]
- Large, T.H.; Bodary, S.C.; Clegg, D.O.; Weskamp, G.; Otten, U.; Reichardt, L.F. Nerve growth factor gene expression in the developing rat brain. Science 1986, 234, 352–355. [Google Scholar] [CrossRef] [PubMed]
- Milner, T.; Loy, R.; Amaral, D.G. An anatomical study of the development of the septo-hippocampal projection in the rat. Dev. Brain Res. 1983, 8, 343–371. [Google Scholar] [CrossRef]
- Whittemore, S.R.; Ebendal, T.; Lärkfors, L.; Olson, L.; Seiger, A.; Strömberg, I.; Persson, H. Development and regional expression of beta nerve growth factor messenger RNA and protein in the rat central nervous system. Proc. Natl. Acad. Sci. USA 1986, 83, 817–821. [Google Scholar] [CrossRef]
- Cuello, A.; Garofalo, L.; Kenigsberg, R.; Maysinger, D. Gangliosides potentiate in vivo and in vitro effects of nerve growth factor on central cholinergic neurons. Proc. Natl. Acad. Sci. USA 1989, 86, 2056–2060. [Google Scholar] [CrossRef]
- Hartikka, J.; Hefti, F. Development of septal cholinergic neurons in culture: Plating density and glial cells modulate effects of NGF on survival, fiber growth, and expression of transmitter-specific enzymes. J. Neurosci. 1988, 8, 2967–2985. [Google Scholar] [CrossRef] [PubMed]
- Hartikka, J.; Hefti, F. Comparison of nerve growth factor’s effects on development of septum, striatum, and nucleus basalis cholinergic neurons in vitro. J. Neurosci. Res. 1988, 21, 352–364. [Google Scholar] [CrossRef]
- Schwab, M.; Otten, U.; Agid, Y.; Thoenen, H. Nerve growth factor (NGF) in the rat CNS: Absence of specific retrograde axonal transport and tyrosine hydroxylase induction in locus coeruleus and substantia nigra. Brain Res. 1979, 168, 473–483. [Google Scholar] [CrossRef]
- Korsching, S.; Auburger, G.; Heumann, R.; Scott, J.; Thoenen, H. Levels of nerve growth factor and its mRNA in the central nervous system of the rat correlate with cholinergic innervation. EMBO J. 1985, 4, 1389–1393. [Google Scholar] [CrossRef]
- Shelton, D.L.; Reichardt, L.F. Studies on the expression of the beta nerve growth factor (NGF) gene in the central nervous system: Level and regional distribution of NGF mRNA suggest that NGF functions as a trophic factor for several distinct populations of neurons. Proc. Natl. Acad. Sci. USA 1986, 83, 2714–2718. [Google Scholar] [CrossRef]
- Whittemore, S.R.; Seiger, Å. The expression, localization and functional significance of β-nerve growth factor in the central nervous system. Brain Res. Rev. 1987, 12, 439–464. [Google Scholar] [CrossRef]
- Hefti, F. Nerve growth factor promotes survival of septal cholinergic neurons after fimbrial transections. J. Neurosci. 1986, 6, 2155–2162. [Google Scholar] [CrossRef]
- Kromer, L.F. Nerve growth factor treatment after brain injury prevents neuronal death. Science 1987, 235, 214–216. [Google Scholar] [CrossRef] [PubMed]
- Williams, L.R.; Varon, S.; Peterson, G.M.; Wictorin, K.; Fischer, W.; Bjorklund, A.; Gage, F.H. Continuous infusion of nerve growth factor prevents basal forebrain neuronal death after fimbria fornix transection. Proc. Natl. Acad. Sci. USA 1986, 83, 9231–9235. [Google Scholar] [CrossRef]
- Sofroniew, M.; Pearson, R.; Eckenstein, F.; Cuello, A.; Powell, T. Retrograde changes in cholinergic neurons in the basal forebrain of the rat following cortical damage. Brain Res. 1983, 289, 370–374. [Google Scholar] [CrossRef]
- Sofroniew, M.V.; Galletly, N.P.; Isacson, O.; Svendsen, C.N. Survival of adult basal forebrain cholinergic neurons after loss of target neurons. Science 1990, 247, 338–342. [Google Scholar] [CrossRef] [PubMed]
- Pearson, R.; Sofroniew, M.; Cuello, A.; Powell, T.; Eckenstein, F.; Esiri, M.; Wilcock, G. Persistence of cholinergic neurons in the basal nucleus in a brain with senile dementia of the Alzheimer’s type demonstrated by immunohistochemical staining for choline acetyltransferase. Brain Res. 1983, 289, 375–379. [Google Scholar] [CrossRef]
- Sofroniew, M.; Cooper, J.; Svendsen, C.; Crossman, P.; Ip, N.; Lindsay, R.; Zafra, F.; Lindholm, D. Atrophy but not death of adult septal cholinergic neurons after ablation of target capacity to produce mRNAs for NGF, BDNF, and NT3. J. Neurosci. 1993, 13, 5263–5276. [Google Scholar] [CrossRef]
