
Current Therapeutic Landscape and Safety Roadmap for Targeting the Aryl Hydrocarbon Receptor in Inflammatory Gastrointestinal Indications

 ,
,
Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Role of AhR within the Gut Microenvironment
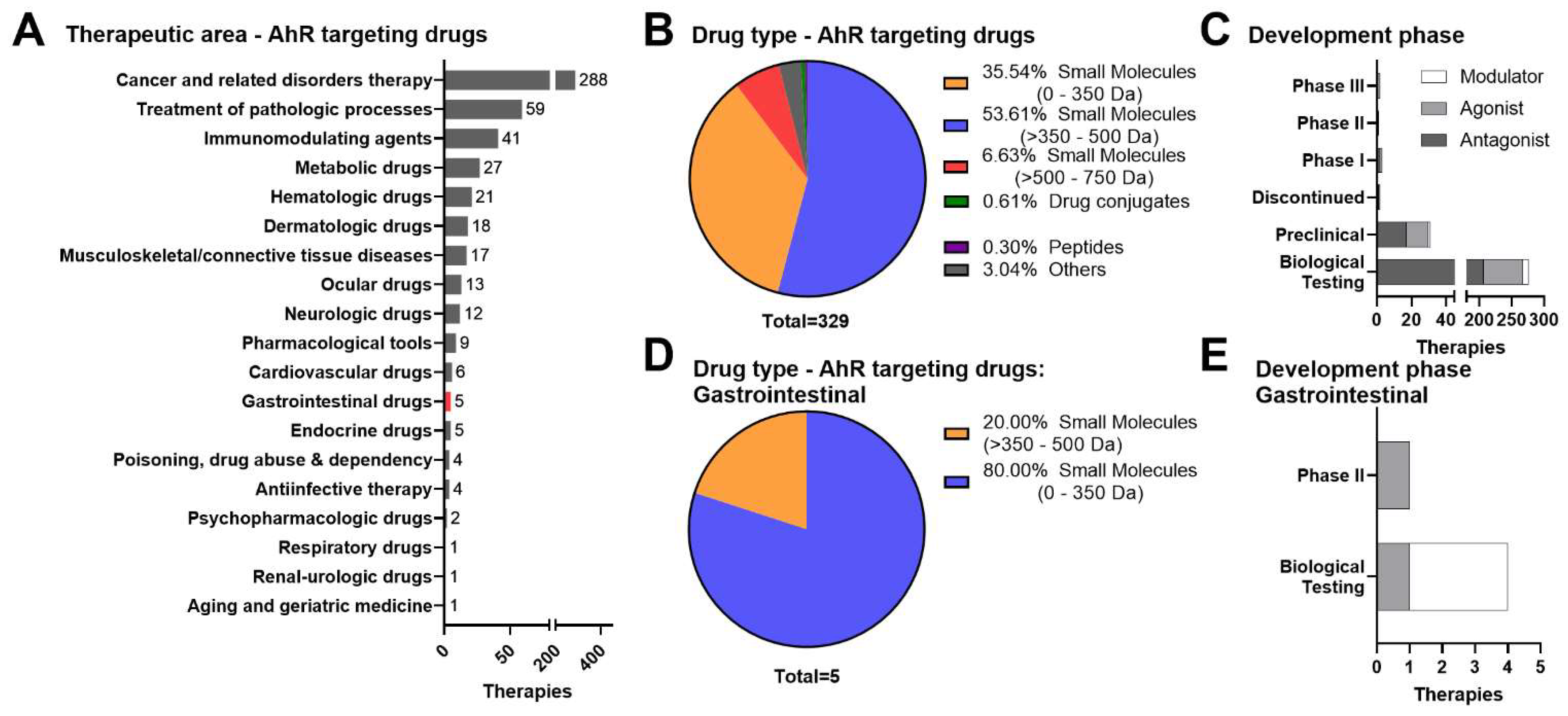
3. Therapeutic Landscape of AhR-Targeted Molecules for Combatting Inflammatory GI Indications
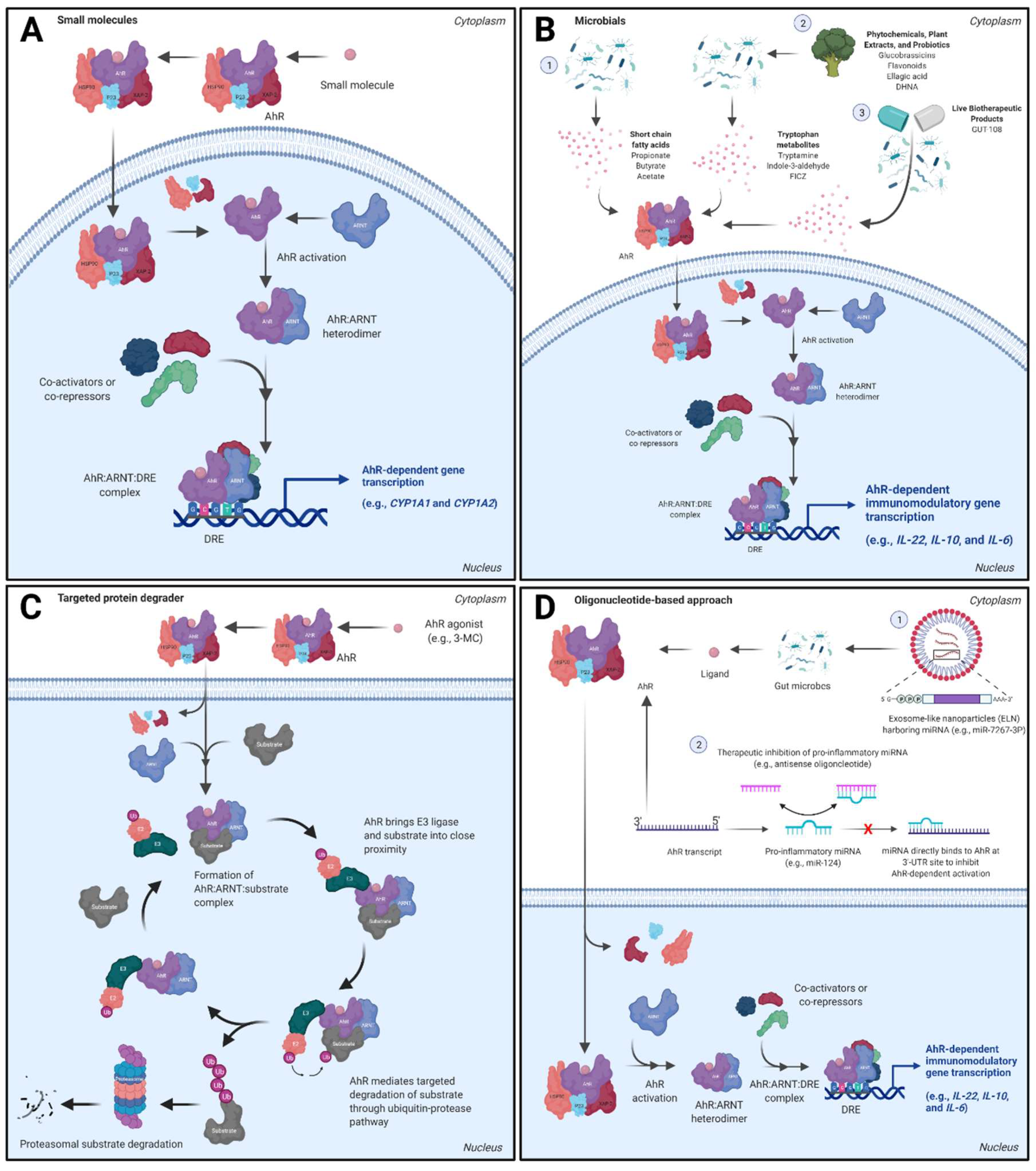
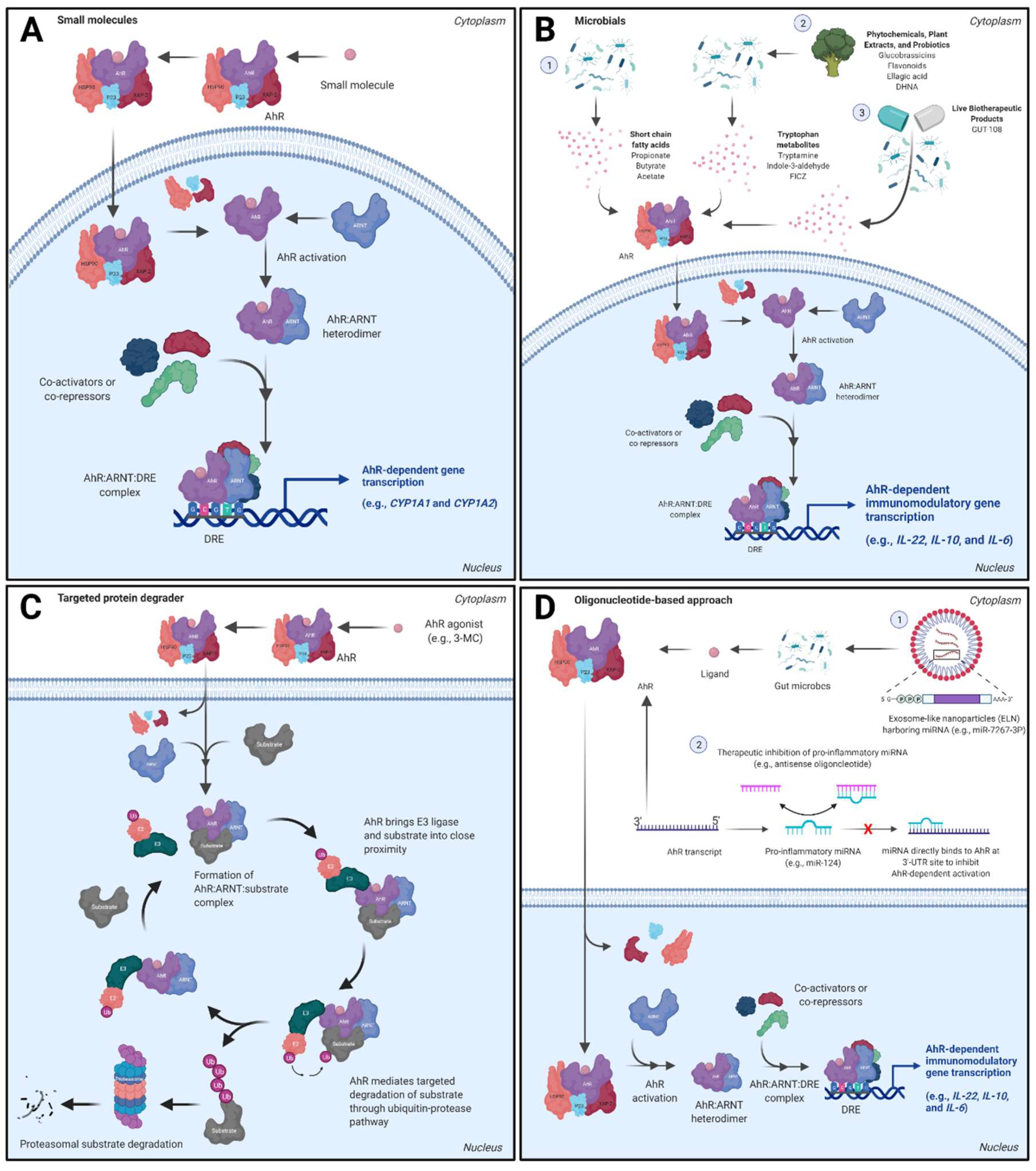
3.1. Small Molecule Approaches
3.2. Dietary Metabolites, Microbials, and Live Biotherapeutic Products
3.3. AhR Proteolysis Targeting Chimeras (PROTACs)
3.4. Oligonucleotide-Based Approaches
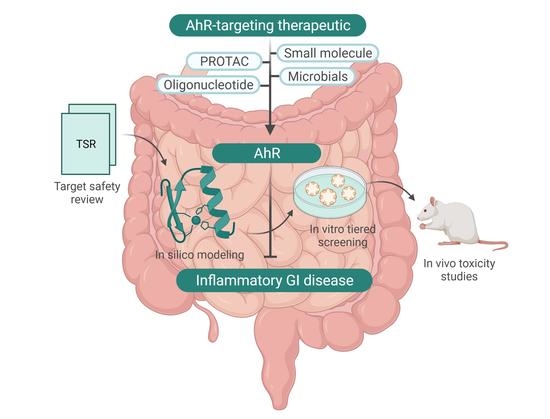
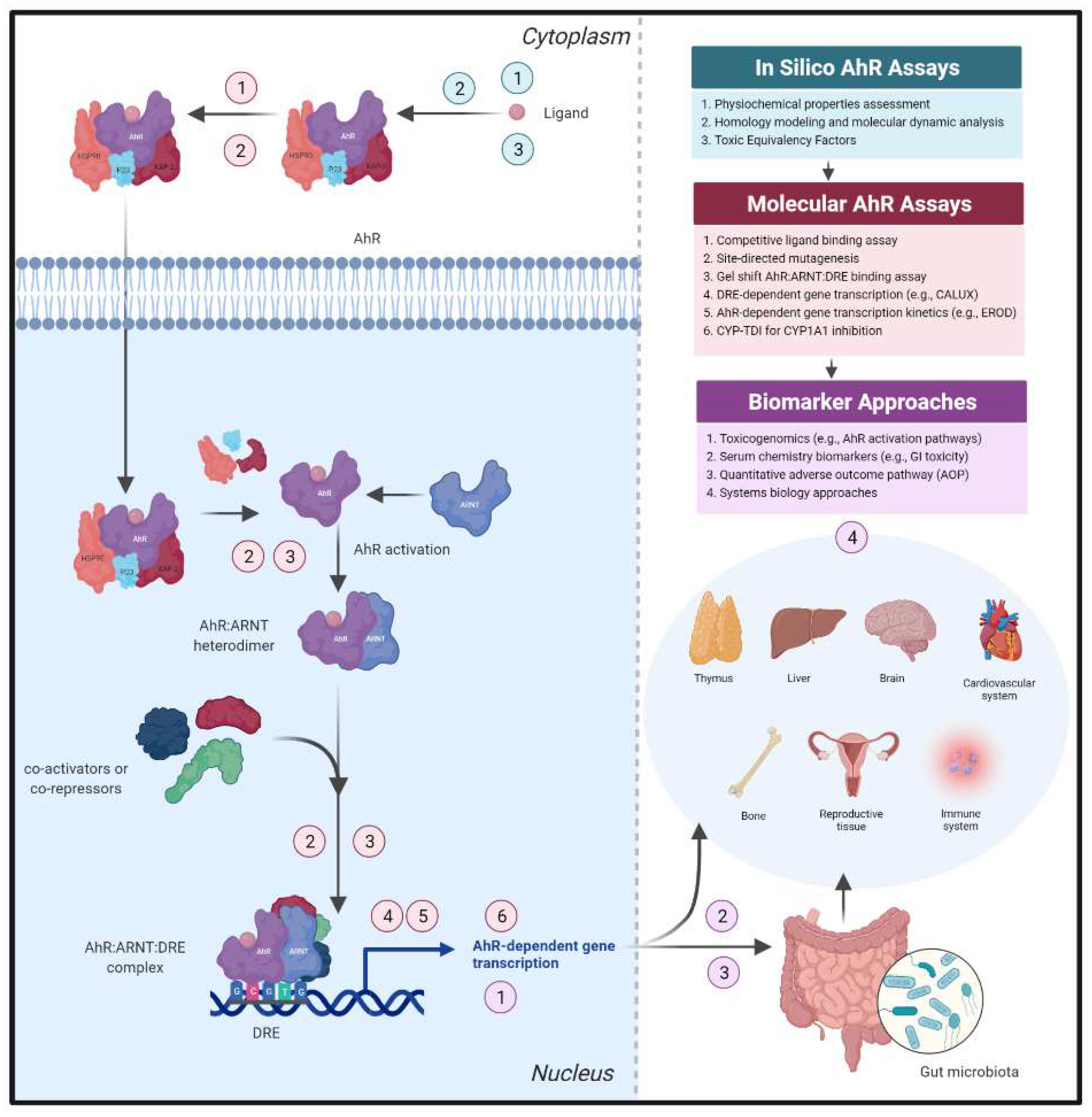
4. Addressing AhR Safety Liabilities Going Forward
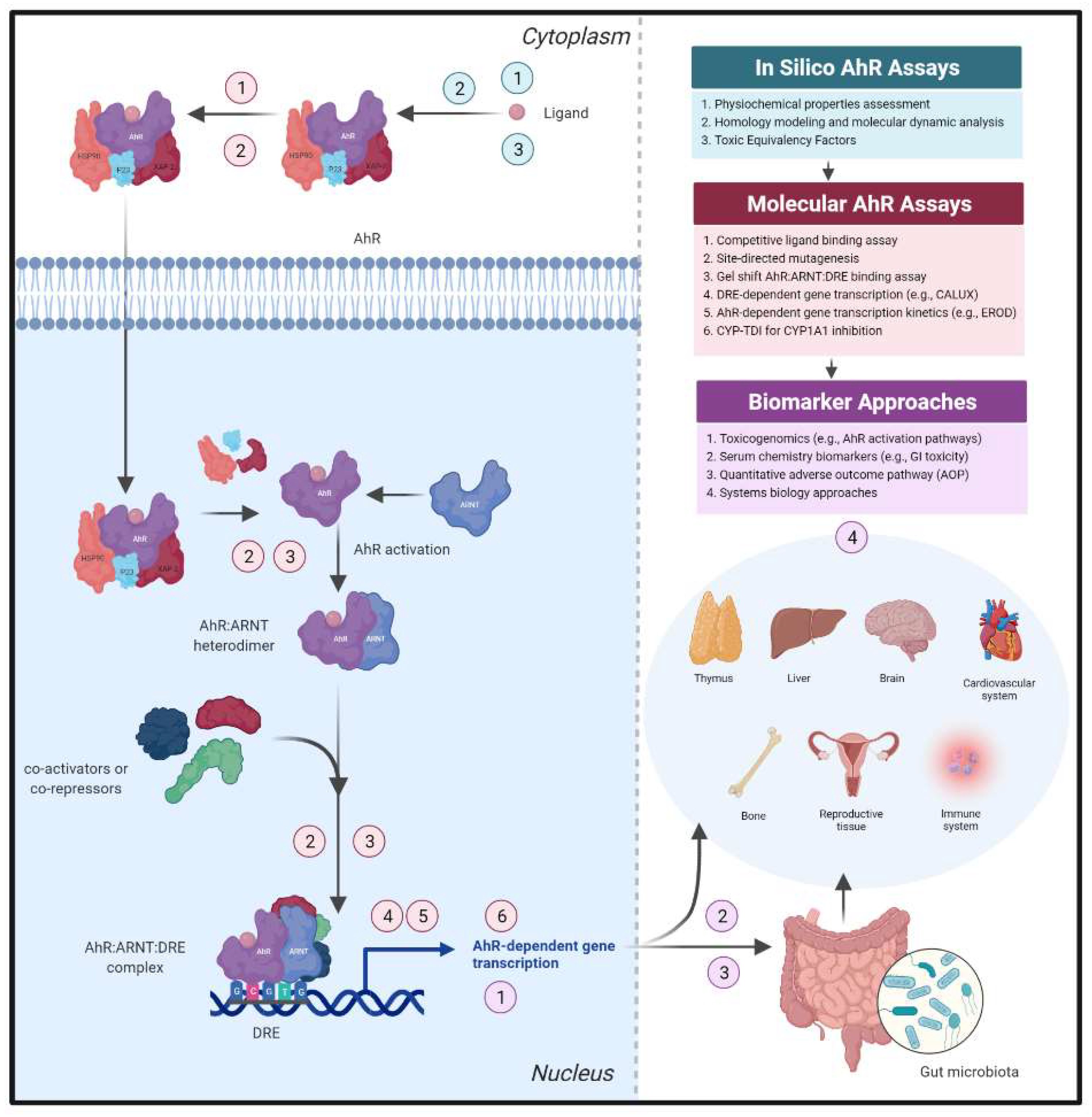
4.1. Confirmation of AhR Canonical Signal Transduction as an Early De-Risking Strategy
4.2. Harnessing AhR-Dependent Transcriptomic Profiles to Identify Safety Thresholds
4.3. Assessment of AhR-Dependent Pathways within Intestinal Microenvironment Cultures
4.4. Safety Assessment Concerns Regarding AhR Target Modulation
5. Summary and Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Safe, S. Polychlorinated biphenyls (PCBs), dibenzo-p-dioxins (PCDDs), dibenzofurans (PCDFs), and related compounds: Environmental and mechanistic considerations which support the development of toxic equivalency factors (TEFs). Crit. Rev. Toxicol. 1990, 21, 51–88. [Google Scholar] [CrossRef] [PubMed]
- Schmidt, J.V.; Bradfield, C.A. Ah receptor signaling pathways. Annu. Rev. Cell Dev. Biol. 1996, 12, 55–89. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gasiewicz, T.A.; Singh, K.P.; Bennett, J.A. The Ah receptor in stem cell cycling, regulation, and quiescence. Ann. N. Y. Acad. Sci. 2014, 1310, 44–50. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fernandez-Salguero, P.M.; Ward, J.M.; Sundberg, J.P.; Gonzalez, F.J. Lesions of aryl-hydrocarbon receptor-deficient mice. Vet. Pathol. 1997, 34, 605–614. [Google Scholar] [CrossRef]
- Monteleone, I.; Rizzo, A.; Sarra, M.; Sica, G.; Sileri, P.; Biancone, L.; MacDonald, T.T.; Pallone, F.; Monteleone, G. Aryl hydrocarbon receptor-induced signals up-regulate IL-22 production and inhibit inflammation in the gastrointestinal tract. Gastroenterology 2011, 141, 237–248.e1. [Google Scholar] [CrossRef]
- Ji, T.; Xu, C.; Sun, L.; Yu, M.; Peng, K.; Qiu, Y.; Xiao, W.; Yang, H. Aryl Hydrocarbon Receptor Activation Down-Regulates IL-7 and Reduces Inflammation in a Mouse Model of DSS-Induced Colitis. Dig. Dis. Sci. 2015, 60, 1958–1966. [Google Scholar] [CrossRef]
- Korecka, A.; Dona, A.; Lahiri, S.; Tett, A.J.; Al-Asmakh, M.; Braniste, V.; D’Arienzo, R.; Abbaspour, A.; Reichardt, N.; Fujii-Kuriyama, Y.; et al. Bidirectional communication between the Aryl hydrocarbon Receptor (AhR) and the microbiome tunes host metabolism. NPJ Biofilms Microbiomes 2016, 2, 16014. [Google Scholar] [CrossRef] [Green Version]
- Furumatsu, K.; Nishiumi, S.; Kawano, Y.; Ooi, M.; Yoshie, T.; Shiomi, Y.; Kutsumi, H.; Ashida, H.; Fujii-Kuriyama, Y.; Azuma, T.; et al. A role of the aryl hydrocarbon receptor in attenuation of colitis. Dig. Dis. Sci. 2011, 56, 2532–2544. [Google Scholar] [CrossRef]
- Kimura, A.; Abe, H.; Tsuruta, S.; Chiba, S.; Fujii-Kuriyama, Y.; Sekiya, T.; Morita, R.; Yoshimura, A. Aryl hydrocarbon receptor protects against bacterial infection by promoting macrophage survival and reactive oxygen species production. Int. Immunol. 2014, 26, 209–220. [Google Scholar] [CrossRef]
- Wagage, S.; John, B.; Krock, B.L.; Hall, A.O.; Randall, L.M.; Karp, C.L.; Simon, M.C.; Hunter, C.A. The aryl hydrocarbon receptor promotes IL-10 production by NK cells. J. Immunol. 2014, 192, 1661–1670. [Google Scholar] [CrossRef] [Green Version]
- Metidji, A.; Omenetti, S.; Crotta, S.; Li, Y.; Nye, E.; Ross, E.; Li, V.; Maradana, M.R.; Schiering, C.; Stockinger, B. The Environmental Sensor AHR Protects from Inflammatory Damage by Maintaining Intestinal Stem Cell Homeostasis and Barrier Integrity. Immunity 2019, 50, 1542. [Google Scholar] [CrossRef] [PubMed]
- Lanis, J.M.; Alexeev, E.E.; Curtis, V.F.; Kitzenberg, D.A.; Kao, D.J.; Battista, K.D.; Gerich, M.E.; Glover, L.E.; Kominsky, D.J.; Colgan, S.P. Tryptophan metabolite activation of the aryl hydrocarbon receptor regulates IL-10 receptor expression on intestinal epithelia. Mucosal Immunol. 2017, 10, 1133–1144. [Google Scholar] [CrossRef] [PubMed]
- Monteleone, I.; Zorzi, F.; Marafini, I.; Di Fusco, D.; Dinallo, V.; Caruso, R.; Izzo, R.; Franze, E.; Colantoni, A.; Pallone, F.; et al. Aryl hydrocarbon receptor-driven signals inhibit collagen synthesis in the gut. Eur. J. Immunol. 2016, 46, 1047–1057. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Haller, C.A.; Jernigan, F.E.; Koerner, S.K.; Wong, D.J.; Wang, Y.; Cheong, J.E.; Kosaraju, R.; Kwan, J.; Park, D.D.; et al. Modulation of lymphocyte-mediated tissue repair by rational design of heterocyclic aryl hydrocarbon receptor agonists. Sci. Adv. 2020, 6, eaay8230. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vogel, C.F.; Sciullo, E.; Li, W.; Wong, P.; Lazennec, G.; Matsumura, F. RelB, a new partner of aryl hydrocarbon receptor-mediated transcription. Mol. Endocrinol. 2007, 21, 2941–2955. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wilson, S.R.; Joshi, A.D.; Elferink, C.J. The tumor suppressor Kruppel-like factor 6 is a novel aryl hydrocarbon receptor DNA binding partner. J. Pharmacol. Exp. Ther. 2013, 345, 419–429. [Google Scholar] [CrossRef] [Green Version]
- Safe, S.; Jin, U.H.; Park, H.; Chapkin, R.S.; Jayaraman, A. Aryl Hydrocarbon Receptor (AHR) Ligands as Selective AHR Modulators (SAhRMs). Int. J. Mol. Sci. 2020, 21, 6654. [Google Scholar] [CrossRef]
- Li, Y.; Innocentin, S.; Withers, D.R.; Roberts, N.A.; Gallagher, A.R.; Grigorieva, E.F.; Wilhelm, C.; Veldhoen, M. Exogenous stimuli maintain intraepithelial lymphocytes via aryl hydrocarbon receptor activation. Cell 2011, 147, 629–640. [Google Scholar] [CrossRef] [Green Version]
- Quintana, F.J.; Basso, A.S.; Iglesias, A.H.; Korn, T.; Farez, M.F.; Bettelli, E.; Caccamo, M.; Oukka, M.; Weiner, H.L. Control of Treg and TH17 cell differentiation by the aryl hydrocarbon receptor. Nature 2008, 453, 65–71. [Google Scholar] [CrossRef]
- Gandhi, R.; Kumar, D.; Burns, E.J.; Nadeau, M.; Dake, B.; Laroni, A.; Kozoriz, D.; Weiner, H.L.; Quintana, F.J. Activation of the aryl hydrocarbon receptor induces human type 1 regulatory T cell-like and Foxp3+ regulatory T cells. Nat. Immunol. 2010, 11, 846–853. [Google Scholar] [CrossRef] [Green Version]
- Zhang, L.; Ma, J.; Takeuchi, M.; Usui, Y.; Hattori, T.; Okunuki, Y.; Yamakawa, N.; Kezuka, T.; Kuroda, M.; Goto, H. Suppression of experimental autoimmune uveoretinitis by inducing differentiation of regulatory T cells via activation of aryl hydrocarbon receptor. Investig. Ophthalmol. Vis. Sci. 2010, 51, 2109–2117. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kerkvliet, N.I.; Steppan, L.B.; Vorachek, W.; Oda, S.; Farrer, D.; Wong, C.P.; Pham, D.; Mourich, D.V. Activation of aryl hydrocarbon receptor by TCDD prevents diabetes in NOD mice and increases Foxp3+ T cells in pancreatic lymph nodes. Immunotherapy 2009, 1, 539–547. [Google Scholar] [CrossRef] [PubMed]
