
New Animal Models for Understanding FMRP Functions and FXS Pathology

{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
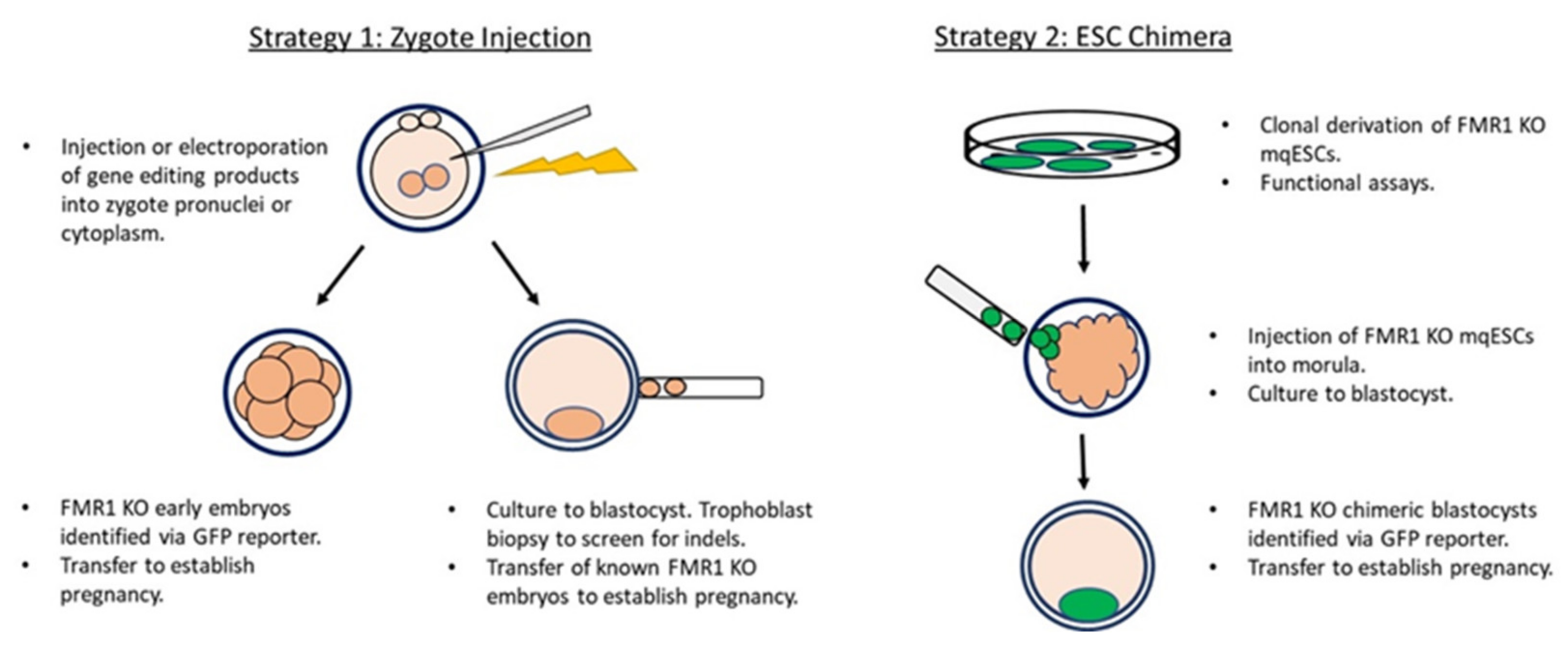
2. Strategies and Current Status in the Development of a Nonhuman Primate Model of FXS
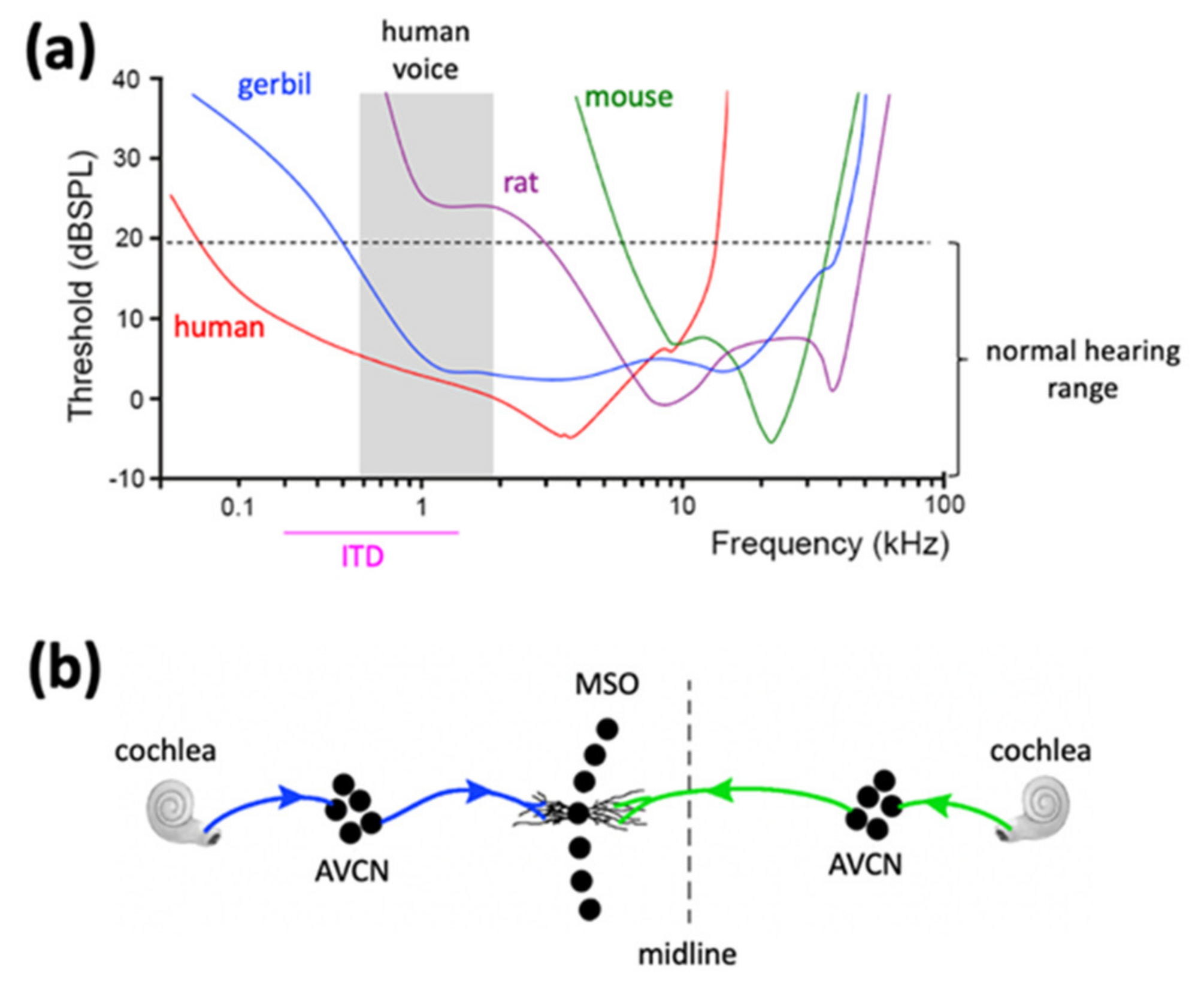
3. The Mongolian Gerbil Is a More Human-like Rodent Species for Modeling Sensory Dysfunction and Social Difficulties in FXS as Compared to Mice and Rats
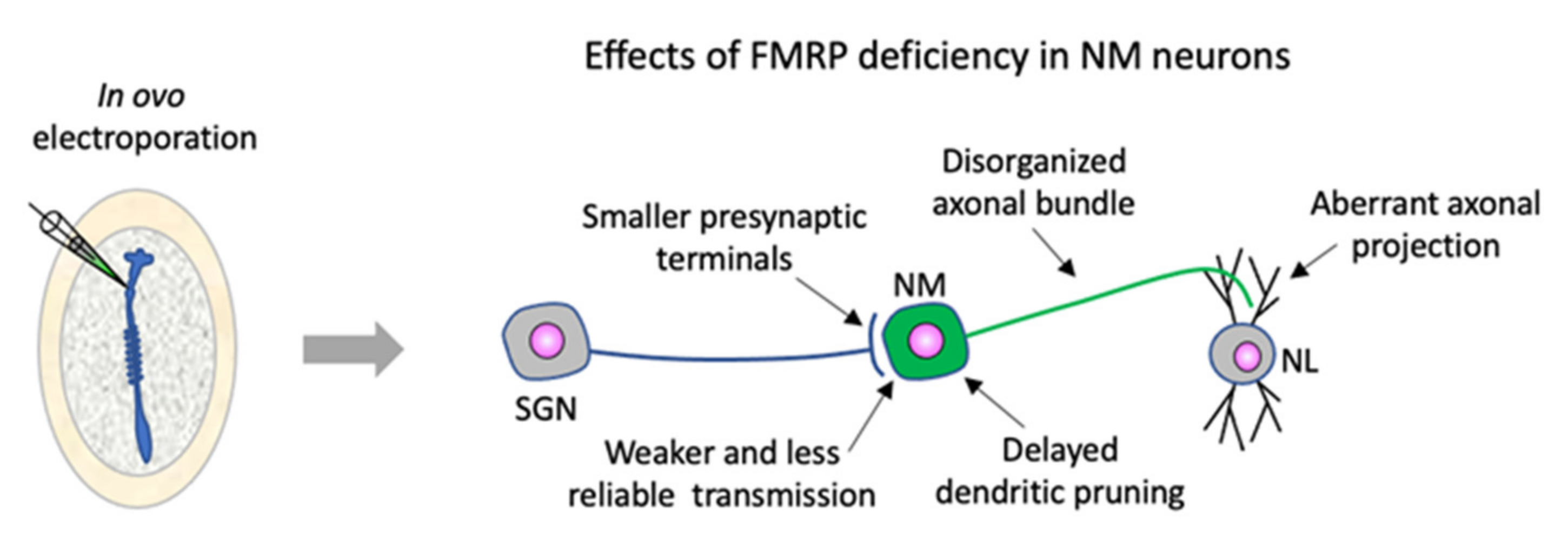
4. Chicken Embryos Are a Useful Model Organism for In-Depth Dissection of FMRP Function in Assembling Neural Circuits and for Identification of the Emergence of FXS Neuropathology
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Pieretti, M.; Zhang, F.P.; Fu, Y.H.; Warren, S.T.; Oostra, B.A.; Caskey, C.T.; Nelson, D.L. Absence of Expression of the FMR-1 Gene in Fragile X Syndrome. Cell 1991, 66, 817–822. [Google Scholar] [CrossRef]
- Verheij, C.; Bakker, C.E.; de Graaff, E.; Keulemans, J.; Willemsen, R.; Verkerk, A.J.; Galjaard, H.; Reuser, A.J.; Hoogeveen, A.T.; Oostra, B.A. Characterization and Localization of the FMR-1 Gene Product Associated with Fragile X Syndrome. Nature 1993, 363, 722–724. [Google Scholar] [CrossRef] [PubMed]
- Zhao, X.; Usdin, K. (Dys)Function Follows Form: Nucleic Acid Structure, Repeat Expansion, and Disease Pathology in FMR1 Disorders. Int. J. Mol. Sci. 2021, 22, 9167. [Google Scholar] [CrossRef] [PubMed]
- Largo, R.H.; Schinzel, A. Developmental and Behavioural Disturbances in 13 Boys with Fragile X Syndrome. Eur. J. Pediatr. 1985, 143, 269–275. [Google Scholar] [CrossRef]
- Hagerman, R.J. Fragile X Syndrome. Curr. Probl. Pediatr. 1987, 17, 621–674. [Google Scholar] [CrossRef]
- Reiss, A.L.; Freund, L.S.; Baumgardner, T.L.; Abrams, M.T.; Denckla, M.B. Contribution of the FMR1 Gene Mutation to Human Intellectual Dysfunction. Nat. Genet. 1995, 11, 331–334. [Google Scholar] [CrossRef]
- Boyle, L.; Kaufmann, W.E. The Behavioral Phenotype of FMR1 Mutations. Am. J. Med. Genet. C. Semin. Med. Genet. 2010, 154C, 469–476. [Google Scholar] [CrossRef]
- Cregenzán-Royo, O.; Brun-Gasca, C.; Fornieles-Deu, A. Behavior Problems and Social Competence in Fragile X Syndrome: A Systematic Review. Genes 2022, 13, 280. [Google Scholar] [CrossRef]
- Hagerman, R.J.; Berry-Kravis, E.; Hazlett, H.C.; Bailey, D.B.; Moine, H.; Kooy, R.F.; Tassone, F.; Gantois, I.; Sonenberg, N.; Mandel, J.L.; et al. Fragile X Syndrome. Nat. Rev. Dis. Primers 2017, 3, 17065. [Google Scholar] [CrossRef]
- Zarnescu, D.C.; Shan, G.; Warren, S.T.; Jin, P. Come FLY with Us: Toward Understanding Fragile X Syndrome. Genes Brain Behav. 2005, 4, 385–392. [Google Scholar] [CrossRef]
- Banerjee, P.; Nayar, S.; Hebbar, S.; Fox, C.F.; Jacobs, M.C.; Park, J.H.; Fernandes, J.J.; Dockendorff, T.C. Substitution of Critical Isoleucines in the KH Domains of Drosophila Fragile X Protein Results in Partial Loss-of-Function Phenotypes. Genetics 2007, 175, 1241–1250. [Google Scholar] [CrossRef] [Green Version]
- Drozd, M.; Bardoni, B.; Capovilla, M. Modeling Fragile X Syndrome in Drosophila. Front. Mol. Neurosci. 2018, 11, 124. [Google Scholar] [CrossRef]
- Bakker, C.; Verheiji, C.; Willemsen, R.; Vanderhelm, R.; Oerlemanns, F.; Vermey, M.; Bygrave, A.; Hoogeveen, A.; Oostra, B.; Reyniers, E.; et al. Fmr1 Knockout Mice: A Model to Study Fragile X Mental Retardation. The Dutch-Belgian Fragile X Consortium. Cell 1994, 78, 23–33. [Google Scholar]
- Mientjes, E.J.; Nieuwenhuizen, I.; Kirkpatrick, L.; Zu, T.; Hoogeveen-Westerveld, M.; Severijnen, L.; Rifé, M.; Willemsen, R.; Nelson, D.L.; Oostra, B.A. The Generation of a Conditional Fmr1 Knock out Mouse Model to Study Fmrp Function in Vivo. Neurobiol. Dis. 2006, 21, 549–555. [Google Scholar] [CrossRef]
- Den Broeder, M.J.; van der Linde, H.; Brouwer, J.R.; Oostra, B.A.; Willemsen, R.; Ketting, R.F. Generation and Characterization of FMR1 Knockout Zebrafish. PLoS ONE 2009, 4, e7910. [Google Scholar] [CrossRef] [Green Version]
- Till, S.M.; Asiminas, A.; Jackson, A.D.; Katsanevaki, D.; Barnes, S.A.; Osterweil, E.K.; Bear, M.F.; Chattarji, S.; Wood, E.R.; Wyllie, D.J.A.; et al. Conserved Hippocampal Cellular Pathophysiology but Distinct Behavioural Deficits in a New Rat Model of FXS. Hum. Mol. Genet. 2015, 24, 5977–5984. [Google Scholar] [CrossRef] [Green Version]
- Tian, Y.; Yang, C.; Shang, S.; Cai, Y.; Deng, X.; Zhang, J.; Shao, F.; Zhu, D.; Liu, Y.; Chen, G.; et al. Loss of FMRP Impaired Hippocampal Long-Term Plasticity and Spatial Learning in Rats. Front. Mol. Neurosci. 2017, 10, 269. [Google Scholar] [CrossRef] [Green Version]
- Dahlhaus, R. Of Men and Mice: Modeling the Fragile X Syndrome. Front. Mol. Neurosci. 2018, 11, 41. [Google Scholar] [CrossRef]
