
Abnormal Epigenetic Regulations in the Immunocytes of Sjögren’s Syndrome Patients and Therapeutic Potentials



{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Epigenetic Regulations in Immune Responses
2.1. DNA Methylation
2.2. Histone Modifications
2.3. ncRNAs
2.3.1. Adaptive Immunity
2.3.2. Innate Immunity
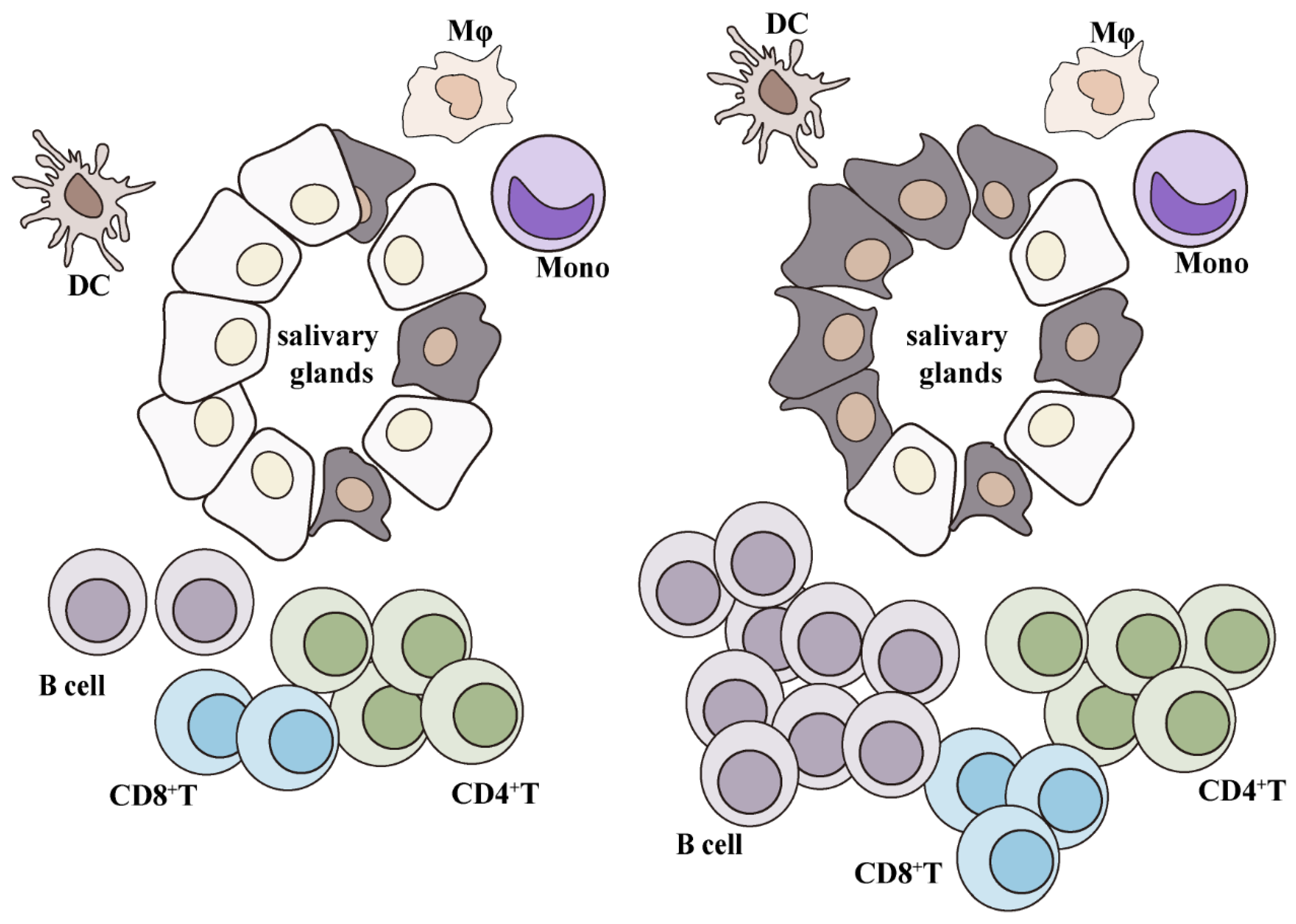
3. Immunocyte Infiltration in SjS
3.1. T Cells
3.1.1. Th1 and Th17
3.1.2. Tfh
3.1.3. Treg
3.1.4. CD8+ T Cells
3.2. B Cells
3.3. Innate Lymphoid Cells
3.4. Antigen-Presenting Cells
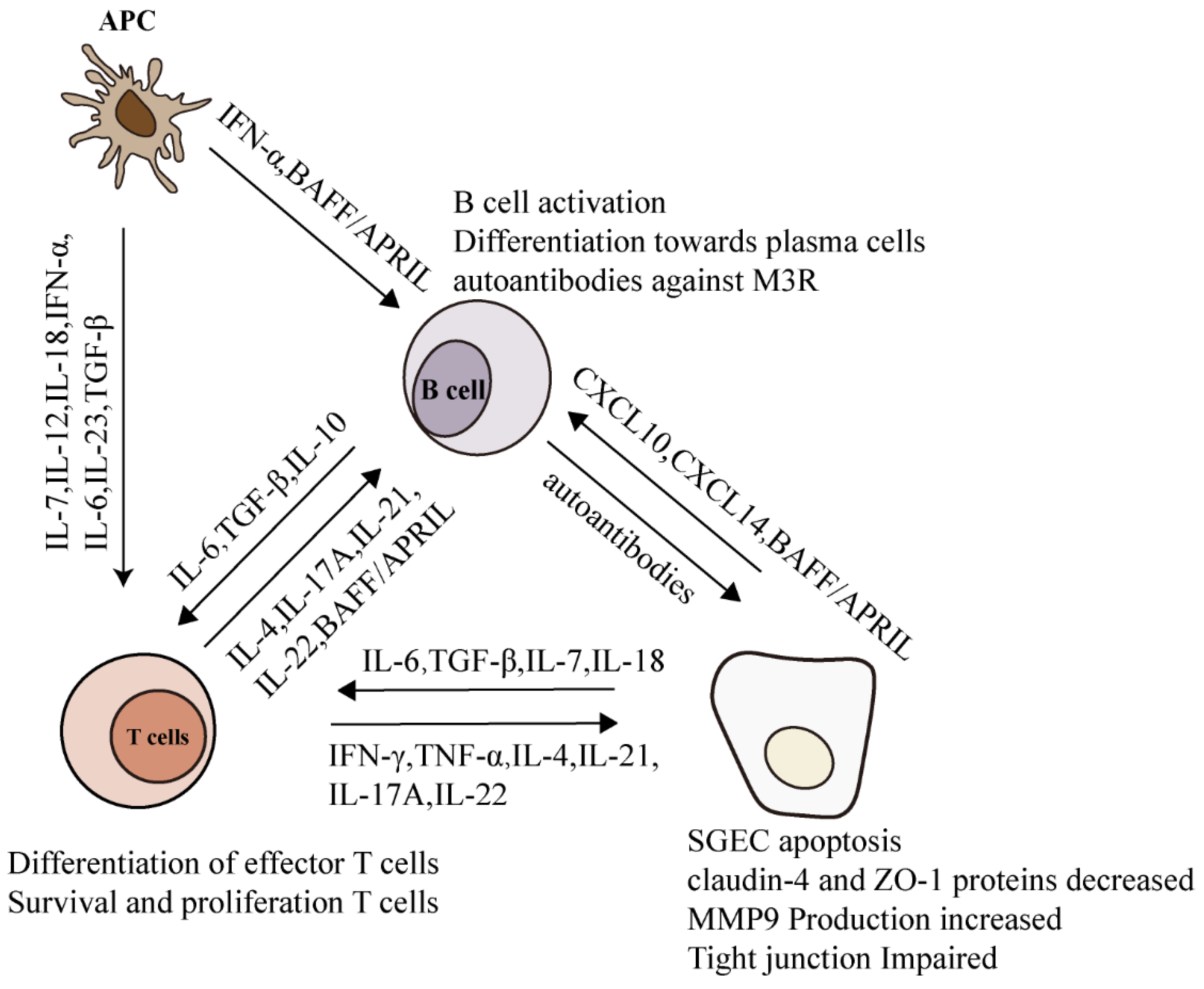
4. Pro-Inflammatory Cytokines in SjS
4.1. Interferons
4.1.1. Type I interferons
4.1.2. Type II interferon
4.1.3. Type III interferon
4.2. Interleukins
4.2.1. IL-17 and IL-22
4.2.2. IL-21
4.2.3. IL-4
4.3. Tumor-Necrosis-Factor Superfamily Members
4.3.1. TNF-α
4.3.2. BAFF and APRIL
5. Epigenetic Modifications in SjS
5.1. Epigenetic Modification of IFN-Related Genes
5.2. Epigenetic Modifications in B Cells
5.3. Epigenetic Modification of Foxp3
5.4. Correlation between SjS-Related SNPs and Epigenetic Changes
6. Epigenetic Therapeutics of SjS
7. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Acknowledgments
Conflicts of Interest
References
- Hayashi, T. Dysfunction of lacrimal and salivary glands in Sjögren’s syndrome: Nonimmunologic injury in preinflammatory phase and mouse model. J. Biomed. Biotechnol. 2011, 2011, 407031. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Theander, E.; Jonsson, R.; Sjöström, B.; Brokstad, K.; Olsson, P.; Henriksson, G. Prediction of Sjögren’s Syndrome Years before Diagnosis and Identification of Patients with Early Onset and Severe Disease Course by Autoantibody Profiling. Arthritis Rheumatol. 2015, 67, 2427–2436. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fragkioudaki, S.; Moutsopoulos, H.M.; Mavragani, C.P. Sjgren’s Syndrome; The Heart in Rheumatic, Autoimmune and Inflammatory Diseases; Academic Press: Cambridge, MA, USA, 2017. [Google Scholar]
- Solans-Laqué, R.; López-Hernandez, A.; Bosch-Gil, J.A.; Palacios, A.; Campillo, M.; Vilardell-Tarres, M. Risk, predictors, and clinical characteristics of lymphoma development in primary Sjögren’s syndrome. Semin. Arthritis Rheum. 2011, 41, 415–423. [Google Scholar] [CrossRef] [PubMed]
