
Adipose-Specific PPARα Knockout Mice Have Increased Lipogenesis by PASK–SREBP1 Signaling and a Polarity Shift to Inflammatory Macrophages in White Adipose Tissue

 ,
, 
 ,
,  and
and
Abstract
:1. Introduction
2. Materials and Methods
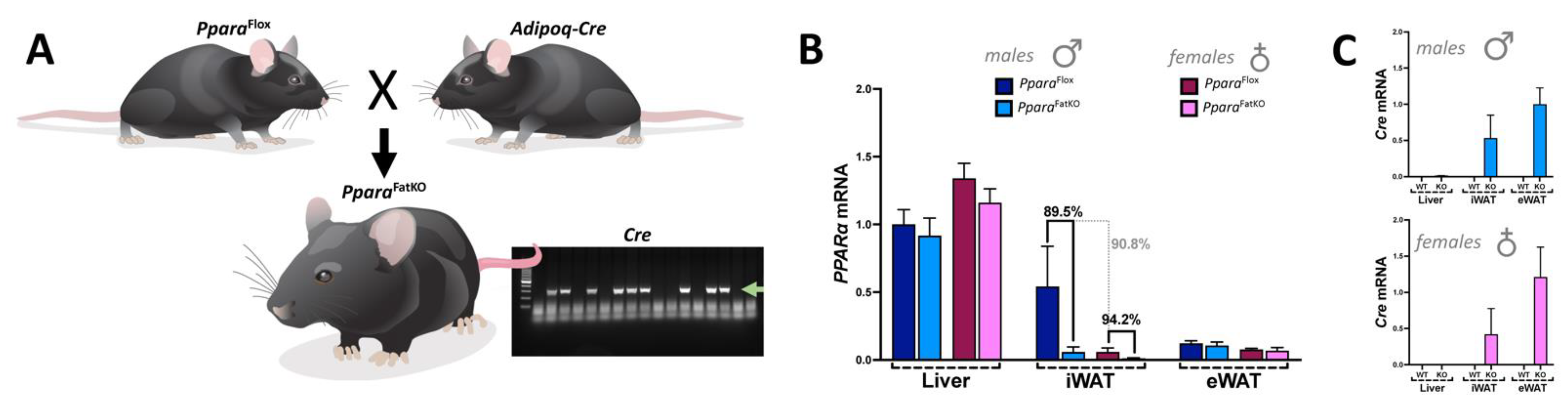
2.1. Animals
2.2. Body Composition
2.3. Analysis of Plasma Lipids and Metabolites
2.4. Lipidomics
2.5. Quantitative Real-Time PCR Analysis
2.6. Gel Electrophoresis and Western Blotting
2.7. Statistical Analysis
3. Results
3.1. Adiposity in Adipocyte-Specific PPARα Knockout and Control Littermate Mice on High-Fat and Regular-Chow Diets
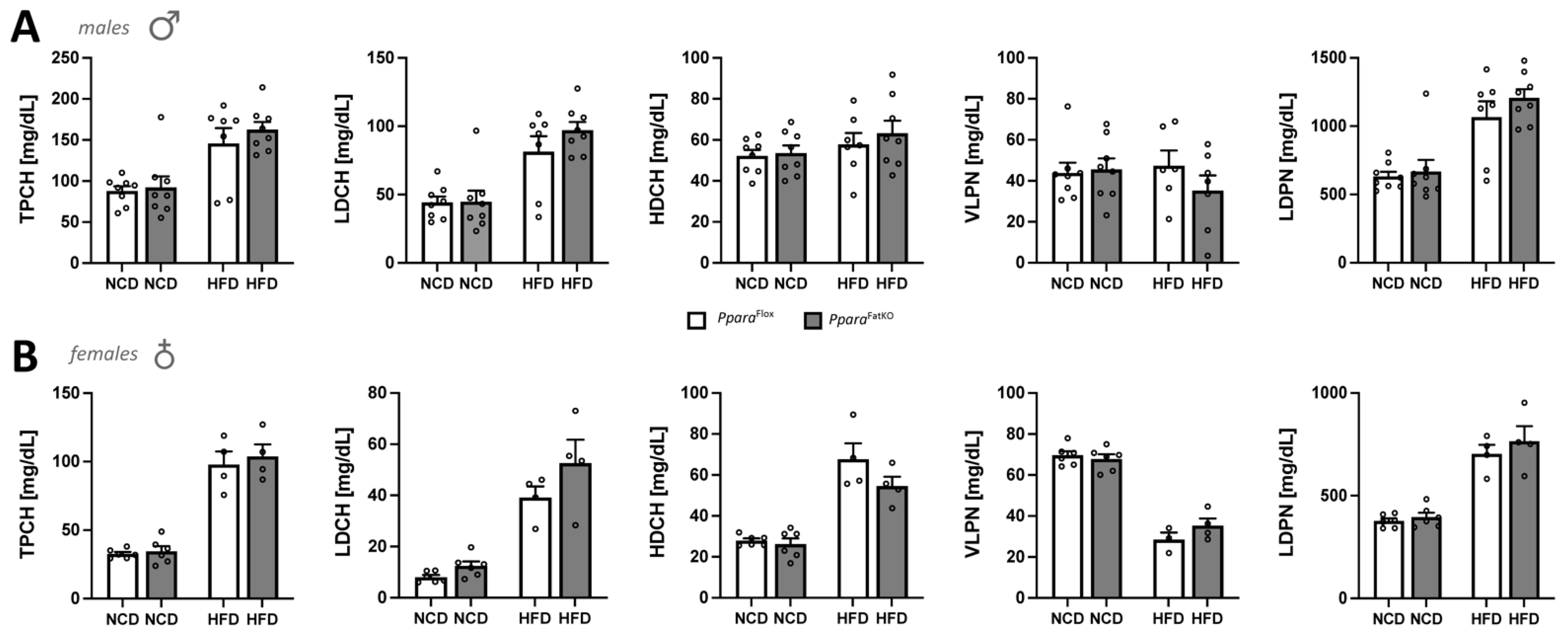
3.2. Analysis of Serum Metabolites and Cholesterol in Adipocyte-Specific PPARα Knockout and Control Littermate Mice
3.3. Lipidomics and Signaling in Adipocyte-Specific PPARα Knockout and Littermate Control Mice
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Issemann, I.; Green, S. Activation of a member of the steroid hormone receptor superfamily by peroxisome proliferators. Nature 1990, 347, 645–650. [Google Scholar] [CrossRef]
- Gordon, D.M.; Neifer, K.L.; Hamoud, A.A.; Hawk, C.F.; Nestor-Kalinoski, A.L.; Miruzzi, S.A.; Morran, M.P.; Adeosun, S.O.; Sarver, J.G.; Erhardt, P.W.; et al. Bilirubin remodels murine white adipose tissue by reshaping mitochondrial activity and the coregulator profile of peroxisome proliferator-activated receptor alpha. J. Biol. Chem. 2020, 295, 9804–9822. [Google Scholar] [CrossRef] [PubMed]
- Gordon, D.M.; Hong, S.H.; Kipp, Z.A.; Hinds, T.D., Jr. Identification of Binding Regions of Bilirubin in the Ligand-Binding Pocket of the Peroxisome Proliferator-Activated Receptor-A (PPARalpha). Molecules 2021, 26, 2975. [Google Scholar] [CrossRef]
- Gordon, D.M.; Blomquist, T.M.; Miruzzi, S.A.; McCullumsmith, R.; Stec, D.E.; Hinds, T.D., Jr. RNA sequencing in human HepG2 hepatocytes reveals PPAR-alpha mediates transcriptome responsiveness of bilirubin. Physiol. Genom. 2019, 51, 234–240. [Google Scholar] [CrossRef] [PubMed]
- Stec, D.E.; John, K.; Trabbic, C.J.; Luniwal, A.; Hankins, M.W.; Baum, J.; Hinds, T.D., Jr. Bilirubin Binding to PPARalpha Inhibits Lipid Accumulation. PLoS ONE 2016, 11, e0153427. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Srivastava, R.A.; Jahagirdar, R.; Azhar, S.; Sharma, S.; Bisgaier, C.L. Peroxisome proliferator-activated receptor-alpha selective ligand reduces adiposity, improves insulin sensitivity and inhibits atherosclerosis in LDL receptor-deficient mice. Mol. Cell. Biochem. 2006, 285, 35–50. [Google Scholar] [CrossRef]
- Hinds, T.D., Jr.; Creeden, J.F.; Gordon, D.M.; Stec, D.F.; Donald, M.C.; Stec, D.E. Bilirubin Nanoparticles Reduce Diet-Induced Hepatic Steatosis, Improve Fat Utilization, and Increase Plasma beta-Hydroxybutyrate. Front. Pharm. 2020, 11, 594574. [Google Scholar] [CrossRef]
- Linden, D.; Lindberg, K.; Oscarsson, J.; Claesson, C.; Asp, L.; Li, L.; Gustafsson, M.; Boren, J.; Olofsson, S.O. Influence of peroxisome proliferator-activated receptor alpha agonists on the intracellular turnover and secretion of apolipoprotein (Apo) B-100 and ApoB-48. J. Biol. Chem. 2002, 277, 23044–23053. [Google Scholar] [CrossRef] [Green Version]
- Shah, A.; Rader, D.J.; Millar, J.S. The effect of PPAR-alpha agonism on apolipoprotein metabolism in humans. Atherosclerosis 2010, 210, 35–40. [Google Scholar] [CrossRef]