- Whitehouse, P.J.; Price, D.L.; Clark, A.W.; Coyle, J.T.; DeLong, M.R. Alzheimer disease: Evidence for selective loss of cholinergic neurons in the nucleus basalis. Ann. Neurol. Off. J. Am. Neurol. Assoc. Child Neurol. Soc. 1981, 10, 122–126. [Google Scholar] [CrossRef] [PubMed]
- Arendt, T.; Bigl, V.; Arendt, A.; Tennstedt, A. Loss of neurons in the nucleus basalis of Meynert in Alzheimer’s disease, paralysis agitans and Korsakoff’s disease. Acta Neuropathol. 1983, 61, 101–108. [Google Scholar] [CrossRef]
- Pearson, R.; Gatter, K.; Powell, T.P. Retrograde cell degeneration in the basal nucleus in monkey and man. Brain Res. 1983, 261, 321–326. [Google Scholar] [CrossRef]
- Chen, K.S.; Nishimura, M.C.; Armanini, M.P.; Crowley, C.; Spencer, S.D.; Phillips, H.S. Disruption of a single allele of the nerve growth factor gene results in atrophy of basal forebrain cholinergic neurons and memory deficits. J. Neurosci. 1997, 17, 7288–7296. [Google Scholar] [CrossRef]
- Cuello, A.C. Effects of trophic factors on the CNS cholinergic phenotype. Prog. Brain Res. 1996, 109, 347–358. [Google Scholar]
- Cuello, A.C.; Pentz, R.; Hall, H. The brain NGF metabolic pathway in health and in Alzheimer’s pathology. Front. Neurosci. 2019, 13, 62. [Google Scholar] [CrossRef] [PubMed]
- Cuello, A.C. Trophic responses of forebrain cholinergic neurons: A discussion. Prog. Brain Res. 1993, 98, 265–277. [Google Scholar] [PubMed]
- Debeir, T.; Saragovi, H.U.; Cuello, A.C. A nerve growth factor mimetic TrkA antagonist causes withdrawal of cortical cholinergic boutons in the adult rat. Proc. Natl. Acad. Sci. USA 1999, 96, 4067–4072. [Google Scholar] [CrossRef]
- Venero, J.; Knüsel, B.; Beck, K.; Hefti, F. Expression of neurotrophin and trk receptor genes in adult rats with fimbria transections: Effect of intraventricular nerve growth factor and brain-derived neurotrophic factor administration. Neuroscience 1994, 59, 797–815. [Google Scholar] [CrossRef]
- Figueiredo, B.; Skup, M.; Bedard, A.; Tetzlaff, W.; Cuello, A. Differential expression of p140trk, p75NGFR and growth-associated phosphoprotein-43 genes in nucleus basalis magnocellularis, thalamus and adjacent cortex following neocortical infarction and nerve growth factor treatment. Neuroscience 1995, 68, 29–45. [Google Scholar] [CrossRef]
- Gibbs, R.B.; Pfaff, D. In situ hybridization detection of trkA mRNA in brain: Distribution, colocalization with p75NGFR and up-regulation by nerve growth factor. J. Comp. Neurol. 1994, 341, 324–339. [Google Scholar] [CrossRef]
- Higgins, G.A.; Koh, S.; Chen, K.S.; Gage, F.H. NGF induction of NGF receptor gene expression and cholinergic neuronal hypertrophy within the basal forebrain of the adult rat. Neuron 1989, 3, 247–256. [Google Scholar] [CrossRef]
- Holtzman, D.M.; Li, Y.; Parada, L.F.; Kinsman, S.; Chen, C.-K.; Valletta, J.S.; Zhou, J.; Long, J.B.; Mobley, W.C. p140trk mRNA marks NGF-responsive forebrain neurons: Evidence that trk gene expression is induced by NGF. Neuron 1992, 9, 465–478. [Google Scholar] [CrossRef]
- Angelastro, J.M.; Klimaschewski, L.; Tang, S.; Vitolo, O.V.; Weissman, T.A.; Donlin, L.T.; Shelanski, M.L.; Greene, L.A. Identification of diverse nerve growth factor-regulated genes by serial analysis of gene expression (SAGE) profiling. Proc. Natl. Acad. Sci. USA 2000, 97, 10424–10429. [Google Scholar] [CrossRef] [PubMed]
- Angelastro, J.M.; Töröcsik, B.; Greene, L.A. Nerve growth factor selectively regulates expression of transcripts encoding ribosomal proteins. BMC Neurosci. 2002, 3, 3. [Google Scholar] [CrossRef] [PubMed]
- D’Onofrio, M.; Paoletti, F.; Arisi, I.; Brandi, R.; Malerba, F.; Fasulo, L.; Cattaneo, A. NGF and proNGF regulate functionally distinct mRNAs in PC12 cells: An early gene expression profiling. PLoS ONE 2011, 6, e20839. [Google Scholar] [CrossRef] [PubMed]
- Groot, M.; Boxer, L.M.; Thiel, G. Nerve growth factor-and epidermal growth factor-regulated gene transcription in PC12 pheochromocytoma and INS-1 insulinoma cells. Eur. J. Cell Biol. 2000, 79, 924–935. [Google Scholar] [CrossRef]
