
Methionine Deprivation Reveals the Pivotal Roles of Cell Cycle Progression in Ferroptosis That Is Induced by Cysteine Starvation


{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Cell Cultures and Chemicals
2.2. Evaluation of Cytotoxicity of Cells
2.3. Hoechst and Propidium Iodide (PI) Double Staining
2.4. Liquid Chromatography-Mass Spectrometry (LC-MS) Analyses
2.5. Flow Cytometry
2.6. Detection of Intracellular Ferrous Iron under Fluorescent Microscopy
2.7. Immunostaining
2.8. Statistical Analysis
3. Results
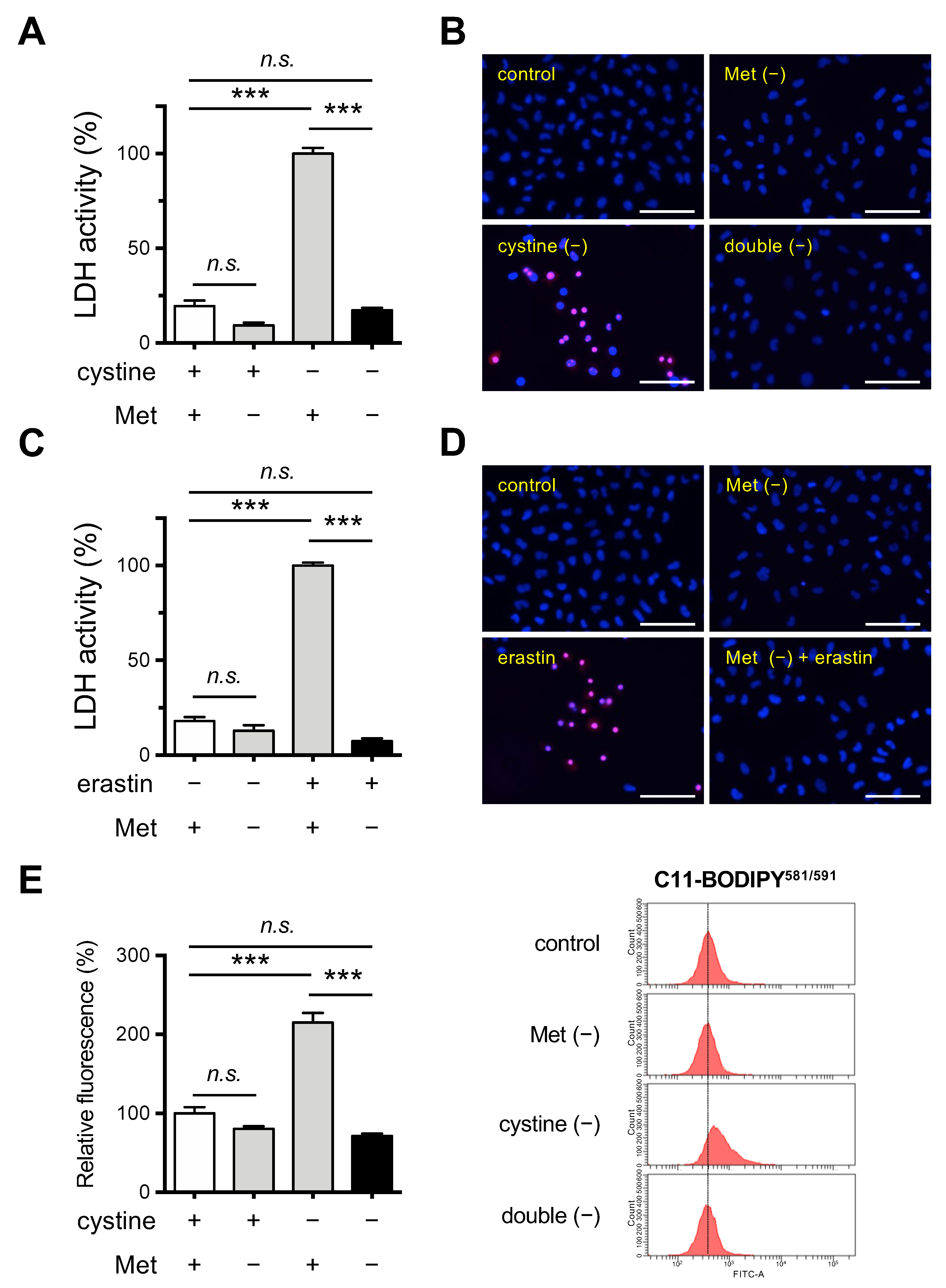
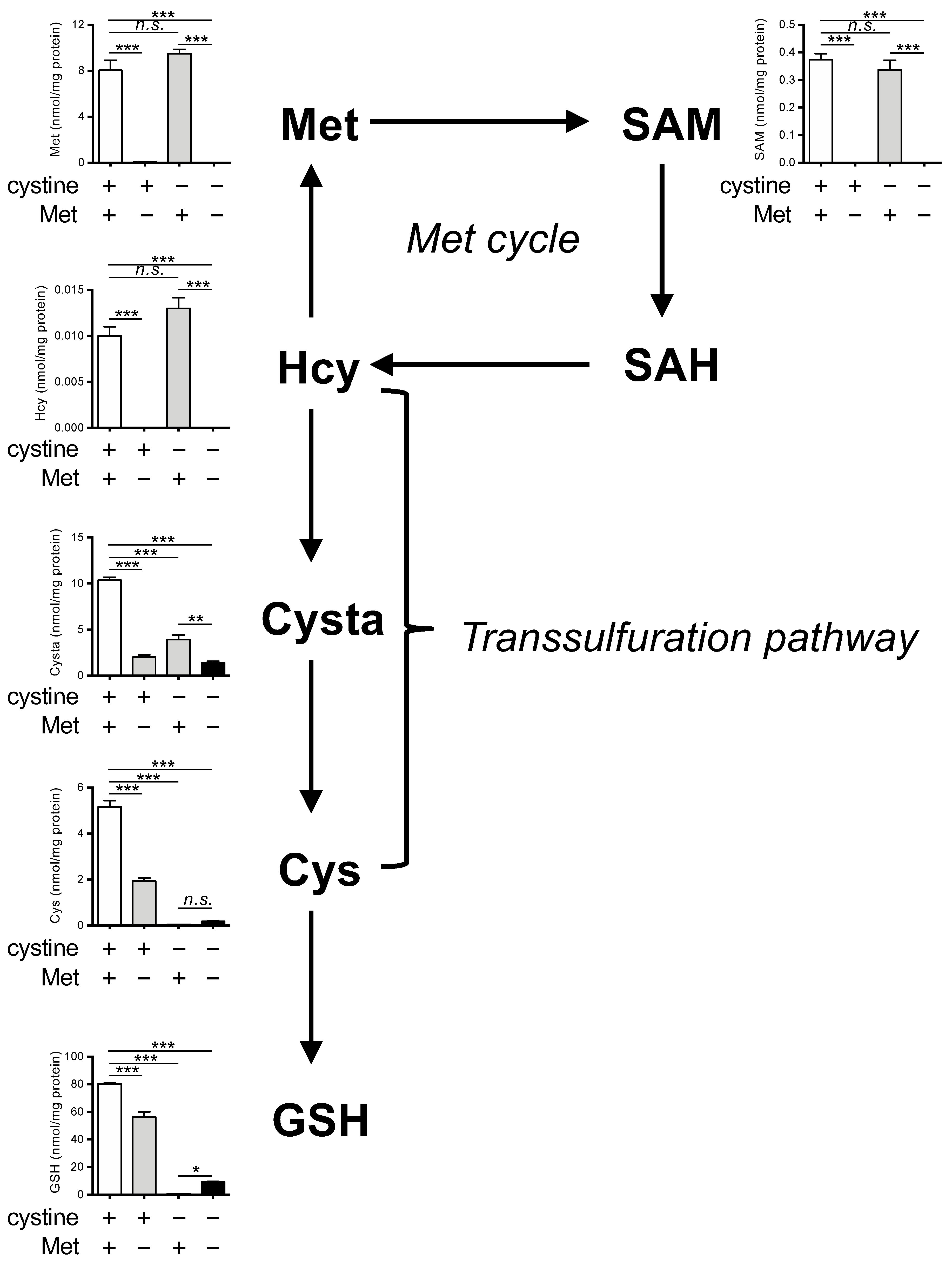
3.1. Methionine Deprivation Suppresses Ferroptosis Induced by Cystine Deprivation
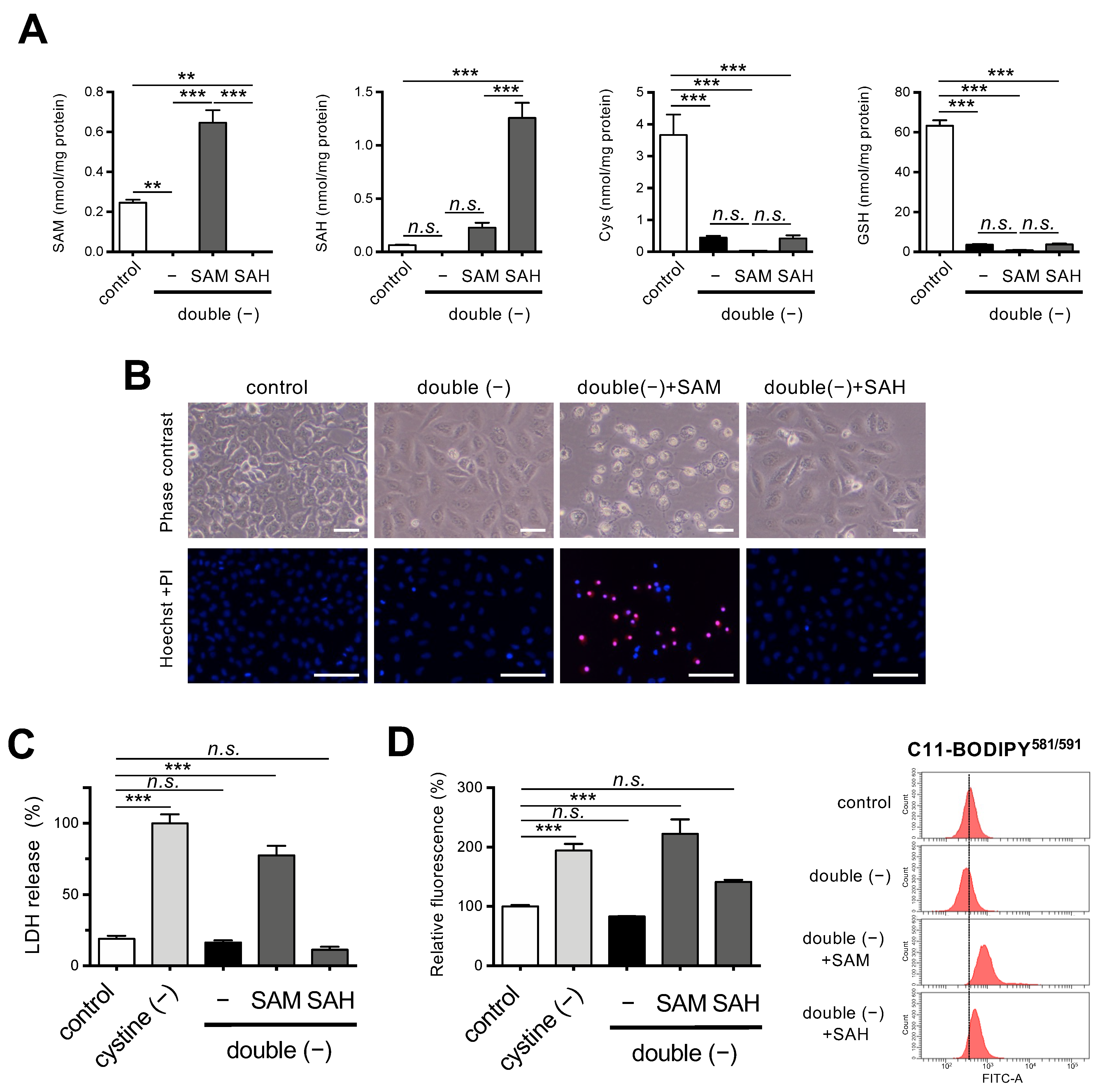
3.2. Supplementation of SAM Restores Ferroptosis Induction under Met/Cystine Double-Free Conditions
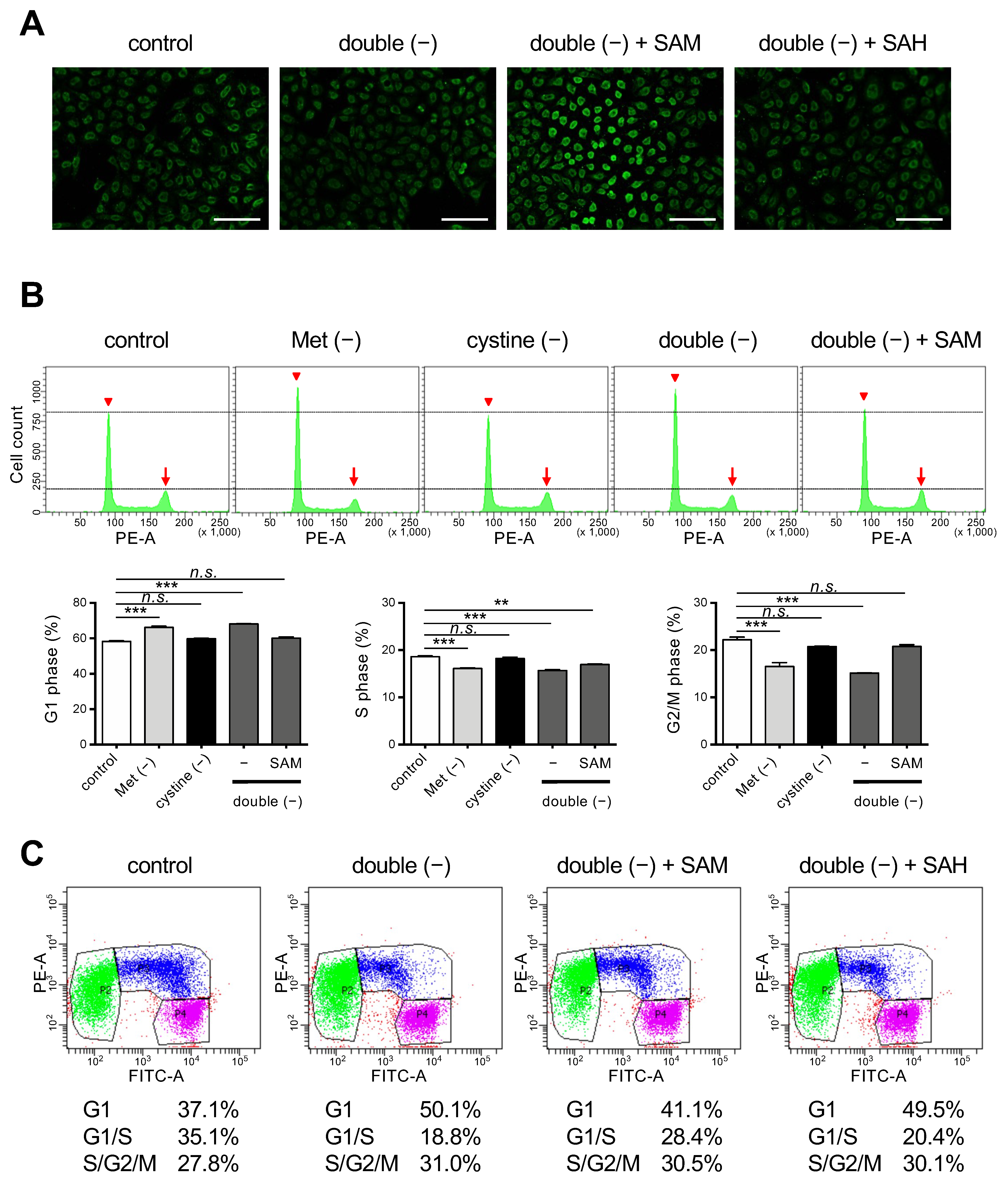
3.3. Supplementation of SAM in a Double-Depleted Environment Induces Ferroptosis as well as Releasing Cell Cycle Arrest in HeLa Cells
4. Discussion
5. Concluding Remarks
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Dixon, S.J.; Lemberg, K.M.; Lamprecht, M.R.; Skouta, R.; Zaitsev, E.M.; Gleason, C.E.; Patel, D.N.; Bauer, A.J.; Cantley, A.M.; Yang, W.S.; et al. Ferroptosis: An Iron-Dependent Form of Nonapoptotic Cell Death. Cell 2012, 149, 1060–1072. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stockwell, B.R.; Friedmann Angeli, J.P.; Bayir, H.; Bush, A.I.; Conrad, M.; Dixon, S.J.; Fulda, S.; Gascón, S.; Hatzios, S.K.; Kagan, V.E.; et al. Ferroptosis: A Regulated Cell Death Nexus Linking Metabolism, Redox Biology, and Disease. Cell 2017, 171, 273–285. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, W.S.; SriRamaratnam, R.; Welsch, M.E.; Shimada, K.; Skouta, R.; Viswanathan, V.S.; Cheah, J.H.; Clemons, P.A.; Shamji, A.F.; Clish, C.B.; et al. Regulation of Ferroptotic Cancer Cell Death by GPX4. Cell 2014, 156, 317–331. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Friedmann Angeli, J.P.; Schneider, M.; Proneth, B.; Tyurina, Y.Y.; Tyurin, V.A.; Hammond, V.J.; Herbach, N.; Aichler, M.; Walch, A.; Eggenhofer, E.; et al. Inactivation of the Ferroptosis Regulator Gpx4 Triggers Acute Renal Failure in Mice. Nat. Cell Biol. 2014, 16, 1180–1191. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Conrad, M.; Sato, H. The Oxidative Stress-Inducible Cystine/Glutamate Antiporter, System x (c) (-) : Cystine Supplier and Beyond. Amino Acids 2012, 42, 231–246. [Google Scholar] [CrossRef]
- McBean, G.J. The Transsulfuration Pathway: A Source of Cysteine for Glutathione in Astrocytes. Amino Acids 2012, 42, 199–205. [Google Scholar] [CrossRef] [Green Version]
- Lee, J.; Kang, E.S.; Kobayashi, S.; Homma, T.; Sato, H.; Seo, H.G.; Fujii, J. The Viability of Primary Hepatocytes Is Maintained under a Low Cysteine-Glutathione Redox State with a Marked Elevation in Ophthalmic Acid Production. Exp. Cell Res. 2017, 361, 178–191. [Google Scholar] [CrossRef]