- Jeong, S.; Yoon, M. Fenofibrate inhibits adipocyte hypertrophy and insulin resistance by activating adipose PPARalpha in high fat diet-induced obese mice. Exp. Mol. Med. 2009, 41, 397–405. [Google Scholar] [CrossRef] [Green Version]
- Yoon, M. PPARalpha in Obesity: Sex Difference and Estrogen Involvement. PPAR Res. 2010, 2010, 584296. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tsuchida, A.; Yamauchi, T.; Takekawa, S.; Hada, Y.; Ito, Y.; Maki, T.; Kadowaki, T. Peroxisome proliferator-activated receptor (PPAR)alpha activation increases adiponectin receptors and reduces obesity-related inflammation in adipose tissue: Comparison of activation of PPARalpha, PPARgamma, and their combination. Diabetes 2005, 54, 3358–3370. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guerre-Millo, M.; Gervois, P.; Raspe, E.; Madsen, L.; Poulain, P.; Derudas, B.; Herbert, J.M.; Winegar, D.A.; Willson, T.M.; Fruchart, J.C.; et al. Peroxisome proliferator-activated receptor alpha activators improve insulin sensitivity and reduce adiposity. J. Biol. Chem. 2000, 275, 16638–16642. [Google Scholar] [CrossRef] [Green Version]
- Lee, S.S.; Pineau, T.; Drago, J.; Lee, E.J.; Owens, J.W.; Kroetz, D.L.; Fernandez-Salguero, P.M.; Westphal, H.; Gonzalez, F.J. Targeted disruption of the alpha isoform of the peroxisome proliferator-activated receptor gene in mice results in abolishment of the pleiotropic effects of peroxisome proliferators. Mol. Cell. Biol. 1995, 15, 3012–3022. [Google Scholar] [CrossRef] [Green Version]
- Kim, B.H.; Won, Y.S.; Kim, E.Y.; Yoon, M.; Nam, K.T.; Oh, G.T.; Kim, D.Y. Phenotype of peroxisome proliferator-activated receptor-alpha(PPARalpha)deficient mice on mixed background fed high fat diet. J. Vet. Sci. 2003, 4, 239–244. [Google Scholar] [CrossRef]
- Costet, P.; Legendre, C.; More, J.; Edgar, A.; Galtier, P.; Pineau, T. Peroxisome proliferator-activated receptor alpha-isoform deficiency leads to progressive dyslipidemia with sexually dimorphic obesity and steatosis. J. Biol. Chem. 1998, 273, 29577–29585. [Google Scholar] [CrossRef] [Green Version]
- Akiyama, T.E.; Nicol, C.J.; Fievet, C.; Staels, B.; Ward, J.M.; Auwerx, J.; Lee, S.S.; Gonzalez, F.J.; Peters, J.M. Peroxisome proliferator-activated receptor-alpha regulates lipid homeostasis, but is not associated with obesity: Studies with congenic mouse lines. J. Biol. Chem. 2001, 276, 39088–39093. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bougarne, N.; Weyers, B.; Desmet, S.J.; Deckers, J.; Ray, D.W.; Staels, B.; De Bosscher, K. Molecular Actions of PPARalpha in Lipid Metabolism and Inflammation. Endocr. Rev. 2018, 39, 760–802. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De Souza, A.T.; Dai, X.; Spencer, A.G.; Reppen, T.; Menzie, A.; Roesch, P.L.; He, Y.; Caguyong, M.J.; Bloomer, S.; Herweijer, H.; et al. Transcriptional and phenotypic comparisons of Ppara knockout and siRNA knockdown mice. Nucleic Acids Res. 2006, 34, 4486–4494. [Google Scholar] [CrossRef]
- Hinds, T.D., Jr.; Sodhi, K.; Meadows, C.; Fedorova, L.; Puri, N.; Kim, D.H.; Peterson, S.J.; Shapiro, J.; Abraham, N.G.; Kappas, A. Increased HO-1 levels ameliorate fatty liver development through a reduction of heme and recruitment of FGF21. Obesity 2014, 22, 705–712. [Google Scholar] [CrossRef] [Green Version]