- Heese, K.; Nagai, Y.; Sawada, T. Nerve growth factor (NGF) induces mRNA expression of the new transcription factor protein p48ZnF. Exp. Mol. Med. 2004, 36, 130–134. [Google Scholar] [CrossRef] [PubMed]
- Dijkmans, T.F.; van Hooijdonk, L.W.; Schouten, T.G.; Kamphorst, J.T.; Vellinga, A.C.; Meerman, J.H.; Fitzsimons, C.P.; de Kloet, E.R.; Vreugdenhil, E. Temporal and functional dynamics of the transcriptome during nerve growth factor-induced differentiation. J. Neurochem. 2008, 105, 2388–2403. [Google Scholar] [CrossRef]
- Hefti, F.; Weiner, W.J. Nerve growth factor and Alzheimer’s disease. Ann. Neurol. Off. J. Am. Neurol. Assoc. Child Neurol. Soc. 1986, 20, 275–281. [Google Scholar] [CrossRef]
- Koliatsos, V.E.; Price, D.L.; Clatterbuck, R.E.; Markowska, A.L.; Olton, D.S.; Wilcox, B.J. Neurotrophic Strategies for Treating Alzheimer’s Disease: Lessons from Basic Neurobiology and Animal Models a. Ann. Acad. Sci. 1993, 695, 292–299. [Google Scholar] [CrossRef]
- Varon, S.; Hagg, T.; Fass, B.; Vahlsing, L.; Manthorpe, M. Neuronotrophic factors in cellular functional and cognitive repair of adult brain. Pharmacopsychiatry 1989, 22, 120–124. [Google Scholar] [CrossRef] [PubMed]
- Tuszyfiski, M.H.; Fred, H. Potential use of neurotrophic agents in the treatment of neurodegenerative disorders. Acta Neurobiol. Exp. 1990, 50, 311–322. [Google Scholar]
- Cuello, A.C. Rita Levi-Montalcini, NGF Metabolism in Health and in the Alzheimer’s Pathology. In Recent Advances in NGF and Related Molecules; Springer: Berlin/Heidelberg, Germany, 2021; pp. 119–144. [Google Scholar]
- Tuszynski, M.H. Growth-factor gene therapy for neurodegenerative disorders. Lancet Neurol. 2002, 1, 51–57. [Google Scholar] [CrossRef]
- Mitra, S.; Gera, R.; Linderoth, B.; Lind, G.; Wahlberg, L.; Almqvist, P.; Behbahani, H.; Eriksdotter, M. A Review of Techniques for Biodelivery of Nerve Growth Factor (NGF) to the Brain in Relation to Alzheimer’s Disease. Recent Adv. NGF Relat. Mol. 2021, 167–191. [Google Scholar]
- Mitra, S.; Behbahani, H.; Eriksdotter, M. Innovative therapy for Alzheimer’s disease-with focus on biodelivery of NGF. Front. Neurosci. 2019, 13, 38. [Google Scholar] [CrossRef]
- Hebb, D.O. Organization of Behavior; Wiley: New York, NY, USA, 1949. [Google Scholar]
- Drachman, D.A.; Leavitt, J. Human memory and the cholinergic system: A relationship to aging? Arch. Neurol. 1974, 30, 113–121. [Google Scholar] [CrossRef]
- Bowen, D.M.; Smith, C.B.; White, P.; Davison, A.N. Neurotransmitter-related enzymes and indices of hypoxia in senile dementia and other abiotrophies. Brain J. Neurol. 1976, 99, 459–496. [Google Scholar] [CrossRef]
- Davies, P.; Maloney, A. Selective loss of central cholinergic neurons in Alzheimer’s disease. Lancet 1976, 308, 1403. [Google Scholar] [CrossRef]
- Bartus, R.T.; Dean, R.r.; Beer, B.; Lippa, A.S. The cholinergic hypothesis of geriatric memory dysfunction. Science 1982, 217, 408–414. [Google Scholar] [CrossRef]
- Whitehouse, P.J.; Price, D.L.; Struble, R.G.; Clark, A.W.; Coyle, J.T.; Delon, M.R. Alzheimer’s disease and senile dementia: Loss of neurons in the basal forebrain. Science 1982, 215, 1237–1239. [Google Scholar] [CrossRef]
- Coyle, J.T.; Price, D.L.; Delong, M.R. Alzheimer’s disease: A disorder of cortical cholinergic innervation. Science 1983, 219, 1184–1190. [Google Scholar] [CrossRef] [PubMed]
- Hampel, H.; Mesulam, M.-M.; Cuello, A.C.; Farlow, M.R.; Giacobini, E.; Grossberg, G.T.; Khachaturian, A.S.; Vergallo, A.; Cavedo, E.; Snyder, P.J. The cholinergic system in the pathophysiology and treatment of Alzheimer’s disease. Brain 2018, 141, 1917–1933. [Google Scholar] [CrossRef] [PubMed]
- Kabir, M.T.; Uddin, M.; Begum, M.; Thangapandiyan, S.; Rahman, M.; Aleya, L.; Mathew, B.; Ahmed, M.; Barreto, G.E.; Ashraf, G.M. Cholinesterase inhibitors for Alzheimer’s disease: Multitargeting strategy based on anti-Alzheimer’s drugs repositioning. Curr. Pharm. Des. 2019, 25, 3519–3535. [Google Scholar] [CrossRef] [PubMed]