- Clarivate. Cortellis Drug Discovery IntelligenceTM. Available online: https://www.cortellis.com/drugdiscovery (accessed on 31 January 2022).
- Denison, M.S.; Nagy, S.R. Activation of the aryl hydrocarbon receptor by structurally diverse exogenous and endogenous chemicals. Annu. Rev. Pharmacol. Toxicol. 2003, 43, 309–334. [Google Scholar] [CrossRef] [PubMed]
- Sugimoto, S.; Naganuma, M.; Kanai, T. Indole compounds may be promising medicines for ulcerative colitis. J. Gastroenterol. 2016, 51, 853–861. [Google Scholar] [CrossRef]
- Naganuma, M. Treatment with indigo naturalis for inflammatory bowel disease and other immune diseases. Immunol. Med. 2019, 42, 16–21. [Google Scholar] [CrossRef] [Green Version]
- Naganuma, M.; Sugimoto, S.; Mitsuyama, K.; Kobayashi, T.; Yoshimura, N.; Ohi, H.; Tanaka, S.; Andoh, A.; Ohmiya, N.; Saigusa, K.; et al. Efficacy of Indigo Naturalis in a Multicenter Randomized Controlled Trial of Patients with Ulcerative Colitis. Gastroenterology 2018, 154, 935–947. [Google Scholar] [CrossRef] [Green Version]
- Saiki, J.P.; Andreasson, J.O.; Grimes, K.V.; Frumkin, L.R.; Sanjines, E.; Davidson, M.G.; Park, K.T.; Limketkai, B. Treatment-refractory ulcerative colitis responsive to indigo naturalis. BMJ Open Gastroenterol. 2021, 8, e000813. [Google Scholar] [CrossRef]
- D’Haens, G.; Sandborn, W.J.; Colombel, J.F.; Rutgeerts, P.; Brown, K.; Barkay, H.; Sakov, A.; Haviv, A.; Feagan, B.G.; Laquinimod for Crohn’s Disease, I. A phase II study of laquinimod in Crohn’s disease. Gut 2015, 64, 1227–1235. [Google Scholar] [CrossRef] [Green Version]
- Chen, Z.Q.; Liu, Y.; Zhao, J.H.; Wang, L.; Feng, N.P. Improved oral bioavailability of poorly water-soluble indirubin by a supersaturatable self-microemulsifying drug delivery system. Int. J. Nanomed. 2012, 7, 1115–1125. [Google Scholar] [CrossRef] [Green Version]
- Benigni, R.; Bossa, C.; Tcheremenskaia, O. Nongenotoxic carcinogenicity of chemicals: Mechanisms of action and early recognition through a new set of structural alerts. Chem. Rev. 2013, 113, 2940–2957. [Google Scholar] [CrossRef]
- Wang, K.; Li, Y.; Jiang, Y.Z.; Dai, C.F.; Patankar, M.S.; Song, J.S.; Zheng, J. An endogenous aryl hydrocarbon receptor ligand inhibits proliferation and migration of human ovarian cancer cells. Cancer Lett. 2013, 340, 63–71. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marafini, I.; Di Fusco, D.; Dinallo, V.; Franze, E.; Stolfi, C.; Sica, G.; Monteleone, G.; Monteleone, I. NPD-0414-2 and NPD-0414-24, Two Chemical Entities Designed as Aryl Hydrocarbon Receptor (AhR) Ligands, Inhibit Gut Inflammatory Signals. Front. Pharmacol. 2019, 10, 380. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wincent, E.; Amini, N.; Luecke, S.; Glatt, H.; Bergman, J.; Crescenzi, C.; Rannug, A.; Rannug, U. The suggested physiologic aryl hydrocarbon receptor activator and cytochrome P4501 substrate 6-formylindolo[3,2-b]carbazole is present in humans. J. Biol. Chem. 2009, 284, 2690–2696. [Google Scholar] [CrossRef] [Green Version]
- Cheng, J.; Li, W.; Kang, B.; Zhou, Y.; Song, J.; Dan, S.; Yang, Y.; Zhang, X.; Li, J.; Yin, S.; et al. Tryptophan derivatives regulate the transcription of Oct4 in stem-like cancer cells. Nat. Commun. 2015, 6, 7209. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fang, Z.Z.; Krausz, K.W.; Nagaoka, K.; Tanaka, N.; Gowda, K.; Amin, S.G.; Perdew, G.H.; Gonzalez, F.J. In vivo effects of the pure aryl hydrocarbon receptor antagonist GNF-351 after oral administration are limited to the gastrointestinal tract. Br. J. Pharmacol. 2014, 171, 1735–1746. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yeste, A.; Nadeau, M.; Burns, E.J.; Weiner, H.L.; Quintana, F.J. Nanoparticle-mediated codelivery of myelin antigen and a tolerogenic small molecule suppresses experimental autoimmune encephalomyelitis. Proc. Natl. Acad. Sci. USA 2012, 109, 11270–11275. [Google Scholar] [CrossRef] [Green Version]
- Rothhammer, V.; Quintana, F.J. Environmental control of autoimmune inflammation in the central nervous system. Curr. Opin. Immunol. 2016, 43, 46–53. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marinelli, L.; Martin-Gallausiaux, C.; Bourhis, J.M.; Beguet-Crespel, F.; Blottiere, H.M.; Lapaque, N. Identification of the novel role of butyrate as AhR ligand in human intestinal epithelial cells. Sci. Rep. 2019, 9, 643. [Google Scholar] [CrossRef] [PubMed]
- Gasaly, N.; de Vos, P.; Hermoso, M.A. Impact of Bacterial Metabolites on Gut Barrier Function and Host Immunity: A Focus on Bacterial Metabolism and Its Relevance for Intestinal Inflammation. Front. Immunol. 2021, 12, 658354. [Google Scholar] [CrossRef]
- Hubbard, T.D.; Liu, Q.; Murray, I.A.; Dong, F.; Miller, C., III; Smith, P.B.; Gowda, K.; Lin, J.M.; Amin, S.; Patterson, A.D.; et al. Microbiota Metabolism Promotes Synthesis of the Human Ah Receptor Agonist 2,8-Dihydroxyquinoline. J. Proteome Res. 2019, 18, 1715–1724. [Google Scholar] [CrossRef]
- Murray, I.A.; Perdew, G.H. How Ah Receptor Ligand Specificity Became Important in Understanding Its Physiological Function. Int. J. Mol. Sci. 2020, 21, 9614. [Google Scholar] [CrossRef] [PubMed]
- Stockinger, B.; Shah, K.; Wincent, E. AHR in the intestinal microenvironment: Safeguarding barrier function. Nat. Rev. Gastroenterol. Hepatol. 2021, 18, 559–570. [Google Scholar] [CrossRef] [PubMed]
- Murray, I.A.; Perdew, G.H. Ligand activation of the Ah receptor contributes to gastrointestinal homeostasis. Curr. Opin. Toxicol. 2017, 2, 15–23. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lamas, B.; Hernandez-Galan, L.; Galipeau, H.J.; Constante, M.; Clarizio, A.; Jury, J.; Breyner, N.M.; Caminero, A.; Rueda, G.; Hayes, C.L.; et al. Aryl hydrocarbon receptor ligand production by the gut microbiota is decreased in celiac disease leading to intestinal inflammation. Sci. Transl. Med. 2020, 12, eaba0624. [Google Scholar] [CrossRef] [PubMed]
- Nikolaus, S.; Schulte, B.; Al-Massad, N.; Thieme, F.; Schulte, D.M.; Bethge, J.; Rehman, A.; Tran, F.; Aden, K.; Hasler, R.; et al. Increased Tryptophan Metabolism Is Associated with Activity of Inflammatory Bowel Diseases. Gastroenterology 2017, 153, 1504–1516.e2. [Google Scholar] [CrossRef] [Green Version]
- Jing, W.; Dong, S.; Luo, X.; Liu, J.; Wei, B.; Du, W.; Yang, L.; Luo, H.; Wang, Y.; Wang, S.; et al. Berberine improves colitis by triggering AhR activation by microbial tryptophan catabolites. Pharmacol. Res. 2021, 164, 105358. [Google Scholar] [CrossRef]
- Scott, S.A.; Fu, J.; Chang, P.V. Microbial tryptophan metabolites regulate gut barrier function via the aryl hydrocarbon receptor. Proc. Natl. Acad. Sci. USA 2020, 117, 19376–19387. [Google Scholar] [CrossRef]
- Muku, G.E.; Murray, I.A.; Espin, J.C.; Perdew, G.H. Urolithin A Is a Dietary Microbiota-Derived Human Aryl Hydrocarbon Receptor Antagonist. Metabolites 2018, 8, 86. [Google Scholar] [CrossRef] [Green Version]