- Hu, J.; Chen, L.; Yin, J.; Yin, H.; Huang, Y.; Tian, J. Hyperactivity, Memory Defects, and Craniofacial Abnormalities in Zebrafish Fmr1 Mutant Larvae. Behav. Genet. 2020, 50, 152–160. [Google Scholar] [CrossRef]
- Musumeci, S.A.; Bosco, P.; Calabrese, G.; Bakker, C.; De Sarro, G.B.; Elia, M.; Ferri, R.; Oostra, B.A. Audiogenic Seizures Susceptibility in Transgenic Mice with Fragile X Syndrome. Epilepsia 2000, 41, 19–23. [Google Scholar] [CrossRef]
- Spencer, C.M.; Alekseyenko, O.; Serysheva, E.; Yuva-Paylor, L.A.; Paylor, R. Altered Anxiety-Related and Social Behaviors in the Fmr1 Knockout Mouse Model of Fragile X Syndrome. Genes Brain. Behav. 2005, 4, 420–430. [Google Scholar] [CrossRef]
- De Vrij, F.M.S.; Levenga, J.; van der Linde, H.C.; Koekkoek, S.K.; De Zeeuw, C.I.; Nelson, D.L.; Oostra, B.A.; Willemsen, R. Rescue of Behavioral Phenotype and Neuronal Protrusion Morphology in Fmr1 KO Mice. Neurobiol. Dis. 2008, 31, 127–132. [Google Scholar] [CrossRef] [Green Version]
- Spencer, C.M.; Alekseyenko, O.; Hamilton, S.M.; Thomas, A.M.; Serysheva, E.; Yuva-Paylor, L.A.; Paylor, R. Modifying Behavioral Phenotypes in Fmr1KO Mice: Genetic Background Differences Reveal Autistic-like Responses. Autism Res. 2011, 4, 40–56. [Google Scholar] [CrossRef] [Green Version]
- Leach, P.T.; Hayes, J.; Pride, M.; Silverman, J.L.; Crawley, J.N. Normal Performance of Fmr1 Mice on a Touchscreen Delayed Nonmatching to Position Working Memory Task. eNeuro 2016, 3, ENEURO.0143-15.2016. [Google Scholar] [CrossRef] [Green Version]
- Pan, L.; Zhang, Y.Q.; Woodruff, E.; Broadie, K. The Drosophila Fragile X Gene Negatively Regulates Neuronal Elaboration and Synaptic Differentiation. Curr. Biol. 2004, 14, 1863–1870. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Y.Q.; Matthies, H.J.G.; Mancuso, J.; Andrews, H.K.; Woodruff, E.; Friedman, D.; Broadie, K. The Drosophila Fragile X-Related Gene Regulates Axoneme Differentiation during Spermatogenesis. Dev. Biol. 2004, 270, 290–307. [Google Scholar] [CrossRef] [Green Version]
- Chang, S.; Bray, S.M.; Li, Z.; Zarnescu, D.C.; He, C.; Jin, P.; Warren, S.T. Identification of Small Molecules Rescuing Fragile X Syndrome Phenotypes in Drosophila. Nat. Chem. Biol. 2008, 4, 256–263. [Google Scholar] [CrossRef]
- Tropepe, V.; Sive, H.L. Can Zebrafish Be Used as a Model to Study the Neurodevelopmental Causes of Autism? Genes Brain Behav. 2003, 2, 268–281. [Google Scholar] [CrossRef]
- Mathur, P.; Guo, S. Use of Zebrafish as a Model to Understand Mechanisms of Addiction and Complex Neurobehavioral Phenotypes. Neurobiol. Dis. 2010, 40, 66–72. [Google Scholar] [CrossRef] [Green Version]
- Kalueff, A.V.; Stewart, A.M.; Gerlai, R. Zebrafish as an Emerging Model for Studying Complex Brain Disorders. Trends Pharmacol. Sci. 2014, 35, 63–75. [Google Scholar] [CrossRef] [Green Version]
- Bear, M.F.; Huber, K.M.; Warren, S.T. The mGluR theory of fragile X mental retardation. Trends Neurosci. 2004, 27, 370–377. [Google Scholar] [CrossRef] [PubMed]
- D’Hulst, C.; Kooy, R.F. The GABAA Receptor: A Novel Target for Treatment of Fragile X? Trends Neurosci. 2007, 30, 425–431. [Google Scholar] [CrossRef] [PubMed]
- Braat, S.; D’Hulst, C.; Heulens, I.; De Rubeis, S.; Mientjes, E.; Nelson, D.L.; Willemsen, R.; Bagni, C.; Van Dam, D.; De Deyn, P.P.; et al. The GABAA Receptor Is an FMRP Target with Therapeutic Potential in Fragile X Syndrome. Cell Cycle 2015, 14, 2985–2995. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Telias, M. Molecular Mechanisms of Synaptic Dysregulation in Fragile X Syndrome and Autism Spectrum Disorders. Front. Mol. Neurosci. 2019, 12, 51. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Richter, J.D.; Zhao, X. The Molecular Biology of FMRP: New Insights into Fragile X Syndrome. Nat. Rev. Neurosci. 2021, 22, 209–222. [Google Scholar] [CrossRef]
- Budimirovic, D.B.; Dominick, K.C.; Gabis, L.V.; Adams, M.; Adera, M.; Huang, L.; Ventola, P.; Tartaglia, N.R.; Berry-Kravis, E. Gaboxadol in Fragile X Syndrome: A 12-Week Randomized, Double-Blind, Parallel-Group, Phase 2a Study. Front. Pharmacol. 2021, 12, 757825. [Google Scholar] [CrossRef]
- Champigny, C.; Morin-Parent, F.; Bellehumeur-Lefebvre, L.; Çaku, A.; Lepage, J.-F.; Corbin, F. Combining Lovastatin and Minocycline for the Treatment of Fragile X Syndrome: Results From the LovaMiX Clinical Trial. Front. Psychiatry 2021, 12, 762967. [Google Scholar] [CrossRef]
- Protic, D.D.; Aishworiya, R.; Salcedo-Arellano, M.J.; Tang, S.J.; Milisavljevic, J.; Mitrovic, F.; Hagerman, R.J.; Budimirovic, D.B. Fragile X Syndrome: From Molecular Aspect to Clinical Treatment. Int. J. Mol. Sci. 2022, 23, 1935. [Google Scholar] [CrossRef]
- Available online: http://www.clinicaltrials.gov (accessed on 17 February 2022).