- Kapsogeorgou, E.K.; Papageorgiou, A.; Protogerou, A.D.; Voulgarelis, M.; Tzioufas, A.G. Low miR200b-5p levels in minor salivary glands: A novel molecular marker predicting lymphoma development in patients with Sjögren’s syndrome. Ann. Rheum. Dis. 2018, 77, 1200–1207. [Google Scholar] [CrossRef] [PubMed]
- Qin, B.; Wang, J.; Yang, Z.; Yang, M.; Ma, N.; Huang, F.; Zhong, R. Epidemiology of primary Sjogren’s syndrome: A systematic review and meta-analysis. Ann. Rheum. Dis. 2015, 74, 1983–1989. [Google Scholar] [CrossRef] [PubMed]
- Harris, V.M.; Sharma, R.; Cavett, J.; Kurien, B.T.; Liu, K.; Koelsch, K.A.; Rasmussen, A.; Radfar, L.; Lewis, D.; Stone, D.U.; et al. Klinefelter’s syndrome (47,XXY) is in excess among men with Sjögren’s syndrome. Clin. Immunol. 2016, 168, 25–29. [Google Scholar] [CrossRef] [Green Version]
- Liu, K.; Kurien, B.T.; Zimmerman, S.L.; Kaufman, K.M.; Taft, D.H.; Kottyan, L.C.; Lazaro, S.; Weaver, C.A.; Ice, J.A.; Adler, A.J.; et al. X Chromosome Dose and Sex Bias in Autoimmune Diseases: Increased Prevalence of 47,XXX in Systemic Lupus Erythematosus and Sjögren’s Syndrome. Arthritis Rheumatol. 2016, 68, 1290–1300. [Google Scholar] [CrossRef]
- Shaw, T.M.; Zhang, W.; McCoy, S.S.; Pagenkopf, A.; Carp, D.M.; Garg, S.; Parker, M.H.; Qiu, X.; Scofield, R.H.; Galipeau, J.; et al. X-linked genes exhibit miR6891-5p-regulated skewing in Sjögren’s syndrome. J. Mol. Med. 2022, 100, 1–13. [Google Scholar] [CrossRef]
- Li, Y.; Zhang, K.; Chen, H.; Sun, F.; Xu, J.; Wu, Z.; Li, P.; Zhang, L.; Du, Y.; Luan, H.; et al. A genome-wide association study in Han Chinese identifies a susceptibility locus for primary Sjögren’s syndrome at 7q11.23. Nat. Genet. 2013, 45, 1361–1365. [Google Scholar] [CrossRef]
- Lessard, C.J.; Li, H.; Adrianto, I.; Ice, J.A.; Rasmussen, A.; Grundahl, K.M.; Kelly, J.A.; Dozmorov, M.G.; Miceli-Richard, C.; Bowman, S.; et al. Variants at multiple loci implicated in both innate and adaptive immune responses are associated with Sjögren’s syndrome. Nat. Genet. 2013, 45, 1284–1292. [Google Scholar] [CrossRef]
- Xuan, J.; Ji, Z.; Wang, B.; Zeng, X.; Shi, G.J.F.I.I. Serological Evidence for the Association between Epstein-Barr Virus Infection and Sjgren’s Syndrome. Front. Immunol. 2020, 11, 590444. [Google Scholar] [CrossRef] [PubMed]