- Lv, Q.; Shi, C.; Qiao, S.; Cao, N.; Guan, C.; Dai, Y.; Wei, Z. Alpinetin exerts anti-colitis efficacy by activating AhR, regulating miR-302/DNMT-1/CREB signals, and therefore promoting Treg differentiation. Cell Death Dis. 2018, 9, 890. [Google Scholar] [CrossRef]
- Goya-Jorge, E.; Jorge Rodriguez, M.E.; Veitia, M.S.; Giner, R.M. Plant Occurring Flavonoids as Modulators of the Aryl Hydrocarbon Receptor. Molecules 2021, 26, 2315. [Google Scholar] [CrossRef]
- Van der Beek, C.M.; Dejong, C.H.C.; Troost, F.J.; Masclee, A.A.M.; Lenaerts, K. Role of short-chain fatty acids in colonic inflammation, carcinogenesis, and mucosal protection and healing. Nutr. Rev. 2017, 75, 286–305. [Google Scholar] [CrossRef] [PubMed]
- Yang, W.; Yu, T.; Huang, X.; Bilotta, A.J.; Xu, L.; Lu, Y.; Sun, J.; Pan, F.; Zhou, J.; Zhang, W.; et al. Intestinal microbiota-derived short-chain fatty acids regulation of immune cell IL-22 production and gut immunity. Nat. Commun. 2020, 11, 4457. [Google Scholar] [CrossRef] [PubMed]
- Iyer, S.S.; Gensollen, T.; Gandhi, A.; Oh, S.F.; Neves, J.F.; Collin, F.; Lavin, R.; Serra, C.; Glickman, J.; de Silva, P.S.A.; et al. Dietary and Microbial Oxazoles Induce Intestinal Inflammation by Modulating Aryl Hydrocarbon Receptor Responses. Cell 2018, 173, 1123–1134.e11. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kawai, S.; Iijima, H.; Shinzaki, S.; Hiyama, S.; Yamaguchi, T.; Araki, M.; Iwatani, S.; Shiraishi, E.; Mukai, A.; Inoue, T.; et al. Indigo Naturalis ameliorates murine dextran sodium sulfate-induced colitis via aryl hydrocarbon receptor activation. J. Gastroenterol. 2017, 52, 904–919. [Google Scholar] [CrossRef]
- Krishnan, S.; Ding, Y.; Saedi, N.; Choi, M.; Sridharan, G.V.; Sherr, D.H.; Yarmush, M.L.; Alaniz, R.C.; Jayaraman, A.; Lee, K. Gut Microbiota-Derived Tryptophan Metabolites Modulate Inflammatory Response in Hepatocytes and Macrophages. Cell. Rep. 2018, 23, 1099–1111. [Google Scholar] [CrossRef]
- Rothhammer, V.; Borucki, D.M.; Tjon, E.C.; Takenaka, M.C.; Chao, C.C.; Ardura-Fabregat, A.; de Lima, K.A.; Gutierrez-Vazquez, C.; Hewson, P.; Staszewski, O.; et al. Microglial control of astrocytes in response to microbial metabolites. Nature 2018, 557, 724–728. [Google Scholar] [CrossRef]
- De Pessemier, B.; Grine, L.; Debaere, M.; Maes, A.; Paetzold, B.; Callewaert, C. Gut-Skin Axis: Current Knowledge of the Interrelationship between Microbial Dysbiosis and Skin Conditions. Microorganisms 2021, 9, 353. [Google Scholar] [CrossRef]
- Yip, W.; Hughes, M.R.; Li, Y.; Cait, A.; Hirst, M.; Mohn, W.W.; McNagny, K.M. Butyrate Shapes Immune Cell Fate and Function in Allergic Asthma. Front. Immunol. 2021, 12, 628453. [Google Scholar] [CrossRef]
- Hezaveh, K.; Shinde, R.S.; Klotgen, A.; Halaby, M.J.; Lamorte, S.; Ciudad, M.T.; Quevedo, R.; Neufeld, L.; Liu, Z.Q.; Jin, R.; et al. Tryptophan-derived microbial metabolites activate the aryl hydrocarbon receptor in tumor-associated macrophages to suppress anti-tumor immunity. Immunity 2022, 55, 324–340.e8. [Google Scholar] [CrossRef]
- Jin, U.H.; Cheng, Y.; Park, H.; Davidson, L.A.; Callaway, E.S.; Chapkin, R.S.; Jayaraman, A.; Asante, A.; Allred, C.; Weaver, E.A.; et al. Short Chain Fatty Acids Enhance Aryl Hydrocarbon (Ah) Responsiveness in Mouse Colonocytes and Caco-2 Human Colon Cancer Cells. Sci. Rep. 2017, 7, 10163. [Google Scholar] [CrossRef] [Green Version]
- Wei, Y.L.; Chen, Y.Q.; Gong, H.; Li, N.; Wu, K.Q.; Hu, W.; Wang, B.; Liu, K.J.; Wen, L.Z.; Xiao, X.; et al. Fecal Microbiota Transplantation Ameliorates Experimentally Induced Colitis in Mice by Upregulating AhR. Front. Microbiol. 2018, 9, 1921. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fukumoto, S.; Toshimitsu, T.; Matsuoka, S.; Maruyama, A.; Oh-Oka, K.; Takamura, T.; Nakamura, Y.; Ishimaru, K.; Fujii-Kuriyama, Y.; Ikegami, S.; et al. Identification of a probiotic bacteria-derived activator of the aryl hydrocarbon receptor that inhibits colitis. Immunol. Cell Biol. 2014, 92, 460–465. [Google Scholar] [CrossRef] [PubMed]
- Shen, J.; Zuo, Z.X.; Mao, A.P. Effect of probiotics on inducing remission and maintaining therapy in ulcerative colitis, Crohn’s disease, and pouchitis: Meta-analysis of randomized controlled trials. Inflamm. Bowel Dis. 2014, 20, 21–35. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Derwa, Y.; Gracie, D.J.; Hamlin, P.J.; Ford, A.C. Systematic review with meta-analysis: The efficacy of probiotics in inflammatory bowel disease. Aliment. Pharmacol. Ther. 2017, 46, 389–400. [Google Scholar] [CrossRef] [PubMed]
- Zelante, T.; Iannitti, R.G.; Cunha, C.; De Luca, A.; Giovannini, G.; Pieraccini, G.; Zecchi, R.; D’Angelo, C.; Massi-Benedetti, C.; Fallarino, F.; et al. Tryptophan catabolites from microbiota engage aryl hydrocarbon receptor and balance mucosal reactivity via interleukin-22. Immunity 2013, 39, 372–385. [Google Scholar] [CrossRef] [Green Version]
- Van der Lelie, D.; Oka, A.; Taghavi, S.; Umeno, J.; Fan, T.J.; Merrell, K.E.; Watson, S.D.; Ouellette, L.; Liu, B.; Awoniyi, M.; et al. Rationally designed bacterial consortia to treat chronic immune-mediated colitis and restore intestinal homeostasis. Nat. Commun. 2021, 12, 3105. [Google Scholar] [CrossRef]
- Lamas, B.; Richard, M.L.; Leducq, V.; Pham, H.P.; Michel, M.L.; Da Costa, G.; Bridonneau, C.; Jegou, S.; Hoffmann, T.W.; Natividad, J.M.; et al. CARD9 impacts colitis by altering gut microbiota metabolism of tryptophan into aryl hydrocarbon receptor ligands. Nat. Med. 2016, 22, 598–605. [Google Scholar] [CrossRef]
- McGovern, D.P.; Gardet, A.; Torkvist, L.; Goyette, P.; Essers, J.; Taylor, K.D.; Neale, B.M.; Ong, R.T.; Lagace, C.; Li, C.; et al. Genome-wide association identifies multiple ulcerative colitis susceptibility loci. Nat. Genet. 2010, 42, 332–337. [Google Scholar] [CrossRef] [Green Version]
- Liu, J.Z.; van Sommeren, S.; Huang, H.; Ng, S.C.; Alberts, R.; Takahashi, A.; Ripke, S.; Lee, J.C.; Jostins, L.; Shah, T.; et al. Association analyses identify 38 susceptibility loci for inflammatory bowel disease and highlight shared genetic risk across populations. Nat. Genet. 2015, 47, 979–986. [Google Scholar] [CrossRef]
- Pradhan, D.; Mallappa, R.H.; Grover, S. Comprehensive approaches for assessing the safety of probiotic bacteria. Food Control. 2020, 108, 106872. [Google Scholar] [CrossRef]
- Rouanet, A.; Bolca, S.; Bru, A.; Claes, I.; Cvejic, H.; Girgis, H.; Harper, A.; Lavergne, S.N.; Mathys, S.; Pane, M.; et al. Live Biotherapeutic Products, A Road Map for Safety Assessment. Front. Med. 2020, 7, 237. [Google Scholar] [CrossRef] [PubMed]
- Van de Wiele, T.; Vanhaecke, L.; Boeckaert, C.; Peru, K.; Headley, J.; Verstraete, W.; Siciliano, S. Human colon microbiota transform polycyclic aromatic hydrocarbons to estrogenic metabolites. Environ. Health Perspect. 2005, 113, 6–10. [Google Scholar] [CrossRef] [PubMed]
- Koduru, L.; Lakshmanan, M.; Hoon, S.; Lee, D.Y.; Lee, Y.K.; Ow, D.S. Systems Biology of Gut Microbiota-Human Receptor Interactions: Toward Anti-inflammatory Probiotics. Front. Microbiol. 2022, 13, 846555. [Google Scholar] [CrossRef] [PubMed]
