
The Anti-Inflammatory Effect of the β1-Adrenergic Receptor Antagonist Metoprolol on High Glucose Treated Human Microvascular Retinal Endothelial Cells

 , , , and
, , , and
Abstract
:1. Introduction
2. Materials and Methods
2.1. Reagents
2.2. HREC Culture and Treatments
2.3. Cell Viability
2.4. Tube Formation Assay
2.5. ROS Measurements
2.6. Western Blot Analysis
2.7. High-Content Screening (HCS) and Image Analysis
2.8. Extraction of Total RNA and Real-Time Reverse Transcriptase-Polymerase Chain Reaction (RT-PCR)
2.9. Statistical Analysis
3. Results
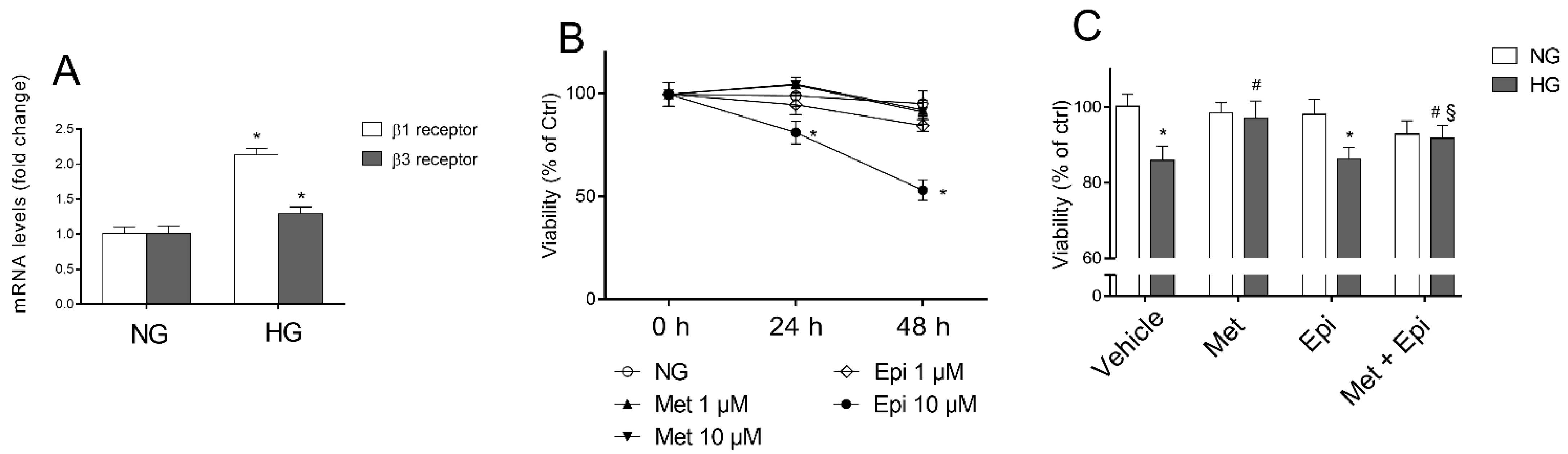
3.1. β1-Adrenergic Receptor Blockade Prevents HG-Induced Effects upon Metabolic Activity and Cell Proliferation in HREC
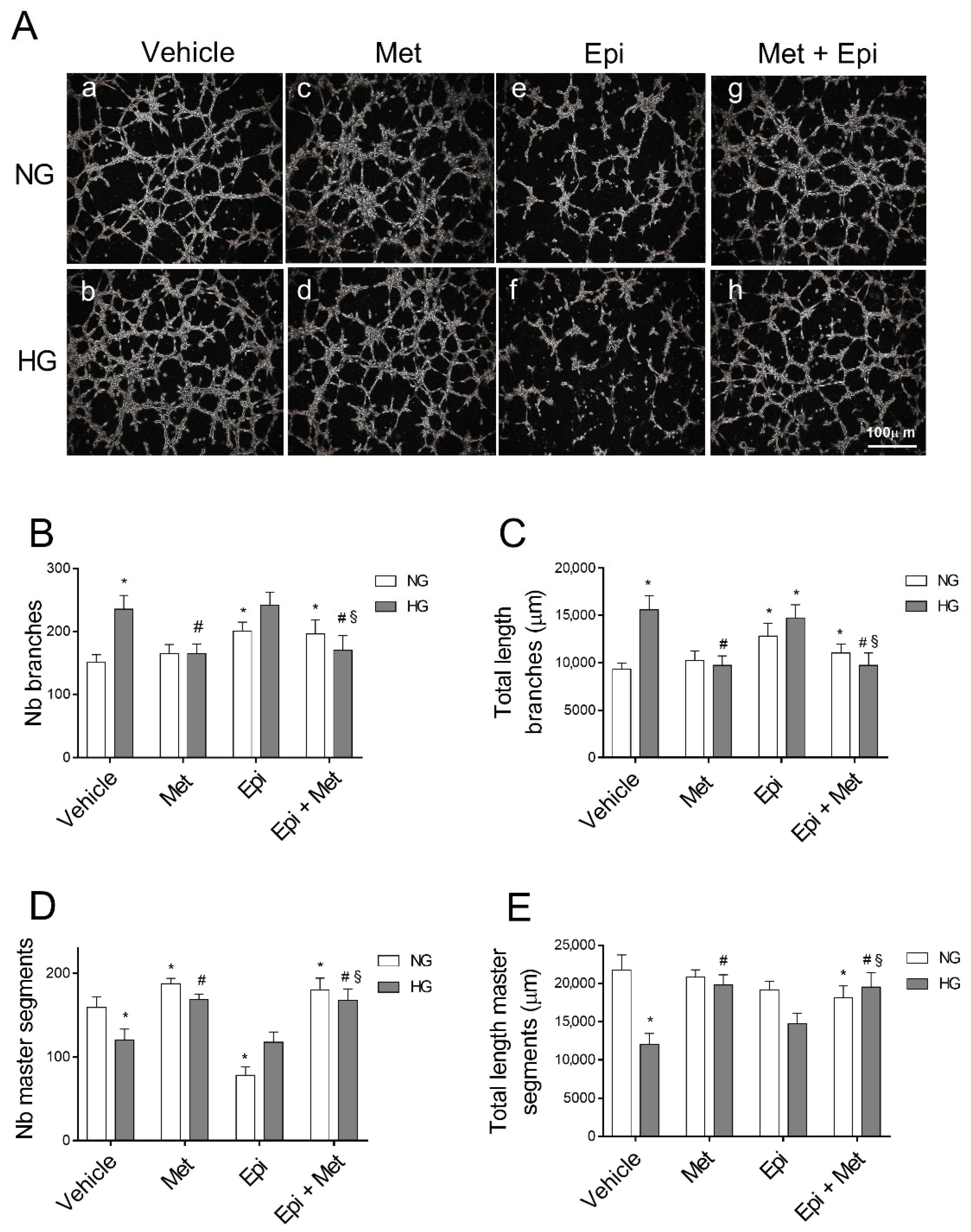
3.2. Metoprolol Counteracts the Increase in the Tube-like Structures of HREC Stimulated with HG
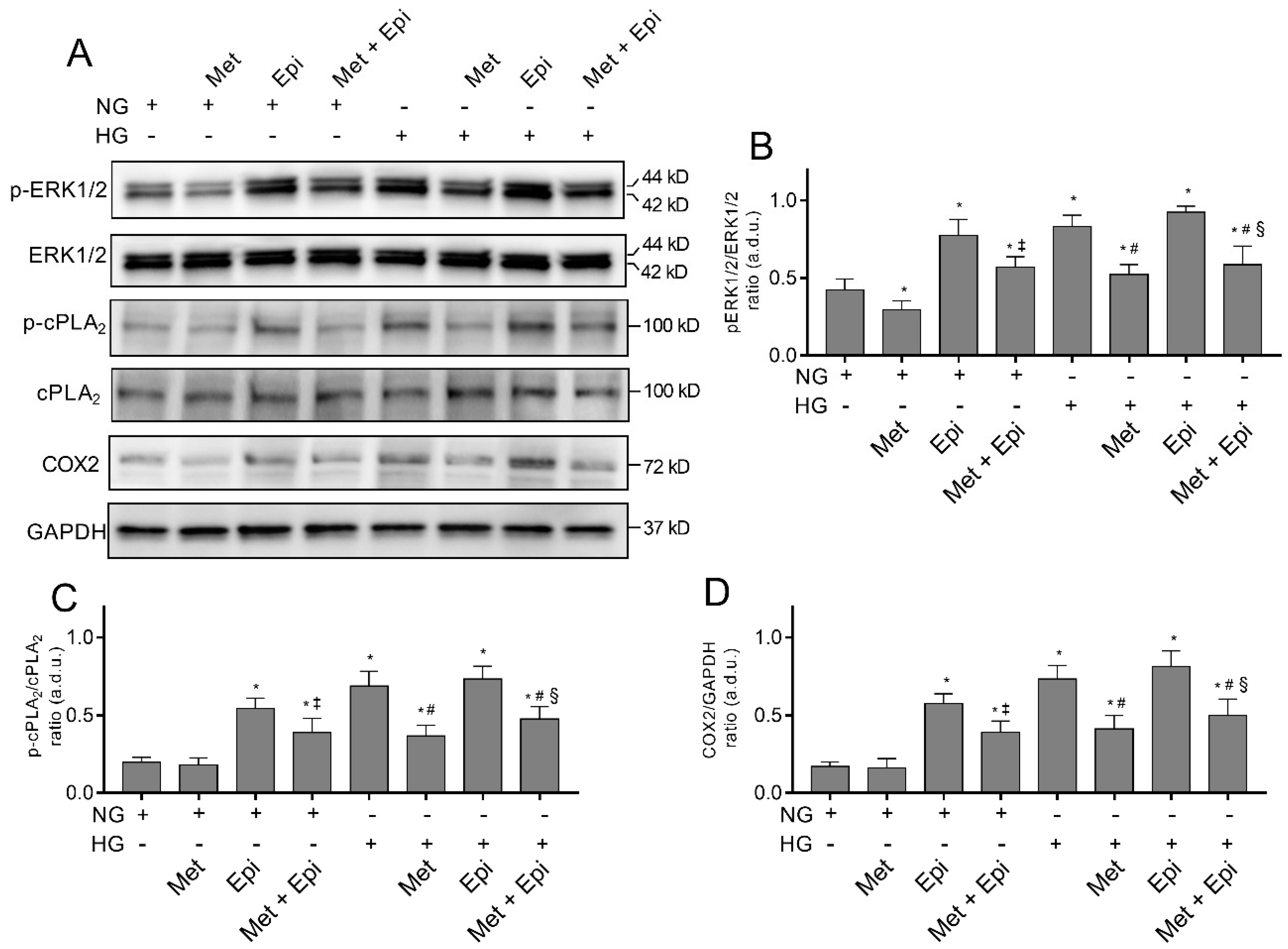
3.3. Metoprolol Down-Regulates the ERK1/2/cPLA2/COX2 Axis in HREC Treated with HG
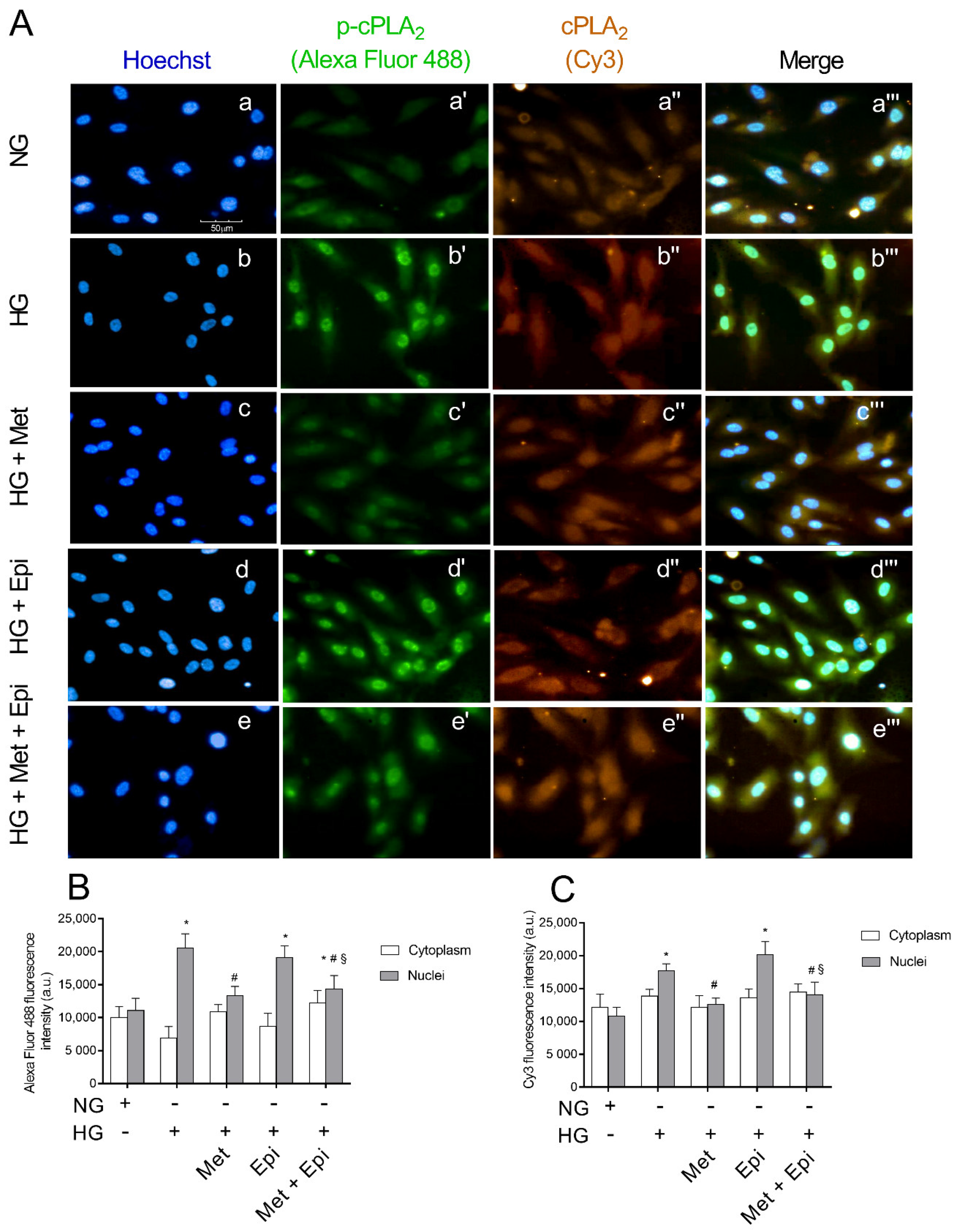
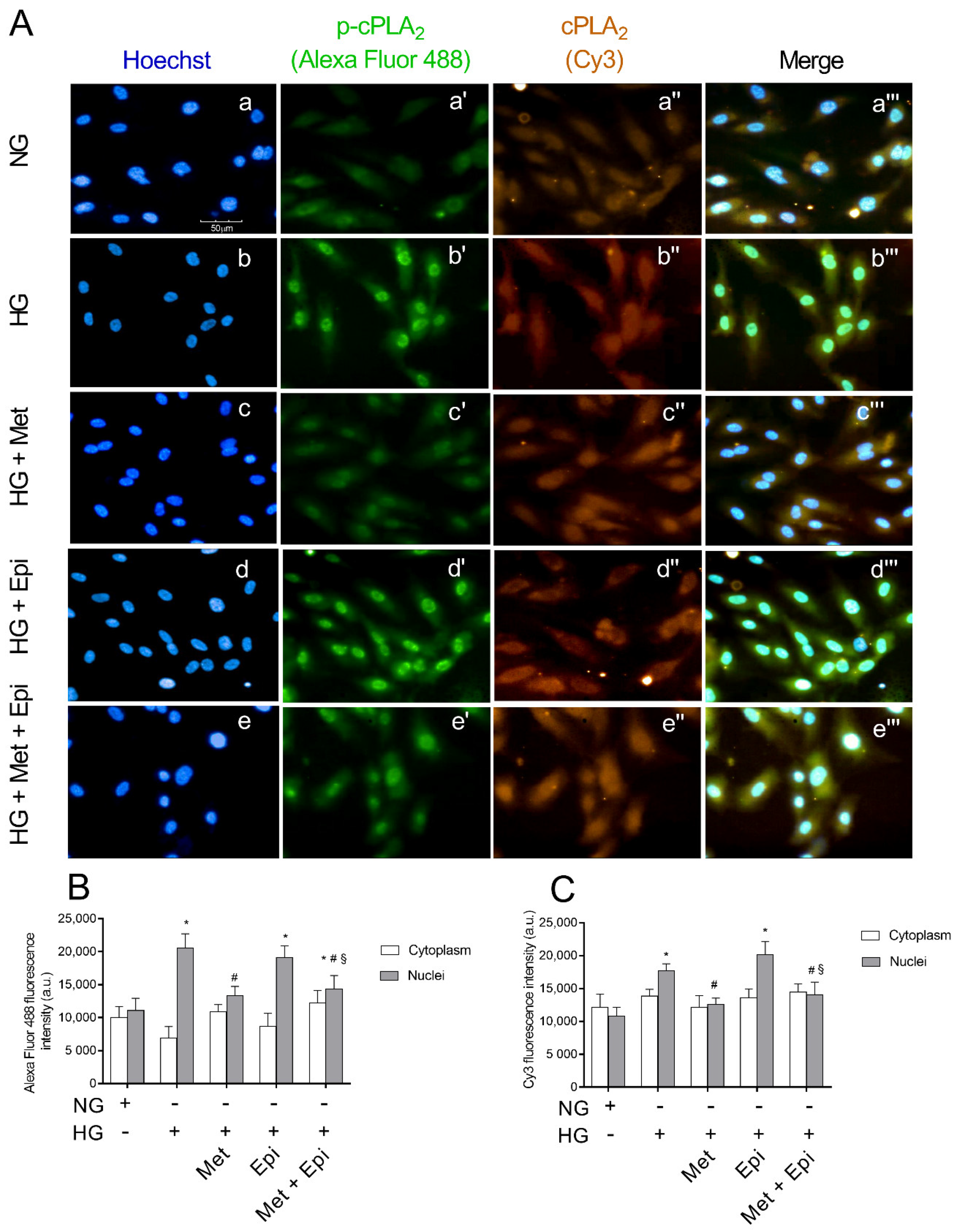
3.4. Metoprolol Reduced cPLA2 Nuclear Translocation in HREC Challenged with HG
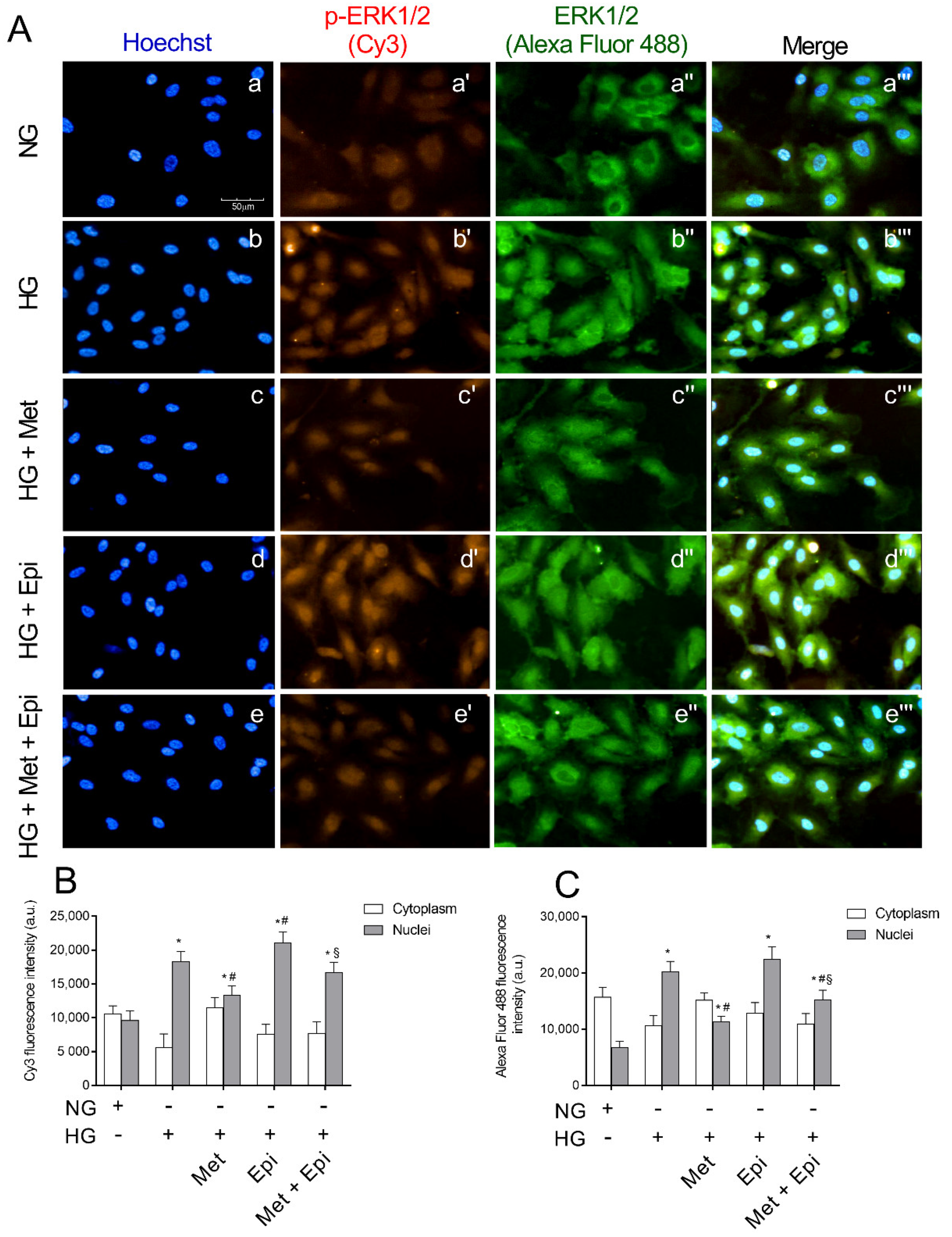
3.5. Metoprolol Reduced ERK 1/2 Phosphorylation in HREC Challenged with HG
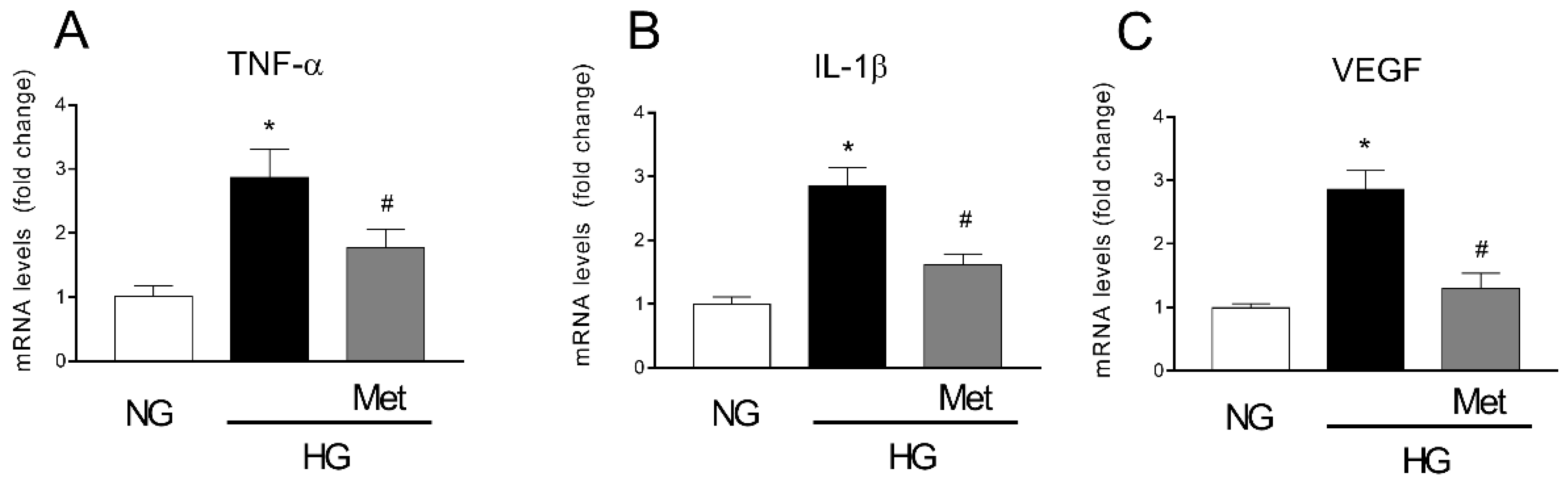
3.6. Metoprolol Down-Regulates the Release of TNF-α, VEGF, and IL-1b in HREC Treated with HG
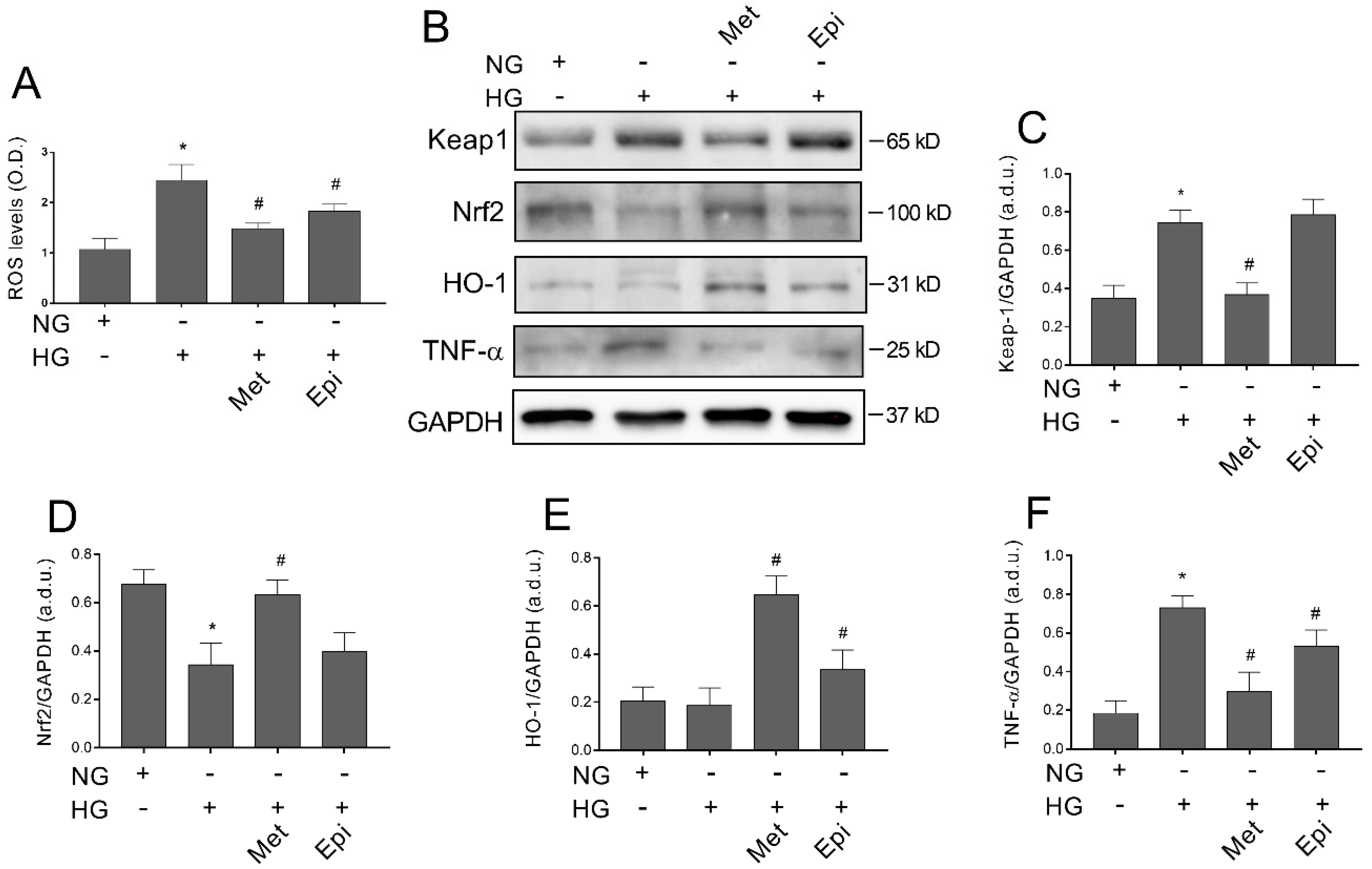
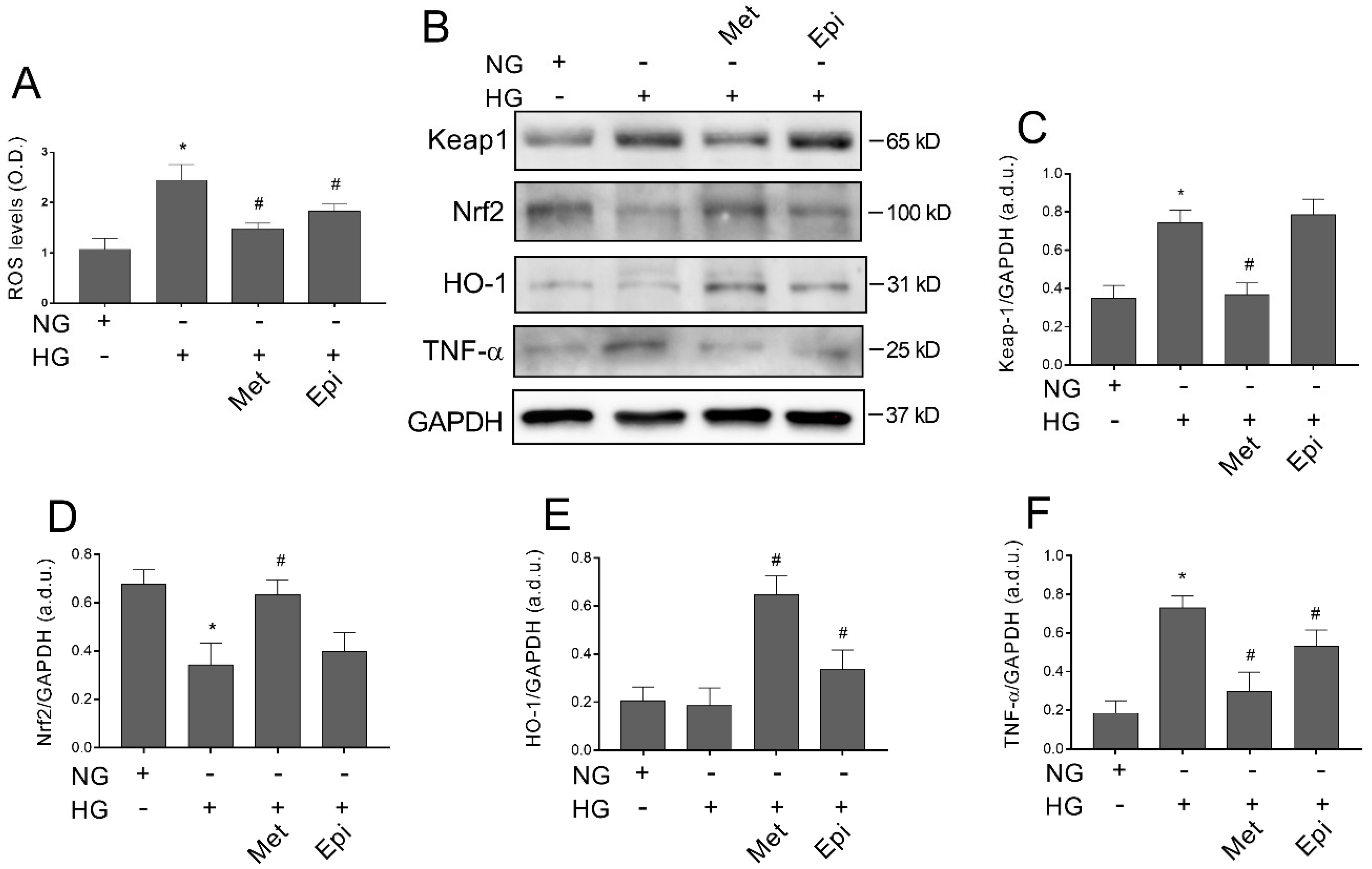
3.7. Metoprolol Counteracts Glucose-Induced ROS Accumulation by Activating the Keap1/Nrf2/HO-1 Pathway in HREC
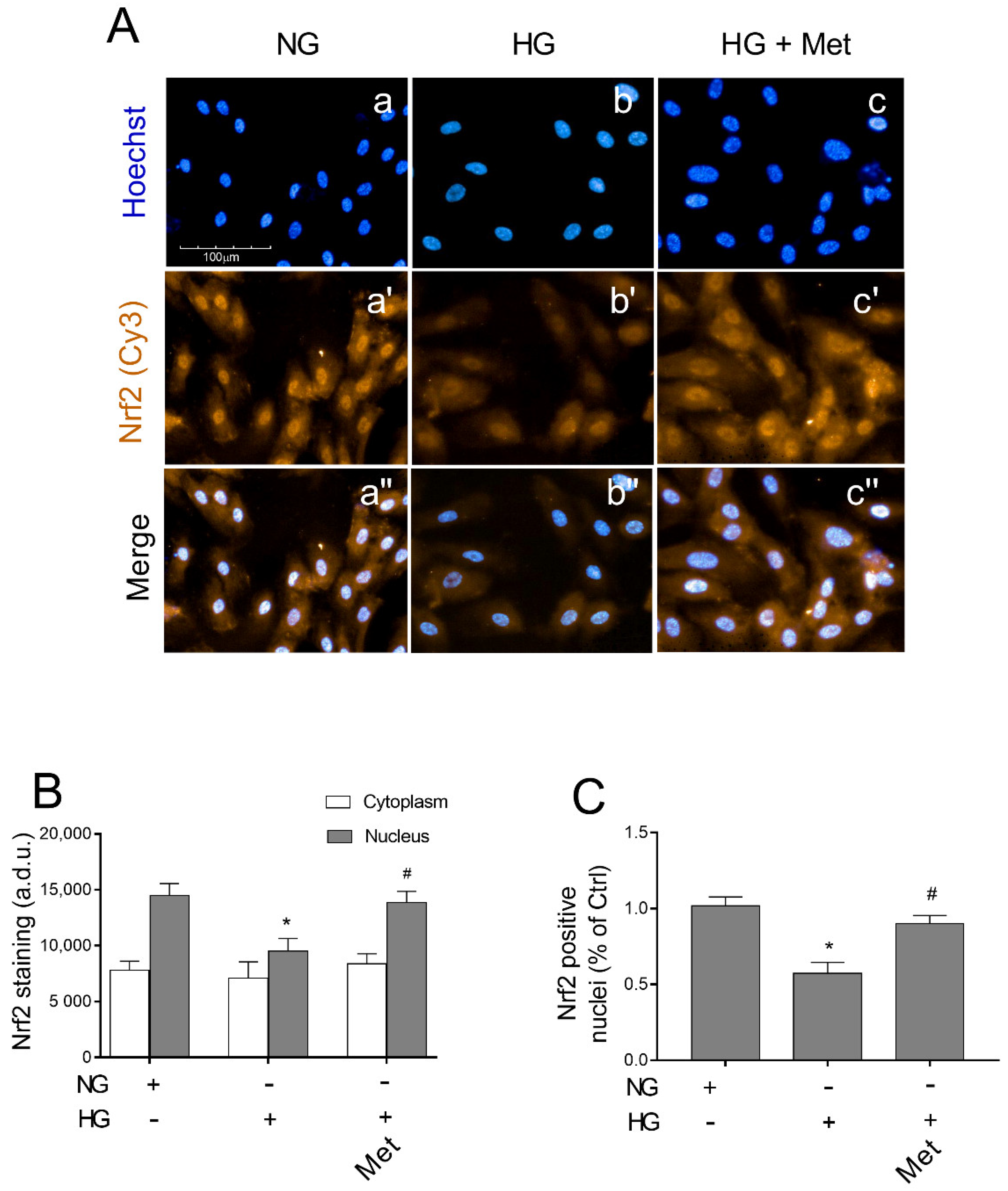
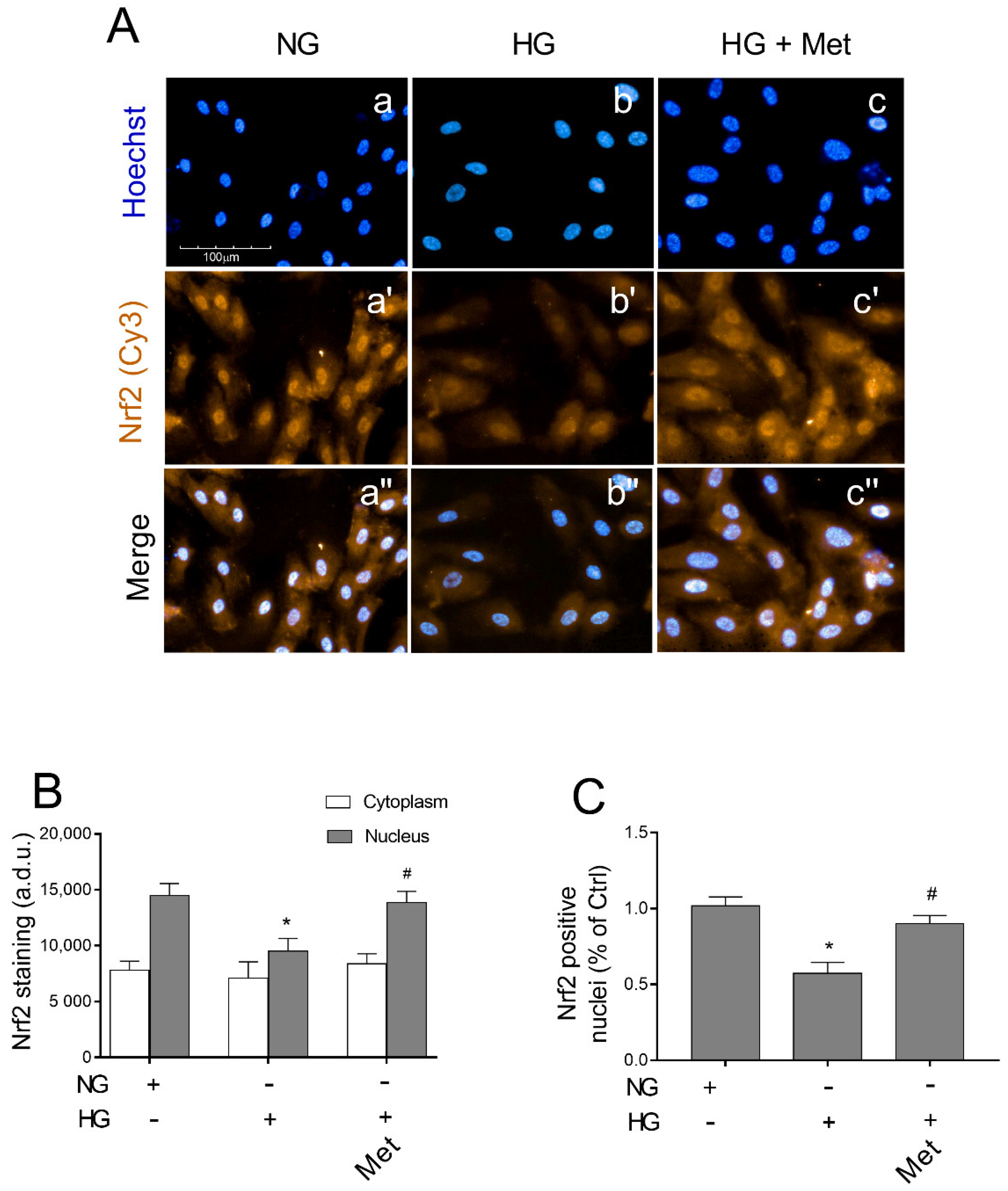
3.8. Metoprolol Induced Nuclear Nrf2 Nuclear Compartmentalization in HG-Treated HREC
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Antonetti, D.A.; Silva, P.S.; Stitt, A.W. Current Understanding of the Molecular and Cellular Pathology of Diabetic Retinopathy. Nat. Rev. Endocrinol. 2021, 17, 195–206. [Google Scholar] [CrossRef]
- Flaxman, S.R.; Bourne, R.R.A.; Resnikoff, S.; Ackland, P.; Braithwaite, T.; Cicinelli, M.V.; Das, A.; Jonas, J.B.; Keeffe, J.; Kempen, J.H.; et al. Global Causes of Blindness and Distance Vision Impairment 1990–2020: A Systematic Review and Meta-Analysis. Lancet Glob. Health 2017, 5, e1221–e1234. [Google Scholar] [CrossRef] [Green Version]