- Hayano, M.; Yang, W.S.; Corn, C.K.; Pagano, N.C.; Stockwell, B.R. Loss of Cysteinyl-TRNA Synthetase (CARS) Induces the Transsulfuration Pathway and Inhibits Ferroptosis Induced by Cystine Deprivation. Cell Death Differ. 2016, 23, 270–278. [Google Scholar] [CrossRef] [Green Version]
- Judde, J.G.; Ellis, M.; Frost, P. Biochemical Analysis of the Role of Transmethylation in the Methionine Dependence of Tumor Cells. Cancer Res. 1989, 49, 4859–4865. [Google Scholar]
- Stern, P.H.; Hoffman, R.M. Elevated Overall Rates of Transmethylation in Cell Lines from Diverse Human Tumors. In Vitro 1984, 20, 663–670. [Google Scholar] [CrossRef]
- Zhu, J.; Berisa, M.; Schwörer, S.; Qin, W.; Cross, J.R.; Thompson, C.B. Transsulfuration Activity Can Support Cell Growth upon Extracellular Cysteine Limitation. Cell Metab. 2019, 30, 865–876.e5. [Google Scholar] [CrossRef] [PubMed]
- Guo, H.Y.; Herrera, H.; Groce, A.; Hoffman, R.M. Expression of the Biochemical Defect of Methionine Dependence in Fresh Patient Tumors in Primary Histoculture. Cancer Res. 1993, 53, 2479–2483. [Google Scholar] [PubMed]
- Halpern, B.C.; Clark, B.R.; Hardy, D.N.; Halpern, R.M.; Smith, R.A. The Effect of Replacement of Methionine by Homocystine on Survival of Malignant and Normal Adult Mammalian Cells in Culture. Proc. Natl. Acad. Sci. USA 1974, 71, 1133–1136. [Google Scholar] [CrossRef] [Green Version]
- Mecham, J.O.; Rowitch, D.; Wallace, C.D.; Stern, P.H.; Hoffman, R.M. The Metabolic Defect of Methionine Dependence Occurs Frequently in Human Tumor Cell Lines. Biochem. Biophys. Res. Commun. 1983, 117, 429–434. [Google Scholar] [CrossRef]