- Marucci, G.; Buccioni, M.; Dal Ben, D.; Lambertucci, C.; Volpini, R.; Amenta, F. Efficacy of acetylcholinesterase inhibitors in Alzheimer’s disease. Neuropharmacology 2020, 108352. [Google Scholar] [CrossRef]
- Schmitz, T.; Nathan Spreng, R. Alzheimer’s Disease Neuroimaging Initiative Basal forebrain degeneration precedes and predicts the cortical spread of Alzheimer’s pathology. Nat. Commun. 2016, 7, 13249. [Google Scholar] [CrossRef]
- Schmitz, T.W.; Mur, M.; Aghourian, M.; Bedard, M.-A.; Spreng, R.N.; Initiative, A.s.D.N. Longitudinal Alzheimer’s degeneration reflects the spatial topography of cholinergic basal forebrain projections. Cell Rep. 2018, 24, 38–46. [Google Scholar] [CrossRef]
- Herdick, M.; Dyrba, M.; Fritz, H.-C.J.; Altenstein, S.; Ballarini, T.; Brosseron, F.; Buerger, K.; Cetindag, A.C.; Dechent, P.; Dobisch, L. Multimodal MRI analysis of basal forebrain structure and function across the Alzheimer’s disease spectrum. NeuroImage Clin. 2020, 28, 102495. [Google Scholar] [CrossRef]
- Hall, A.M.; Moore, R.Y.; Lopez, O.L.; Kuller, L.; Becker, J.T. Basal forebrain atrophy is a presymptomatic marker for Alzheimer’s disease. Alzheimer’s Dement. 2008, 4, 271–279. [Google Scholar] [CrossRef]
- Bruno, M.A.; Cuello, A.C. Activity-dependent release of precursor nerve growth factor, conversion to mature nerve growth factor, and its degradation by a protease cascade. Proc. Natl. Acad. Sci. USA 2006, 103, 6735–6740. [Google Scholar] [CrossRef]
- Goedert, M.; Fine, A.; Hunt, S.; Ullrich, A. Nerve growth factor mRNA in peripheral and central rat tissues and in the human central nervous system: Lesion effects in the rat brain and levels in Alzheimer’s disease. Mol. Brain Res. 1986, 1, 85–92. [Google Scholar] [CrossRef]
- Jette, N.; Cole, M.; Fahnestock, M. NGF mRNA is not decreased in frontal cortex from Alzheimer’s disease patients. Mol. Brain Res. 1994, 25, 242–250. [Google Scholar] [CrossRef]
- Fahnestock, M.; Scott, S.A.; Jetté, N.; Weingartner, J.A.; Crutcher, K.A. Nerve growth factor mRNA and protein levels measured in the same tissue from normal and Alzheimer’s disease parietal cortex. Mol. Brain Res. 1996, 42, 175–178. [Google Scholar] [CrossRef]
- Fahnestock, M.; Yu, G.; Michalski, B.; Mathew, S.; Colquhoun, A.; Ross, G.M.; Coughlin, M.D. The nerve growth factor precursor proNGF exhibits neurotrophic activity but is less active than mature nerve growth factor. J. Neurochem. 2004, 89, 581–592. [Google Scholar] [CrossRef]
- Peng, S.; Wuu, J.; Mufson, E.J.; Fahnestock, M. Increased proNGF levels in subjects with mild cognitive impairment and mild Alzheimer disease. J. Neuropathol. Exp. Neurol. 2004, 63, 641–649. [Google Scholar] [CrossRef]
- Pedraza, C.E.; Podlesniy, P.; Vidal, N.; Arévalo, J.C.; Lee, R.; Hempstead, B.; Ferrer, I.; Iglesias, M.; Espinet, C. Pro-NGF isolated from the human brain affected by Alzheimer’s disease induces neuronal apoptosis mediated by p75NTR. Am. J. Pathol. 2005, 166, 533–543. [Google Scholar] [CrossRef]
- Bruno, M.A.; Leon, W.C.; Fragoso, G.; Mushynski, W.E.; Almazan, G.; Cuello, A.C. Amyloid β-induced nerve growth factor dysmetabolism in Alzheimer disease. J. Neuropathol. Exp. Neurol. 2009, 68, 857–869. [Google Scholar] [CrossRef] [PubMed]
- Al-Shawi, R.; Hafner, A.; Olson, J.; Chun, S.; Raza, S.; Thrasivoulou, C.; Lovestone, S.; Killick, R.; Simons, P.; Cowen, T. Neurotoxic and neurotrophic roles of proNGF and the receptor sortilin in the adult and ageing nervous system. Eur. J. Neurosci. 2008, 27, 2103–2114. [Google Scholar] [CrossRef] [PubMed]
- Mufson, E.J.; He, B.; Nadeem, M.; Perez, S.E.; Counts, S.E.; Leurgans, S.; Fritz, J.; Lah, J.; Ginsberg, S.D.; Wuu, J. Hippocampal proNGF signaling pathways and β-amyloid levels in mild cognitive impairment and Alzheimer disease. J. Neuropathol. Exp. Neurol. 2012, 71, 1018–1029. [Google Scholar] [CrossRef] [PubMed]
- Pentz, R.; Iulita, M.F.; Mikutra-Cencora, M.; Ducatenzeiler, A.; Bennett, D.A.; Cuello, A.C. A new role for matrix metalloproteinase-3 in the NGF metabolic pathway: Proteolysis of mature NGF and sex-specific differences in the continuum of Alzheimer’s pathology. Neurobiol. Dis. 2021, 148, 105150. [Google Scholar] [CrossRef] [PubMed]