- Rudraraju, M.; Narayanan, S.P.; Somanath, P.R. Regulation of Blood-Retinal Barrier Cell-Junctions in Diabetic Retinopathy. Pharmacol. Res. 2020, 161, 105115. [Google Scholar] [CrossRef]
- Wang, W.; Lo, A. Diabetic Retinopathy: Pathophysiology and Treatments. IJMS 2018, 19, 1816. [Google Scholar] [CrossRef] [Green Version]
- Gonzalez, V.H.; Campbell, J.; Holekamp, N.M.; Kiss, S.; Loewenstein, A.; Augustin, A.J.; Ma, J.; Ho, A.C.; Patel, V.; Whitcup, S.M.; et al. Early and Long-Term Responses to Anti–Vascular Endothelial Growth Factor Therapy in Diabetic Macular Edema: Analysis of Protocol I Data. Am. J. Ophthalmol. 2016, 172, 72–79. [Google Scholar] [CrossRef] [Green Version]
- Casini, G.; Dal Monte, M.; Fornaciari, I.; Filippi, L.; Bagnoli, P. The β-Adrenergic System as a Possible New Target for Pharmacologic Treatment of Neovascular Retinal Diseases. Prog. Retin. Eye Res. 2014, 42, 103–129. [Google Scholar] [CrossRef]
- Laverty, R. Catecholamines: Role in Health and Disease. Drugs 1978, 16, 418–440. [Google Scholar] [CrossRef]
- Sorriento, D.; Santulli, G.; Del Giudice, C.; Anastasio, A.; Trimarco, B.; Iaccarino, G. Endothelial Cells Are Able to Synthesize and Release Catecholamines Both In Vitro and In Vivo. Hypertension 2012, 60, 129–136. [Google Scholar] [CrossRef] [Green Version]
- Steinle, J.J.; Chin, V.C.; Williams, K.P.; Panjala, S.R. Beta-Adrenergic Receptor Stimulation Modulates INOS Protein Levels through P38 and ERK1/2 Signaling in Human Retinal Endothelial Cells. Exp. Eye Res. 2008, 87, 30–34. [Google Scholar] [CrossRef]
- Steinle, J.J. Sympathetic Neurotransmission Modulates Expression of Inflammatory Markers in the Rat Retina. Exp. Eye Res. 2007, 84, 118–125. [Google Scholar] [CrossRef]
- Forrester, J.V.; Kuffova, L.; Delibegovic, M. The Role of Inflammation in Diabetic Retinopathy. Front. Immunol. 2020, 11, 583687. [Google Scholar] [CrossRef] [PubMed]
- Murakami, M.; Nakatani, Y.; Atsumi, G.; Inoue, K.; Kudo, I. Regulatory Functions of Phospholipase A2. Crit. Rev. Immunol. 2017, 37, 121–179. [Google Scholar] [CrossRef] [PubMed]
- Lupo, G.; Motta, C.; Giurdanella, G.; Anfuso, C.D.; Alberghina, M.; Drago, F.; Salomone, S.; Bucolo, C. Role of Phospholipases A2 in Diabetic Retinopathy: In Vitro and in Vivo Studies. Biochem. Pharmacol. 2013, 86, 1603–1613. [Google Scholar] [CrossRef]
- Leslie, C.C. Cytosolic Phospholipase A2: Physiological Function and Role in Disease. J. Lipid Res. 2015, 56, 1386–1402. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Murakami, M.; Kudo, I. Phospholipase A2. J. Biochem. 2002, 131, 285–292. [Google Scholar] [CrossRef]