- Lockhead, S.; Moskaleva, A.; Kamenz, J.; Chen, Y.; Kang, M.; Reddy, A.R.; Santos, S.D.M.; Ferrell, J.E. The Apparent Requirement for Protein Synthesis during G2 Phase Is Due to Checkpoint Activation. Cell Rep. 2020, 32, 107901. [Google Scholar] [CrossRef] [PubMed]
- Chen, Z.; Zhang, Y. Role of Mammalian DNA Methyltransferases in Development. Annu. Rev. Biochem. 2020, 89, 135–158. [Google Scholar] [CrossRef]
- Chen, T.; Hevi, S.; Gay, F.; Tsujimoto, N.; He, T.; Zhang, B.; Ueda, Y.; Li, E. Complete Inactivation of DNMT1 Leads to Mitotic Catastrophe in Human Cancer Cells. Nat. Genet. 2007, 39, 391–396. [Google Scholar] [CrossRef]
- Komninou, D.; Leutzinger, Y.; Reddy, B.S.; Richie, J.P. Methionine Restriction Inhibits Colon Carcinogenesis. Nutr. Cancer 2006, 54, 202–208. [Google Scholar] [CrossRef]
- Guo, H.; Lishko, V.K.; Herrera, H.; Groce, A.; Kubota, T.; Hoffman, R.M. Therapeutic Tumor-Specific Cell Cycle Block Induced by Methionine Starvation in Vivo. Cancer Res. 1993, 53, 5676–5679. [Google Scholar]
- Xu, Q.; Li, Y.; Gao, X.; Kang, K.; Williams, J.G.; Tong, L.; Liu, J.; Ji, M.; Deterding, L.J.; Tong, X.; et al. HNF4α Regulates Sulfur Amino Acid Metabolism and Confers Sensitivity to Methionine Restriction in Liver Cancer. Nat. Commun. 2020, 11, 3978. [Google Scholar] [CrossRef]
- Friedmann Angeli, J.P.; Krysko, D.V.; Conrad, M. Ferroptosis at the Crossroads of Cancer-Acquired Drug Resistance and Immune Evasion. Nat. Rev. Cancer 2019, 19, 405–414. [Google Scholar] [CrossRef] [PubMed]