- Allard, S.; Leon, W.C.; Pakavathkumar, P.; Bruno, M.A.; Ribeiro-da-Silva, A.; Cuello, A.C. Impact of the NGF maturation and degradation pathway on the cortical cholinergic system phenotype. J. Neurosci. 2012, 32, 2002–2012. [Google Scholar] [CrossRef] [PubMed]
- Allard, S.; Jacobs, M.L.; Do Carmo, S.; Cuello, A.C. Compromise of cortical proNGF maturation causes selective retrograde atrophy in cholinergic nucleus basalis neurons. Neurobiol. Aging 2018, 67, 10–20. [Google Scholar] [CrossRef] [PubMed]
- Bruno, M.A.; Mufson, E.J.; Wuu, J.; Cuello, A.C. Increased matrix metalloproteinase 9 activity in mild cognitive impairment. J. Neuropathol. Exp. Neurol. 2009, 68, 1309–1318. [Google Scholar] [CrossRef] [PubMed]
- Hanzel, C.E.; Iulita, M.F.; Eyjolfsdottir, H.; Hjorth, E.; Schultzberg, M.; Eriksdotter, M.; Cuello, A.C. Analysis of matrix metallo-proteases and the plasminogen system in mild cognitive impairment and Alzheimer’s disease cerebrospinal fluid. J. Alzheimer’s Dis. 2014, 40, 667–678. [Google Scholar] [CrossRef]
- Pentz, R.; Iulita, M.F.; Ducatenzeiler, A.; Bennett, D.A.; Cuello, A.C. The human brain NGF metabolic pathway is impaired in the pre-clinical and clinical continuum of Alzheimers disease. Mol. Psychiatry 2020. [Google Scholar] [CrossRef] [PubMed]
- Iulita, M.F.; Cuello, A.C. Nerve growth factor metabolic dysfunction in Alzheimer’s disease and Down syndrome. Trends Pharmacol. Sci. 2014, 35, 338–348. [Google Scholar] [CrossRef] [PubMed]
- Iulita, M.F.; Do Carmo, S.; Ower, A.K.; Fortress, A.M.; Aguilar, L.F.; Hanna, M.; Wisniewski, T.; Granholm, A.-C.; Buhusi, M.; Busciglio, J. Nerve growth factor metabolic dysfunction in Down’s syndrome brains. Brain 2014, 137, 860–872. [Google Scholar] [CrossRef] [PubMed]
- Cuello, A.C.; Bruno, M.A.; Allard, S.; Leon, W.; Iulita, M.F. Cholinergic involvement in Alzheimer’s disease. A link with NGF maturation and degradation. J. Mol. Neurosci. 2010, 40, 230–235. [Google Scholar] [CrossRef]
- Pentz, R.; Iulita, M.F.; Ducatenzeiler, A.; Videla, L.; Benejam, B.; Iragui, M.C.; Blesa, R.; Lleó, A.; Fortea, J.; Cuello, A.C. Nerve growth factor (NGF) pathway biomarkers in Down syndrome prior to and after the onset of clinical Alzheimer’s disease: A paired CSF and plasma study. Alzheimer’s Dement. 2021, 17, 605–617. [Google Scholar] [CrossRef]
- Iulita, M.F.; Ower, A.; Barone, C.; Pentz, R.; Gubert, P.; Romano, C.; Cantarella, R.A.; Elia, F.; Buono, S.; Recupero, M. An inflammatory and trophic disconnect biomarker profile revealed in Down syndrome plasma: Relation to cognitive decline and longitudinal evaluation. Alzheimer’s Dement. 2016, 12, 1132–1148. [Google Scholar] [CrossRef]
- Florencia Iulita, M.; Claudio Cuello, A. The NGF metabolic pathway in the CNS and its dysregulation in Down syndrome and Alzheimer’s disease. Curr. Alzheimer Res. 2016, 13, 53–67. [Google Scholar] [CrossRef]
- Florencia Iulita, M.; Caraci, F.; Claudio Cuello, A. A link between nerve growth factor metabolic deregulation and amyloid-β-driven inflammation in down syndrome. CNS Neurol. Disord. Drug Targets 2016, 15, 434–447. [Google Scholar] [CrossRef] [PubMed]
- Caraci, F.; Iulita, M.F.; Pentz, R.; Aguilar, L.F.; Orciani, C.; Barone, C.; Romano, C.; Drago, F.; Cuello, A.C. Searching for new pharmacological targets for the treatment of Alzheimer’s disease in Down syndrome. Eur. J. Pharmacol. 2017, 817, 7–19. [Google Scholar] [CrossRef] [PubMed]
- Lott, I.T.; Lai, F. Dementia in Down’s syndrome: Observations from a neurology clinic. Appl. Res. Ment. Retard. 1982, 3, 233–239. [Google Scholar] [CrossRef]
- Zigman, W.B.; Lott, I.T. Alzheimer’s disease in Down syndrome: Neurobiology and risk. Ment. Retard. Dev. Disabil. Res. Rev. 2007, 13, 237–246. [Google Scholar] [CrossRef]
- Davidson, Y.S.; Robinson, A.; Prasher, V.P.; Mann, D.M. The age of onset and evolution of Braak tangle stage and Thal amyloid pathology of Alzheimer’s disease in individuals with Down syndrome. Acta Neuropathol. Commun. 2018, 6, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Strydom, A.; Coppus, A.; Blesa, R.; Danek, A.; Fortea, J.; Hardy, J.; Levin, J.; Nuebling, G.; Rebillat, A.-S.; Ritchie, C. Alzheimer’s disease in Down syndrome: An overlooked population for prevention trials. Alzheimer’s Dement. Transl. Res. Clin. Interv. 2018, 4, 703–713. [Google Scholar] [CrossRef]