- Donath, M.Y. Targeting Inflammation in the Treatment of Type 2 Diabetes: Time to Start. Nat. Rev. Drug Discov. 2014, 13, 465–476. [Google Scholar] [CrossRef]
- Hotamisligil, G.S. Inflammation and Metabolic Disorders. Nature 2006, 444, 860–867. [Google Scholar] [CrossRef]
- Kensler, T.W.; Wakabayashi, N.; Biswal, S. Cell Survival Responses to Environmental Stresses via the Keap1-Nrf2-ARE Pathway. Annu. Rev. Pharmacol. Toxicol. 2007, 47, 89–116. [Google Scholar] [CrossRef]
- Mitsuishi, Y.; Motohashi, H.; Yamamoto, M. The Keap1–Nrf2 System in Cancers: Stress Response and Anabolic Metabolism. Front. Oncol. 2012, 2, 200. [Google Scholar] [CrossRef] [Green Version]
- Kansanen, E.; Kuosmanen, S.M.; Leinonen, H.; Levonen, A.-L. The Keap1-Nrf2 Pathway: Mechanisms of Activation and Dysregulation in Cancer. Redox Biol. 2013, 1, 45–49. [Google Scholar] [CrossRef] [Green Version]
- Zhang, D.D.; Hannink, M. Distinct Cysteine Residues in Keap1 Are Required for Keap1-Dependent Ubiquitination of Nrf2 and for Stabilization of Nrf2 by Chemopreventive Agents and Oxidative Stress. Mol. Cell Biol. 2003, 23, 8137–8151. [Google Scholar] [CrossRef] [Green Version]
- Chen, X.; Qi, J.; Wu, Q.; Jiang, H.; Wang, J.; Chen, W.; Mao, A.; Zhu, M. High Glucose Inhibits Vascular Endothelial Keap1/Nrf2/ARE Signal Pathway via Downregulation of Monomethyltransferase SET8 Expression. Acta Biochim. Biophys. Sin. 2020, 52, 506–516. [Google Scholar] [CrossRef]
- Chen, G.; Chen, X.; Niu, C.; Huang, X.; An, N.; Sun, J.; Huang, S.; Ye, W.; Li, S.; Shen, Y.; et al. Baicalin Alleviates Hyperglycemia-Induced Endothelial Impairment via Nrf2. J. Endocrinol. 2019, 240, 81–98. [Google Scholar] [CrossRef] [PubMed]
- Giurdanella, G.; Anfuso, C.D.; Olivieri, M.; Lupo, G.; Caporarello, N.; Eandi, C.M.; Drago, F.; Bucolo, C.; Salomone, S. Aflibercept, Bevacizumab and Ranibizumab Prevent Glucose-Induced Damage in Human Retinal Pericytes in Vitro, through a PLA2/COX-2/VEGF-A Pathway. Biochem. Pharmacol. 2015, 96, 278–287. [Google Scholar] [CrossRef] [PubMed]
- Lupo, G.; Cambria, M.T.; Olivieri, M.; Rocco, C.; Caporarello, N.; Longo, A.; Zanghì, G.; Salmeri, M.; Foti, M.C.; Anfuso, C.D. Anti-angiogenic Effect of Quercetin and Its 8-methyl Pentamethyl Ether Derivative in Human Microvascular Endothelial Cells. J. Cell. Mol. Med. 2019, 23, 6565–6577. [Google Scholar] [CrossRef] [Green Version]