- Dong, F.; Hao, F.; Murray, I.A.; Smith, P.B.; Koo, I.; Tindall, A.M.; Kris-Etherton, P.M.; Gowda, K.; Amin, S.G.; Patterson, A.D.; et al. Intestinal microbiota-derived tryptophan metabolites are predictive of Ah receptor activity. Gut Microbes 2020, 12, 1788899. [Google Scholar] [CrossRef]
- Sakamoto, K.M.; Kim, K.B.; Kumagai, A.; Mercurio, F.; Crews, C.M.; Deshaies, R.J. Protacs: Chimeric molecules that target proteins to the Skp1-Cullin-F box complex for ubiquitination and degradation. Proc. Natl. Acad. Sci. USA 2001, 98, 8554–8559. [Google Scholar] [CrossRef] [Green Version]
- Ishida, T.; Ciulli, A. E3 Ligase Ligands for PROTACs: How They Were Found and How to Discover New Ones. SLAS Discov. 2021, 26, 484–502. [Google Scholar] [CrossRef]
- Burslem, G.M.; Crews, C.M. Proteolysis-Targeting Chimeras as Therapeutics and Tools for Biological Discovery. Cell 2020, 181, 102–114. [Google Scholar] [CrossRef]
- Kleiger, G.; Mayor, T. Perilous journey: A tour of the ubiquitin-proteasome system. Trends Cell Biol. 2014, 24, 352–359. [Google Scholar] [CrossRef] [Green Version]
- Halford, B. Arvinas unveils PROTAC structures. CEN Glob. Enterp. 2021, 99, 5. [Google Scholar] [CrossRef]
- ClinicalTrials. Study to Evaluate the Safety, Tolerability, Pharmacokinetics, and Pharmacodynamics of CC-94676 in Subjects with Metastatic Castration-Resistant Prostate Cancer; National Library of Medicine: Bethesda, MD, USA, 2021. [Google Scholar]
- Puppala, D.; Lee, H.; Kim, K.B.; Swanson, H.I. Development of an aryl hydrocarbon receptor antagonist using the proteolysis-targeting chimeric molecules approach: A potential tool for chemoprevention. Mol. Pharmacol. 2008, 73, 1064–1071. [Google Scholar] [CrossRef]
- Lee, H.; Puppala, D.; Choi, E.Y.; Swanson, H.; Kim, K.B. Targeted degradation of the aryl hydrocarbon receptor by the PROTAC approach: A useful chemical genetic tool. ChemBioChem 2007, 8, 2058–2062. [Google Scholar] [CrossRef] [PubMed]
- Ohtake, F.; Baba, A.; Takada, I.; Okada, M.; Iwasaki, K.; Miki, H.; Takahashi, S.; Kouzmenko, A.; Nohara, K.; Chiba, T.; et al. Dioxin receptor is a ligand-dependent E3 ubiquitin ligase. Nature 2007, 446, 562–566. [Google Scholar] [CrossRef] [PubMed]
- Wormke, M.; Stoner, M.; Saville, B.; Walker, K.; Abdelrahim, M.; Burghardt, R.; Safe, S. The aryl hydrocarbon receptor mediates degradation of estrogen receptor alpha through activation of proteasomes. Mol. Cell. Biol. 2003, 23, 1843–1855. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ohtake, F.; Takeyama, K.; Matsumoto, T.; Kitagawa, H.; Yamamoto, Y.; Nohara, K.; Tohyama, C.; Krust, A.; Mimura, J.; Chambon, P.; et al. Modulation of oestrogen receptor signalling by association with the activated dioxin receptor. Nature 2003, 423, 545–550. [Google Scholar] [CrossRef] [PubMed]
- Ohtake, F.; Fujii-Kuriyama, Y.; Kawajiri, K.; Kato, S. Cross-talk of dioxin and estrogen receptor signals through the ubiquitin system. J. Steroid Biochem. Mol. Biol. 2011, 127, 102–107. [Google Scholar] [CrossRef] [PubMed]
- Ma, Q.; Baldwin, K.T. 2,3,7,8-tetrachlorodibenzo-p-dioxin-induced degradation of aryl hydrocarbon receptor (AhR) by the ubiquitin-proteasome pathway. Role of the transcription activaton and DNA binding of AhR. J. Biol. Chem. 2000, 275, 8432–8438. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Luecke-Johansson, S.; Gralla, M.; Rundqvist, H.; Ho, J.C.; Johnson, R.S.; Gradin, K.; Poellinger, L. A Molecular Mechanism to Switch the Aryl Hydrocarbon Receptor from a Transcription Factor to an E3 Ubiquitin Ligase. Mol. Cell. Biol. 2017, 37, e00630-16. [Google Scholar] [CrossRef] [Green Version]
- Ohoka, N.; Tsuji, G.; Shoda, T.; Fujisato, T.; Kurihara, M.; Demizu, Y.; Naito, M. Development of Small Molecule Chimeras That Recruit AhR E3 Ligase to Target Proteins. ACS Chem. Biol. 2019, 14, 2822–2832. [Google Scholar] [CrossRef]
- Neavin, D.R.; Liu, D.; Ray, B.; Weinshilboum, R.M. The Role of the Aryl Hydrocarbon Receptor (AHR) in Immune and Inflammatory Diseases. Int. J. Mol. Sci. 2018, 19, 3851. [Google Scholar] [CrossRef] [Green Version]
- Lahoti, T.S.; John, K.; Hughes, J.M.; Kusnadi, A.; Murray, I.A.; Krishnegowda, G.; Amin, S.; Perdew, G.H. Aryl hydrocarbon receptor antagonism mitigates cytokine-mediated inflammatory signalling in primary human fibroblast-like synoviocytes. Ann. Rheum. Dis. 2013, 72, 1708–1716. [Google Scholar] [CrossRef]
- Murray, I.A.; Patterson, A.D.; Perdew, G.H. Aryl hydrocarbon receptor ligands in cancer: Friend and foe. Nat. Rev. Cancer 2014, 14, 801–814. [Google Scholar] [CrossRef] [PubMed]
- Chamberlain, P.P.; D’Agostino, L.A.; Ellis, J.M.; Hansen, J.D.; Matyskiela, M.E.; McDonald, J.J.; Riggs, J.R.; Hamann, L.G. Evolution of Cereblon-Mediated Protein Degradation as a Therapeutic Modality. ACS Med. Chem. Lett. 2019, 10, 1592–1602. [Google Scholar] [CrossRef] [PubMed]
- Kuijper, E.C.; Bergsma, A.J.; Pijnappel, W.; Aartsma-Rus, A. Opportunities and challenges for antisense oligonucleotide therapies. J. Inherit. Metab. Dis. 2021, 44, 72–87. [Google Scholar] [CrossRef] [PubMed]
- Marafini, I.; Monteleone, G. Inflammatory bowel disease: New therapies from antisense oligonucleotides. Ann. Med. 2018, 50, 361–370. [Google Scholar] [CrossRef]
- Di Fusco, D.; Dinallo, V.; Marafini, I.; Figliuzzi, M.M.; Romano, B.; Monteleone, G. Antisense Oligonucleotide: Basic Concepts and Therapeutic Application in Inflammatory Bowel Disease. Front. Pharmacol. 2019, 10, 305. [Google Scholar] [CrossRef] [Green Version]
- Scarozza, P.; Schmitt, H.; Monteleone, G.; Neurath, M.F.; Atreya, R. Oligonucleotides-A Novel Promising Therapeutic Option for IBD. Front. Pharmacol. 2019, 10, 314. [Google Scholar] [CrossRef]
- Gareb, B.; Otten, A.T.; Frijlink, H.W.; Dijkstra, G.; Kosterink, J.G.W. Review: Local Tumor Necrosis Factor-alpha Inhibition in Inflammatory Bowel Disease. Pharmaceutics 2020, 12, 539. [Google Scholar] [CrossRef]
- Marafini, I.; Monteleone, G. Therapeutic Oligonucleotides for Patients with Inflammatory Bowel Diseases. Biologics 2020, 14, 47–51. [Google Scholar] [CrossRef]
- Schmidt, C.; Grunert, P.C.; Stallmach, A. An Update for Pharmacologists on New Treatment Options for Inflammatory Bowel Disease: The Clinicians’ Perspective. Front. Pharmacol. 2021, 12, 655054. [Google Scholar] [CrossRef]
- Monteleone, G.; Neurath, M.F.; Ardizzone, S.; Di Sabatino, A.; Fantini, M.C.; Castiglione, F.; Scribano, M.L.; Armuzzi, A.; Caprioli, F.; Sturniolo, G.C.; et al. Mongersen, an oral SMAD7 antisense oligonucleotide, and Crohn’s disease. N. Engl. J. Med. 2015, 372, 1104–1113. [Google Scholar] [CrossRef] [Green Version]
- Duan, B.; Li, M.; Sun, Y.; Zou, S.; Xu, X. Orally Delivered Antisense Oligodeoxyribonucleotides of TNF-alpha via Polysaccharide-Based Nanocomposites Targeting Intestinal Inflammation. Adv. Healthc. Mater. 2019, 8, e1801389. [Google Scholar] [CrossRef] [PubMed]
- Huang, Z.; Gan, J.; Jia, L.; Guo, G.; Wang, C.; Zang, Y.; Ding, Z.; Chen, J.; Zhang, J.; Dong, L. An orally administrated nucleotide-delivery vehicle targeting colonic macrophages for the treatment of inflammatory bowel disease. Biomaterials 2015, 48, 26–36. [Google Scholar] [CrossRef] [PubMed]