- Kaas, J.H. The Evolution of Brains from Early Mammals to Humans. Wiley Interdiscip. Rev. Cogn. Sci. 2013, 4, 33–45. [Google Scholar] [CrossRef]
- Anacker, A.M.J.; Beery, A.K. Life in Groups: The Roles of Oxytocin in Mammalian Sociality. Front. Behav. Neurosci. 2013, 7, 185. [Google Scholar] [CrossRef] [Green Version]
- Freeman, S.M.; Young, L.J. Comparative Perspectives on Oxytocin and Vasopressin Receptor Research in Rodents and Primates: Translational Implications. J. Neuroendocrinol. 2016, 28. [Google Scholar] [CrossRef]
- Quik, M.; Polonskaya, Y.; Gillespie, A.; Jakowec, M.; Lloyd, G.K.; Langston, J.W. Localization of Nicotinic Receptor Subunit MRNAs in Monkey Brain by in Situ Hybridization. J. Comp. Neurol. 2000, 425, 58–69. [Google Scholar] [CrossRef]
- Van der Staay, F.J. Animal Models of Behavioral Dysfunctions: Basic Concepts and Classifications, and an Evaluation Strategy. Brain Res. Rev. 2006, 52, 131–159. [Google Scholar] [CrossRef]
- Ventura-Antunes, L.; Mota, B.; Herculano-Houzel, S. Different Scaling of White Matter Volume, Cortical Connectivity, and Gyrification across Rodent and Primate Brains. Front. Neuroanat. 2013, 7, 3. [Google Scholar] [CrossRef] [Green Version]
- Liu, C.; Tian, X.; Liu, H.; Mo, Y.; Bai, F.; Zhao, X.; Ma, Y.; Wang, J. Rhesus Monkey Brain Development during Late Infancy and the Effect of Phencyclidine: A Longitudinal MRI and DTI Study. Neuroimage 2015, 107, 65–75. [Google Scholar] [CrossRef]
- Otani, T.; Marchetto, M.C.; Gage, F.H.; Simons, B.D.; Livesey, F.J. 2D and 3D Stem Cell Models of Primate Cortical Development Identify Species-Specific Differences in Progenitor Behavior Contributing to Brain Size. Cell Stem Cell 2016, 18, 467–480. [Google Scholar] [CrossRef] [Green Version]
- Chan, A.W.; Chong, K.Y.; Martinovich, C.; Simerly, C.; Schatten, G. Transgenic Monkeys Produced by Retroviral Gene Transfer into Mature Oocytes. Science 2001, 291, 309–312. [Google Scholar] [CrossRef]
- Yang, S.-H.; Cheng, P.-H.; Banta, H.; Piotrowska-Nitsche, K.; Yang, J.-J.; Cheng, E.C.H.; Snyder, B.; Larkin, K.; Liu, J.; Orkin, J.; et al. Towards a Transgenic Model of Huntington’s Disease in a Non-Human Primate. Nature 2008, 453, 921–924. [Google Scholar] [CrossRef] [Green Version]
- Liu, Z.; Li, X.; Zhang, J.-T.; Cai, Y.-J.; Cheng, T.-L.; Cheng, C.; Wang, Y.; Zhang, C.-C.; Nie, Y.-H.; Chen, Z.-F.; et al. Autism-like Behaviours and Germline Transmission in Transgenic Monkeys Overexpressing MeCP2. Nature 2016, 530, 98–102. [Google Scholar] [CrossRef]
- Park, J.E.; Zhang, X.F.; Choi, S.-H.; Okahara, J.; Sasaki, E.; Silva, A.C. Generation of Transgenic Marmosets Expressing Genetically Encoded Calcium Indicators. Sci. Rep. 2016, 6, 34931. [Google Scholar] [CrossRef]
- Eichler, E.E.; Kunst, C.B.; Lugenbeel, K.A.; Ryder, O.A.; Davison, D.; Warren, S.T.; Nelson, D.L. Evolution of the Cryptic FMR1 CGG Repeat. Nat. Genet. 1995, 11, 301–308. [Google Scholar] [CrossRef]
- Eichler, E.E.; Hammond, H.A.; Macpherson, J.N.; Ward, P.A.; Nelson, D.L. Population Survey of the Human FMR1 CGG Repeat Substructure Suggests Biased Polarity for the Loss of AGG Interruptions. Hum. Mol. Genet. 1995, 4, 2199–2208. [Google Scholar] [CrossRef]
- Garcia Arocena, D.; Breece, K.E.; Hagerman, P.J. Distribution of CGG Repeat Sizes within the Fragile X Mental Retardation 1 (FMR1) Homologue in a Non-Human Primate Population. Hum. Genet. 2003, 113, 371–376. [Google Scholar] [CrossRef]
- Namdar-Aligoodarzi, P.; Mohammadparast, S.; Zaker-Kandjani, B.; Talebi Kakroodi, S.; Jafari Vesiehsari, M.; Ohadi, M. Exceptionally Long 5′ UTR Short Tandem Repeats Specifically Linked to Primates. Gene 2015, 569, 88–94. [Google Scholar] [CrossRef]
- Brouwer, J.R.; Mientjes, E.J.; Bakker, C.E.; Nieuwenhuizen, I.M.; Severijnen, L.A.; Van der Linde, H.C.; Nelson, D.L.; Oostra, B.A.; Willemsen, R. Elevated Fmr1 MRNA Levels and Reduced Protein Expression in a Mouse Model with an Unmethylated Fragile X Full Mutation. Exp. Cell Res. 2007, 313, 244–253. [Google Scholar] [CrossRef] [Green Version]
- Entezam, A.; Biacsi, R.; Orrison, B.; Saha, T.; Hoffman, G.E.; Grabczyk, E.; Nussbaum, R.L.; Usdin, K. Regional FMRP Deficits and Large Repeat Expansions into the Full Mutation Range in a New Fragile X Premutation Mouse Model. Gene 2007, 395, 125–134. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Siomi, H.; Choi, M.; Siomi, M.C.; Nussbaum, R.L.; Dreyfuss, G. Essential Role for KH Domains in RNA Binding: Impaired RNA Binding by a Mutation in the KH Domain of FMR1 That Causes Fragile X Syndrome. Cell 1994, 77, 33–39. [Google Scholar] [CrossRef]
- Handt, M.; Epplen, A.; Hoffjan, S.; Mese, K.; Epplen, J.T.; Dekomien, G. Point Mutation Frequency in the FMR1 Gene as Revealed by Fragile X Syndrome Screening. Mol. Cell Probes 2014, 28, 279–283. [Google Scholar] [CrossRef] [PubMed]
- Quartier, A.; Poquet, H.; Gilbert-Dussardier, B.; Rossi, M.; Casteleyn, A.-S.; des Portes, V.; Feger, C.; Nourisson, E.; Kuentz, P.; Redin, C.; et al. Intragenic FMR1 Disease-Causing Variants: A Significant Mutational Mechanism Leading to Fragile-X Syndrome. Eur. J. Hum. Genet. 2017, 25, 423–431. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tekendo-Ngongang, C.; Grochowsky, A.; Solomon, B.D.; Yano, S.T. Beyond Trinucleotide Repeat Expansion in Fragile X Syndrome: Rare Coding and Noncoding Variants in FMR1 and Associated Phenotypes. Genes 2021, 12, 1669. [Google Scholar] [CrossRef]
- Prieto, M.; Folci, A.; Poupon, G.; Schiavi, S.; Buzzelli, V.; Pronot, M.; François, U.; Pousinha, P.; Lattuada, N.; Abelanet, S.; et al. Missense Mutation of Fmr1 Results in Impaired AMPAR-Mediated Plasticity and Socio-Cognitive Deficits in Mice. Nat. Commun. 2021, 12, 1557. [Google Scholar] [CrossRef]
- Zhang, T.; Yin, Y.; Liu, H.; Du, W.; Ren, C.; Wang, L.; Lu, H.; Zhang, Z. Generation of VDR Knock-Out Mice via Zygote Injection of CRISPR/Cas9 System. PLoS ONE 2016, 11, e0163551. [Google Scholar] [CrossRef]
- Zuo, E.; Cai, Y.-J.; Li, K.; Wei, Y.; Wang, B.-A.; Sun, Y.; Liu, Z.; Liu, J.; Hu, X.; Wei, W.; et al. One-Step Generation of Complete Gene Knockout Mice and Monkeys by CRISPR/Cas9-Mediated Gene Editing with Multiple SgRNAs. Cell Res. 2017, 27, 933–945. [Google Scholar] [CrossRef]
- Leidy-Davis, T.; Cheng, K.; Goodwin, L.O.; Morgan, J.L.; Juan, W.C.; Roca, X.; Ong, S.T.; Bergstrom, D.E. Viable Mice with Extensive Gene Humanization (25-Kbp) Created Using Embryonic Stem Cell/Blastocyst and CRISPR/Zygote Injection Approaches. Sci. Rep. 2018, 8, 15028. [Google Scholar] [CrossRef]
- Hashimoto, M.; Yamashita, Y.; Takemoto, T. Electroporation of Cas9 Protein/SgRNA into Early Pronuclear Zygotes Generates Non-Mosaic Mutants in the Mouse. Dev. Biol 2016, 418, 1–9. [Google Scholar] [CrossRef]
- Challa, A.K.; Stanford, D.; Allen, A.; Rasmussen, L.; Amanor, F.K.; Raju, S.V. Validation of Gene Editing Efficiency with CRISPR-Cas9 System Directly in Rat Zygotes Using Electroporation Mediated Delivery and Embryo Culture. MethodsX 2021, 8, 101419. [Google Scholar] [CrossRef]