- Head, E.; Lott, I.T. Down syndrome and beta-amyloid deposition. Curr. Opin. Neurol. 2004, 17, 95–100. [Google Scholar] [CrossRef] [PubMed]
- Lott, I.T.; Head, E. Dementia in Down syndrome: Unique insights for Alzheimer disease research. Nat. Rev. Neurol. 2019, 15, 135–147. [Google Scholar] [CrossRef]
- Head, E.; Silverman, W.; Patterson, D.; Lott, I.T. Aging and Down Syndrome; Hindawi: London, UK, 2012. [Google Scholar]
- Handen, B.L.; Lott, I.T.; Christian, B.T.; Schupf, N.; OBryant, S.; Mapstone, M.; Fagan, A.M.; Lee, J.H.; Tudorascu, D.; Wang, M.C. The Alzheimer’s Biomarker Consortium-Down Syndrome: Rationale and methodology. Alzheimer’s Dement. Diagn. Assess. Dis. Monit. 2020, 12, e12065. [Google Scholar] [CrossRef]
- Fortea, J.; Carmona-Iragui, M.; Benejam, B.; Fernández, S.; Videla, L.; Barroeta, I.; Alcolea, D.; Pegueroles, J.; Muñoz, L.; Belbin, O. Plasma and CSF biomarkers for the diagnosis of Alzheimer’s disease in adults with Down syndrome: A cross-sectional study. Lancet Neurol. 2018, 17, 860–869. [Google Scholar] [CrossRef]
- Fortea, J.; Vilaplana, E.; Carmona-Iragui, M.; Benejam, B.; Videla, L.; Barroeta, I.; Fernández, S.; Altuna, M.; Pegueroles, J.; Montal, V. Clinical and biomarker changes of Alzheimer’s disease in adults with Down syndrome: A cross-sectional study. Lancet 2020, 395, 1988–1997. [Google Scholar] [CrossRef]
- Yates, C.; Simpson, J.; Gordon, A.; Maloney, A.; Allison, Y.; Ritchie, I.; Urquhart, A. Catecholamines and cholinergic enzymes in pre-senile and senile Alzheimer-type dementia and Down’s syndrome. Brain Res. 1983, 280, 119–126. [Google Scholar] [CrossRef]
- Kish, S.; Karlinsky, H.; Becker, L.; Gilbert, J.; Rebbetoy, M.; Chang, L.J.; DiStefano, L.; Hornykiewicz, O. Down’s syndrome individuals begin life with normal levels of brain cholinergic markers. J. Neurochem. 1989, 52, 1183–1187. [Google Scholar] [CrossRef] [PubMed]
- Lubec, B.; Yoo, B.; Dierssen, M.; Balic, N.; Lubec, G. Down syndrome patients start early prenatal life with normal cholinergic, monoaminergic and serotoninergic innervation. In Protein Expression in Down Syndrome Brain; Springer: Berlin/Heidelberg, Germany, 2001; pp. 303–310. [Google Scholar]
- Busciglio, J.; Pelsman, A.; Wong, C.; Pigino, G.; Yuan, M.; Mori, H.; Yankner, B.A. Altered metabolism of the amyloid β precursor protein is associated with mitochondrial dysfunction in Down’s syndrome. Neuron 2002, 33, 677–688. [Google Scholar] [CrossRef]
- Iulita, M.F.; Millón, M.B.B.; Pentz, R.; Aguilar, L.F.; Do Carmo, S.; Allard, S.; Michalski, B.; Wilson, E.N.; Ducatenzeiler, A.; Bruno, M.A. Differential deregulation of NGF and BDNF neurotrophins in a transgenic rat model of Alzheimer’s disease. Neurobiol. Dis. 2017, 108, 307–323. [Google Scholar] [CrossRef] [PubMed]
- Cuello, A.; Ferretti, M.; Iulita, M. Preplaque (‘preclinical’) Aβ-induced inflammation and nerve growth factor deregulation in transgenic models of Alzheimer’s disease-like amyloid pathology. Neurodegener. Dis. 2012, 10, 104–107. [Google Scholar] [CrossRef] [PubMed]
- Granholm, A.C.; Sanders, L.; Seo, H.; Lin, L.; Ford, K.; Isacson, O. Estrogen alters amyloid precursor protein as well as dendritic and cholinergic markers in a mouse model of Down syndrome. Hippocampus 2003, 13, 905–914. [Google Scholar] [CrossRef]
- Hunter, C.L.; Bimonte-Nelson, H.A.; Nelson, M.; Eckman, C.B.; Granholm, A.-C. Behavioral and neurobiological markers of Alzheimer’s disease in Ts65Dn mice: Effects of estrogen. Neurobiol. Aging 2004, 25, 873–884. [Google Scholar] [CrossRef]
- Hunter, C.L.; Isacson, O.; Nelson, M.; Bimonte-Nelson, H.; Seo, H.; Lin, L.; Ford, K.; Kindy, M.S.; Granholm, A.-C. Regional alterations in amyloid precursor protein and nerve growth factor across age in a mouse model of Down’s syndrome. Neurosci. Res. 2003, 45, 437–445. [Google Scholar] [CrossRef]