- Suri, K.; Bubier, J.A.; Wiles, M.V.; Shultz, L.D.; Amiji, M.M.; Hosur, V. Role of MicroRNA in Inflammatory Bowel Disease: Clinical Evidence and the Development of Preclinical Animal Models. Cells 2021, 10, 2204. [Google Scholar] [CrossRef]
- Zhao, Y.; Ma, T.; Chen, W.; Chen, Y.; Li, M.; Ren, L.; Chen, J.; Cao, R.; Feng, Y.; Zhang, H.; et al. MicroRNA-124 Promotes Intestinal Inflammation by Targeting Aryl Hydrocarbon Receptor in Crohn’s Disease. J. Crohn’s Colitis 2016, 10, 703–712. [Google Scholar] [CrossRef] [Green Version]
- Chu, Y.; Zhu, Y.; Zhang, Y.; Liu, X.; Guo, Y.; Chang, L.; Yun, X.; Wei, Z.; Xia, Y.; Dai, Y. Tetrandrine attenuates intestinal epithelial barrier defects caused by colitis through promoting the expression of Occludin via the AhR-miR-429 pathway. FASEB J. 2021, 35, e21502. [Google Scholar] [CrossRef]
- Chinen, I.; Nakahama, T.; Kimura, A.; Nguyen, N.T.; Takemori, H.; Kumagai, A.; Kayama, H.; Takeda, K.; Lee, S.; Hanieh, H.; et al. The aryl hydrocarbon receptor/microRNA-212/132 axis in T cells regulates IL-10 production to maintain intestinal homeostasis. Int. Immunol. 2015, 27, 405–415. [Google Scholar] [CrossRef]
- Yu, M.; Luo, Y.; Cong, Z.; Mu, Y.; Qiu, Y.; Zhong, M. MicroRNA-590-5p Inhibits Intestinal Inflammation by Targeting YAP. J. Crohn’s Colitis 2018, 12, 993–1004. [Google Scholar] [CrossRef] [PubMed]
- Cheng, X.; Zhang, X.; Su, J.; Zhang, Y.; Zhou, W.; Zhou, J.; Wang, C.; Liang, H.; Chen, X.; Shi, R.; et al. miR-19b downregulates intestinal SOCS3 to reduce intestinal inflammation in Crohn’s disease. Sci. Rep. 2015, 5, 10397. [Google Scholar] [CrossRef] [Green Version]
- Ren, L.; Zhang, Z.; Feng, Y.; Luo, M.; Hao, Z. MicroRNA-876-5p represses the cell proliferation and invasion of colorectal cancer through suppressing YAP signalling via targeting RASAL2. Clin. Exp. Pharmacol. Physiol. 2020, 47, 867–876. [Google Scholar] [CrossRef]
- Deng, Z.; Rong, Y.; Teng, Y.; Mu, J.; Zhuang, X.; Tseng, M.; Samykutty, A.; Zhang, L.; Yan, J.; Miller, D.; et al. Broccoli-Derived Nanoparticle Inhibits Mouse Colitis by Activating Dendritic Cell AMP-Activated Protein Kinase. Mol. Ther. 2017, 25, 1641–1654. [Google Scholar] [CrossRef] [Green Version]
- Teng, Y.; Ren, Y.; Sayed, M.; Hu, X.; Lei, C.; Kumar, A.; Hutchins, E.; Mu, J.; Deng, Z.; Luo, C.; et al. Plant-Derived Exosomal MicroRNAs Shape the Gut Microbiota. Cell Host Microbe 2018, 24, 637–652.e8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mahiout, S.; Linden, J.; Esteban, J.; Sanchez-Perez, I.; Sankari, S.; Pettersson, L.; Hakansson, H.; Pohjanvirta, R. Toxicological characterisation of two novel selective aryl hydrocarbon receptor modulators in Sprague-Dawley rats. Toxicol. Appl. Pharmacol. 2017, 326, 54–65. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yu, T.Y.; Kondo, T.; Matsumoto, T.; Fujii-Kuriyama, Y.; Imai, Y. Aryl hydrocarbon receptor catabolic activity in bone metabolism is osteoclast dependent in vivo. Biochem. Biophys. Res. Commun. 2014, 450, 416–422. [Google Scholar] [CrossRef] [PubMed]
- Schulz, V.J.; Smit, J.J.; Bol-Schoenmakers, M.; van Duursen, M.B.; van den Berg, M.; Pieters, R.H. Activation of the aryl hydrocarbon receptor reduces the number of precursor and effector T cells, but preserves thymic CD4+CD25+Foxp3+ regulatory T cells. Toxicol. Lett. 2012, 215, 100–109. [Google Scholar] [CrossRef]
- Wu, D.; Nishimura, N.; Kuo, V.; Fiehn, O.; Shahbaz, S.; Van Winkle, L.; Matsumura, F.; Vogel, C.F. Activation of aryl hydrocarbon receptor induces vascular inflammation and promotes atherosclerosis in apolipoprotein E−/− mice. Arter. Thromb. Vasc. Biol. 2011, 31, 1260–1267. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Q.; Kurita, H.; Carreira, V.; Ko, C.I.; Fan, Y.; Zhang, X.; Biesiada, J.; Medvedovic, M.; Puga, A. Ah Receptor Activation by Dioxin Disrupts Activin, BMP, and WNT Signals during the Early Differentiation of Mouse Embryonic Stem Cells and Inhibits Cardiomyocyte Functions. Toxicol. Sci. 2016, 149, 346–357. [Google Scholar] [CrossRef] [Green Version]
- Safe, S.; Lee, S.O.; Jin, U.H. Role of the aryl hydrocarbon receptor in carcinogenesis and potential as a drug target. Toxicol. Sci. 2013, 135, 1–16. [Google Scholar] [CrossRef] [Green Version]
- Schiering, C.; Wincent, E.; Metidji, A.; Iseppon, A.; Li, Y.; Potocnik, A.J.; Omenetti, S.; Henderson, C.J.; Wolf, C.R.; Nebert, D.W.; et al. Feedback control of AHR signalling regulates intestinal immunity. Nature 2017, 542, 242–245. [Google Scholar] [CrossRef] [Green Version]
- Kinoshita, H.; Abe, J.; Akadegawa, K.; Yurino, H.; Uchida, T.; Ikeda, S.; Matsushima, K.; Ishikawa, S. Breakdown of mucosal immunity in gut by 2,3,7,8-tetraclorodibenzo-p-dioxin (TCDD). Environ. Health Prev. Med. 2006, 11, 256–263. [Google Scholar] [CrossRef]
- Chmill, S.; Kadow, S.; Winter, M.; Weighardt, H.; Esser, C. 2,3,7,8-Tetrachlorodibenzo-p-dioxin impairs stable establishment of oral tolerance in mice. Toxicol. Sci. 2010, 118, 98–107. [Google Scholar] [CrossRef] [Green Version]
- Stedtfeld, R.D.; Stedtfeld, T.M.; Fader, K.A.; Williams, M.R.; Bhaduri, P.; Quensen, J.; Zacharewski, T.R.; Tiedje, J.M.; Hashsham, S.A. TCDD influences reservoir of antibiotic resistance genes in murine gut microbiome. FEMS Microbiol. Ecol. 2017, 93, fix058. [Google Scholar] [CrossRef] [PubMed]
- Cheng, Y.; Jin, U.H.; Davidson, L.A.; Chapkin, R.S.; Jayaraman, A.; Tamamis, P.; Orr, A.; Allred, C.; Denison, M.S.; Soshilov, A.; et al. Editor’s Highlight: Microbial-Derived 1,4-Dihydroxy-2-naphthoic Acid and Related Compounds as Aryl Hydrocarbon Receptor Agonists/Antagonists: Structure-Activity Relationships and Receptor Modeling. Toxicol. Sci. 2017, 155, 458–473. [Google Scholar] [CrossRef] [Green Version]
- Androutsopoulos, V.P.; Tsatsakis, A.M.; Spandidos, D.A. Cytochrome P450 CYP1A1: Wider roles in cancer progression and prevention. BMC Cancer 2009, 9, 187. [Google Scholar] [CrossRef] [Green Version]
- White, S.S.; Fenton, S.E.; Birnbaum, L.S. Adverse Health Outcomes Caused by Dioxin-Activated AHR in Humans. In The AH Receptor in Biology and Toxicology; Pohjanvirta, R., Ed.; John Wiley & Sons, Inc.: Hoboken, NJ, USA, 2011; pp. 307–316. [Google Scholar] [CrossRef]
- Liu, D.; Ray, B.; Neavin, D.R.; Zhang, J.; Athreya, A.P.; Biernacka, J.M.; Bobo, W.V.; Hall-Flavin, D.K.; Skime, M.K.; Zhu, H.; et al. Beta-defensin 1, aryl hydrocarbon receptor and plasma kynurenine in major depressive disorder: Metabolomics-informed genomics. Transl. Psychiatry 2018, 8, 10. [Google Scholar] [CrossRef] [Green Version]
- Kim, J.B.; Pjanic, M.; Nguyen, T.; Miller, C.L.; Iyer, D.; Liu, B.; Wang, T.; Sazonova, O.; Carcamo-Orive, I.; Matic, L.P.; et al. TCF21 and the environmental sensor aryl-hydrocarbon receptor cooperate to activate a pro-inflammatory gene expression program in coronary artery smooth muscle cells. PLoS Genet. 2017, 13, e1006750. [Google Scholar] [CrossRef] [PubMed]
- Stockinger, B.; Hirota, K.; Duarte, J.; Veldhoen, M. External influences on the immune system via activation of the aryl hydrocarbon receptor. Semin. Immunol. 2011, 23, 99–105. [Google Scholar] [CrossRef] [PubMed]