- Tanihara, F.; Hirata, M.; Nguyen, N.T.; Sawamoto, O.; Kikuchi, T.; Otoi, T. One-Step Generation of Multiple Gene-Edited Pigs by Electroporation of the CRISPR/Cas9 System into Zygotes to Reduce Xenoantigen Biosynthesis. Int. J. Mol. Sci. 2021, 22, 2249. [Google Scholar] [CrossRef]
- Schmidt, J.K.; Strelchenko, N.; Park, M.A.; Kim, Y.H.; Mean, K.D.; Schotzko, M.L.; Kang, H.J.; Golos, T.G.; Slukvin, I.I. Genome Editing of CCR5 by CRISPR-Cas9 in Mauritian Cynomolgus Macaque Embryos. Sci. Rep. 2020, 10, 18457. [Google Scholar] [CrossRef]
- Rubinstein, C.D.; McLean, D.T.; Lehman, B.P.; Meudt, J.J.; Schomberg, D.T.; Krentz, K.J.; Reichert, J.L.; Meyer, M.B.; Adams, M.; Konsitzke, C.M.; et al. Assessment of Mosaicism and Detection of Cryptic Alleles in CRISPR/Cas9-Engineered Neurofibromatosis Type 1 and TP53 Mutant Porcine Models Reveals Overlooked Challenges in Precision Modeling of Human Diseases. Front. Genet. 2021, 12, 721045. [Google Scholar] [CrossRef]
- Parker-Thornburg, J.V.; Alana, J.L.; Smith, C.N.; Detry, M.; Rojas, M.L.; Baskin, K.K. Cryopreserved Morulae Can Be Used to Efficiently Generate Germline-Transmitting Chimeras by Blastocyst Injection. Transgenic Res. 2005, 14, 685–690. [Google Scholar] [CrossRef]
- Yamazaki, K.; Kubara, K.; Ishii, S.; Li, P.; Dairiki, R.; Hihara, T.; Ishizuka, Y.; Izumi, Y.; Kumai, M.; Kamisako, T.; et al. In Vitro and in Vivo Functions of T Cells Produced in Complemented Thymi of Chimeric Mice Generated by Blastocyst Complementation. Sci. Rep. 2022, 12, 3242. [Google Scholar] [CrossRef]
- Tachibana, M.; Sparman, M.; Ramsey, C.; Ma, H.; Lee, H.-S.; Penedo, M.C.T.; Mitalipov, S. Generation of Chimeric Rhesus Monkeys. Cell 2012, 148, 285–295. [Google Scholar] [CrossRef] [Green Version]
- Cockburn, K.; Rossant, J. Making the Blastocyst: Lessons from the Mouse. J. Clin. Investig. 2010, 120, 995–1003. [Google Scholar] [CrossRef] [Green Version]
- Niakan, K.K.; Han, J.; Pedersen, R.A.; Simon, C.; Pera, R.A.R. Human Pre-Implantation Embryo Development. Development 2012, 139, 829–841. [Google Scholar] [CrossRef] [Green Version]
- Chen, Y.; Niu, Y.; Li, Y.; Ai, Z.; Kang, Y.; Shi, H.; Xiang, Z.; Yang, Z.; Tan, T.; Si, W.; et al. Generation of Cynomolgus Monkey Chimeric Fetuses Using Embryonic Stem Cells. Cell Stem Cell 2015, 17, 116–124. [Google Scholar] [CrossRef] [Green Version]
- Kang, Y.; Ai, Z.; Duan, K.; Si, C.; Wang, Y.; Zheng, Y.; He, J.; Yin, Y.; Zhao, S.; Niu, B.; et al. Improving Cell Survival in Injected Embryos Allows Primed Pluripotent Stem Cells to Generate Chimeric Cynomolgus Monkeys. Cell Rep. 2018, 25, 2563–2576.e9. [Google Scholar] [CrossRef] [Green Version]
- Park, C.-H.; Jeoung, Y.-H.; Uh, K.-J.; Park, K.-E.; Bridge, J.; Powell, A.; Li, J.; Pence, L.; Zhang, L.; Liu, T.; et al. Extraembryonic Endoderm (XEN) Cells Capable of Contributing to Embryonic Chimeras Established from Pig Embryos. Stem Cell Rep. 2021, 16, 212–223. [Google Scholar] [CrossRef]
- Tan, T.; Wu, J.; Si, C.; Dai, S.; Zhang, Y.; Sun, N.; Zhang, E.; Shao, H.; Si, W.; Yang, P.; et al. Chimeric Contribution of Human Extended Pluripotent Stem Cells to Monkey Embryos Ex Vivo. Cell 2021, 184, 2020–2032.e14. [Google Scholar] [CrossRef]
- Zhao, L.; Gao, X.; Zheng, Y.; Wang, Z.; Zhao, G.; Ren, J.; Zhang, J.; Wu, J.; Wu, B.; Chen, Y.; et al. Establishment of Bovine Expanded Potential Stem Cells. Proc. Natl. Acad. Sci. USA 2021, 118, e2018505118. [Google Scholar] [CrossRef]
- De Boulle, K.; Verkerk, A.J.; Reyniers, E.; Vits, L.; Hendrickx, J.; Van Roy, B.; Van den Bos, F.; de Graaff, E.; Oostra, B.A.; Willems, P.J. A Point Mutation in the FMR-1 Gene Associated with Fragile X Mental Retardation. Nat. Genet. 1993, 3, 31–35. [Google Scholar] [CrossRef]
- Lugenbeel, K.A.; Peier, A.M.; Carson, N.L.; Chudley, A.E.; Nelson, D.L. Intragenic Loss of Function Mutations Demonstrate the Primary Role of FMR1 in Fragile X Syndrome. Nat. Genet. 1995, 10, 483–485. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.C.; Lin, M.L.; Lin, S.J.; Li, Y.C.; Li, S.Y. Novel Point Mutation within Intron 10 of FMR-1 Gene Causing Fragile X Syndrome. Hum. Mutat. 1997, 10, 393–399. [Google Scholar] [CrossRef]
- Myrick, L.K.; Hashimoto, H.; Cheng, X.; Warren, S.T. Human FMRP Contains an Integral Tandem Agenet (Tudor) and KH Motif in the Amino Terminal Domain. Hum. Mol. Genet. 2015, 24, 1733–1740. [Google Scholar] [CrossRef] [PubMed]
- Fu, X.; Yan, A.; Xu, Y.; Liao, J.; Guo, X.; Zhang, D.; Yang, W.; Zheng, D.; Lan, F. Splicing of Exon 9a in FMR1 Transcripts Results in a Truncated FMRP with Altered Subcellular Distribution. Gene 2020, 731, 144359. [Google Scholar] [CrossRef]
- Abe, Y.; Nakao, H.; Goto, M.; Tamano, M.; Koebis, M.; Nakao, K.; Aiba, A. Efficient Marmoset Genome Engineering by Autologous Embryo Transfer and CRISPR/Cas9 Technology. Sci. Rep. 2021, 11, 20234. [Google Scholar] [CrossRef]
- Willemsen, R.; Bontekoe, C.J.M.; Severijnen, L.-A.; Oostra, B.A. Timing of the Absence of FMR1 Expression in Full Mutation Chorionic Villi. Hum. Genet. 2002, 110, 601–605. [Google Scholar] [CrossRef]
- Sutcliffe, J.S.; Nelson, D.L.; Zhang, F.; Pieretti, M.; Caskey, C.T.; Saxe, D.; Warren, S.T. DNA Methylation Represses FMR-1 Transcription in Fragile X Syndrome. Hum. Mol. Genet. 1992, 1, 397–400. [Google Scholar] [CrossRef]
- Kidd, S.A.; Lachiewicz, A.; Barbouth, D.; Blitz, R.K.; Delahunty, C.; McBrien, D.; Visootsak, J.; Berry-Kravis, E. Fragile X Syndrome: A Review of Associated Medical Problems. Pediatrics 2014, 134, 995–1005. [Google Scholar] [CrossRef] [Green Version]
- Maestripieri, D.; Carroll, K.A. Causes and consequences of infant abuse and neglect in monkeys. Aggress. Violent Behav. 2000, 5, 245–254. [Google Scholar] [CrossRef]
- Burbacher, T.M.; Grant, K.S.; Worlein, J.; Ha, J.; Curnow, E.; Juul, S.; Sackett, G.P. Four Decades of Leading-Edge Research in the Reproductive and Developmental Sciences: The Infant Primate Research Laboratory at the University of Washington National Primate Research Center. Am. J. Primatol. 2013, 75, 1063–1083. [Google Scholar] [CrossRef] [Green Version]
- Baranek, G.T.; Roberts, J.E.; David, F.J.; Sideris, J.; Mirrett, P.L.; Hatton, D.D.; Bailey, D.B. Developmental Trajectories and Correlates of Sensory Processing in Young Boys with Fragile X Syndrome. Phys. Occup. Ther. Pediatr. 2008, 28, 79–98. [Google Scholar] [CrossRef]
- Rais, M.; Binder, D.K.; Razak, K.A.; Ethell, I.M. Sensory Processing Phenotypes in Fragile X Syndrome. ASN Neuro 2018, 10, 1759091418801092. [Google Scholar] [CrossRef] [Green Version]
- Moore, T.; Zirnsak, M. Neural Mechanisms of Selective Visual Attention. Annu. Rev. Psychol. 2017, 68, 47–72. [Google Scholar] [CrossRef]
- Petersen, C.C.H. Sensorimotor Processing in the Rodent Barrel Cortex. Nat. Rev. Neurosci. 2019, 20, 533–546. [Google Scholar] [CrossRef]
- Flossmann, T.; Rochefort, N.L. Spatial Navigation Signals in Rodent Visual Cortex. Curr. Opin. Neurobiol. 2021, 67, 163–173. [Google Scholar] [CrossRef]
- Samuelsen, C.L.; Vincis, R. Cortical Hub for Flavor Sensation in Rodents. Front. Syst Neurosci. 2021, 15, 772286. [Google Scholar] [CrossRef]
