
Targeting PGM3 as a Novel Therapeutic Strategy in KRAS/LKB1 Co-Mutant Lung Cancer

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Cell Lines, Culture, and Reagents
2.2. Western Blot Analysis
2.3. Lectin Binding Analysis
2.4. Wheat Germ Agglutinin (WGA) Pull down Assay
2.5. Cell Proliferation and Death
2.6. RNA Interference
2.7. Soft-Agar Colony-Formation Assay
2.8. Mouse Xenografts and Tumor Tissue Analysis
2.9. 15N-Glutamine Labeling
2.10. Metabolomics
2.11. Patient Survival Data
2.12. Statistics
3. Results
3.1. GFPT2 Inhibition Selectively Reduces Glycosylation of KL Co-Mutant Cells
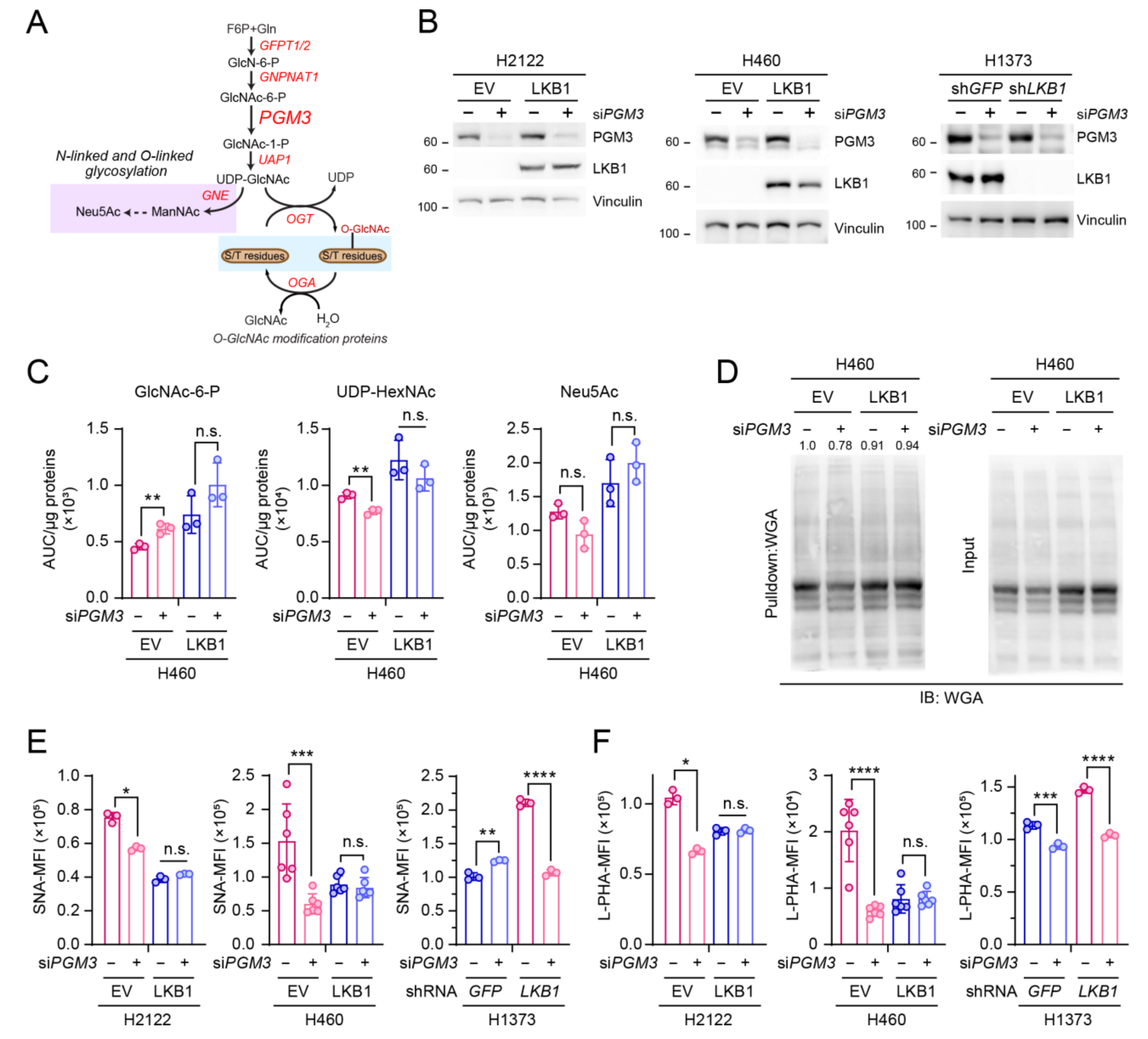
3.2. PGM3 Inhibition Also Selectively Reduces Glycosylation of KL Co-Mutant Cells
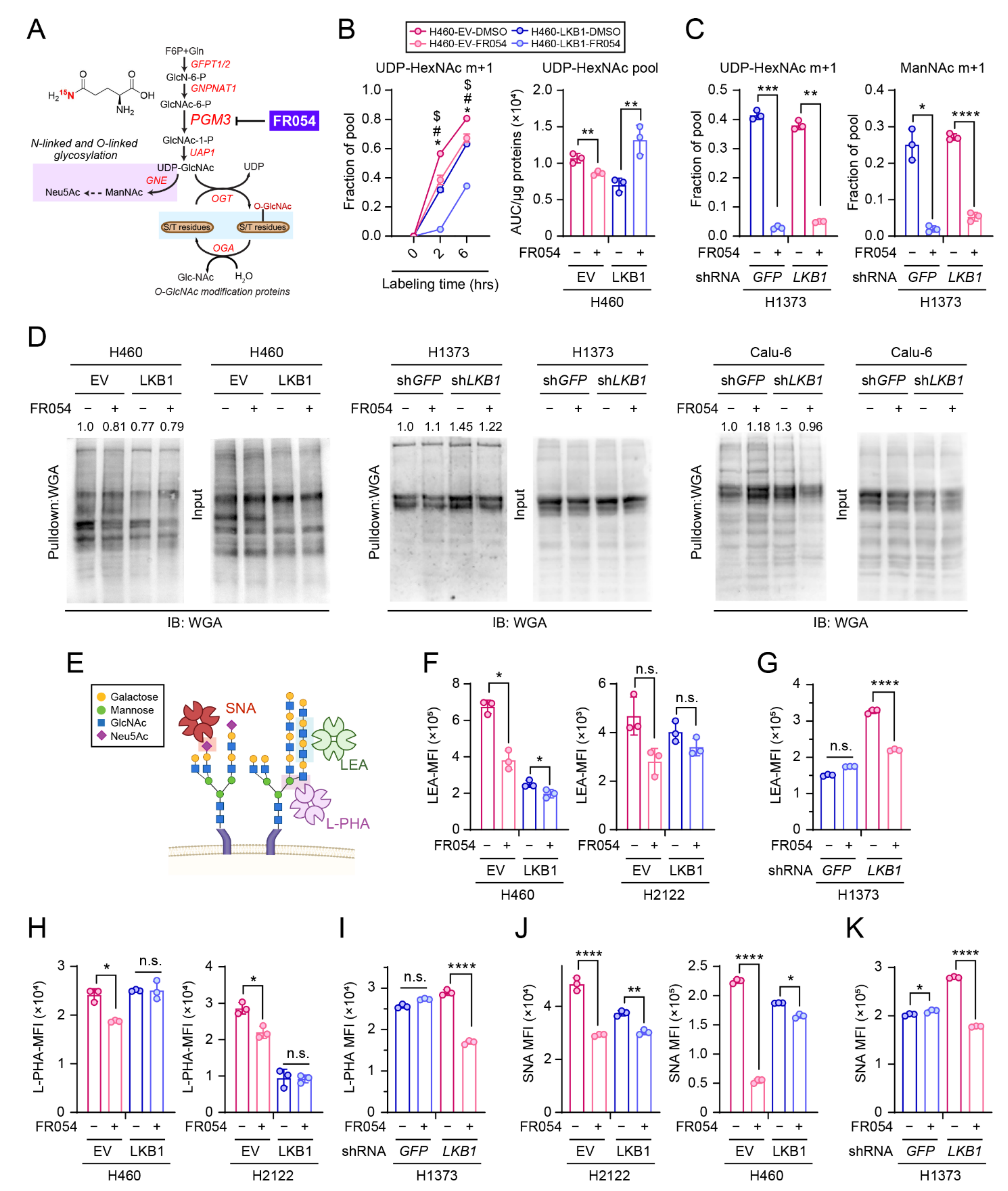
3.3. LKB1 Loss Confers PGM3 Dependence in KRAS-Mutant Lung Cancer Cells
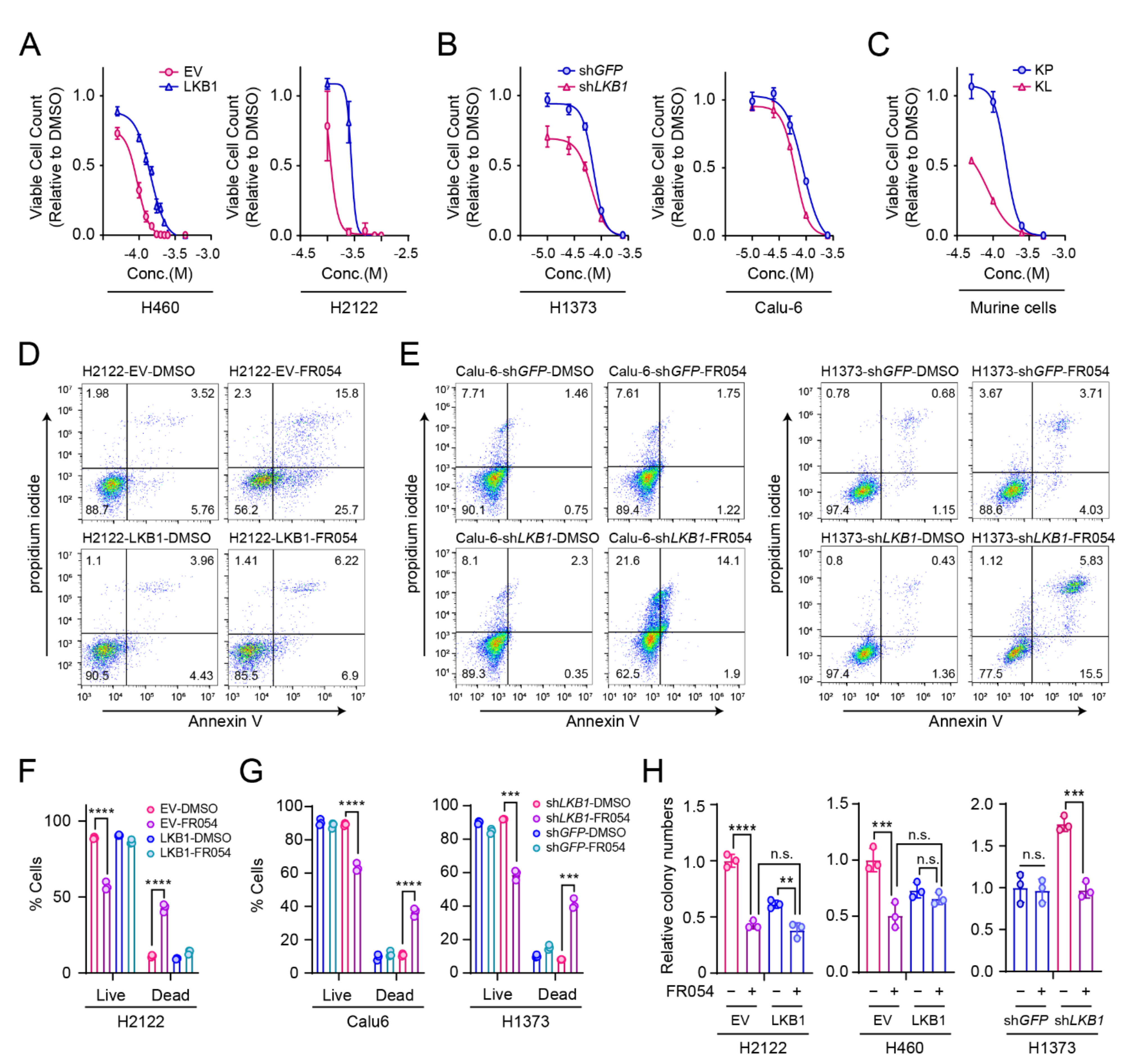
3.4. PGM3 Inhibitor FR054 Reduces the HBP Flow in Both KL and K Cells but Selectively Suppresses Viability of KL co-Mutant Cells
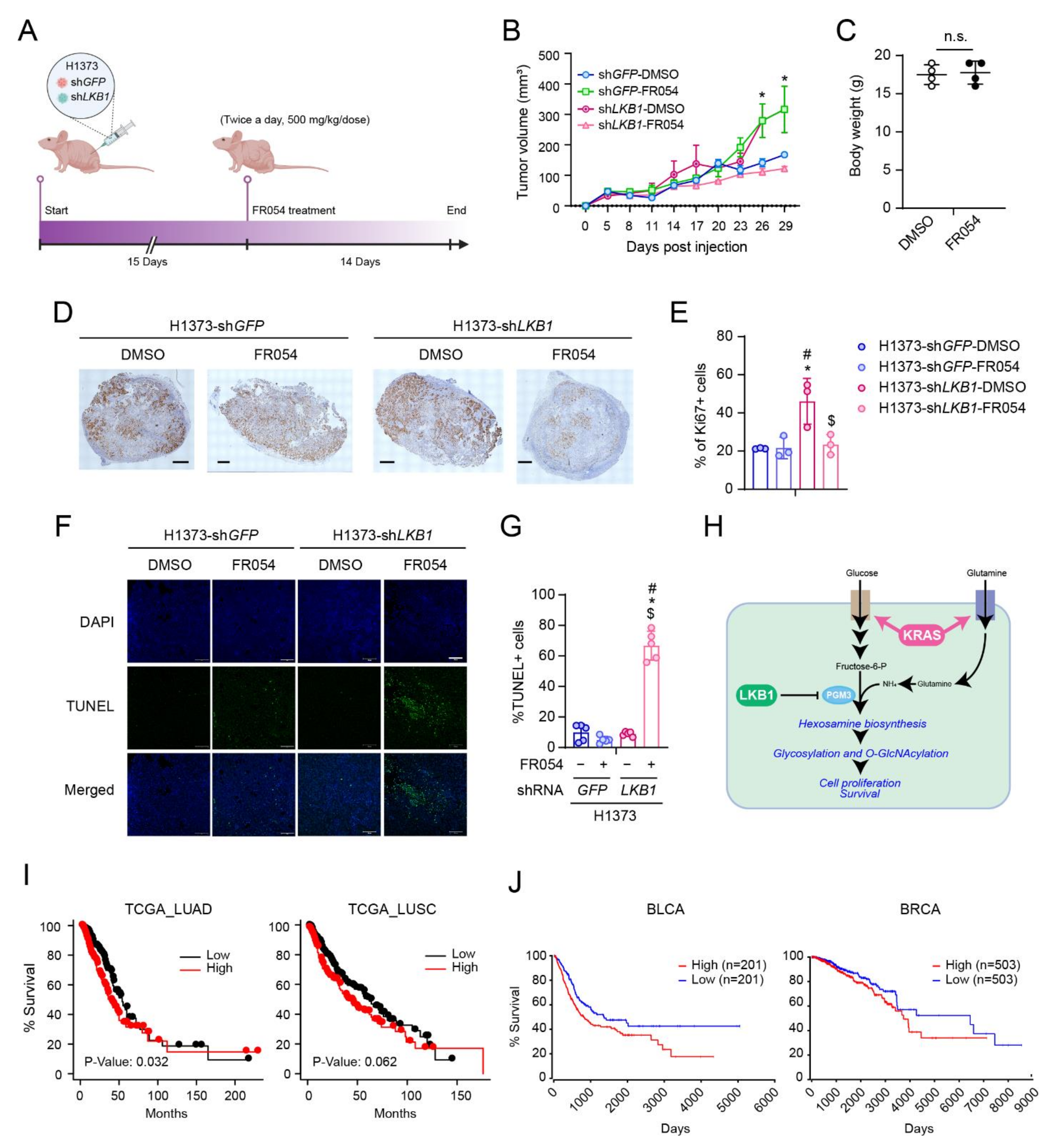
3.5. PGM3 Inhibition Reduces KL Co-Mutant Tumor Growth In Vivo
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Sung, H.; Ferlay, J.; Siegel, R.L.; Laversanne, M.; Soerjomataram, I.; Jemal, A.; Bray, F. Global cancer statistics 2020: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA: A Cancer J. Clin. 2021, 71, 209–249. [Google Scholar] [CrossRef]
- Herbst, R.S.; Heymach, J.V.; Lippman, S.M. Lung cancer. N. Engl. J. Med. 2008, 359, 1367–1380. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Travis, W.D. Pathology of lung cancer. Clin. Chest Med. 2002, 23, 65–81. [Google Scholar] [CrossRef]