- Li, R.; Shugart, Y.Y.; Zhou, W.; An, Y.; Yang, Y.; Zhou, Y.; Zhang, B.; Lu, D.; Wang, H.; Qian, J.; et al. Common genetic variations of the cytochrome P450 1A1 gene and risk of hepatocellular carcinoma in a Chinese population. Eur. J. Cancer 2009, 45, 1239–1247. [Google Scholar] [CrossRef] [PubMed]
- Dogan, M.V.; Shields, B.; Cutrona, C.; Gao, L.; Gibbons, F.X.; Simons, R.; Monick, M.; Brody, G.H.; Tan, K.; Beach, S.R.; et al. The effect of smoking on DNA methylation of peripheral blood mononuclear cells from African American women. BMC Genom. 2014, 15, 151. [Google Scholar] [CrossRef] [Green Version]
- Liu, D.; Qin, S.; Ray, B.; Kalari, K.R.; Wang, L.; Weinshilboum, R.M. Single Nucleotide Polymorphisms (SNPs) Distant from Xenobiotic Response Elements Can Modulate Aryl Hydrocarbon Receptor Function: SNP-Dependent CYP1A1 Induction. Drug Metab. Dispos. 2018, 46, 1372–1381. [Google Scholar] [CrossRef]
- Soshilov, A.A.; Motta, S.; Bonati, L.; Denison, M.S. Transitional States in Ligand-Dependent Transformation of the Aryl Hydrocarbon Receptor into Its DNA-Binding Form. Int. J. Mol. Sci. 2020, 21, 2474. [Google Scholar] [CrossRef] [Green Version]
- Flaveny, C.; Reen, R.K.; Kusnadi, A.; Perdew, G.H. The mouse and human Ah receptor differ in recognition of LXXLL motifs. Arch. Biochem. Biophys. 2008, 471, 215–223. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Whitlock, J.P., Jr. Induction of cytochrome P4501A1. Annu. Rev. Pharmacol. Toxicol. 1999, 39, 103–125. [Google Scholar] [CrossRef] [PubMed]
- Hankinson, O. The aryl hydrocarbon receptor complex. Annu. Rev. Pharmacol. Toxicol. 1995, 35, 307–340. [Google Scholar] [CrossRef] [PubMed]
- Gasiewicz, T.A.; Henry, E.C.; Collins, L.L. Expression and activity of aryl hydrocarbon receptors in development and cancer. Crit. Rev. Eukaryot. Gene Expr. 2008, 18, 279–321. [Google Scholar] [CrossRef] [PubMed]
- Bradfield, C.A.; Kende, A.S.; Poland, A. Kinetic and equilibrium studies of Ah receptor-ligand binding: Use of [125I]2-iodo-7,8-dibromodibenzo-p-dioxin. Mol. Pharmacol. 1988, 34, 229–237. [Google Scholar]
- Bohonowych, J.E.; Denison, M.S. Persistent binding of ligands to the aryl hydrocarbon receptor. Toxicol. Sci. 2007, 98, 99–109. [Google Scholar] [CrossRef]
- Stinn, A.; Furkert, J.; Kaufmann, S.H.E.; Moura-Alves, P.; Kolbe, M. Novel Method for Quantifying AhR-Ligand Binding Affinities Using Microscale Thermophoresis. Biosensors 2021, 11, 60. [Google Scholar] [CrossRef]
- Soshilov, A.A.; Denison, M.S. Ligand promiscuity of aryl hydrocarbon receptor agonists and antagonists revealed by site-directed mutagenesis. Mol. Cell. Biol. 2014, 34, 1707–1719. [Google Scholar] [CrossRef] [Green Version]
- DeGroot, D.E.; Denison, M.S. Nucleotide specificity of DNA binding of the aryl hydrocarbon receptor:ARNT complex is unaffected by ligand structure. Toxicol. Sci. 2014, 137, 102–113. [Google Scholar] [CrossRef] [Green Version]
- DeGroot, D.E.; Hayashi, A.; Denison, M.S. Lack of ligand-selective binding of the aryl hydrocarbon receptor to putative DNA binding sites regulating expression of Bax and paraoxonase 1 genes. Arch. Biochem. Biophys. 2014, 541, 13–20. [Google Scholar] [CrossRef] [Green Version]
- Pandini, A.; Soshilov, A.A.; Song, Y.; Zhao, J.; Bonati, L.; Denison, M.S. Detection of the TCDD binding-fingerprint within the Ah receptor ligand binding domain by structurally driven mutagenesis and functional analysis. Biochemistry 2009, 48, 5972–5983. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Giani Tagliabue, S.; Faber, S.C.; Motta, S.; Denison, M.S.; Bonati, L. Modeling the binding of diverse ligands within the Ah receptor ligand binding domain. Sci. Rep. 2019, 9, 10693. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Denison, M.S.; Faber, S.C. And Now for Something Completely Different: Diversity in Ligand-Dependent Activation of Ah Receptor Responses. Curr. Opin. Toxicol. 2017, 2, 124–131. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fraccalvieri, D.; Soshilov, A.A.; Karchner, S.I.; Franks, D.G.; Pandini, A.; Bonati, L.; Hahn, M.E.; Denison, M.S. Comparative analysis of homology models of the AH receptor ligand binding domain: Verification of structure-function predictions by site-directed mutagenesis of a nonfunctional receptor. Biochemistry 2013, 52, 714–725. [Google Scholar] [CrossRef] [PubMed]
- Corrada, D.; Denison, M.S.; Bonati, L. Structural modeling of the AhR:ARNT complex in the bHLH-PASA-PASB region elucidates the key determinants of dimerization. Mol. Biosyst. 2017, 13, 981–990. [Google Scholar] [CrossRef] [Green Version]
- Corrada, D.; Soshilov, A.A.; Denison, M.S.; Bonati, L. Deciphering Dimerization Modes of PAS Domains: Computational and Experimental Analyses of the AhR:ARNT Complex Reveal New Insights into the Mechanisms of AhR Transformation. PLoS Comput. Biol. 2016, 12, e1004981. [Google Scholar] [CrossRef]
- Bonati, L.; Corrada, D.; Tagliabue, S.G.; Motta, S. Molecular modeling of the AhR structure and interactions can shed light on ligand-dependent activation and transformation mechanisms. Curr. Opin. Toxicol. 2017, 2, 42–49. [Google Scholar] [CrossRef] [Green Version]
- Bisson, W.H.; Koch, D.C.; O’Donnell, E.F.; Khalil, S.M.; Kerkvliet, N.I.; Tanguay, R.L.; Abagyan, R.; Kolluri, S.K. Modeling of the aryl hydrocarbon receptor (AhR) ligand binding domain and its utility in virtual ligand screening to predict new AhR ligands. J. Med. Chem. 2009, 52, 5635–5641. [Google Scholar] [CrossRef] [Green Version]
- Pandini, A.; Denison, M.S.; Song, Y.; Soshilov, A.A.; Bonati, L. Structural and functional characterization of the aryl hydrocarbon receptor ligand binding domain by homology modeling and mutational analysis. Biochemistry 2007, 46, 696–708. [Google Scholar] [CrossRef] [Green Version]
- Seok, S.H.; Lee, W.; Jiang, L.; Molugu, K.; Zheng, A.; Li, Y.; Park, S.; Bradfield, C.A.; Xing, Y. Structural hierarchy controlling dimerization and target DNA recognition in the AHR transcriptional complex. Proc. Natl. Acad. Sci. USA 2017, 114, 5431–5436. [Google Scholar] [CrossRef] [Green Version]
- Faber, S.C.; Soshilov, A.A.; Giani Tagliabue, S.; Bonati, L.; Denison, M.S. Comparative In Vitro and In Silico Analysis of the Selectivity of Indirubin as a Human Ah Receptor Agonist. Int. J. Mol. Sci. 2018, 19, 2692. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Flaveny, C.A.; Murray, I.A.; Perdew, G.H. Differential gene regulation by the human and mouse aryl hydrocarbon receptor. Toxicol. Sci. 2010, 114, 217–225. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Flaveny, C.A.; Perdew, G.H. Transgenic Humanized AHR Mouse Reveals Differences between Human and Mouse AHR Ligand Selectivity. Mol. Cell Pharmacol. 2009, 1, 119–123. [Google Scholar] [CrossRef] [PubMed]
- Faber, S.C.; Giani Tagliabue, S.; Bonati, L.; Denison, M.S. The Cellular and Molecular Determinants of Naphthoquinone-Dependent Activation of the Aryl Hydrocarbon Receptor. Int. J. Mol. Sci. 2020, 21, 4111. [Google Scholar] [CrossRef] [PubMed]
- He, G.; Tsutsumi, T.; Zhao, B.; Baston, D.S.; Zhao, J.; Heath-Pagliuso, S.; Denison, M.S. Third-generation Ah receptor-responsive luciferase reporter plasmids: Amplification of dioxin-responsive elements dramatically increases CALUX bioassay sensitivity and responsiveness. Toxicol. Sci. 2011, 123, 511–522. [Google Scholar] [CrossRef] [Green Version]