- Stec, D.E.; Gordon, D.M.; Hipp, J.A.; Hong, S.; Mitchell, Z.L.; Franco, N.R.; Robison, J.W.; Anderson, C.D.; Stec, D.F.; Hinds, T.D., Jr. The loss of hepatic PPARalpha promotes inflammation and serum hyperlipidemia in diet-induced obesity. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2019, 317, R733–R745. [Google Scholar] [CrossRef] [PubMed]
- Montagner, A.; Polizzi, A.; Fouche, E.; Ducheix, S.; Lippi, Y.; Lasserre, F.; Barquissau, V.; Regnier, M.; Lukowicz, C.; Benhamed, F.; et al. Liver PPARalpha is crucial for whole-body fatty acid homeostasis and is protective against NAFLD. Gut 2016, 65, 1202–1214. [Google Scholar] [CrossRef] [Green Version]
- Brocker, C.N.; Yue, J.; Kim, D.; Qu, A.; Bonzo, J.A.; Gonzalez, F.J. Hepatocyte-specific PPARA expression exclusively promotes agonist-induced cell proliferation without influence from nonparenchymal cells. Am. J. Physiol. Gastrointest. Liver Physiol. 2017, 312, G283–G299. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Zhu, X.X.; Jiao, S.Y.; Qi, D.; Yu, B.Q.; Xie, G.M.; Liu, Y.; Song, Y.T.; Xu, Q.; Xu, Q.B.; et al. Cardiomyocyte peroxisome proliferator-activated receptor alpha is essential for energy metabolism and extracellular matrix homeostasis during pressure overload-induced cardiac remodeling. Acta Pharmacol. Sin. 2021, 1–12. [Google Scholar] [CrossRef]
- Regnier, M.; Polizzi, A.; Smati, S.; Lukowicz, C.; Fougerat, A.; Lippi, Y.; Fouche, E.; Lasserre, F.; Naylies, C.; Betoulieres, C.; et al. Hepatocyte-specific deletion of Pparalpha promotes NAFLD in the context of obesity. Sci. Rep. 2020, 10, 6489. [Google Scholar] [CrossRef]
- Gordon, D.M.; Adeosun, S.O.; Ngwudike, S.I.; Anderson, C.D.; Hall, J.E.; Hinds, T.D., Jr.; Stec, D.E. CRISPR Cas9-mediated deletion of biliverdin reductase A (BVRA) in mouse liver cells induces oxidative stress and lipid accumulation. Arch. Biochem. Biophys. 2019, 672, 108072. [Google Scholar] [CrossRef]
- Hinds, T.D., Jr.; Hosick, P.A.; Hankins, M.W.; Nestor-Kalinoski, A.; Stec, D.E. Mice with hyperbilirubinemia due to Gilbert’s Syndrome polymorphism are resistant to hepatic steatosis by decreased serine 73 phosphorylation of PPARalpha. Am. J. Physiol. Endocrinol. Metab. 2017, 312, E244–E252. [Google Scholar] [CrossRef] [Green Version]
- Hinds, T.D., Jr.; Adeosun, S.O.; Alamodi, A.A.; Stec, D.E. Does bilirubin prevent hepatic steatosis through activation of the PPARalpha nuclear receptor? Med. Hypotheses 2016, 95, 54–57. [Google Scholar] [CrossRef] [Green Version]
- Creeden, J.F.; Gordon, D.M.; Stec, D.E.; Hinds, T.D., Jr. Bilirubin as a metabolic hormone: The physiological relevance of low levels. Am. J. Physiol. Endocrinol. Metab. 2021, 320, E191–E207. [Google Scholar] [CrossRef]
- Hinds, T.D., Jr.; Burns, K.A.; Hosick, P.A.; McBeth, L.; Nestor-Kalinoski, A.; Drummond, H.A.; AlAmodi, A.A.; Hankins, M.W.; Vanden Heuvel, J.P.; Stec, D.E. Biliverdin reductase A attenuates hepatic steatosis by inhibition of glycogen synthase kinase (GSK) 3beta phosphorylation of serine 73 of peroxisome proliferator-activated receptor (PPAR) alpha. J. Biol. Chem. 2016, 291, 25179–25191. [Google Scholar] [CrossRef] [Green Version]
- Mueller, P.A.; Yang, L.; Ubele, M.; Mao, G.; Brandon, J.; Vandra, J.; Nichols, T.C.; Escalante-Alcalde, D.; Morris, A.J.; Smyth, S.S. Coronary Artery Disease Risk-Associated Plpp3 Gene and Its Product Lipid Phosphate Phosphatase 3 Regulate Experimental Atherosclerosis. Arter. Thromb. Vasc. Biol. 2019, 39, 2261–2272. [Google Scholar] [CrossRef] [PubMed]