- Heffner, R.S.; Koay, G.; Heffner, H.E. Audiograms of Five Species of Rodents: Implications for the Evolution of Hearing and the Perception of Pitch. Hear. Res. 2001, 157, 138–152. [Google Scholar] [CrossRef]
- Zwicker, E.; Fastl, H. Psychoacoustics: Facts and Models; Springer Science & Business Media: New York, NY, USA, 2013; ISBN 978-3-662-09562-1. [Google Scholar]
- Hillenbrand, J.; Getty, L.A.; Clark, M.J.; Wheeler, K. Acoustic Characteristics of American English Vowels. J. Acoust. Soc. Am. 1995, 97, 3099–3111. [Google Scholar] [CrossRef] [Green Version]
- Brughera, A.; Dunai, L.; Hartmann, W.M. Human Interaural Time Difference Thresholds for Sine Tones: The High-Frequency Limit. J. Acoust. Soc. Am. 2013, 133, 2839–2855. [Google Scholar] [CrossRef] [Green Version]
- Hartmann, W.M.; Dunai, L.; Qu, T. Interaural Time Difference Thresholds as a Function of Frequency. Adv. Exp. Med. Biol. 2013, 787, 239–246. [Google Scholar] [CrossRef]
- Batchelder, M.; Keller, L.S.; Ball Sauer, M.; West, W.L. The Laboratory Rabbit, Guinea Pig, Hamster, and Other Rodents; Academic Press: Boston, MA, USA,, 2012; pp. 1131–1155. [Google Scholar] [CrossRef]
- Steppan, S.; Adkins, R.; Anderson, J. Phylogeny and Divergence-Date Estimates of Rapid Radiations in Muroid Rodents Based on Multiple Nuclear Genes. Syst. Biol. 2004, 53, 533–553. [Google Scholar] [CrossRef]
- Chevret, P.; Dobigny, G. Systematics and Evolution of the Subfamily Gerbillinae (Mammalia, Rodentia, Muridae). Mol. Phylogenetics Evol. 2005, 35, 674–688. [Google Scholar] [CrossRef]
- Lay, D.M. The Anatomy, Physiology, Functional Significance and Evolution of Specialized Hearing Organs of Gerbilline Rodents. J. Morphol. 1972, 138, 41–120. [Google Scholar] [CrossRef] [Green Version]
- Ryan, A. Hearing Sensitivity of the Mongolian Gerbil, Meriones Unguiculatis. J. Acoust. Soc. Am. 1976, 59, 1222. [Google Scholar] [CrossRef]
- Grothe, B. The Evolution of Temporal Processing in the Medial Superior Olive, an Auditory Brainstem Structure. Prog. Neurobiol. 2000, 61, 581–610. [Google Scholar] [CrossRef]
- Grothe, B.; Pecka, M. The Natural History of Sound Localization in Mammals—A Story of Neuronal Inhibition. Front. Neural. Circuits. 2014, 8, 116. [Google Scholar] [CrossRef] [Green Version]
- Ollo, C.; Schwartz, I.R. The superior olivary complex in C57BL/6 mice. Am. J. Anat. 1979, 155, 349–373. [Google Scholar] [CrossRef]
- Fischl, M.J.; Burger, R.M.; Schmidt-Pauly, M.; Alexandrova, O.; Sinclair, J.L.; Grothe, B.; Forsythe, I.D.; Kopp-Scheinpflug, C. Physiology and Anatomy of Neurons in the Medial Superior Olive of the Mouse. J. Neurophysiol. 2016, 116, 2676–2688. [Google Scholar] [CrossRef] [Green Version]
- Moore, J.K. Organization of the Human Superior Olivary Complex. Microsc. Res. Tech. 2000, 51, 403–412. [Google Scholar] [CrossRef]
- Kulesza, R.J. Cytoarchitecture of the Human Superior Olivary Complex: Medial and Lateral Superior Olive. Hear. Res. 2007, 225, 80–90. [Google Scholar] [CrossRef]
- Wagner, E.; Klump, G.M.; Hamann, I. Gap Detection in Mongolian Gerbils (Meriones unguiculatus). Hear. Res. 2003, 176, 11–16. [Google Scholar] [CrossRef]
- Gleich, O.; Hamann, I.; Kittel, M.C.; Klump, G.M.; Strutz, J. A Quantitative Analysis of Psychometric Functions for Different Auditory Tasks in Gerbils. Hear. Res. 2006, 220, 27–37. [Google Scholar] [CrossRef] [PubMed]
- Rosowski, J.J.; Ravicz, M.E.; Teoh, S.W.; Flandermeyer, D. Measurements of Middle-Ear Function in the Mongolian Gerbil, a Specialized Mammalian Ear. Audiol. Neurotol. 1999, 4, 129–136. [Google Scholar] [CrossRef] [PubMed]
- Hermann, J.; Pecka, M.; von Gersdorff, H.; Grothe, B.; Klug, A. Synaptic Transmission at the Calyx of Held under in Vivo like Activity Levels. J. Neurophysiol. 2007, 98, 807–820. [Google Scholar] [CrossRef] [Green Version]
- Cant, N.B. Patterns of Convergence in the Central Nucleus of the Inferior Colliculus of the Mongolian Gerbil: Organization of Inputs from the Superior Olivary Complex in the Low Frequency Representation. Front. Neural. Circuits 2013, 7, 29. [Google Scholar] [CrossRef] [Green Version]
- Roberts, M.T.; Seeman, S.C.; Golding, N.L. A Mechanistic Understanding of the Role of Feedforward Inhibition in the Mammalian Sound Localization Circuitry. Neuron 2013, 78, 923–935. [Google Scholar] [CrossRef] [Green Version]
- Mayer, F.; Albrecht, O.; Dondzillo, A.; Klug, A. Glycinergic Inhibition to the Medial Nucleus of the Trapezoid Body Shows Prominent Facilitation and Can Sustain High Levels of Ongoing Activity. J. Neurophysiol. 2014, 112, 2901–2915. [Google Scholar] [CrossRef] [Green Version]
- Kotak, V.C.; Mowery, T.M.; Sanes, D.H. Characterization of Auditory Synaptic Inputs to Gerbil Perirhinal Cortex. Front. Neural Circuits 2015, 9, 40. [Google Scholar] [CrossRef] [Green Version]
- Ko, K.W.; Rasband, M.N.; Meseguer, V.; Kramer, R.H.; Golding, N.L. Serotonin Modulates Spike Probability in the Axon Initial Segment through HCN Channels. Nat. NeuroSci. 2016, 19, 826–834. [Google Scholar] [CrossRef] [Green Version]
- Stange-Marten, A.; Nabel, A.L.; Sinclair, J.L.; Fischl, M.; Alexandrova, O.; Wohlfrom, H.; Kopp-Scheinpflug, C.; Pecka, M.; Grothe, B. Input Timing for Spatial Processing Is Precisely Tuned via Constant Synaptic Delays and Myelination Patterns in the Auditory Brainstem. Proc. Natl. Acad. Sci. USA 2017, 114, E4851–E4858. [Google Scholar] [CrossRef] [Green Version]
- Wei, L.; Karino, S.; Verschooten, E.; Joris, P.X. Enhancement of Phase-Locking in Rodents. I. An Axonal Recording Study in Gerbil. J. Neurophysiol. 2017, 118, 2009–2023. [Google Scholar] [CrossRef]
- Paraouty, N.; Rizzuto, C.R.; Sanes, D.H. Dopaminergic Signaling Supports Auditory Social Learning. Sci. Rep. 2021, 11, 13117. [Google Scholar] [CrossRef]
- Wang, Y.; Sakano, H.; Beebe, K.; Brown, M.R.; de Laat, R.; Bothwell, M.; Kulesza, R.J.; Rubel, E.W. Intense and specialized dendritic localization of the fragile X mental retardation protein in binaural brainstem neurons: A comparative study in the alligator, chicken, gerbil, and human. J. Comp. Neurol. 2014, 522, 2107–2128. [Google Scholar] [CrossRef] [Green Version]
- Kulesza, R.J.; Mangunay, K. Morphological Features of the Medial Superior Olive in Autism. Brain Res. 2008, 1200, 132–137. [Google Scholar] [CrossRef]
- Beebe, K.; Wang, Y.; Kulesza, R. Distribution of fragile X mental retardation protein in the human auditory brainstem. Neuroscience 2014, 273, 79–91. [Google Scholar] [CrossRef]
- McCullagh, E.A.; Rotschafer, S.E.; Auerbach, B.D.; Klug, A.; Kaczmarek, L.K.; Cramer, K.S.; Kulesza, R.J.; Razak, K.A.; Lovelace, J.W.; Lu, Y.; et al. Mechanisms underlying auditory processing deficits in Fragile X syndrome. FASEB J. 2020, 34, 3501–3518. [Google Scholar] [CrossRef] [Green Version]
- Kelly, J.B.; Masterton, B. Auditory Sensitivity of the Albino Rat. J. Comp. Physiol. Psychol. 1977, 91, 930–936. [Google Scholar] [CrossRef]
- Gleich, O.; Strutz, J. The Mongolian Gerbil as a model for the analysis of peripheral and central age-dependent hearing loss. In Hearing Loss; Naz, S., Ed.; InTechOpen: London, UK, 2012; ISBN 978-953-51-0366-0. [Google Scholar]