- Canon, J.; Rex, K.; Saiki, A.Y.; Mohr, C.; Cooke, K.; Bagal, D.; Gaida, K.; Holt, T.; Knutson, C.G.; Koppada, N.; et al. The clinical KRAS(G12C) inhibitor AMG 510 drives anti-tumour immunity. Nature 2019, 575, 217–223. [Google Scholar] [CrossRef] [PubMed]
- Ding, L.; Getz, G.; Wheeler, D.A.; Mardis, E.R.; McLellan, M.D.; Cibulskis, K.; Sougnez, C.; Greulich, H.; Muzny, D.M.; Morgan, M.B.; et al. Somatic mutations affect key pathways in lung adenocarcinoma. Nature 2008, 455, 1069–1075. [Google Scholar] [CrossRef]
- Imielinski, M.; Berger, A.H.; Hammerman, P.S.; Hernandez, B.; Pugh, T.J.; Hodis, E.; Cho, J.; Suh, J.; Capelletti, M.; Sivachenko, A.; et al. Mapping the hallmarks of lung adenocarcinoma with massively parallel sequencing. Cell 2012, 150, 1107–1120. [Google Scholar] [CrossRef] [Green Version]
- Biancur, D.E.; Paulo, J.A.; Malachowska, B.; Del Rey, M.Q.; Sousa, C.M.; Wang, X.; Sohn, A.S.W.; Chu, G.C.; Gygi, S.P.; Harper, J.W.; et al. Compensatory metabolic networks in pancreatic cancers upon perturbation of glutamine metabolism. Nat. Commun. 2017, 8, 15965. [Google Scholar] [CrossRef]
- Skoulidis, F.; Goldberg, M.E.; Greenawalt, D.M.; Hellmann, M.D.; Awad, M.M.; Gainor, J.F.; Schrock, A.B.; Hartmaier, R.J.; Trabucco, S.E.; Gay, L.; et al. STK11/LKB1 mutations and PD-1 inhibitor resistance in KRAS-mutant lung adenocarcinoma. Cancer Discov. 2018, 8, 822–835. [Google Scholar] [CrossRef] [Green Version]