- Fujii, J.; Homma, T.; Kobayashi, S. Ferroptosis Caused by Cysteine Insufficiency and Oxidative Insult. Free Radic. Res. 2020, 54, 969–980. [Google Scholar] [CrossRef] [PubMed]
- Sakaue-Sawano, A.; Kurokawa, H.; Morimura, T.; Hanyu, A.; Hama, H.; Osawa, H.; Kashiwagi, S.; Fukami, K.; Miyata, T.; Miyoshi, H.; et al. Visualizing Spatiotemporal Dynamics of Multicellular Cell-Cycle Progression. Cell 2008, 132, 487–498. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Homma, T.; Kobayashi, S.; Sato, H.; Fujii, J. Edaravone, a Free Radical Scavenger, Protects against Ferroptotic Cell Death in Vitro. Exp. Cell Res. 2019, 384, 111592. [Google Scholar] [CrossRef]
- Homma, T.; Kobayashi, S.; Sato, H.; Fujii, J. Superoxide Produced by Mitochondrial Complex III Plays a Pivotal Role in the Execution of Ferroptosis Induced by Cysteine Starvation. Arch. Biochem. Biophys. 2021, 700, 108775. [Google Scholar] [CrossRef]
- Mukaide, T.; Hattori, Y.; Misawa, N.; Funahashi, S.; Jiang, L.; Hirayama, T.; Nagasawa, H.; Toyokuni, S. Histological Detection of Catalytic Ferrous Iron with the Selective Turn-on Fluorescent Probe RhoNox-1 in a Fenton Reaction-Based Rat Renal Carcinogenesis Model. Free Radic. Res. 2014, 48, 990–995. [Google Scholar] [CrossRef]
- Jin, B.; Robertson, K.D. DNA Methyltransferases, DNA Damage Repair, and Cancer. Adv. Exp. Med. Biol. 2013, 754, 3–29. [Google Scholar] [CrossRef] [Green Version]
- Codenotti, S.; Poli, M.; Asperti, M.; Zizioli, D.; Marampon, F.; Fanzani, A. Cell Growth Potential Drives Ferroptosis Susceptibility in Rhabdomyosarcoma and Myoblast Cell Lines. J. Cancer Res. Clin. Oncol. 2018, 144, 1717–1730. [Google Scholar] [CrossRef]