- Seo, H.; Isacson, O. Abnormal APP, cholinergic and cognitive function in Ts65Dn Down’s model mice. Exp. Neurol. 2005, 193, 469–480. [Google Scholar] [CrossRef]
- Salehi, A.; Delcroix, J.-D.; Belichenko, P.V.; Zhan, K.; Wu, C.; Valletta, J.S.; Takimoto-Kimura, R.; Kleschevnikov, A.M.; Sambamurti, K.; Chung, P.P. Increased App expression in a mouse model of Down’s syndrome disrupts NGF transport and causes cholinergic neuron degeneration. Neuron 2006, 51, 29–42. [Google Scholar] [CrossRef] [PubMed]
- Choi, J.H.; Berger, J.D.; Mazzella, M.J.; Morales-Corraliza, J.; Cataldo, A.M.; Nixon, R.A.; Ginsberg, S.D.; Levy, E.; Mathews, P.M. Age-dependent dysregulation of brain amyloid precursor protein in the Ts65Dn Down syndrome mouse model. J. Neurochem. 2009, 110, 1818–1827. [Google Scholar] [CrossRef]
- Lockrow, J.; Prakasam, A.; Huang, P.; Bimonte-Nelson, H.; Sambamurti, K.; Granholm, A.-C. Cholinergic degeneration and memory loss delayed by vitamin E in a Down syndrome mouse model. Exp. Neurol. 2009, 216, 278–289. [Google Scholar] [CrossRef]
- Reeves, R.; Irving, N.G.; Moran, T.H.; Wohn, A.; Kitt, C.; Sisodia, S.S.; Schmidt, C.; Bronson, R.T.; Davisson, M.T. A mouse model for Down syndrome exhibits learning and behaviour deficits. Nat. Genet. 1995, 11, 177–184. [Google Scholar] [CrossRef] [PubMed]
- Mesulam, M.; Shaw, P.; Mash, D.; Weintraub, S. Cholinergic nucleus basalis tauopathy emerges early in the aging-MCI-AD continuum. Ann. Neurol. Off. J. Am. Neurol. Assoc. Child Neurol. Soc. 2004, 55, 815–828. [Google Scholar] [CrossRef] [PubMed]
- Geula, C.; Dunlop, S.R.; Ayala, I.; Kawles, A.S.; Flanagan, M.E.; Gefen, T.; Mesulam, M.M. Basal forebrain cholinergic system in the dementias: Vulnerability, resilience, and resistance. J. Neurochem. 2021, 158, 1394–1411. [Google Scholar] [CrossRef] [PubMed]
- Counts, S.E.; Mufson, E.J. The role of nerve growth factor receptors in cholinergic basal forebrain degeneration in prodromal Alzheimer disease. J. Neuropathol. Exp. Neurol. 2005, 64, 263–272. [Google Scholar] [CrossRef] [PubMed]
- Counts, S.E.; Nadeem, M.; Wuu, J.; Ginsberg, S.D.; Saragovi, H.U.; Mufson, E.J. Reduction of cortical TrkA but not p75NTR protein in early-stage Alzheimer’s disease. Ann. Neurol. Off. J. Am. Neurol. Assoc. Child Neurol. Soc. 2004, 56, 520–531. [Google Scholar] [CrossRef]
- Mufson, E.J.; Ma, S.Y.; Cochran, E.J.; Bennett, D.A.; Beckett, L.A.; Jaffar, S.; Saragovi, H.U.; Kordower, J.H. Loss of nucleus basalis neurons containing trkA immunoreactivity in individuals with mild cognitive impairment and early Alzheimer’s disease. J. Comp. Neurol. 2000, 427, 19–30. [Google Scholar] [CrossRef]
- Parikh, V.; Howe, W.M.; Welchko, R.M.; Naughton, S.X.; D’Amore, D.E.; Han, D.H.; Deo, M.; Turner, D.L.; Sarter, M. Diminished trk A receptor signaling reveals cholinergic-attentional vulnerability of aging. Eur. J. Neurosci. 2013, 37, 278–293. [Google Scholar] [CrossRef]
- Ginsberg, S.D.; Che, S.; Wuu, J.; Counts, S.E.; Mufson, E.J. Down regulation of trk but not p75NTR gene expression in single cholinergic basal forebrain neurons mark the progression of Alzheimer’s disease. J. Neurochem. 2006, 97, 475–487. [Google Scholar] [CrossRef]
- Parks, W.C.; Wilson, C.L.; López-Boado, Y.S. Matrix metalloproteinases as modulators of inflammation and innate immunity. Nat. Rev. Immunol. 2004, 4, 617–629. [Google Scholar] [CrossRef] [PubMed]
- Khokha, R.; Murthy, A.; Weiss, A. Metalloproteinases and their natural inhibitors in inflammation and immunity. Nat. Rev. Immunol. 2013, 13, 649–665. [Google Scholar] [CrossRef] [PubMed]
- Nissinen, L.; Kähäri, V.-M. Matrix metalloproteinases in inflammation. Biochim. Biophys. Acta Gen. Subj. 2014, 1840, 2571–2580. [Google Scholar] [CrossRef] [PubMed]
- Rozemuller, A.J.; van Gool, W.A.; Eikelenboom, P. The neuroinflammatory response in plaques and amyloid angiopathy in Alzheimer’s disease: Therapeutic implications. Curr. Drug Targets-CNS Neurol. Disord. 2005, 4, 223–233. [Google Scholar] [CrossRef]
- McGeer, P.L.; Rogers, J.; McGeer, E.G. Inflammation, antiinflammatory agents, and Alzheimer’s disease: The last 22 years. J. Alzheimer’s Dis. 2016, 54, 853–857. [Google Scholar] [CrossRef]