- Altorok, N.; Coit, P.; Hughes, T.; Koelsch, K.A.; Stone, D.U.; Rasmussen, A.; Radfar, L.; Scofield, R.H.; Sivils, K.L.; Farris, A.D.; et al. Genome-Wide DNA Methylation Patterns in Naive CD4+ T Cells from Patients with Primary Sjogren’s Syndrome. Arthritis Rheumatol. 2013, 66, 731–739. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Arvaniti, P.; Zachou, K.; Lyberopoulou, A.; Gatselis, N.K.; Brooks, W.H.; Dalekos, G.N.; Renaudineau, Y. Epigenetic Modifications in Generalized Autoimmune Epithelitis: Sjogren’s Syndrome and Primary Biliary Cholangitis. Epigenomes 2019, 3, 15. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mazzone, R.; Zwergel, C.; Artico, M.; Taurone, S.; Ralli, M.; Greco, A.; Mai, A. The emerging role of epigenetics in human autoimmune disorders. Clin. Epigenetics 2019, 11, 34. [Google Scholar] [CrossRef] [Green Version]
- Schmolka, N.; Silva-Santos, B.; Gomes, A.Q. Epigenetic mechanisms in the regulation of lymphocyte differentiation. Epigenetics Immune Syst. 2020, 16, 77–116. [Google Scholar]
- Imgenberg-Kreuz, J.; Sandling, J.K.; Nordmark, G. Epigenetic alterations in primary Sjogren’s syndrome—An overview. Clin. Immunol. 2018, 196, 12–20. [Google Scholar] [CrossRef]
- Correa, L.O.; Jordan, M.S.; Carty, S.A. DNA Methylation in T-Cell Development and Differentiation. Crit. Rev. Immunol. 2020, 40, 135–156. [Google Scholar] [CrossRef]
- Thomas, R.M.; Gamper, C.J.; Ladle, B.H.; Powell, J.D.; Wells, A.D. De novo DNA methylation is required to restrict T helper lineage plasticity. J. Biol. Chem. 2012, 287, 22900–22909. [Google Scholar] [CrossRef] [Green Version]
- Wang, L.; Liu, Y.; Beier, U.H.; Han, R.; Bhatti, T.R.; Akimova, T.; Hancock, W.W. Foxp3+ T-regulatory cells require DNA methyltransferase 1 expression to prevent development of lethal autoimmunity. Blood 2013, 121, 3631–3639. [Google Scholar] [CrossRef] [Green Version]
- Ichiyama, K.; Chen, T.; Wang, X.; Yan, X.; Kim, B.S.; Tanaka, S.; Ndiaye-Lobry, D.; Deng, Y.; Zou, Y.; Zheng, P.; et al. The methylcytosine dioxygenase Tet2 promotes DNA demethylation and activation of cytokine gene expression in T cells. Immunity 2015, 42, 613–626. [Google Scholar] [CrossRef] [Green Version]