- Petrulis, J.R.; Chen, G.; Benn, S.; LaMarre, J.; Bunce, N.J. Application of the ethoxyresorufin-O-deethylase (EROD) assay to mixtures of halogenated aromatic compounds. Environ. Toxicol. 2001, 16, 177–184. [Google Scholar] [CrossRef]
- Mahiout, S.; Tagliabue, S.G.; Nasri, A.; Omoruyi, I.M.; Pettersson, L.; Bonati, L.; Pohjanvirta, R. In vitro toxicity and in silico docking analysis of two novel selective AH-receptor modulators. Toxicol. In Vitro 2018, 52, 178–188. [Google Scholar] [CrossRef]
- Smirnova, A.; Wincent, E.; Vikstrom Bergander, L.; Alsberg, T.; Bergman, J.; Rannug, A.; Rannug, U. Evidence for New Light-Independent Pathways for Generation of the Endogenous Aryl Hydrocarbon Receptor Agonist FICZ. Chem. Res. Toxicol. 2016, 29, 75–86. [Google Scholar] [CrossRef]
- Klinge, C.M.; Bowers, J.L.; Kulakosky, P.C.; Kamboj, K.K.; Swanson, H.I. The aryl hydrocarbon receptor (AHR)/AHR nuclear translocator (ARNT) heterodimer interacts with naturally occurring estrogen response elements. Mol. Cell. Endocrinol. 1999, 157, 105–119. [Google Scholar] [CrossRef]
- Taylor, S.J.; Demont, E.H.; Gray, J.; Deeks, N.; Patel, A.; Nguyen, D.; Taylor, M.; Hood, S.; Watson, R.J.; Bit, R.A.; et al. Navigating CYP1A Induction and Arylhydrocarbon Receptor Agonism in Drug Discovery. A Case History with S1P1 Agonists. J. Med. Chem. 2015, 58, 8236–8256. [Google Scholar] [CrossRef]
- Sistare, F.D.; Morton, D.; Alden, C.; Christensen, J.; Keller, D.; Jonghe, S.D.; Storer, R.D.; Reddy, M.V.; Kraynak, A.; Trela, B.; et al. An analysis of pharmaceutical experience with decades of rat carcinogenicity testing: Support for a proposal to modify current regulatory guidelines. Toxicol. Pathol. 2011, 39, 716–744. [Google Scholar] [CrossRef] [PubMed]
- Hill, T., III; Nelms, M.D.; Edwards, S.W.; Martin, M.; Judson, R.; Corton, J.C.; Wood, C.E. Editor’s Highlight: Negative Predictors of Carcinogenicity for Environmental Chemicals. Toxicol. Sci. 2017, 155, 157–169. [Google Scholar] [CrossRef] [PubMed]
- Rooney, J.; Hill, T., III; Qin, C.; Sistare, F.D.; Corton, J.C. Adverse outcome pathway-driven identification of rat liver tumorigens in short-term assays. Toxicol. Appl. Pharmacol. 2018, 356, 99–113. [Google Scholar] [CrossRef] [PubMed]
- Igarashi, Y.; Nakatsu, N.; Yamashita, T.; Ono, A.; Ohno, Y.; Urushidani, T.; Yamada, H. Open TG-GATEs: A large-scale toxicogenomics database. Nucleic Acids Res. 2015, 43, D921–D927. [Google Scholar] [CrossRef] [PubMed]
- Qin, C.; Aslamkhan, A.G.; Pearson, K.; Tanis, K.Q.; Podtelezhnikov, A.; Frank, E.; Pacchione, S.; Pippert, T.; Glaab, W.E.; Sistare, F.D. AhR Activation in Pharmaceutical Development: Applying Liver Gene Expression Biomarker Thresholds to Identify Doses Associated with Tumorigenic Risks in Rats. Toxicol. Sci. 2019, 171, 46–55. [Google Scholar] [CrossRef]
- Glaab, W.E.; Holder, D.; He, Y.D.; Bailey, W.J.; Gerhold, D.L.; Beare, C.; Erdos, Z.; Lane, P.; Michna, L.; Muniappa, N.; et al. Universal Toxicity Gene Signatures for Early Identification of Drug-Induced Tissue Injuries in Rats. Toxicol. Sci. 2021, 181, 148–159. [Google Scholar] [CrossRef]
- Podtelezhnikov, A.A.; Monroe, J.J.; Aslamkhan, A.G.; Pearson, K.; Qin, C.; Tamburino, A.M.; Loboda, A.P.; Glaab, W.E.; Sistare, F.D.; Tanis, K.Q. Quantitative Transcriptional Biomarkers of Xenobiotic Receptor Activation in Rat Liver for the Early Assessment of Drug Safety Liabilities. Toxicol. Sci. 2020, 175, 98–112. [Google Scholar] [CrossRef]
- Monroe, J.J.; Tanis, K.Q.; Podtelezhnikov, A.A.; Nguyen, T.; Machotka, S.V.; Lynch, D.; Evers, R.; Palamanda, J.; Miller, R.R.; Pippert, T.; et al. Application of a Rat Liver Drug Bioactivation Transcriptional Response Assay Early in Drug Development That Informs Chemically Reactive Metabolite Formation and Potential for Drug-induced Liver Injury. Toxicol. Sci. 2020, 177, 281–299. [Google Scholar] [CrossRef]
- Buick, J.K.; Moffat, I.; Williams, A.; Swartz, C.D.; Recio, L.; Hyduke, D.R.; Li, H.H.; Fornace, A.J., Jr.; Aubrecht, J.; Yauk, C.L. Integration of metabolic activation with a predictive toxicogenomics signature to classify genotoxic versus nongenotoxic chemicals in human TK6 cells. Environ. Mol. Mutagen. 2015, 56, 520–534. [Google Scholar] [CrossRef]
- Yauk, C.L.; Buick, J.K.; Williams, A.; Swartz, C.D.; Recio, L.; Li, H.H.; Fornace, A.J., Jr.; Thomson, E.M.; Aubrecht, J. Application of the TGx-28.65 transcriptomic biomarker to classify genotoxic and non-genotoxic chemicals in human TK6 cells in the presence of rat liver S9. Environ. Mol. Mutagen. 2016, 57, 243–260. [Google Scholar] [CrossRef] [Green Version]
- Li, H.H.; Chen, R.; Hyduke, D.R.; Williams, A.; Frotschl, R.; Ellinger-Ziegelbauer, H.; O’Lone, R.; Yauk, C.L.; Aubrecht, J.; Fornace, A.J., Jr. Development and validation of a high-throughput transcriptomic biomarker to address 21st century genetic toxicology needs. Proc. Natl. Acad. Sci. USA 2017, 114, E10881–E10889. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, H.H.; Hyduke, D.R.; Chen, R.; Heard, P.; Yauk, C.L.; Aubrecht, J.; Fornace, A.J., Jr. Development of a toxicogenomics signature for genotoxicity using a dose-optimization and informatics strategy in human cells. Environ. Mol. Mutagen. 2015, 56, 505–519. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moffat, I.; Chepelev, N.; Labib, S.; Bourdon-Lacombe, J.; Kuo, B.; Buick, J.K.; Lemieux, F.; Williams, A.; Halappanavar, S.; Malik, A.; et al. Comparison of toxicogenomics and traditional approaches to inform mode of action and points of departure in human health risk assessment of benzo[a]pyrene in drinking water. Crit. Rev. Toxicol. 2015, 45, 1–43. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shi, L.; Kusko, R.; Wolfinger, R.D.; Haibe-Kains, B.; Fischer, M.; Sansone, S.A.; Mason, C.E.; Furlanello, C.; Jones, W.D.; Ning, B.; et al. The international MAQC Society launches to enhance reproducibility of high-throughput technologies. Nat. Biotechnol. 2017, 35, 1127–1128. [Google Scholar] [CrossRef] [PubMed]
- Foran, C.M.; Rycroft, T.; Keisler, J.; Perkins, E.J.; Linkov, I.; Garcia-Reyero, N. A modular approach for assembly of quantitative adverse outcome pathways. ALTEX Altern. Anim. Exp. 2019, 36, 353–362. [Google Scholar] [CrossRef] [Green Version]
- Perkins, E.J.; Gayen, K.; Shoemaker, J.E.; Antczak, P.; Burgoon, L.; Falciani, F.; Gutsell, S.; Hodges, G.; Kienzler, A.; Knapen, D.; et al. Chemical hazard prediction and hypothesis testing using quantitative adverse outcome pathways. ALTEX Altern. Anim. Exp. 2019, 36, 91–102. [Google Scholar] [CrossRef]
- Wittwehr, C.; Aladjov, H.; Ankley, G.; Byrne, H.J.; de Knecht, J.; Heinzle, E.; Klambauer, G.; Landesmann, B.; Luijten, M.; MacKay, C.; et al. How Adverse Outcome Pathways Can Aid the Development and Use of Computational Prediction Models for Regulatory Toxicology. Toxicol. Sci. 2017, 155, 326–336. [Google Scholar] [CrossRef] [Green Version]