- Kraemer, M.P.; Mao, G.; Hammill, C.; Yan, B.; Li, Y.; Onono, F.; Smyth, S.S.; Morris, A.J. Effects of diet and hyperlipidemia on levels and distribution of circulating lysophosphatidic acid. J. Lipid Res. 2019, 60, 1818–1828. [Google Scholar] [CrossRef]
- Khan, M.J.; Codreanu, S.G.; Goyal, S.; Wages, P.A.; Gorti, S.K.K.; Pearson, M.J.; Uribe, I.; Sherrod, S.D.; McLean, J.A.; Porter, N.A.; et al. Evaluating a targeted multiple reaction monitoring approach to global untargeted lipidomic analyses of human plasma. Rapid Commun. Mass Spectrom. 2020, 34, e8911. [Google Scholar] [CrossRef]
- Marino, J.S.; Stechschulte, L.A.; Stec, D.E.; Nestor-Kalinoski, A.; Coleman, S.; Hinds, T.D., Jr. Glucocorticoid receptor beta induces hepatic steatosis by augmenting inflammation and inhibition of the peroxisome proliferator-activated receptor (PPAR) alpha. J. Biol. Chem. 2016, 291, 25776–25788. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hinds, T.D., Jr.; Stechschulte, L.A.; Cash, H.A.; Whisler, D.; Banerjee, A.; Yong, W.; Khuder, S.S.; Kaw, M.K.; Shou, W.; Najjar, S.M.; et al. Protein phosphatase 5 mediates lipid metabolism through reciprocal control of glucocorticoid receptor and peroxisome proliferator-activated receptor-gamma (PPARgamma). J. Biol. Chem. 2011, 286, 42911–42922. [Google Scholar] [CrossRef] [Green Version]
- Stec, D.E.; Gordon, D.M.; Nestor-Kalinoski, A.L.; Donald, M.C.; Mitchell, Z.L.; Creeden, J.F.; Hinds, T.D., Jr. Biliverdin Reductase A (BVRA) Knockout in Adipocytes Induces Hypertrophy and Reduces Mitochondria in White Fat of Obese Mice. Biomolecules 2020, 10, 387. [Google Scholar] [CrossRef] [Green Version]
- Shen, Y.; Su, Y.; Silva, F.J.; Weller, A.H.; Sostre-Colon, J.; Titchenell, P.M.; Steger, D.J.; Seale, P.; Soccio, R.E. Shared PPARalpha/gamma Target Genes Regulate Brown Adipocyte Thermogenic Function. Cell Rep. 2020, 30, 3079–3091.e5. [Google Scholar] [CrossRef] [Green Version]
- Goto, T.; Lee, J.Y.; Teraminami, A.; Kim, Y.I.; Hirai, S.; Uemura, T.; Inoue, H.; Takahashi, N.; Kawada, T. Activation of peroxisome proliferator-activated receptor-alpha stimulates both differentiation and fatty acid oxidation in adipocytes. J. Lipid Res. 2011, 52, 873–884. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jeong, S.; Yoon, M. Inhibition of the actions of peroxisome proliferator-activated receptor alpha on obesity by estrogen. Obesity 2007, 15, 1430–1440. [Google Scholar] [CrossRef] [PubMed]
- Yoon, M. The role of PPARalpha in lipid metabolism and obesity: Focusing on the effects of estrogen on PPARalpha actions. Pharmacol. Res. 2009, 60, 151–159. [Google Scholar] [CrossRef]
- Lee, H.; Yoon, M. 17β-estradiol inhibits PPARα of skeletal muscle. Anim. Cells Syst. 2013, 17, 331–340. [Google Scholar] [CrossRef]
- Wang, Y.; Shoemaker, R.; Thatcher, S.E.; Batifoulier-Yiannikouris, F.; English, V.L.; Cassis, L.A. Administration of 17beta-estradiol to ovariectomized obese female mice reverses obesity-hypertension through an ACE2-dependent mechanism. Am. J. Physiol. Endocrinol. Metab. 2015, 308, E1066–E1075. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kersten, S.; Seydoux, J.; Peters, J.M.; Gonzalez, F.J.; Desvergne, B.; Wahli, W. Peroxisome proliferator-activated receptor alpha mediates the adaptive response to fasting. J. Clin. Investig. 1999, 103, 1489–1498. [Google Scholar] [CrossRef] [Green Version]