- Yang, R.; Qu, C.; Zhou, Y.; Konkel, J.E.; Shi, S.; Liu, Y.; Chen, C.; Liu, S.; Liu, D.; Chen, Y.; et al. Hydrogen Sulfide Promotes Tet1- and Tet2-Mediated Foxp3 Demethylation to Drive Regulatory T Cell Differentiation and Maintain Immune Homeostasis. Immunity 2015, 43, 251–263. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yue, X.; Trifari, S.; Äijö, T.; Tsagaratou, A.; Pastor, W.A.; Zepeda-Martínez, J.A.; Lio, C.W.; Li, X.; Huang, Y.; Vijayanand, P.; et al. Control of Foxp3 stability through modulation of TET activity. J. Exp. Med. 2016, 213, 377–397. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nair, V.S.; Oh, K.I. Vitamin C and DNA Demethylation in Regulatory T Cells. In Handbook of Nutrition, Diet, and Epigenetics; Patel, V., Preedy, V., Eds.; Springer International Publishing: Cham, Switzerland, 2017; pp. 1–15. [Google Scholar]
- Barwick, B.G.; Scharer, C.D.; Martinez, R.J.; Price, M.J.; Wein, A.N.; Haines, R.R.; Bally, A.P.R.; Kohlmeier, J.E.; Boss, J.M. B cell activation and plasma cell differentiation are inhibited by de novo DNA methylation. Nat. Commun. 2018, 9, 1900. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lio, C.J.; Shukla, V.; Samaniego-Castruita, D.; González-Avalos, E.; Chakraborty, A.; Yue, X.; Schatz, D.G.; Ay, F.; Rao, A. TET enzymes augment activation-induced deaminase (AID) expression via 5-hydroxymethylcytosine modifications at the Aicda superenhancer. Sci. Immunol. 2019, 4, eaau7523. [Google Scholar] [CrossRef]
- Li, J.; Ding, Y.; Ling, Z.J.T.E. Histone-Mediated Transgenerational Epigenetics; Academic Press: Cambridge, MA, USA, 2014; pp. 87–103. [Google Scholar]
- Ellmeier, W.; Seiser, C. Histone deacetylase function in CD4(+) T cells. Nat. Rev. Immunol. 2018, 18, 617–634. [Google Scholar] [CrossRef]
- Grausenburger, R.; Bilic, I.; Boucheron, N.; Zupkovitz, G.; El-Housseiny, L.; Tschismarov, R.; Zhang, Y.; Rembold, M.; Gaisberger, M.; Hartl, A.; et al. Conditional deletion of histone deacetylase 1 in T cells leads to enhanced airway inflammation and increased Th2 cytokine production. J. Immunol. 2010, 185, 3489–3497. [Google Scholar] [CrossRef]
- Woods, D.M.; Woan, K.V.; Cheng, F.; Sodré, A.L.; Wang, D.; Wu, Y.; Wang, Z.; Chen, J.; Powers, J.; Pinilla-Ibarz, J.; et al. T cells lacking HDAC11 have increased effector functions and mediate enhanced alloreactivity in a murine model. Blood 2017, 130, 146–155. [Google Scholar] [CrossRef] [Green Version]
- Lim, H.W.; Kang, S.G.; Ryu, J.K.; Schilling, B.; Fei, M.; Lee, I.S.; Kehasse, A.; Shirakawa, K.; Yokoyama, M.; Schnölzer, M.; et al. SIRT1 deacetylates RORγt and enhances Th17 cell generation. J. Exp. Med. 2015, 212, 607–617. [Google Scholar] [CrossRef]