- Song, X.; Liu, J.; Kuang, F.; Chen, X.; Zeh, H.J.; Kang, R.; Kroemer, G.; Xie, Y.; Tang, D. PDK4 Dictates Metabolic Resistance to Ferroptosis by Suppressing Pyruvate Oxidation and Fatty Acid Synthesis. Cell Rep. 2021, 34, 108767. [Google Scholar] [CrossRef]
- Lee, H.; Zandkarimi, F.; Zhang, Y.; Meena, J.K.; Kim, J.; Zhuang, L.; Tyagi, S.; Ma, L.; Westbrook, T.F.; Steinberg, G.R.; et al. Energy-Stress-Mediated AMPK Activation Inhibits Ferroptosis. Nat. Cell Biol. 2020, 22, 225–234. [Google Scholar] [CrossRef]
- Gao, M.; Monian, P.; Quadri, N.; Ramasamy, R.; Jiang, X. Glutaminolysis and Transferrin Regulate Ferroptosis. Mol. Cell 2015, 59, 298–308. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gao, M.; Yi, J.; Zhu, J.; Minikes, A.M.; Monian, P.; Thompson, C.B.; Jiang, X. Role of Mitochondria in Ferroptosis. Mol. Cell 2019, 73, 354–363.e3. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Verbon, E.H.; Post, J.A.; Boonstra, J. The Influence of Reactive Oxygen Species on Cell Cycle Progression in Mammalian Cells. Gene 2012, 511, 1–6. [Google Scholar] [CrossRef] [PubMed]
- Boonstra, J.; Post, J.A. Molecular Events Associated with Reactive Oxygen Species and Cell Cycle Progression in Mammalian Cells. Gene 2004, 337, 1–13. [Google Scholar] [CrossRef] [Green Version]
- Patterson, J.C.; Joughin, B.A.; van de Kooij, B.; Lim, D.C.; Lauffenburger, D.A.; Yaffe, M.B. ROS and Oxidative Stress Are Elevated in Mitosis during Asynchronous Cell Cycle Progression and Are Exacerbated by Mitotic Arrest. Cell Syst. 2019, 8, 163–167.e2. [Google Scholar] [CrossRef] [Green Version]