- Grabacka, M.; Pierzchalska, M.; Dean, M.; Reiss, K. Regulation of Ketone Body Metabolism and the Role of PPARalpha. Int. J. Mol. Sci. 2016, 17, 2093. [Google Scholar] [CrossRef] [Green Version]
- Hopkins, T.A.; Sugden, M.C.; Holness, M.J.; Kozak, R.; Dyck, J.R.; Lopaschuk, G.D. Control of cardiac pyruvate dehydrogenase activity in peroxisome proliferator-activated receptor-alpha transgenic mice. Am. J. Physiol. Heart Circ. Physiol. 2003, 285, H270–H276. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Krause, B.R.; Hartman, A.D. Adipose tissue and cholesterol metabolism. J. Lipid Res. 1984, 25, 97–110. [Google Scholar] [CrossRef]
- Goldstein, J.L.; Brown, M.S. A century of cholesterol and coronaries: From plaques to genes to statins. Cell 2015, 161, 161–172. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sekiya, M.; Yahagi, N.; Matsuzaka, T.; Takeuchi, Y.; Nakagawa, Y.; Takahashi, H.; Okazaki, H.; Iizuka, Y.; Ohashi, K.; Gotoda, T.; et al. SREBP-1-independent regulation of lipogenic gene expression in adipocytes. J. Lipid Res. 2007, 48, 1581–1591. [Google Scholar] [CrossRef] [Green Version]
- Wu, X.; Romero, D.; Swiatek, W.I.; Dorweiler, I.; Kikani, C.K.; Sabic, H.; Zweifel, B.S.; McKearn, J.; Blitzer, J.T.; Nickols, G.A.; et al. PAS kinase drives lipogenesis through SREBP-1 maturation. Cell Rep. 2014, 8, 242–255. [Google Scholar] [CrossRef] [Green Version]
- Ebke, L.A.; Nestor-Kalinoski, A.L.; Slotterbeck, B.D.; Al-Dieri, A.G.; Ghosh-Lester, S.; Russo, L.; Najjar, S.M.; von Grafenstein, H.; McInerney, M.F. Tight association between macrophages and adipocytes in obesity: Implications for adipocyte preparation. Obesity 2014, 22, 1246–1255. [Google Scholar] [CrossRef]
- Zhang, F.; Hao, G.; Shao, M.; Nham, K.; An, Y.; Wang, Q.; Zhu, Y.; Kusminski, C.M.; Hassan, G.; Gupta, R.K.; et al. An Adipose Tissue Atlas: An Image-Guided Identification of Human-like BAT and Beige Depots in Rodents. Cell Metab. 2018, 27, 252–262.e3. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- MacCannell, A.D.V.; Futers, T.S.; Whitehead, A.; Moran, A.; Witte, K.K.; Roberts, L.D. Sexual dimorphism in adipose tissue mitochondrial function and metabolic flexibility in obesity. Int. J. Obes. 2021, 45, 1773–1781. [Google Scholar] [CrossRef]
- Chait, A.; den Hartigh, L.J. Adipose Tissue Distribution, Inflammation and Its Metabolic Consequences, Including Diabetes and Cardiovascular Disease. Front. Cardiovasc. Med. 2020, 7, 22. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sam, S. Differential effect of subcutaneous abdominal and visceral adipose tissue on cardiometabolic risk. Horm. Mol. Biol. Clin. Investig. 2018, 33, 20180014. [Google Scholar] [CrossRef]
- O’Brien, L.; Hosick, P.A.; John, K.; Stec, D.E.; Hinds, T.D., Jr. Biliverdin reductase isozymes in metabolism. Trends Endocrinol. Metab. 2015, 26, 212–220. [Google Scholar] [CrossRef] [Green Version]
- John, K.; Marino, J.S.; Sanchez, E.R.; Hinds, T.D., Jr. The glucocorticoid receptor: Cause of or cure for obesity? Am. J. Physiol. Endocrinol. Metab. 2016, 310, E249–E257. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yu, B.L.; Zhao, S.P.; Hu, J.R. Cholesterol imbalance in adipocytes: A possible mechanism of adipocytes dysfunction in obesity. Obes. Rev. Off. J. Int. Assoc. Study Obes. 2010, 11, 560–567. [Google Scholar] [CrossRef]