- El Kaoutari, A.; Armougom, F.; Gordon, J.I.; Raoult, D.; Henrissat, B. The abundance and variety of carbohydrate-active enzymes in the human gut microbiota. Nat. Rev. Microbiol. 2013, 11, 497–504. [Google Scholar] [CrossRef]
- Kim, C.H.; Park, J.; Kim, M. Gut microbiota-derived short-chain Fatty acids, T cells, and inflammation. Immune Netw. 2014, 14, 277–288. [Google Scholar] [CrossRef] [Green Version]
- Tanoue, T.; Atarashi, K.; Honda, K. Development and maintenance of intestinal regulatory T cells. Nat. Rev. Immunol. 2016, 16, 295–309. [Google Scholar] [CrossRef] [PubMed]
- Arpaia, N.; Campbell, C.; Fan, X.; Dikiy, S.; van der Veeken, J.; de Roos, P.; Liu, H.; Cross, J.R.; Pfeffer, K.; Coffer, P.J.; et al. Metabolites produced by commensal bacteria promote peripheral regulatory T-cell generation. Nature 2013, 504, 451–455. [Google Scholar] [CrossRef] [PubMed]
- Sun, M.; Wu, W.; Chen, L.; Yang, W.; Huang, X.; Ma, C.; Chen, F.; Xiao, Y.; Zhao, Y.; Ma, C.; et al. Microbiota-derived short-chain fatty acids promote Th1 cell IL-10 production to maintain intestinal homeostasis. Nat. Commun. 2018, 9, 3555. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Luu, M.; Pautz, S.; Kohl, V.; Singh, R.; Romero, R.; Lucas, S.; Hofmann, J.; Raifer, H.; Vachharajani, N.; Carrascosa, L.C.; et al. The short-chain fatty acid pentanoate suppresses autoimmunity by modulating the metabolic-epigenetic crosstalk in lymphocytes. Nat. Commun. 2019, 10, 760. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lemercier, C.; Brocard, M.P.; Puvion-Dutilleul, F.; Kao, H.Y.; Albagli, O.; Khochbin, S. Class II histone deacetylases are directly recruited by BCL6 transcriptional repressor. J. Biol. Chem. 2002, 277, 22045–22052. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, S.C.; Bottaro, A.; Insel, R.A. Activation of terminal B cell differentiation by inhibition of histone deacetylation. Mol. Immunol. 2003, 39, 923–932. [Google Scholar] [CrossRef]
- White, C.A.; Pone, E.J.; Lam, T.; Tat, C.; Hayama, K.L.; Li, G.; Zan, H.; Casali, P. Histone deacetylase inhibitors upregulate B cell microRNAs that silence AID and Blimp-1 expression for epigenetic modulation of antibody and autoantibody responses. J. Immunol. 2014, 193, 5933–5950. [Google Scholar] [CrossRef]