- Cuello, A.C. Early and late CNS inflammation in Alzheimer’s disease: Two extremes of a continuum? Trends Pharmacol. Sci. 2017, 38, 956–966. [Google Scholar] [CrossRef]
- Gamage, R.; Wagnon, I.; Rossetti, I.; Childs, R.; Niedermayer, G.; Chesworth, R.; Gyengesi, E. Cholinergic modulation of glial function during aging and chronic neuroinflammation. Front. Cell. Neurosci. 2020, 14. [Google Scholar] [CrossRef]
- Lehner, K.R.; Silverman, H.A.; Addorisio, M.E.; Roy, A.; Al-Onaizi, M.A.; Levine, Y.; Olofsson, P.S.; Chavan, S.S.; Gros, R.; Nathanson, N.M. Forebrain cholinergic signaling regulates innate immune responses and inflammation. Front. Immunol. 2019, 10, 585. [Google Scholar] [CrossRef]
- Field, R.H.; Gossen, A.; Cunningham, C. Prior pathology in the basal forebrain cholinergic system predisposes to inflammation-induced working memory deficits: Reconciling inflammatory and cholinergic hypotheses of delirium. J. Neurosci. 2012, 32, 6288–6294. [Google Scholar] [CrossRef]
- Giovannini, M.G.; Scali, C.; Prosperi, C.; Bellucci, A.; Vannucchi, M.G.; Rosi, S.; Pepeu, G.; Casamenti, F. β-Amyloid-induced inflammation and cholinergic hypofunction in the rat brain in vivo: Involvement of the p38MAPK pathway. Neurobiol. Dis. 2002, 11, 257–274. [Google Scholar] [CrossRef]
- Vacher, M.; Porter, T.; Villemagne, V.L.; Milicic, L.; Peretti, M.; Fowler, C.; Martins, R.; Rainey-Smith, S.; Ames, D.; Masters, C.L.; et al. Validation of a priori candidate Alzheimer’s disease SNPs with brain amyloid-beta deposition. Sci. Rep. 2019, 9, 17069. [Google Scholar] [CrossRef]
- McDade, E.; Bateman, R.J. Stop Alzheimer’s before it starts. Nat. News 2017, 547, 153. [Google Scholar] [CrossRef]
- Winblad, B.; Amouyel, P.; Andrieu, S.; Ballard, C.; Brayne, C.; Brodaty, H.; Cedazo-Minguez, A.; Dubois, B.; Edvardsson, D.; Feldman, H. Defeating Alzheimer’s disease and other dementias: A priority for European science and society. Lancet Neurol. 2016, 15, 455–532. [Google Scholar] [CrossRef]
- Gandy, S. Perspective: Prevention is better than cure. Nature 2011, 475, S15. [Google Scholar] [CrossRef]
- Aisen, P.; Cummings, J.; Jack, C., Jr.; Morris, J.C.; Sperling, R.; Frolich, L.; Jones, R.W.; Dowsett, S.A.; Matthews, B.R.; Raskin, J.; et al. On the path to 2025: Understanding the alzheimer’s disease continuum. Alzheimers Res. 2017, 9, 60. [Google Scholar] [CrossRef] [PubMed]
- Sperling, R.; Mormino, E.; Johnson, K. The evolution of preclinical Alzheimer’s disease: Implications for prevention trials. Neuron 2014, 84, 608–622. [Google Scholar] [CrossRef]
- Iulita, M.F.; Ganesh, A.; Pentz, R.; Flores Aguilar, L.; Gubert, P.; Ducatenzeiler, A.; Christie, S.; Wilcock, G.K.; Cuello, A.C. Identification and preliminary validation of a plasma profile associated with cognitive decline in dementia and at-risk individuals: A retrospective cohort analysis. J. Alzheimer’s Dis. 2019, 67, 327–341. [Google Scholar] [CrossRef] [PubMed]
- Counts, S.E.; He, B.; Prout, J.G.; Michalski, B.; Farotti, L.; Fahnestock, M.; Mufson, E.J. Cerebrospinal fluid proNGF: A putative biomarker for early Alzheimer’s disease. Curr. Alzheimer Res. 2016, 13, 800–808. [Google Scholar] [CrossRef]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Do Carmo, S.; Kannel, B.; Cuello, A.C. The Nerve Growth Factor Metabolic Pathway Dysregulation as Cause of Alzheimer’s Cholinergic Atrophy. Cells 2022, 11, 16. https://doi.org/10.3390/cells11010016
Do Carmo S, Kannel B, Cuello AC. The Nerve Growth Factor Metabolic Pathway Dysregulation as Cause of Alzheimer’s Cholinergic Atrophy. Cells. 2022; 11(1):16. https://doi.org/10.3390/cells11010016
Chicago/Turabian StyleDo Carmo, Sonia, Benjamin Kannel, and A. Claudio Cuello. 2022. "The Nerve Growth Factor Metabolic Pathway Dysregulation as Cause of Alzheimer’s Cholinergic Atrophy" Cells 11, no. 1: 16. https://doi.org/10.3390/cells11010016
APA StyleDo Carmo, S., Kannel, B., & Cuello, A. C. (2022). The Nerve Growth Factor Metabolic Pathway Dysregulation as Cause of Alzheimer’s Cholinergic Atrophy. Cells, 11(1), 16. https://doi.org/10.3390/cells11010016