- Yauk, C.L.; Harrill, A.H.; Ellinger-Ziegelbauer, H.; van der Laan, J.W.; Moggs, J.; Froetschl, R.; Sistare, F.; Pettit, S. A cross-sector call to improve carcinogenicity risk assessment through use of genomic methodologies. Regul. Toxicol. Pharmacol. 2020, 110, 104526. [Google Scholar] [CrossRef]
- Peters, M.F.; Landry, T.; Pin, C.; Maratea, K.; Dick, C.; Wagoner, M.P.; Choy, A.L.; Barthlow, H.; Snow, D.; Stevens, Z.; et al. Human 3D Gastrointestinal Microtissue Barrier Function as a Predictor of Drug-Induced Diarrhea. Toxicol. Sci. 2019, 168, 3–17. [Google Scholar] [CrossRef] [Green Version]
- Belair, D.G.; Visconti, R.J.; Hong, M.; Marella, M.; Peters, M.F.; Scott, C.W.; Kolaja, K.L. Human ileal organoid model recapitulates clinical incidence of diarrhea associated with small molecule drugs. Toxicol. In Vitro 2020, 68, 104928. [Google Scholar] [CrossRef]
- Peters, M.F.; Choy, A.L.; Pin, C.; Leishman, D.J.; Moisan, A.; Ewart, L.; Guzzie-Peck, P.J.; Sura, R.; Keller, D.A.; Scott, C.W.; et al. Developing in vitro assays to transform gastrointestinal safety assessment: Potential for microphysiological systems. Lab Chip 2020, 20, 1177–1190. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dekkers, B.J.; Pearce, S.; van Bolderen-Veldkamp, R.P.; Marshall, A.; Widera, P.; Gilbert, J.; Drost, H.G.; Bassel, G.W.; Muller, K.; King, J.R.; et al. Transcriptional dynamics of two seed compartments with opposing roles in Arabidopsis seed germination. Plant Physiol. 2013, 163, 205–215. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jana, N.R.; Sarkar, S.; Yonemoto, J.; Tohyama, C.; Sone, H. Strain differences in cytochrome P4501A1 gene expression caused by 2,3,7,8-tetrachlorodibenzo-p-dioxin in the rat liver: Role of the aryl hydrocarbon receptor and its nuclear translocator. Biochem. Biophys. Res. Commun. 1998, 248, 554–558. [Google Scholar] [CrossRef]
- Okey, A.B.; Vella, L.M.; Harper, P.A. Detection and characterization of a low affinity form of cytosolic Ah receptor in livers of mice nonresponsive to induction of cytochrome P1-450 by 3-methylcholanthrene. Mol. Pharmacol. 1989, 35, 823–830. [Google Scholar]
- Poland, A.; Glover, E. Characterization and strain distribution pattern of the murine Ah receptor specified by the Ahd and Ahb-3 alleles. Mol. Pharmacol. 1990, 38, 306–312. [Google Scholar] [PubMed]
- Pohjanvirta, R.; Miettinen, H.; Sankari, S.; Hegde, N.; Linden, J. Unexpected gender difference in sensitivity to the acute toxicity of dioxin in mice. Toxicol. Appl. Pharmacol. 2012, 262, 167–176. [Google Scholar] [CrossRef] [PubMed]
- Flaveny, C.A.; Murray, I.A.; Chiaro, C.R.; Perdew, G.H. Ligand selectivity and gene regulation by the human aryl hydrocarbon receptor in transgenic mice. Mol. Pharmacol. 2009, 75, 1412–1420. [Google Scholar] [CrossRef] [Green Version]
- Nebert, D.W.; Dalton, T.P.; Okey, A.B.; Gonzalez, F.J. Role of aryl hydrocarbon receptor-mediated induction of the CYP1 enzymes in environmental toxicity and cancer. J. Biol. Chem. 2004, 279, 23847–23850. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Miyamoto, M.; Iwasaki, S.; Chisaki, I.; Nakagawa, S.; Amano, N.; Hirabayashi, H. Comparison of predictability for human pharmacokinetics parameters among monkeys, rats, and chimeric mice with humanised liver. Xenobiotica 2017, 47, 1052–1063. [Google Scholar] [CrossRef]
- Sameshima, T.; Yukawa, T.; Hirozane, Y.; Yoshikawa, M.; Katoh, T.; Hara, H.; Yogo, T.; Miyahisa, I.; Okuda, T.; Miyamoto, M.; et al. Small-Scale Panel Comprising Diverse Gene Family Targets to Evaluate Compound Promiscuity. Chem. Res. Toxicol. 2020, 33, 154–161. [Google Scholar] [CrossRef]
- Harada, K.; Kohara, H.; Yukawa, T.; Matsumiya, K.; Shinozawa, T. Cell-based high-throughput screening for the evaluation of reactive metabolite formation potential. Toxicol. In Vitro 2021, 74, 105159. [Google Scholar] [CrossRef] [PubMed]
- Zheng, J.; Ma, L.; Xin, B.; Olah, T.; Humphreys, W.G.; Zhu, M. Screening and identification of GSH-trapped reactive metabolites using hybrid triple quadruple linear ion trap mass spectrometry. Chem. Res. Toxicol. 2007, 20, 757–766. [Google Scholar] [CrossRef] [PubMed]
- Li, F.; Lu, J.; Ma, X. Profiling the reactive metabolites of xenobiotics using metabolomic technologies. Chem. Res. Toxicol. 2011, 24, 744–751. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Simpson, K.; Bevan, N.; Hastwell, P.; Eidam, P.; Shah, P.; Gogo, E.; Rees, S.; Brown, A. The BlueScreen-384 assay as an indicator of genotoxic hazard potential in early-stage drug discovery. J. Biomol. Screen 2013, 18, 441–452. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lutz, J.D.; Fujioka, Y.; Isoherranen, N. Rationalization and prediction of in vivo metabolite exposures: The role of metabolite kinetics, clearance predictions and in vitro parameters. Expert Opin. Drug Metab. Toxicol. 2010, 6, 1095–1109. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Safe, S.; Wormke, M. Inhibitory aryl hydrocarbon receptor-estrogen receptor alpha cross-talk and mechanisms of action. Chem. Res. Toxicol. 2003, 16, 807–816. [Google Scholar] [CrossRef] [PubMed]
- Hu, W.; Sorrentino, C.; Denison, M.S.; Kolaja, K.; Fielden, M.R. Induction of cyp1a1 is a nonspecific biomarker of aryl hydrocarbon receptor activation: Results of large scale screening of pharmaceuticals and toxicants in vivo and in vitro. Mol. Pharmacol. 2007, 71, 1475–1486. [Google Scholar] [CrossRef] [PubMed]
- O’Donnell, E.F.; Koch, D.C.; Bisson, W.H.; Jang, H.S.; Kolluri, S.K. The aryl hydrocarbon receptor mediates raloxifene-induced apoptosis in estrogen receptor-negative hepatoma and breast cancer cells. Cell Death Dis. 2014, 5, e1038. [Google Scholar] [CrossRef]
- Ehrlich, A.K.; Kerkvliet, N.I. Is chronic AhR activation by rapidly metabolized ligands safe for the treatment of immune-mediated diseases? Curr. Opin. Toxicol. 2017, 2, 72–78. [Google Scholar] [CrossRef] [Green Version]
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Faber, S.C.; Lahoti, T.S.; Taylor, E.R.; Lewis, L.; Sapiro, J.M.; Toledo Sales, V.; Dragan, Y.P.; Jeffy, B.D. Current Therapeutic Landscape and Safety Roadmap for Targeting the Aryl Hydrocarbon Receptor in Inflammatory Gastrointestinal Indications. Cells 2022, 11, 1708. https://doi.org/10.3390/cells11101708
Faber SC, Lahoti TS, Taylor ER, Lewis L, Sapiro JM, Toledo Sales V, Dragan YP, Jeffy BD. Current Therapeutic Landscape and Safety Roadmap for Targeting the Aryl Hydrocarbon Receptor in Inflammatory Gastrointestinal Indications. Cells. 2022; 11(10):1708. https://doi.org/10.3390/cells11101708
Chicago/Turabian StyleFaber, Samantha C., Tejas S. Lahoti, Ewan R. Taylor, Lauren Lewis, Jessica M. Sapiro, Vicencia Toledo Sales, Yvonne P. Dragan, and Brandon D. Jeffy. 2022. "Current Therapeutic Landscape and Safety Roadmap for Targeting the Aryl Hydrocarbon Receptor in Inflammatory Gastrointestinal Indications" Cells 11, no. 10: 1708. https://doi.org/10.3390/cells11101708
APA StyleFaber, S. C., Lahoti, T. S., Taylor, E. R., Lewis, L., Sapiro, J. M., Toledo Sales, V., Dragan, Y. P., & Jeffy, B. D. (2022). Current Therapeutic Landscape and Safety Roadmap for Targeting the Aryl Hydrocarbon Receptor in Inflammatory Gastrointestinal Indications. Cells, 11(10), 1708. https://doi.org/10.3390/cells11101708