- la Rose, A.M.; Bazioti, V.; Westerterp, M. Adipocyte Membrane Cholesterol Regulates Obesity. Arter. Thromb. Vasc. Biol. 2018, 38, 687–689. [Google Scholar] [CrossRef] [Green Version]
- Saraswathi, V.; Gao, L.; Morrow, J.D.; Chait, A.; Niswender, K.D.; Hasty, A.H. Fish oil increases cholesterol storage in white adipose tissue with concomitant decreases in inflammation, hepatic steatosis, and atherosclerosis in mice. J. Nutr. 2007, 137, 1776–1782. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chui, P.C.; Guan, H.P.; Lehrke, M.; Lazar, M.A. PPARgamma regulates adipocyte cholesterol metabolism via oxidized LDL receptor 1. J. Clin. Investig. 2005, 115, 2244–2256. [Google Scholar] [CrossRef] [Green Version]
- Chinetti, G.; Lestavel, S.; Fruchart, J.C.; Clavey, V.; Staels, B. Peroxisome proliferator-activated receptor alpha reduces cholesterol esterification in macrophages. Circ. Res. 2003, 92, 212–217. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Smedlund, K.B.; Sanchez, E.R.; Hinds, T.D., Jr. FKBP51 and the molecular chaperoning of metabolism. Trends Endocrinol. Metab. 2021, 32, 862–874. [Google Scholar] [CrossRef]
- Chiu, M.; McBeth, L.; Sindhwani, P.; Hinds, T.D. Deciphering the Roles of Thiazolidinediones and PPARgamma in Bladder Cancer. PPAR Res. 2017, 2017, 4810672. [Google Scholar] [CrossRef] [Green Version]
- Kim, J.B.; Spiegelman, B.M. ADD1/SREBP1 promotes adipocyte differentiation and gene expression linked to fatty acid metabolism. Genes Dev. 1996, 10, 1096–1107. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Crewe, C.; Zhu, Y.; Paschoal, V.A.; Joffin, N.; Ghaben, A.L.; Gordillo, R.; Oh, D.Y.; Liang, G.; Horton, J.D.; Scherer, P.E. SREBP-regulated adipocyte lipogenesis is dependent on substrate availability and redox modulation of mTORC1. JCI Insight 2019, 5, e129397. [Google Scholar] [CrossRef]
- Miyazaki, M.; Kim, Y.C.; Gray-Keller, M.P.; Attie, A.D.; Ntambi, J.M. The biosynthesis of hepatic cholesterol esters and triglycerides is impaired in mice with a disruption of the gene for stearoyl-CoA desaturase 1. J. Biol. Chem. 2000, 275, 30132–30138. [Google Scholar] [CrossRef] [Green Version]
- Ntambi, J.M.; Miyazaki, M.; Stoehr, J.P.; Lan, H.; Kendziorski, C.M.; Yandell, B.S.; Song, Y.; Cohen, P.; Friedman, J.M.; Attie, A.D. Loss of stearoyl-CoA desaturase-1 function protects mice against adiposity. Proc. Natl. Acad. Sci. USA 2002, 99, 11482–11486. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dongil, P.; Perez-Garcia, A.; Hurtado-Carneiro, V.; Herrero-de-Dios, C.; Blazquez, E.; Alvarez, E.; Sanz, C. Pas Kinase Deficiency Triggers Antioxidant Mechanisms in the Liver. Sci. Rep. 2018, 8, 13810. [Google Scholar] [CrossRef] [Green Version]
- Adeosun, S.O.; Moore, K.H.; Lang, D.M.; Nwaneri, A.C.; Hinds, T.D., Jr.; Stec, D.E. A Novel Fluorescence-Based Assay for the Measurement of Biliverdin Reductase Activity. React. Oxyg. Species 2018, 5, 35–45. [Google Scholar] [CrossRef] [Green Version]
- Adeosun, S.O.; Gordon, D.M.; Weeks, M.F.; Moore, K.H.; Hall, J.E.; Hinds, T.D., Jr.; Stec, D.E. Loss of biliverdin reductase-A promotes lipid accumulation and lipotoxicity in mouse proximal tubule cells. Am. J. Physiol. Ren. Physiol. 2018, 315, F323–F331. [Google Scholar] [CrossRef]
- Dongil, P.; Perez-Garcia, A.; Hurtado-Carneiro, V.; Herrero-de-Dios, C.; Alvarez, E.; Sanz, C. PAS kinase deficiency reduces aging effects in mice. Aging 2020, 12, 2275–2301. [Google Scholar] [CrossRef]