- Havens, C.G.; Ho, A.; Yoshioka, N.; Dowdy, S.F. Regulation of Late G 1 /S Phase Transition and APC Cdh1 by Reactive Oxygen Species. Mol. Cell. Biol. 2006, 26, 4701–4711. [Google Scholar] [CrossRef] [Green Version]
- Lim, J.M.; Lee, K.S.; Woo, H.A.; Kang, D.; Rhee, S.G. Control of the Pericentrosomal H2O2 Level by Peroxiredoxin I Is Critical for Mitotic Progression. J. Cell Biol. 2015, 210, 23–33. [Google Scholar] [CrossRef] [Green Version]
- Li, N.; Oberley, T.D. Modulation of Antioxidant Enzymes, Reactive Oxygen Species, and Glutathione Levels in Manganese Superoxide Dismutaseoverexpressing NIH/3T3 Fibroblasts during the Cell Cycle. J. Cell. Physiol. 1998, 177, 148–160. [Google Scholar] [CrossRef]
- Diaz Vivancos, P.; Wolff, T.; Markovic, J.; Pallardó, F.V.; Foyer, C.H. A Nuclear Glutathione Cycle within the Cell Cycle. Biochem. J. 2010, 431, 169–178. [Google Scholar] [CrossRef] [Green Version]
- Sanz, A.; Caro, P.; Ayala, V.; Portero-Otin, M.; Pamplona, R.; Barja, G. Methionine Restriction Decreases Mitochondrial Oxygen Radical Generation and Leak as Well as Oxidative Damage to Mitochondrial DNA and Proteins. FASEB J. 2006, 20, 1064–1073. [Google Scholar] [CrossRef]
- Sanchez-Roman, I.; Gomez, A.; Gomez, J.; Suarez, H.; Sanchez, C.; Naudi, A.; Ayala, V.; Portero-Otin, M.; Lopez-Torres, M.; Pamplona, R.; et al. Forty Percent Methionine Restriction Lowers DNAmethylation, Complex i ROS Generation, and Oxidative Damage to MtDNA and Mitochondrial Proteins in Rat Heart. J. Bioenerg. Biomembr. 2011, 43, 699–708. [Google Scholar] [CrossRef] [PubMed]