- Baker, A.G.; Emerson, V.F. Grating Acuity of the Mongolian Gerbil (Meriones unguiculatus). Behav. Brain Res. 1983, 8, 195–209. [Google Scholar] [CrossRef]
- Yang, S.; Luo, X.; Xiong, G.; So, K.-F.; Yang, H.; Xu, Y. The Electroretinogram of Mongolian Gerbil (Meriones unguiculatus): Comparison to Mouse. Neurosci. Lett. 2015, 589, 7–12. [Google Scholar] [CrossRef]
- Govardovskii, V.I.; Ro¨hlich, P.; Sze’l, A.; Khokhlova, T.V. Cones in the Retina of the Mongolian Gerbil, Meriones unguiculatus: An Immunocytochemical and Electrophysiological Study. Vis. Res. 1992, 32, 19–27. [Google Scholar] [CrossRef]
- Bytyqi, A.H.; Layer, P.G. Lamina Formation in the Mongolian Gerbil Retina (Meriones unguiculatus). Anat. Embryol. 2005, 209, 217–225. [Google Scholar] [CrossRef] [PubMed]
- Garbers, C.; Henke, J.; Leibold, C.; Wachtler, T.; Thurley, K. Contextual Processing of Brightness and Color in Mongolian Gerbils. J. Vis. 2015, 15, 13. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, T.; Huang, L.; Zhang, L.; Tan, M.; Pu, M.; Pickard, G.E.; So, K.-F.; Ren, C. ON and OFF Retinal Ganglion Cells Differentially Regulate Serotonergic and GABAergic Activity in the Dorsal Raphe Nucleus. Sci. Rep. 2016, 6, 26060. [Google Scholar] [CrossRef]
- Delbarre, B.; Delbarre, G.; Rochat, C.; Calinon, F. Effect of Piribedil, a D-2 Dopaminergic Agonist, on Dopamine, Amino Acids, and Free Radicals in Gerbil Brain after Cerebral Ischemia. Mol. Chem. Neuropathol. 1995, 26, 43–52. [Google Scholar] [CrossRef]
- Takayanagi, T.H.; Akao, N.; Suzuki, R.; Tomoda, M.; Tsukidate, S.; Fujita, K. New Animal Model for Human Ocular Toxocariasis: Ophthalmoscopic Observation. Br. J. Ophthalmol. 1999, 83, 967–972. [Google Scholar] [CrossRef] [Green Version]
- Akao, N.; Hayashi, E.; Sato, H.; Fujita, K.; Furuoka, H. Diffuse Retinochoroiditis Due to Baylisascaris Procyonis in Monglian Gerbils. J. Parasitol. 2003, 89, 174–175. [Google Scholar] [CrossRef]
- Mauck, M.C.; Mancuso, K.; Kuchenbecker, J.A.; Connor, T.B.; Hauswirth, W.W.; Neitz, J.; Neitz, M. Longitudinal Evaluation of Expression of Virally Delivered Transgenes in Gerbil Cone Photoreceptors. Vis. Neurosci. 2008, 25, 273–282. [Google Scholar] [CrossRef] [Green Version]
- Abitbol, M.; Menini, C.; Delezoide, A.L.; Rhyner, T.; Vekemans, M.; Mallet, J. Nucleus Basalis Magnocellularis and Hippocampus Are the Major Sites of FMR-1 Expression in the Human Fetal Brain. Nat. Genet. 1993, 4, 147–153. [Google Scholar] [CrossRef]
- Guimarães-Souza, E.M.; Perche, O.; Morgans, C.W.; Duvoisin, R.M.; Calaza, K.C. Fragile X Mental Retardation Protein Expression in the Retina Is Regulated by Light. Exp. Eye Res. 2016, 146, 72–82. [Google Scholar] [CrossRef] [Green Version]
- Zhang, P.-P.; Yao, H.-H.; Zha, A.-H.; Liu, X.-Y.; Fan, K.-Y.; Xu, Y.; Yuan, H.-Y.; Li, L.; Wang, L.-C. Cellular Localization of the FMRP in Rat Retina. Biosci. Rep. 2020, 40, BSR20200570. [Google Scholar] [CrossRef]
- Kogan, C.S.; Boutet, I.; Cornish, K.; Zangenehpour, S.; Mullen, K.T.; Holden, J.J.A.; Der Kaloustian, V.M.; Andermann, E.; Chaudhuri, A. Differential Impact of the FMR1 Gene on Visual Processing in Fragile X Syndrome. Brain 2004, 127, 591–601. [Google Scholar] [CrossRef] [Green Version]
- Zorio, D.A.R.; Jackson, C.M.; Liu, Y.; Rubel, E.W.; Wang, Y. Cellular distribution of the fragile X mental retardation protein in the mouse brain. J. Comp. Neurol. 2017, 525, 818–849. [Google Scholar] [CrossRef] [Green Version]
- Kranjc, B.S.; Brezigar, A.; Peterlin, B. Bilateral Macular Dysplasia in Fragile X Syndrome. Optom. Vis. Sci. 1998, 75, 856–859. [Google Scholar] [CrossRef]
- Kéri, S.; Benedek, G. Fragile X Protein Expression Is Linked to Visual Functions in Healthy Male Volunteers. Neuroscience 2011, 192, 345–350. [Google Scholar] [CrossRef]
- Kéri, S.; Benedek, G. Visual Pathway Deficit in Female Fragile X Premutation Carriers: A Potential Endophenotype. Brain Cogn. 2009, 69, 291–295. [Google Scholar] [CrossRef]
- Rossignol, R.; Ranchon-Cole, I.; Pâris, A.; Herzine, A.; Perche, A.; Laurenceau, D.; Bertrand, P.; Cercy, C.; Pichon, J.; Mortaud, S.; et al. Visual Sensorial Impairments in Neurodevelopmental Disorders: Evidence for a Retinal Phenotype in Fragile X Syndrome. PLoS ONE 2014, 9, e105996. [Google Scholar] [CrossRef] [Green Version]
- Perche, O.; Felgerolle, C.; Ardourel, M.; Bazinet, A.; Pâris, A.; Rossignol, R.; Meyer-Dilhet, G.; Mausset-Bonnefont, A.-L.; Hébert, B.; Laurenceau, D.; et al. Early Retinal Defects in Fmr1-/y Mice: Toward a Critical Role of Visual Dys-Sensitivity in the Fragile X Syndrome Phenotype? Front. Cell Neurosci. 2018, 12, 96. [Google Scholar] [CrossRef] [Green Version]
- Kay, R.B.; Gabreski, N.A.; Triplett, J.W. Visual Subcircuit-Specific Dysfunction and Input-Specific Mispatterning in the Superior Colliculus of Fragile X Mice. J. Neurodevelop. Disord. 2018, 10, 23. [Google Scholar] [CrossRef] [Green Version]
- Piovanotti, M.R.A.; Vieira, M.L. Presence of the Father and Parental Experience Have Differentiated Effects on Pup Development in Mongolian Gerbils (Meriones Unguiculatus). Behav. Processes 2004, 66, 107–117. [Google Scholar] [CrossRef]
- Hendrie, C.A.; Pickles, A.R.; Duxon, M.S.; Riley, G.; Hagan, J.J. Effects of Fluoxetine on Social Behaviour and Plasma Corticosteroid Levels in Female Mongolian Gerbils. Behav. Pharmacol. 2003, 14, 545–550. [Google Scholar] [CrossRef]
- Pickles, A.R.; Hagan, J.J.; Jones, D.N.C.; Hendrie, C.A. Short-Term Individual Housing Induced Social Deficits in Female Mongolian Gerbils: Attenuation by Chronic but Not Acute Imipramine. Neuropharmacology 2012, 62, 1993–1998. [Google Scholar] [CrossRef]
- Rico, J.L.; Penagos-Gil, M.; Castañeda, A.F.; Corredor, K. Gerbils Exhibit Stable Open-Arms Exploration across Repeated Testing on the Elevated plus-Maze. Behav. Processes 2016, 122, 104–109. [Google Scholar] [CrossRef]
- Bechara, E.G.; Didiot, M.C.; Melko, M.; Davidovic, L.; Bensaid, M.; Martin, P.; Castets, M.; Pognonec, P.; Khandjian, E.W.; Moine, H.; et al. A Novel Function for Fragile X Mental Retardation Protein in Translational Activation. PLoS Biol. 2009, 7, e16. [Google Scholar] [CrossRef] [Green Version]
- Mithal, D.S.; Chandel, N.S. Mitochondrial Dysfunction in Fragile-X Syndrome: Plugging the Leak May Save the Ship. Mol. Cell 2020, 80, 381–383. [Google Scholar] [CrossRef]
- Licznerski, P.; Park, H.-A.; Rolyan, H.; Chen, R.; Mnatsakanyan, N.; Miranda, P.; Graham, M.; Wu, J.; Cruz-Reyes, N.; Mehta, N.; et al. ATP Synthase C-Subunit Leak Causes Aberrant Cellular Metabolism in Fragile X Syndrome. Cell 2020, 182, 1170–1185.e9. [Google Scholar] [CrossRef]
- Bach, S.; Shovlin, S.; Moriarty, M.; Bardoni, B.; Tropea, D. Rett Syndrome and Fragile X Syndrome: Different Etiology With Common Molecular Dysfunctions. Front. Cell Neurosci. 2021, 15, 764761. [Google Scholar] [CrossRef]
- D’Antoni, S.; de Bari, L.; Valenti, D.; Borro, M.; Bonaccorso, C.M.; Simmaco, M.; Vacca, R.A.; Catania, M.V. Aberrant Mitochondrial Bioenergetics in the Cerebral Cortex of the Fmr1 Knockout Mouse Model of Fragile X Syndrome. Biol. Chem. 2020, 401, 497–503. [Google Scholar] [CrossRef]