- Perez-Garcia, A.; Dongil, P.; Hurtado-Carneiro, V.; Blazquez, E.; Sanz, C.; Alvarez, E. PAS Kinase deficiency alters the glucokinase function and hepatic metabolism. Sci. Rep. 2018, 8, 11091. [Google Scholar] [CrossRef]
- Perez-Garcia, A.; Dongil, P.; Hurtado-Carneiro, V.; Blazquez, E.; Sanz, C.; Alvarez, E. High-fat diet alters PAS kinase regulation by fasting and feeding in liver. J. Nutr. Biochem. 2018, 57, 14–25. [Google Scholar] [CrossRef] [PubMed]
- Hao, H.X.; Cardon, C.M.; Swiatek, W.; Cooksey, R.C.; Smith, T.L.; Wilde, J.; Boudina, S.; Abel, E.D.; McClain, D.A.; Rutter, J. PAS kinase is required for normal cellular energy balance. Proc. Natl. Acad. Sci. USA 2007, 104, 15466–15471. [Google Scholar] [CrossRef] [Green Version]
- Zhang, D.D.; Zhang, J.G.; Wang, Y.Z.; Liu, Y.; Liu, G.L.; Li, X.Y. Per-Arnt-Sim Kinase (PASK): An Emerging Regulator of Mammalian Glucose and Lipid Metabolism. Nutrients 2015, 7, 7437–7450. [Google Scholar] [CrossRef] [Green Version]
- Grabacka, M.; Reiss, K. Anticancer Properties of PPARalpha-Effects on Cellular Metabolism and Inflammation. PPAR Res. 2008, 2008, 930705. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stec, D.E.; Hinds, T.D., Jr. Natural Product Heme Oxygenase Inducers as Treatment for Nonalcoholic Fatty Liver Disease. Int. J. Mol. Sci. 2020, 21, 9493. [Google Scholar] [CrossRef]
- Hinds, T.D., Jr.; Stec, D.E. Bilirubin Safeguards Cardiorenal and Metabolic Diseases: A Protective Role in Health. Curr. Hypertens. Rep. 2019, 21, 87. [Google Scholar] [CrossRef] [PubMed]
- Weaver, L.; Hamoud, A.R.; Stec, D.E.; Hinds, T.D., Jr. Biliverdin reductase and bilirubin in hepatic disease. Am. J. Physiol. Gastrointest. Liver Physiol. 2018, 314, G668–G676. [Google Scholar] [CrossRef]
- Sundararaghavan, V.L.; Binepal, S.; Stec, D.E.; Sindhwani, P.; Hinds, T.D., Jr. Bilirubin, a new therapeutic for kidney transplant? Transpl. Rev. 2018, 32, 234–240. [Google Scholar] [CrossRef]
- Hinds, T.D., Jr.; Stec, D.E. Bilirubin, a Cardiometabolic Signaling Molecule. Hypertension 2018, 72, 788–795. [Google Scholar] [CrossRef]
- Hamoud, A.R.; Weaver, L.; Stec, D.E.; Hinds, T.D., Jr. Bilirubin in the Liver-Gut Signaling Axis. Trends Endocrinol. Metab. 2018, 29, 140–150. [Google Scholar] [CrossRef] [PubMed]
- Hinds, T.D., Jr.; Creeden, J.F.; Gordon, D.M.; Spegele, A.C.; Britton, S.L.; Koch, L.G.; Stec, D.E. Rats Genetically Selected for High Aerobic Exercise Capacity Have Elevated Plasma Bilirubin by Upregulation of Hepatic Biliverdin Reductase-A (BVRA) and Suppression of UGT1A1. Antioxidants 2020, 9, 889. [Google Scholar] [CrossRef]
- Lumeng, C.N.; Bodzin, J.L.; Saltiel, A.R. Obesity induces a phenotypic switch in adipose tissue macrophage polarization. J. Clin. Invest. 2007, 117, 175–184. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Becerril, S.; Rodriguez, A.; Catalan, V.; Mendez-Gimenez, L.; Ramirez, B.; Sainz, N.; Llorente, M.; Unamuno, X.; Gomez-Ambrosi, J.; Fruhbeck, G. Targeted disruption of the iNOS gene improves adipose tissue inflammation and fibrosis in leptin-deficient ob/ob mice: Role of tenascin C. Int. J. Obes. 2018, 42, 1458–1470. [Google Scholar] [CrossRef] [Green Version]