- Giurdanella, G.; Lupo, G.; Gennuso, F.; Conti, F.; Furno, D.L.; Mannino, G.; Anfuso, C.D.; Drago, F.; Salomone, S.; Bucolo, C. Activation of the VEGF-A/ERK/PLA2 Axis Mediates Early Retinal Endothelial Cell Damage Induced by High Glucose: New Insight from an In Vitro Model of Diabetic Retinopathy. IJMS 2020, 21, 7528. [Google Scholar] [CrossRef]
- Mannino, G.; Longo, A.; Gennuso, F.; Anfuso, C.D.; Lupo, G.; Giurdanella, G.; Giuffrida, R.; Lo Furno, D. Effects of High Glucose Concentration on Pericyte-Like Differentiated Human Adipose-Derived Mesenchymal Stem Cells. IJMS 2021, 22, 4604. [Google Scholar] [CrossRef] [PubMed]
- Anfuso, C.D.; Giurdanella, G.; Motta, C.; Muriana, S.; Lupo, G.; Ragusa, N.; Alberghina, M. PKCα-MAPK/ERK-Phospholipase A2 Signaling Is Required for Human Melanoma-Enhanced Brain Endothelial Cell Proliferation and Motility. Microvasc. Res. 2009, 78, 338–357. [Google Scholar] [CrossRef]
- Anfuso, C.D.; Longo, A.; Distefano, A.; Amorini, A.M.; Salmeri, M.; Zanghì, G.; Giallongo, C.; Giurdanella, G.; Lupo, G. Uveal Melanoma Cells Elicit Retinal Pericyte Phenotypical and Biochemical Changes in an in Vitro Model of Coculture. IJMS 2020, 21, 5557. [Google Scholar] [CrossRef]
- Yu, Z.; Zhang, T.; Gong, C.; Sheng, Y.; Lu, B.; Zhou, L.; Ji, L.; Wang, Z. Erianin Inhibits High Glucose-Induced Retinal Angiogenesis via Blocking ERK1/2-Regulated HIF-1α-VEGF/VEGFR2 Signaling Pathway. Sci. Rep. 2016, 6, 34306. [Google Scholar] [CrossRef] [Green Version]
- Tan, A.; Li, T.; Ruan, L.; Yang, J.; Luo, Y.; Li, L.; Wu, X. Knockdown of Malat1 Alleviates High-Glucose-Induced Angiogenesis through Regulating MiR-205-5p/VEGF-A Axis. Exp. Eye Res. 2021, 207, 108585. [Google Scholar] [CrossRef]
- Wang, M.; Wang, Y.; Xie, T.; Zhan, P.; Zou, J.; Nie, X.; Shao, J.; Zhuang, M.; Tan, C.; Tan, J.; et al. Prostaglandin E2/EP2 Receptor Signalling Pathway Promotes Diabetic Retinopathy in a Rat Model of Diabetes. Diabetologia 2019, 62, 335–348. [Google Scholar] [CrossRef] [Green Version]
- Giurdanella, G.; Lazzara, F.; Caporarello, N.; Lupo, G.; Anfuso, C.D.; Eandi, C.M.; Leggio, G.M.; Drago, F.; Bucolo, C.; Salomone, S. Sulodexide Prevents Activation of the PLA2/COX-2/VEGF Inflammatory Pathway in Human Retinal Endothelial Cells by Blocking the Effect of AGE/RAGE. Biochem. Pharmacol. 2017, 142, 145–154. [Google Scholar] [CrossRef]