- Shen, M.; Wang, F.; Li, M.; Sah, N.; Stockton, M.E.; Tidei, J.J.; Gao, Y.; Korabelnikov, T.; Kannan, S.; Vevea, J.D.; et al. Reduced Mitochondrial Fusion and Huntingtin Levels Contribute to Impaired Dendritic Maturation and Behavioral Deficits in Fmr1-Mutant Mice. Nat. Neurosci. 2019, 22, 386–400. [Google Scholar] [CrossRef]
- Robinson, P.F. Metabolism of the Gerbil, Meriones unguiculatus. Science 1959, 130, 502–503. [Google Scholar] [CrossRef]
- Khakisahneh, S.; Zhang, X.-Y.; Nouri, Z.; Wang, D.-H. Cecal Microbial Transplantation Attenuates Hyperthyroid-Induced Thermogenesis in Mongolian Gerbils. Microb. Biotechnol. 2022, 15, 817–831. [Google Scholar] [CrossRef]
- Guo, Y.-Y.; Hao, S.; Zhang, M.; Zhang, X.; Wang, D. Aquaporins, Evaporative Water Loss and Thermoregulation in Heat-Acclimated Mongolian Gerbils (Meriones unguiculatus). J. Therm. Biol. 2020, 91, 102641. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.-C.; Won, M.-H. Neuroprotection of Antioxidant Enzymes against Transient Global Cerebral Ischemia in Gerbils. Anat. Cell Biol. 2014, 47, 149–156. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nouri, Z.; Zhang, X.-Y.; Khakisahneh, S.; Degen, A.A.; Wang, D.-H. The Microbiota-Gut-Kidney Axis Mediates Host Osmoregulation in a Small Desert Mammal. NPJ Biofilms Microbiomes 2022, 8, 16. [Google Scholar] [CrossRef] [PubMed]
- Lee, C.M.; Lederman, J.D.; Hofmann, N.E.; Erdman, J.W. The Mongolian Gerbil (Meriones unguiculatus) Is an Appropriate Animal Model for Evaluation of the Conversion of Beta-Carotene to Vitamin A. J. Nutr. 1998, 128, 280–286. [Google Scholar] [CrossRef] [Green Version]
- Thiessen, D.D.; Graham, M.; Perkins, J.; Marcks, S. Temperature Regulation and Social Grooming in the Mongolian Gerbil (Meriones unguiculatus). Behav. Biol. 1977, 19, 279–288. [Google Scholar] [CrossRef]
- Thiessen, D.D. Body Temperature and Grooming in the Mongolian Gerbil. Ann. N. Y. Acad. Sci. 1988, 525, 27–39. [Google Scholar] [CrossRef]
- Musumeci, S.A.; Hagerman, R.J.; Ferri, R.; Bosco, P.; Dalla Bernardina, B.; Tassinari, C.A.; De Sarro, G.B.; Elia, M. Epilepsy and EEG Findings in Males with Fragile X Syndrome. Epilepsia 1999, 40, 1092–1099. [Google Scholar] [CrossRef]
- Berry-Kravis, E.; Raspa, M.; Loggin-Hester, L.; Bishop, E.; Holiday, D.; Bailey, D.B. Seizures in Fragile X Syndrome: Characteristics and Comorbid Diagnoses. Am. J. Intellect. Dev. Disabil. 2010, 115, 461–472. [Google Scholar] [CrossRef]
- Loskota, W.J.; Lomax, P.; Rich, S.T. The Gerbil as a Model for the Study of the Epilepsies. Epilepsia 1974, 15, 109–119. [Google Scholar] [CrossRef]
- Ludvig, N.; Farias, P.A.; Ribak, C.E. An Analysis of Various Environmental and Specific Sensory Stimuli on the Seizure Activity of the Mongolian Gerbil. Epilepsy Res. 1991, 8, 30–35. [Google Scholar] [CrossRef] [Green Version]
- Kang, T.-C.; Park, S.-K.; Bahn, J.H.; Jeon, S.G.; Jo, S.M.; Cho, S.-W.; Choi, S.Y.; Won, M.H. The Alteration of G-Aminobutyric Acid-Transaminase Expression in the Gerbil Hippocampus Induced by Seizure. Neurochem. Int. 2001, 6, 609–614. [Google Scholar] [CrossRef]
- Hwang, I.K.; Park, S.-K.; An, S.-J.; Yoo, K.-Y.; Kim, D.-S.; Jung, J.-Y.; Won, M.H.; Choi, S.-Y.; Kwon, O.-S.; Kang, T.-C. GABAA, Not GABAB, Receptor Shows Subunit- and Spatial-Specific Alterations in the Hippocampus of Seizure Prone Gerbils. Brain Res. 2004, 1003, 98–107. [Google Scholar] [CrossRef]
- Kwak, S.-E.; Kim, J.-E.; Kim, D.-S.; Jung, J.-Y.; Ho Won, M.; Kwon, O.-S.; Choi, S.-Y.; Kang, T.-C. Effects of GABAergic Transmissions on the Immunoreactivities of Calcium Binding Proteins in the Gerbil Hippocampus. J. Comp. Neurol. 2005, 485, 153–164. [Google Scholar] [CrossRef]
- Hodges, S.L.; Reynolds, C.D.; Nolan, S.O.; Huebschman, J.L.; Okoh, J.T.; Binder, M.S.; Lugo, J.N. A Single Early-Life Seizure Results in Long-Term Behavioral Changes in the Adult Fmr1 Knockout Mouse. Epilepsy Res. 2019, 157, 106193. [Google Scholar] [CrossRef]
- Liang, P.; Zhang, X.; Chen, Y.; Huang, J. Developmental History and Application of CRISPR in Human Disease. J. Gene Med. 2017, 19. [Google Scholar] [CrossRef] [Green Version]
- Wu, J.; Vilarino, M.; Suzuki, K.; Okamura, D.; Bogliotti, Y.S.; Park, I.; Rowe, J.; McNabb, B.; Ross, P.J.; Belmonte, J.C.I. CRISPR-Cas9 Mediated One-Step Disabling of Pancreatogenesis in Pigs. Sci. Rep. 2017, 7, 10487. [Google Scholar] [CrossRef] [Green Version]
- Cheng, S.; Fu, Y.; Zhang, Y.; Xian, W.; Wang, H.; Grothe, B.; Liu, X.; Xu, X.; Klug, A.; McCullagh, E.A. Enhancement of de Novo Sequen.ncing, Assembly and Annotation of the Mongolian Gerbil Genome with Transcriptome Sequencing and Assembly from Several Different Tissues. BMC Genom. 2019, 20, 903. [Google Scholar] [CrossRef] [Green Version]
- Zorio, D.A.R.; Monsma, S.; Sanes, D.H.; Golding, N.L.; Rubel, E.W.; Wang, Y. De novo sequencing and initial annotation of the Mongolian gerbil (Meriones unguiculatus) genome. Genomics 2019, 111, 441–449. [Google Scholar] [CrossRef]
- Wang, Y.; Zhao, P.; Song, Z.; Du, X.; Huo, X.; Lu, J.; Liu, X.; Lv, J.; Li, C.; Guo, M.; et al. Generation of Gene-Knockout Mongolian Gerbils via CRISPR/Cas9 System. Front. Bioeng. Biotechnol. 2020, 8, 780. [Google Scholar] [CrossRef]
- Davis, J.K.; Broadie, K. Multifarious Functions of the Fragile X Mental Retardation Protein. Trends Genet. 2017, 33, 703–714. [Google Scholar] [CrossRef]
- Wong, H.H.-W.; Lin, J.Q.; Ströhl, F.; Roque, C.G.; Cioni, J.-M.; Cagnetta, R.; Turner-Bridger, B.; Laine, R.F.; Harris, W.A.; Kaminski, C.F.; et al. RNA Docking and Local Translation Regulate Site-Specific Axon Remodeling In Vivo. Neuron 2017, 95, 852–868.e8. [Google Scholar] [CrossRef] [Green Version]
- Gatto, C.L.; Broadie, K. Temporal Requirements of the Fragile x Mental Retardation Protein in Modulating Circadian Clock Circuit Synaptic Architecture. Front. Neural Circuits 2009, 3, 8. [Google Scholar] [CrossRef] [Green Version]
- Doll, C.A.; Broadie, K. Activity-Dependent FMRP Requirements in Development of the Neural Circuitry of Learning and Memory. Development 2015, 142, 1346–1356. [Google Scholar] [CrossRef] [Green Version]
- Sears, J.C.; Broadie, K. Fragile X Mental Retardation Protein Regulates Activity-Dependent Membrane Trafficking and Trans-Synaptic Signaling Mediating Synaptic Remodeling. Front. Mol. Neurosci. 2018, 10, 440. [Google Scholar] [CrossRef] [Green Version]
- Klin, A.; Shultz, S.; Jones, W. Social Visual Engagement in Infants and Toddlers with Autism: Early Developmental Transitions and a Model of Pathogenesis. Neurosci. Biobehav. Rev. 2015, 50, 189–203. [Google Scholar] [CrossRef] [Green Version]
- Ben-Ari, Y. Is birth a critical period in the pathogenesis of autism spectrum disorders? Nat. Rev. Neurosci. 2015, 16, 498–505. [Google Scholar] [CrossRef]
- Price, D.K.; Zhang, F.; Ashley, C.T.; Warren, S.T. The Chicken FMR1 Gene Is Highly Conserved with a CCT 5′-Untranslated Repeat and Encodes an RNA-Binding Protein. Genomics 1996, 31, 3–12. [Google Scholar] [CrossRef] [Green Version]
- Sakano, H.; Zorio, D.A.R.; Wang, X.; Ting, Y.S.; Noble, W.S.; MacCoss, M.J.; Rubel, E.W.; Wang, Y. Proteomic analyses of nucleus laminaris identified candidate targets of the fragile X mental retardation protein. J. Comp. Neurol. 2017, 525, 3341–3359. [Google Scholar] [CrossRef] [Green Version]