- Wei, J.; Bhattacharyya, S.; Tourtellotte, W.G.; Varga, J. Fibrosis in systemic sclerosis: Emerging concepts and implications for targeted therapy. Autoimmun. Rev. 2011, 10, 267–275. [Google Scholar] [CrossRef] [Green Version]
- Scrivo, R.; Vasile, M.; Bartosiewicz, I.; Valesini, G. Inflammation as “common soil” of the multifactorial diseases. Autoimmun. Rev. 2011, 10, 369–374. [Google Scholar] [CrossRef]
- Shu, J.; Silva, B.; Gao, T.; Xu, Z.; Cui, J. Dynamic and Modularized MicroRNA Regulation and Its Implication in Human Cancers. Sci. Rep. 2017, 7, 13356. [Google Scholar] [CrossRef] [Green Version]
- Schickel, R.; Boyerinas, B.; Park, S.M.; Peter, M.E. MicroRNAs: Key players in the immune system, differentiation, tumorigenesis and cell death. Oncogene 2008, 27, 5959–5974. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jeker, L.T.; Bluestone, J.A. MicroRNA regulation of T-cell differentiation and function. Immunol. Rev. 2013, 253, 65–81. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Danger, R.; Braza, F.; Giral, M.; Soulillou, J.P.; Brouard, S. MicroRNAs, Major Players in B Cells Homeostasis and Function. Front. Immunol. 2014, 5, 98. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, B.; Wang, X.; Choi, I.Y.; Wang, Y.C.; Liu, S.; Pham, A.T.; Moon, H.; Smith, D.J.; Rao, D.S.; Boldin, M.P.; et al. miR-146a modulates autoreactive Th17 cell differentiation and regulates organ-specific autoimmunity. J. Clin. Investig. 2017, 127, 3702–3716. [Google Scholar] [CrossRef] [Green Version]
- Basak, J.; Majsterek, I. miRNA-Dependent CD4(+) T Cell Differentiation in the Pathogenesis of Multiple Sclerosis. Mult. Scler. Int. 2021, 2021, 8825588. [Google Scholar] [CrossRef]
- Mohnle, P.; Schutz, S.V.; van der Heide, V.; Hubner, M.; Luchting, B.; Sedlbauer, J.; Limbeck, E.; Hinske, L.C.; Briegel, J.; Kreth, S. MicroRNA-146a controls Th1-cell differentiation of human CD4+ T lymphocytes by targeting PRKCepsilon. Eur. J. Immunol. 2015, 45, 260–272. [Google Scholar] [CrossRef]
- Lu, L.F.; Boldin, M.P.; Chaudhry, A.; Lin, L.L.; Taganov, K.D.; Hanada, T.; Yoshimura, A.; Baltimore, D.; Rudensky, A.Y. Function of miR-146a in controlling Treg cell-mediated regulation of Th1 responses. Cell 2010, 142, 914–929. [Google Scholar] [CrossRef] [Green Version]