- Shackelford, D.B.; Abt, E.; Gerken, L.; Vasquez, D.S.; Seki, A.; Leblanc, M.; Wei, L.; Fishbein, M.C.; Czernin, J.; Mischel, P.S.; et al. LKB1 inactivation dictates therapeutic response of non-small cell lung cancer to the metabolism drug phenformin. Cancer Cell 2013, 23, 143–158. [Google Scholar] [CrossRef] [Green Version]
- Liu, Y.; Marks, K.; Cowley, G.S.; Carretero, J.; Liu, Q.; Nieland, T.J.; Xu, C.; Cohoon, T.J.; Gao, P.; Zhang, Y.; et al. Metabolic and functional genomic studies identify deoxythymidylate kinase as a target in LKB1-mutant lung cancer. Cancer Discov. 2013, 3, 870–879. [Google Scholar] [CrossRef] [Green Version]
- Bryant, K.L.; Mancias, J.D.; Kimmelman, A.C.; Der, C.J. KRAS: Feeding pancreatic cancer proliferation. Trends Biochem. Sci. 2014, 39, 91–100. [Google Scholar] [CrossRef] [Green Version]
- Kim, J.; Hu, Z.; Cai, L.; Li, K.; Choi, E.; Faubert, B.; Bezwada, D.; Rodriguez-Canales, J.; Villalobos, P.; Lin, Y.F.; et al. CPS1 maintains pyrimidine pools and DNA synthesis in KRAS/LKB1-mutant lung cancer cells. Nature 2017, 546, 168–172. [Google Scholar] [CrossRef]
- Kim, J.; Lee, H.M.; Cai, F.; Ko, B.; Yang, C.; Lieu, E.L.; Muhammad, N.; Rhyne, S.; Li, K.; Haloul, M.; et al. The hexosamine biosynthesis pathway is a targetable liability in KRAS/LKB1 mutant lung cancer. Nat. Metab. 2020, 2, 1401–1412. [Google Scholar] [CrossRef]
- Hershfield, M.S.; Seegmiller, J.E. Regulation of de novo purine biosynthesis in human lymphoblasts. Coordinate control of proximal (rate-determining) steps and the inosinic acid branch point. J. Biol. Chem. 1976, 251, 7348–7354. [Google Scholar] [CrossRef]