- Hinds, T.D.; Peck, B.; Shek, E.; Stroup, S.; Hinson, J.; Arthur, S.; Marino, J.S. Overexpression of Glucocorticoid Receptor beta Enhances Myogenesis and Reduces Catabolic Gene Expression. Int. J. Mol. Sci. 2016, 17, 232. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hinds, T.D., Jr.; Ramakrishnan, S.; Cash, H.A.; Stechschulte, L.A.; Heinrich, G.; Najjar, S.M.; Sanchez, E.R. Discovery of glucocorticoid receptor-beta in mice with a role in metabolism. Mol. Endocrinol. 2010, 24, 1715–1727. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stechschulte, L.A.; Wuescher, L.; Marino, J.S.; Hill, J.W.; Eng, C.; Hinds, T.D., Jr. Glucocorticoid receptor beta stimulates Akt1 growth pathway by attenuation of PTEN. J. Biol. Chem. 2014, 289, 17885–17894. [Google Scholar] [CrossRef] [Green Version]
- McBeth, L.; Nwaneri, A.C.; Grabnar, M.; Demeter, J.; Nestor-Kalinoski, A.; Hinds, T.D., Jr. Glucocorticoid receptor beta increases migration of human bladder cancer cells. Oncotarget 2016, 7, 27313–27324. [Google Scholar] [CrossRef] [Green Version]
- McBeth, L.; Grabnar, M.; Selman, S.; Hinds, T.D., Jr. Involvement of the Androgen and Glucocorticoid Receptors in Bladder Cancer. Int. J. Endocrinol. 2015, 2015, 384860. [Google Scholar] [CrossRef] [Green Version]
- Nguyen, Q.T.; Kim, D.; Iamsawat, S.; Le, H.T.; Kim, S.; Qiu, K.T.; Hinds, T.D.; Bazeley, P.; O’Shea, J.J.; Choi, J.; et al. Cutting Edge: Steroid Responsiveness in Foxp3(+) Regulatory T Cells Determines Steroid Sensitivity during Allergic Airway Inflammation in Mice. J. Immunol. 2021, 207, 765–770. [Google Scholar] [CrossRef] [PubMed]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene | Forward Primer | Reverse Primer |
|---|---|---|
| 36B4 | CACTCTCGCTTTCTGGAGGG | ACGCGCTTGTACCCATTGAT |
| Cre | GAACCTGATGGACATGTTCAGG | AGTGCGTTCGAACGCTAGAGCCTGT |
| Ppara | AGAAGTTGCAGGAGGGGATT | TTGAAGGAGCTTTGGGAAGA |
| Pask | GAATCCGACTGAGACTTGCG | TAACTAACACTCGCCGCCAC |
| Scarb1 | CCCCAGGTTCTTCACTACGC | TCCTTATCCTGGGAGCCCTT |
| Abca1 | GGCAATGAGTGTGCCAGAGTTA | TAGTCACATGTGGCACCGTTTT |
| Abcg1 | TCCCCACCTGTAAGTAATTGCA | TCGGACCCTTATCATTCTCTACAGA |
| Adgre1 | GCCCAGGAGTGGAATGTCAA | GCAGACTGAGTTAGGACCACA |
| Nos2 | CCTTGGTGAAGGGACTGAGC | TCCGTGGAGTGAACAAGACC |
| Fizz1 | GGGATGACTGCTACTGGGTG | TCAACGAGTAAGCACAGGCA |
| Arg1 | AAGAGTCAGTGTGGTGCTGG | TGGTTGTCAGGGGAGTGTTG |
| Tnfa | GACTCAAATGGGCTTTCCGA | TCCAGCCTCATTCTGAGACAGAG |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hinds, T.D., Jr.; Kipp, Z.A.; Xu, M.; Yiannikouris, F.B.; Morris, A.J.; Stec, D.F.; Wahli, W.; Stec, D.E. Adipose-Specific PPARα Knockout Mice Have Increased Lipogenesis by PASK–SREBP1 Signaling and a Polarity Shift to Inflammatory Macrophages in White Adipose Tissue. Cells 2022, 11, 4. https://doi.org/10.3390/cells11010004
Hinds TD Jr., Kipp ZA, Xu M, Yiannikouris FB, Morris AJ, Stec DF, Wahli W, Stec DE. Adipose-Specific PPARα Knockout Mice Have Increased Lipogenesis by PASK–SREBP1 Signaling and a Polarity Shift to Inflammatory Macrophages in White Adipose Tissue. Cells. 2022; 11(1):4. https://doi.org/10.3390/cells11010004
Chicago/Turabian StyleHinds, Terry D., Jr., Zachary A. Kipp, Mei Xu, Frederique B. Yiannikouris, Andrew J. Morris, Donald F. Stec, Walter Wahli, and David E. Stec. 2022. "Adipose-Specific PPARα Knockout Mice Have Increased Lipogenesis by PASK–SREBP1 Signaling and a Polarity Shift to Inflammatory Macrophages in White Adipose Tissue" Cells 11, no. 1: 4. https://doi.org/10.3390/cells11010004