- Pratama, A.; Srivastava, M.; Williams, N.J.; Papa, I.; Lee, S.K.; Dinh, X.T.; Hutloff, A.; Jordan, M.A.; Zhao, J.L.; Casellas, R.; et al. MicroRNA-146a regulates ICOS-ICOSL signalling to limit accumulation of T follicular helper cells and germinal centres. Nat. Commun. 2015, 6, 6436. [Google Scholar] [CrossRef]
- King, J.K.; Ung, N.M.; Paing, M.H.; Contreras, J.R.; Alberti, M.O.; Fernando, T.R.; Zhang, K.; Pellegrini, M.; Rao, D.S. Regulation of Marginal Zone B-Cell Differentiation by MicroRNA-146a. Front. Immunol. 2016, 7, 670. [Google Scholar] [CrossRef] [Green Version]
- Gururajan, M.; Haga, C.L.; Das, S.; Leu, C.M.; Hodson, D.; Josson, S.; Turner, M.; Cooper, M.D. MicroRNA 125b inhibition of B cell differentiation in germinal centers. Int. Immunol. 2010, 22, 583–592. [Google Scholar] [CrossRef]
- Nie, K.; Zhang, T.; Allawi, H.; Gomez, M.; Liu, Y.; Chadburn, A.; Wang, Y.L.; Knowles, D.M.; Tam, W. Epigenetic down-regulation of the tumor suppressor gene PRDM1/Blimp-1 in diffuse large B cell lymphomas: A potential role of the microRNA let-7. Am. J. Pathol. 2010, 177, 1470–1479. [Google Scholar] [CrossRef] [PubMed]
- Baulina, N.M.; Kulakova, O.G.; Favorova, O.O. MicroRNAs: The Role in Autoimmune Inflammation. Acta Nat. 2016, 8, 21–33. [Google Scholar] [CrossRef]
- Weissman, R.; Pilar, N.; Durham, B.H.; Ki, M.; Mazor, R.D.; Abdel-Wahab, O.I.; Shomron, N.; Shpilberg, O.; Diamond, E.L.; Rokah, O. The Role of microRNAs in the Pathogenesis of Erdheim-Chester Disease and Their Potential Use As Biomarkers for Diagnosis and Prognosis of the Disease. Blood 2018, 132, 2397. [Google Scholar] [CrossRef]
- Garo, L.P.; Murugaiyan, G. Contribution of MicroRNAs to autoimmune diseases. Cell Mol. Life Sci. 2016, 73, 2041–2051. [Google Scholar] [CrossRef] [PubMed]
- Zilahi, E.; Tarr, T.; Papp, G.; Griger, Z.; Sipka, S.; Zeher, M. Increased microRNA-146a/b, TRAF6 gene and decreased IRAK1 gene expressions in the peripheral mononuclear cells of patients with Sjogren’s syndrome. Immunol. Lett. 2012, 141, 165–168. [Google Scholar] [CrossRef]
- Shi, H.; Zheng, L.Y.; Zhang, P.; Yu, C.Q. miR-146a and miR-155 expression in PBMCs from patients with Sjögren’s syndrome. J. Oral Pathol. Med. 2014, 43, 792–797. [Google Scholar] [CrossRef]