- Roebuck, B.D.; Yager, J.D.; Longnecker, D.S.; Wilpone, S.A. Promotion by unsaturated fat of azaserine-induced pancreatic carcinogenesis in the rat. Cancer Res. 1981, 41, 3961–3966. [Google Scholar]
- Longnecker, D.S.; Roebuck, B.D.; Yager, J.D., Jr.; Lilja, H.S.; Siegmund, B. Pancreatic carcinoma in azaserine-treated rats: Induction, classification and dietary modulation of incidence. Cancer 1981, 47, 1562–1572. [Google Scholar] [CrossRef]
- Dennis, J.W.; Nabi, I.R.; Demetriou, M. Metabolism, cell surface organization, and disease. Cell 2009, 139, 1229–1241. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kornfeld, R.; Kornfeld, S. Assembly of asparagine-linked oligosaccharides. Annu. Rev. Biochem. 1985, 54, 631–664. [Google Scholar] [CrossRef] [PubMed]
- Lau, K.S.; Partridge, E.A.; Grigorian, A.; Silvescu, C.I.; Reinhold, V.N.; Demetriou, M.; Dennis, J.W. Complex N-glycan number and degree of branching cooperate to regulate cell proliferation and differentiation. Cell 2007, 129, 123–134. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mehta, A.; Norton, P.; Liang, H.; Comunale, M.A.; Wang, M.; Rodemich-Betesh, L.; Koszycki, A.; Noda, K.; Miyoshi, E.; Block, T. Increased levels of tetra-antennary N-linked glycan but not core fucosylation are associated with hepatocellular carcinoma tissue. Cancer Epidemiol. Prev. Biomark. 2012, 21, 925–933. [Google Scholar] [CrossRef] [Green Version]