- Álvarez-Diduk, R.; Galano, A. Adrenaline and Noradrenaline: Protectors against Oxidative Stress or Molecular Targets? J. Phys. Chem. B 2015, 119, 3479–3491. [Google Scholar] [CrossRef]
- Hammes, H.-P.; Lin, J.; Renner, O.; Shani, M.; Lundqvist, A.; Betsholtz, C.; Brownlee, M.; Deutsch, U. Pericytes and the Pathogenesis of Diabetic Retinopathy. Diabetes 2002, 51, 3107–3112. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yun, J.H.; Jeong, H.S.; Kim, K.J.; Han, M.H.; Lee, E.H.; Lee, K.; Cho, C.H. B-Adrenergic Receptor Agonists Attenuate Pericyte Loss in Diabetic Retinas through Akt Activation. FASEB J. 2018, 32, 2324–2338. [Google Scholar] [CrossRef] [Green Version]
- Nassiri, S.; Houshmand, G.; Feghhi, M.; Kheirollah, A.; Bahadoram, M.; Nassiri, N. Effect of Periocular Injection of Celecoxib and Propranolol on Ocular Level of Vascular Endothelial Growth Factor in a Diabetic Mouse Model. Int. J. Ophthalmol. 2016, 9, 821. [Google Scholar] [CrossRef]
- Cammalleri, M.; Locri, F.; Catalani, E.; Filippi, L.; Cervia, D.; Dal Monte, M.; Bagnoli, P. The Beta Adrenergic Receptor Blocker Propranolol Counteracts Retinal Dysfunction in a Mouse Model of Oxygen Induced Retinopathy: Restoring the Balance between Apoptosis and Autophagy. Front. Cell. Neurosci. 2017, 11, 395. [Google Scholar] [CrossRef] [PubMed]
- Nourinia, R.; Rezaei Kanavi, M.; Kaharkaboudi, A.; Taghavi, S.I.; Aldavood, S.J.; Darjatmoko, S.R.; Wang, S.; Gurel, Z.; Lavine, J.A.; Safi, S.; et al. Ocular Safety of Intravitreal Propranolol and Its Efficacy in Attenuation of Choroidal Neovascularization. Invest. Ophthalmol. Vis. Sci. 2015, 56, 8228. [Google Scholar] [CrossRef]
- Dal Monte, M.; Casini, G.; la Marca, G.; Isacchi, B.; Filippi, L.; Bagnoli, P. Eye Drop Propranolol Administration Promotes the Recovery of Oxygen-Induced Retinopathy in Mice. Exp. Eye Res. 2013, 111, 27–35. [Google Scholar] [CrossRef] [PubMed]
- Dal Monte, M.; Cammalleri, M.; Mattei, E.; Filippi, L.; Bagnoli, P. Protective Effects of 1/2 Adrenergic Receptor Deletion in a Model of Oxygen-Induced Retinopathy. Investig. Ophthalmol. Vis. Sci. 2015, 56, 59–73. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ristori, C.; Filippi, L.; Monte, M.D.; Martini, D.; Cammalleri, M.; Fortunato, P.; Bagnoli, P. Role of the Adrenergic System in a Mouse Model of Oxygen-Induced Retinopathy: Antiangiogenic Effects of β-adrenoreceptor blockade. Investig. Ophthalmol. Vis. Sci. 2011, 52, 16. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Safi, S.Z.; Qvist, R.; Yan, G.O.S.; Ismail, I.S.B. Differential Expression and Role of Hyperglycemia Induced Oxidative Stress in Epigenetic Regulation of Β1, Β2 and Β3-Adrenergic Receptors in Retinal Endothelial Cells. BMC Med. Genom. 2014, 7, 29. [Google Scholar] [CrossRef] [Green Version]
- Phipps, J.A.; Wilkinson-Berka, J.L.; Fletcher, E.L. Retinal Dysfunction in Diabetic Ren-2 Rats Is Ameliorated by Treatment with Valsartan but Not Atenolol. Invest. Ophthalmol. Vis. Sci. 2007, 48, 927. [Google Scholar] [CrossRef] [Green Version]
- Wilkinsonberka, J.; Tan, G.; Jaworski, K.; Ninkovic, S. Valsartan but Not Atenolol Improves Vascular Pathology in Diabetic Ren-2 Rat Retina. Am. J. Hypertens. 2007, 20, 423–430. [Google Scholar] [CrossRef] [Green Version]
- Yan, L.; Dong, Y.; Qing, T.; Deng, Y.; Han, X.; Shi, W.; Li, J.; Gao, F.; Zhang, X.; Tian, Y.; et al. Metoprolol Rescues Endothelial Progenitor Cell Dysfunction in Diabetes. PeerJ 2020, 8, e9306. [Google Scholar] [CrossRef] [PubMed]
- Wiktorowska-Owczarek, A.; Namiecińska, M.; Berezińska, M.; Nowak, J.Z. Characteristics of Adrenaline-Driven Receptor-Mediated Signals in Human Microvessel-Derived Endothelial Cells. Pharm. Rep. 2008, 60, 950–956. [Google Scholar]
- Warne, T.; Moukhametzianov, R.; Baker, J.G.; Nehmé, R.; Edwards, P.C.; Leslie, A.G.W.; Schertler, G.F.X.; Tate, C.G. The Structural Basis for Agonist and Partial Agonist Action on a β(1)-Adrenergic Receptor. Nature 2011, 469, 241–244. [Google Scholar] [CrossRef] [Green Version]
- Ji, Y.; Chen, S.; Li, K.; Xiao, X.; Zheng, S.; Xu, T. The Role of β-Adrenergic Receptor Signaling in the Proliferation of Hemangioma-Derived Endothelial Cells. Cell Div. 2013, 8, 1. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhao, F.; Yang, X.; Xu, G.; Bi, J.; Lv, R.; Huo, R. Propranolol Suppresses HUVEC Viability, Migration, VEGF Expression, and Promotes Apoptosis by Downregulation of MiR-4295. J. Cell. Biochem. 2019, 120, 6614–6623. [Google Scholar] [CrossRef]
- Andreeva, A.; Piontek, J.; Blasig, I.; Utepbergenov, D. Assembly of Tight Junction Is Regulated by the Antagonism of Conventional and Novel Protein Kinase C Isoforms. Int. J. Biochem. Cell Biol. 2005, 38, 222–223. [Google Scholar] [CrossRef]