- Ying, Y.; Yun, J.; Guoyao, W.; Kaiji, S.; Zhaolai, D.; Zhenlong, W. Dietary L-Methionine Restriction Decreases Oxidative Stress in Porcine Liver Mitochondria. Exp. Gerontol. 2015, 65, 35–41. [Google Scholar] [CrossRef] [PubMed]
- Lin, X.; Ping, J.; Wen, Y.; Wu, Y. The Mechanism of Ferroptosis and Applications in Tumor Treatment. Front. Pharmacol. 2020, 11, 1–15. [Google Scholar] [CrossRef] [PubMed]
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Homma, T.; Kobayashi, S.; Fujii, J. Methionine Deprivation Reveals the Pivotal Roles of Cell Cycle Progression in Ferroptosis That Is Induced by Cysteine Starvation. Cells 2022, 11, 1603. https://doi.org/10.3390/cells11101603
Homma T, Kobayashi S, Fujii J. Methionine Deprivation Reveals the Pivotal Roles of Cell Cycle Progression in Ferroptosis That Is Induced by Cysteine Starvation. Cells. 2022; 11(10):1603. https://doi.org/10.3390/cells11101603
Chicago/Turabian StyleHomma, Takujiro, Sho Kobayashi, and Junichi Fujii. 2022. "Methionine Deprivation Reveals the Pivotal Roles of Cell Cycle Progression in Ferroptosis That Is Induced by Cysteine Starvation" Cells 11, no. 10: 1603. https://doi.org/10.3390/cells11101603
APA StyleHomma, T., Kobayashi, S., & Fujii, J. (2022). Methionine Deprivation Reveals the Pivotal Roles of Cell Cycle Progression in Ferroptosis That Is Induced by Cysteine Starvation. Cells, 11(10), 1603. https://doi.org/10.3390/cells11101603