- Schaeffer, C.; Bardoni, B.; Mandel, J.L.; Ehresmann, B.; Ehresmann, C.; Moine, H. The Fragile X Mental Retardation Protein Binds Specifically to Its MRNA via a Purine Quartet Motif. EMBO J. 2001, 20, 4803–4813. [Google Scholar] [CrossRef]
- Xu, Z.; Che, T.; Li, F.; Tian, K.; Zhu, Q.; Mishra, S.K.; Dai, Y.; Li, M.; Li, D. The Temporal Expression Patterns of Brain Transcriptome during Chicken Development and Ageing. BMC Genom. 2018, 19, 917. [Google Scholar] [CrossRef]
- Nothwang, H.G. Evolution of Mammalian Sound Localization Circuits: A Developmental Perspective. Prog. Neurobiol. 2016, 141, 1–24. [Google Scholar] [CrossRef] [PubMed]
- Hirsch, D.; Kohl, A.; Wang, Y.; Sela-Donenfeld, D. Axonal Projection Patterns of the Dorsal Interneuron Populations in the Embryonic Hindbrain. Front. Neuroanat. 2021, 15, 793161. [Google Scholar] [CrossRef] [PubMed]
- Ashida, G.; Carr, C.E. Sound Localization: Jeffress and Beyond. Curr. Opin. Neurobiol. 2011, 21, 745–751. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sanchez, J.T.; Wang, Y.; Lu, Y.; Burger, R.M.; Seidl, A.H.; Rubel, E.W. Nucleus laminaris. In Handbook of Brain Microcircuits; Oxford University Press: New York, NY, USA, 2017; pp. 425–436. ISBN 978-0-19-063614-2. [Google Scholar]
- Parks, T.N.; Rubel, E.W. Organization and Development of Brain Stem Auditory Nuclei of the Chicken: Organization of Projections from N. Magnocellularis to N. Laminaris. J. Comp. Neurol. 1975, 164, 435–448. [Google Scholar] [CrossRef]
- Rubel, E.W.; Smith, Z.D.; Steward, O. Sprouting in the Avian Brainstem Auditory Pathway: Dependence on Dendritic Integrity. J. Comp. Neurol. 1981, 202, 397–414. [Google Scholar] [CrossRef]
- Jhaveri, S.; Morest, D.K. Neuronal Architecture in Nucleus Magnocellularis of the Chicken Auditory System with Observations on Nucleus Laminaris: A Light and Electron Microscope Study. Neuroscience 1982, 7, 809–836. [Google Scholar] [CrossRef]
- Kuba, H.; Koyano, K.; Ohmori, H. Development of Membrane Conductance Improves Coincidence Detection in the Nucleus Laminaris of the Chicken. J. Physiol. 2002, 540, 529–542. [Google Scholar] [CrossRef]
- Kuba, H. Evaluation of the Limiting Acuity of Coincidence Detection in Nucleus Laminaris of the Chicken. J. Physiol. 2003, 552, 611–620. [Google Scholar] [CrossRef]
- Kuba, H. Tonotopic Specialization of Auditory Coincidence Detection in Nucleus Laminaris of the Chick. J. Neurosci. 2005, 25, 1924–1934. [Google Scholar] [CrossRef] [Green Version]
- Burger, R.M.; Cramer, K.S.; Pfeiffer, J.D.; Rubel, E.W. Avian Superior Olivary Nucleus Provides Divergent Inhibitory Input to Parallel Auditory Pathways. J. Comp. Neurol. 2005, 481, 6–18. [Google Scholar] [CrossRef]
- Blackmer, T.; Kuo, S.P.; Bender, K.J.; Apostolides, P.F.; Trussell, L.O. Dendritic Calcium Channels and Their Activation by Synaptic Signals in Auditory Coincidence Detector Neurons. J. Neurophysiol. 2009, 102, 1218–1226. [Google Scholar] [CrossRef] [Green Version]
- Sanchez, J.T.; Seidl, A.H.; Rubel, E.W.; Barria, A. Control of Neuronal Excitability by NMDA-Type Glutamate Receptors in Early Developing Binaural Auditory Neurons: Early Control of Neuronal Excitability by NMDA-Rs. J. Physiol. 2012, 590, 4801–4818. [Google Scholar] [CrossRef]
- Hong, H.; Lu, T.; Wang, X.; Wang, Y.; Sanchez, J.T. Resurgent sodium current promotes action potential firing in the avian auditory brainstem. J. Physiol. 2018, 596, 423–443. [Google Scholar] [CrossRef] [Green Version]
- Wang, X.; Hong, H.; Brown, D.H.; Sanchez, J.T.; Wang, Y. Distinct Neural Properties in the Low-Frequency Region of the Chicken Cochlear Nucleus Magnocellularis. eNeuro 2017, 4, ENEURO.0016-17.2017. [Google Scholar] [CrossRef] [Green Version]
- Gao, M.; Maynard, K.R.; Chokshi, V.; Song, L.; Jacobs, C.; Wang, H.; Tran, T.; Martinowich, K.; Lee, H.-K. Rebound Potentiation of Inhibition in Juvenile Visual Cortex Requires Vision-Induced BDNF Expression. J. Neurosci. 2014, 34, 10770–10779. [Google Scholar] [CrossRef] [Green Version]
- Wang, X.; Zorio, D.A.R.; Schecterson, L.; Lu, Y.; Wang, Y. Postsynaptic FMRP Regulates Synaptogenesis In Vivo in the Developing Cochlear Nucleus. J. Neurosci. 2018, 38, 6445–6460. [Google Scholar] [CrossRef] [Green Version]
- Wang, X.; Kohl, A.; Yu, X.; Zorio, D.A.R.; Klar, A.; Sela-Donenfeld, D.; Wang, Y. Temporal-specific roles of fragile X mental retardation protein in the development of the hindbrain auditory circuit. Development 2020, 147, dev188797. [Google Scholar] [CrossRef]
- Schecterson, L.C.; Sanchez, J.T.; Rubel, E.W.; Bothwell, M. TrkB Downregulation Is Required for Dendrite Retraction in Developing Neuron.ns of Chicken Nucleus Magnocellularis. J. Neurosci. 2012, 32, 14000–14009. [Google Scholar] [CrossRef] [Green Version]
- Petazzi, P.; Akizu, N.; García, A.; Estarás, C.; Martínez de Paz, A.; Rodríguez-Paredes, M.; Martínez-Balbás, M.A.; Huertas, D.; Esteller, M. An Increase in MECP2 Dosage Impairs Neural Tube Formation. Neurobiol. Dis. 2014, 67, 49–56. [Google Scholar] [CrossRef] [Green Version]
- Garic, A.; Berres, M.E.; Smith, S.M. High-Throughput Transcriptome Sequencing Identifies Candidate Genetic Modifiers of Vulnerability to Fetal Alcohol Spectrum Disorders. Alcohol Clin. Exp. Res. 2014, 38, 1874–1882. [Google Scholar] [CrossRef] [Green Version]
- Flentke, G.R.; Smith, S.M. The Avian Embryo as a Model for Fetal Alcohol Spectrum Disorder. Biochem. Cell Biol. 2018, 96, 98–106. [Google Scholar] [CrossRef] [Green Version]
- Stephen, L.A.; Tawamie, H.; Davis, G.M.; Tebbe, L.; Nürnberg, P.; Nürnberg, G.; Thiele, H.; Thoenes, M.; Boltshauser, E.; Uebe, S.; et al. TALPID3 Controls Centrosome and Cell Polarity and the Human Ortholog KIAA0586 Is Mutated in Joubert Syndrome (JBTS23). eLife 2015, 4, e08077. [Google Scholar] [CrossRef]
- Thawani, A.; Sirohi, D.; Kuhn, R.J.; Fekete, D.M. Zika Virus Can Strongly Infect and Disrupt Secondary Organizers in the Ventricular Zone of the Embryonic Chicken Brain. Cell Rep. 2018, 23, 692–700. [Google Scholar] [CrossRef] [Green Version]
- Zosen, D.; Hadera, M.G.; Lumor, J.S.; Andersen, J.M.; Paulsen, R.E. Chicken Embryo as Animal Model to Study Drug Distribution to the Developing Brain. J. Pharm. Toxicol. Methods 2021, 112, 107105. [Google Scholar] [CrossRef]

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Curnow, E.; Wang, Y. New Animal Models for Understanding FMRP Functions and FXS Pathology. Cells 2022, 11, 1628. https://doi.org/10.3390/cells11101628
Curnow E, Wang Y. New Animal Models for Understanding FMRP Functions and FXS Pathology. Cells. 2022; 11(10):1628. https://doi.org/10.3390/cells11101628
Chicago/Turabian StyleCurnow, Eliza, and Yuan Wang. 2022. "New Animal Models for Understanding FMRP Functions and FXS Pathology" Cells 11, no. 10: 1628. https://doi.org/10.3390/cells11101628
APA StyleCurnow, E., & Wang, Y. (2022). New Animal Models for Understanding FMRP Functions and FXS Pathology. Cells, 11(10), 1628. https://doi.org/10.3390/cells11101628