- Harhaj, N.S.; Felinski, E.A.; Wolpert, E.B.; Sundstrom, J.M.; Gardner, T.W.; Antonetti, D.A. VEGF Activation of Protein Kinase C Stimulates Occludin Phosphorylation and Contributes to Endothelial Permeability. Invest. Ophthalmol. Vis. Sci. 2006, 47, 5106. [Google Scholar] [CrossRef]
- Shi, Y.; Vanhoutte, P.M. Macro- and Microvascular Endothelial Dysfunction in Diabetes. J. Diabetes 2017, 9, 434–449. [Google Scholar] [CrossRef] [Green Version]
- Kowluru, R.A. Role of Interleukin-1 in the Pathogenesis of Diabetic Retinopathy. Br. J. Ophthalmol. 2004, 88, 1343–1347. [Google Scholar] [CrossRef] [Green Version]
- Yao, Y.; Li, R.; Du, J.; Li, X.; Zhao, L.; Long, L.; Li, D.; Lu, S. Tumor Necrosis Factor-α and Diabetic Retinopathy: Review and Meta-Analysis. Clin. Chim. Acta 2018, 485, 210–217. [Google Scholar] [CrossRef]
- Nishikawa, T.; Edelstein, D.; Du, X.L.; Yamagishi, S.; Matsumura, T.; Kaneda, Y.; Yorek, M.A.; Beebe, D.; Oates, P.J.; Hammes, H.-P.; et al. Normalizing Mitochondrial Superoxide Production Blocks Three Pathways of Hyperglycaemic Damage. Nature 2000, 404, 787–790. [Google Scholar] [CrossRef] [PubMed]
- Calderon, G.D.; Juarez, O.H.; Hernandez, G.E.; Punzo, S.M.; De la Cruz, Z.D. Oxidative Stress and Diabetic Retinopathy: Development and Treatment. Eye 2017, 31, 1122–1130. [Google Scholar] [CrossRef]
- Luo, P.; Wang, M.-H. Eicosanoids, β-Cell Function, and Diabetes. Prostaglandins Other Lipid Mediat. 2011, 95, 1–10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dobrian, A.D.; Lieb, D.C.; Cole, B.K.; Taylor-Fishwick, D.A.; Chakrabarti, S.K.; Nadler, J.L. Functional and Pathological Roles of the 12- and 15-Lipoxygenases. Prog. Lipid Res. 2011, 50, 115–131. [Google Scholar] [CrossRef] [Green Version]
- Huang, H.; Al-Shabrawey, M.; Wang, M.-H. Cyclooxygenase- and Cytochrome P450-Derived Eicosanoids in Stroke. Prostaglandins Other Lipid Mediat. 2016, 122, 45–53. [Google Scholar] [CrossRef] [Green Version]
- Li, Y.-B.; Han, J.-Y.; Jiang, W.; Wang, J. Selenium Inhibits High Glucose-Induced Cyclooxygenase-2 and P-Selectin Expression in Vascular Endothelial Cells. Mol. Biol. Rep. 2011, 38, 2301–2306. [Google Scholar] [CrossRef]
- Muzaffar, S.; Shukla, N.; Lobo, C.; Angelini, G.D.; Jeremy, J.Y. Iloprost Inhibits Superoxide Formation and Gp91 phox Expression Induced by the Thromboxane A2 Analogue U46619, 8-Isoprostane F2α, Prostaglandin F2α, Cytokines and Endotoxin in the Pig Pulmonary Artery: Superoxide and Eicosanoids in Pig Pulmonary Artery. Br. J. Pharmacol. 2004, 141, 488–496. [Google Scholar] [CrossRef] [Green Version]