- Zhang, H.; Meng, F.; Wu, S.; Kreike, B.; Sethi, S.; Chen, W.; Miller, F.R.; Wu, G. Engagement of I-Branching β-1, 6-N-acetylglucosaminyltransferase 2 in breast cancer metastasis and TGF-β signaling. Cancer Res. 2011, 71, 4846–4856. [Google Scholar] [CrossRef] [Green Version]
- Mikami, J.; Tobisawa, Y.; Yoneyama, T.; Hatakeyama, S.; Mori, K.; Hashimoto, Y.; Koie, T.; Ohyama, C.; Fukuda, M. I-branching N-acetylglucosaminyltransferase regulates prostate cancer invasiveness by enhancing α5β1 integrin signaling. Cancer Sci. 2016, 107, 359–368. [Google Scholar] [CrossRef]
- Li, C.; Simeone, D.M.; Brenner, D.E.; Anderson, M.A.; Shedden, K.A.; Ruffin, M.T.; Lubman, D.M. Pancreatic cancer serum detection using a lectin/glyco-antibody array method. J. Proteome Res. 2009, 8, 483–492. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, Y.; Yu, X.; Ichikawa, M.; Lyons, J.J.; Datta, S.; Lamborn, I.T.; Jing, H.; Kim, E.S.; Biancalana, M.; Wolfe, L.A.; et al. Autosomal recessive phosphoglucomutase 3 (PGM3) mutations link glycosylation defects to atopy, immune deficiency, autoimmunity, and neurocognitive impairment. J. Allergy Clin. Immunol. 2014, 133, 1400–1409.e5. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sassi, A.; Lazaroski, S.; Wu, G.; Haslam, S.M.; Fliegauf, M.; Mellouli, F.; Patiroglu, T.; Unal, E.; Ozdemir, M.A.; Jouhadi, Z.; et al. Hypomorphic homozygous mutations in phosphoglucomutase 3 (PGM3) impair immunity and increase serum IgE levels. J. Allergy Clin. Immunol. 2014, 133, 1410–1419.e13. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ricciardiello, F.; Votta, G.; Palorini, R.; Raccagni, I.; Brunelli, L.; Paiotta, A.; Tinelli, F.; D’Orazio, G.; Valtorta, S.; De Gioia, L.; et al. Inhibition of the hexosamine biosynthetic pathway by targeting PGM3 causes breast cancer growth arrest and apoptosis. Cell Death Dis. 2018, 9, 377. [Google Scholar] [CrossRef] [PubMed]
- Ricciardiello, F.; Gang, Y.; Palorini, R.; Li, Q.; Giampà, M.; Zhao, F.; You, L.; La Ferla, B.; De Vitto, H.; Guan, W.; et al. Hexosamine pathway inhibition overcomes pancreatic cancer resistance to gemcitabine through unfolded protein response and EGFR-Akt pathway modulation. Oncogene 2020, 39, 4103–4117. [Google Scholar] [CrossRef]
- Ricciardiello, F.; Bergamaschi, L.; De Vitto, H.; Gang, Y.; Zhang, T.; Palorini, R.; Chiaradonna, F. Suppression of the HBP function increases pancreatic cancer cell sensitivity to a Pan-RAS inhibitor. Cells 2021, 10, 431. [Google Scholar] [CrossRef]
- Ji, H.; Ramsey, M.R.; Hayes, D.N.; Fan, C.; McNamara, K.; Kozlowski, P.; Torrice, C.; Wu, M.C.; Shimamura, T.; Perera, S.A.; et al. LKB1 modulates lung cancer differentiation and metastasis. Nature 2007, 448, 807–810. [Google Scholar] [CrossRef]
- Murray, C.W.; Brady, J.J.; Tsai, M.K.; Li, C.; Winters, I.P.; Tang, R.; Andrejka, L.; Ma, R.K.; Kunder, C.A.; Chu, P.; et al. An LKB1-SIK axis suppresses lung tumor growth and controls differentiation. Cancer Discov. 2019, 9, 1590–1605. [Google Scholar] [CrossRef] [Green Version]