- Capone, C.; Faraco, G.; Anrather, J.; Zhou, P.; Iadecola, C. Cyclooxygenase 1–Derived Prostaglandin E 2 and EP1 Receptors Are Required for the Cerebrovascular Dysfunction Induced by Angiotensin II. Hypertension 2010, 55, 911–917. [Google Scholar] [CrossRef] [Green Version]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene | Sequence (5′–3′) | Amplicon (bp) | Accession n. |
|---|---|---|---|
| VEGFA | Fw: ATCTTCAAGCCATCCTGTGTGC | 121 | NM_001025366.3 |
| Rv: GAGGTTTGATCCGCATAATCTG | |||
| IL-1β | Fw: AGCTACGAATCTCCGACCAC | 186 | NM_000576.3 |
| Rv: CGTTATCCCATGTGTCGAAGAA | |||
| TNF-α | Fw: AGCCCATGTTGTAGCAAA CC | 134 | NM_000594.4 |
| Rv: TGAGGTACAGGCCCTCTGAT | |||
| 18S rRNA | Fw: TAAGTCCCTGCCCTTTGTACACA | 69 | NR 146119 |
| Rv: GATCCGAGGGCCTCACTAAAC |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Giurdanella, G.; Longo, A.; Distefano, A.; Olivieri, M.; Cristaldi, M.; Cosentino, A.; Agafonova, A.; Caporarello, N.; Lupo, G.; Anfuso, C.D. The Anti-Inflammatory Effect of the β1-Adrenergic Receptor Antagonist Metoprolol on High Glucose Treated Human Microvascular Retinal Endothelial Cells. Cells 2022, 11, 51. https://doi.org/10.3390/cells11010051
Giurdanella G, Longo A, Distefano A, Olivieri M, Cristaldi M, Cosentino A, Agafonova A, Caporarello N, Lupo G, Anfuso CD. The Anti-Inflammatory Effect of the β1-Adrenergic Receptor Antagonist Metoprolol on High Glucose Treated Human Microvascular Retinal Endothelial Cells. Cells. 2022; 11(1):51. https://doi.org/10.3390/cells11010051
Chicago/Turabian StyleGiurdanella, Giovanni, Anna Longo, Alfio Distefano, Melania Olivieri, Martina Cristaldi, Alessia Cosentino, Aleksandra Agafonova, Nunzia Caporarello, Gabriella Lupo, and Carmelina Daniela Anfuso. 2022. "The Anti-Inflammatory Effect of the β1-Adrenergic Receptor Antagonist Metoprolol on High Glucose Treated Human Microvascular Retinal Endothelial Cells" Cells 11, no. 1: 51. https://doi.org/10.3390/cells11010051
APA StyleGiurdanella, G., Longo, A., Distefano, A., Olivieri, M., Cristaldi, M., Cosentino, A., Agafonova, A., Caporarello, N., Lupo, G., & Anfuso, C. D. (2022). The Anti-Inflammatory Effect of the β1-Adrenergic Receptor Antagonist Metoprolol on High Glucose Treated Human Microvascular Retinal Endothelial Cells. Cells, 11(1), 51. https://doi.org/10.3390/cells11010051