- Hollstein, P.E.; Eichner, L.J.; Brun, S.N.; Kamireddy, A.; Svensson, R.U.; Vera, L.I.; Ross, D.S.; Rymoff, T.J.; Hutchins, A.; Galvez, H.M.; et al. The AMPK-related kinases SIK1 and SIK3 mediate key tumor-suppressive effects of LKB1 in NSCLC. Cancer Discov. 2019, 9, 1606–1627. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Goodwin, J.M.; Svensson, R.U.; Lou, H.J.; Winslow, M.M.; Turk, B.E.; Shaw, R.J. An AMPK-independent signaling pathway downstream of the LKB1 tumor suppressor controls Snail1 and metastatic potential. Mol. Cell 2014, 55, 436–450. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hanover, J.A.; Krause, M.W.; Love, D.C. Linking metabolism to epigenetics through O-GlcNAcylation. Nat. Rev. Mol. Cell Biol. 2012, 13, 312–321. [Google Scholar] [CrossRef] [PubMed]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lee, H.; Cai, F.; Kelekar, N.; Velupally, N.K.; Kim, J. Targeting PGM3 as a Novel Therapeutic Strategy in KRAS/LKB1 Co-Mutant Lung Cancer. Cells 2022, 11, 176. https://doi.org/10.3390/cells11010176
Lee H, Cai F, Kelekar N, Velupally NK, Kim J. Targeting PGM3 as a Novel Therapeutic Strategy in KRAS/LKB1 Co-Mutant Lung Cancer. Cells. 2022; 11(1):176. https://doi.org/10.3390/cells11010176
Chicago/Turabian StyleLee, Hyunmin, Feng Cai, Neil Kelekar, Nipun K. Velupally, and Jiyeon Kim. 2022. "Targeting PGM3 as a Novel Therapeutic Strategy in KRAS/LKB1 Co-Mutant Lung Cancer" Cells 11, no. 1: 176. https://doi.org/10.3390/cells11010176
APA StyleLee, H., Cai, F., Kelekar, N., Velupally, N. K., & Kim, J. (2022). Targeting PGM3 as a Novel Therapeutic Strategy in KRAS/LKB1 Co-Mutant Lung Cancer. Cells, 11(1), 176. https://doi.org/10.3390/cells11010176