- Taganov, K.D.; Boldin, M.P.; Chang, K.J.; Baltimore, D. NF-kappaB-dependent induction of microRNA miR-146, an inhibitor targeted to signaling proteins of innate immune responses. Proc. Natl. Acad. Sci. USA 2006, 103, 12481–12486. [Google Scholar] [CrossRef] [Green Version]
- Boldin, M.P.; Taganov, K.D.; Rao, D.S.; Yang, L.L.; Zhao, J.L.; Kalwani, M.; Garcia-Flores, Y.; Luong, M.; Devrekanli, A.; Xu, J.; et al. miR-146a is a significant brake on autoimmunity, myeloproliferation, and cancer in mice. J. Exp. Med. 2011, 208, 1189–1201. [Google Scholar] [CrossRef]
- Zhao, J.L.; Rao, D.S.; Boldin, M.P.; Taganov, K.D.; O’Connell, R.M.; Baltimore, D. NF-kappa B dysregulation in microRNA-146a-deficient mice drives the development of myeloid malignancies. Proc. Natl. Acad. Sci. USA 2011, 108, 9184–9189. [Google Scholar] [CrossRef] [Green Version]
- Yang, L.; Boldin, M.P.; Yu, Y.; Liu, C.S.; Ea, C.K.; Ramakrishnan, P.; Taganov, K.D.; Zhao, J.L.; Baltimore, D. miR-146a controls the resolution of T cell responses in mice. J. Exp. Med. 2012, 209, 1655–1670. [Google Scholar] [CrossRef] [Green Version]
- Fulci, V.; Chiaretti, S.; Goldoni, M.; Azzalin, G.; Carucci, N.; Tavolaro, S.; Castellano, L.; Magrelli, A.; Citarella, F.; Messina, M.; et al. Quantitative technologies establish a novel microRNA profile of chronic lymphocytic leukemia. Blood 2007, 109, 4944–4951. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Peng, L.; Ma, W.; Yi, F.; Yang, Y.J.; Lin, W.; Chen, H.; Zhang, X.; Zhang, L.H.; Zhang, F.; Du, Q. MicroRNA profiling in Chinese patients with primary Sjögren syndrome reveals elevated miRNA-181a in peripheral blood mononuclear cells. J. Rheumatol. 2014, 41, 2208–2213. [Google Scholar] [CrossRef]
- Benhamou, D.; Labi, V.; Getahun, A.; Benchetrit, E.; Dowery, R.; Rajewsky, K.; Cambier, J.C.; Melamed, D. The c-Myc/miR17-92/PTEN Axis Tunes PI3K Activity to Control Expression of Recombination Activating Genes in Early B Cell Development. Front. Immunol. 2018, 9, 2715. [Google Scholar] [CrossRef] [PubMed]
- Ventura, A.; Young, A.G.; Winslow, M.M.; Lintault, L.; Meissner, A.; Erkeland, S.J.; Newman, J.; Bronson, R.T.; Crowley, D.; Stone, J.R.; et al. Targeted deletion reveals essential and overlapping functions of the miR-17 through 92 family of miRNA clusters. Cell 2008, 132, 875–886. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gonzalez-Martin, A.; Adams, B.D.; Lai, M.; Shepherd, J.; Salvador-Bernaldez, M.; Salvador, J.M.; Lu, J.; Nemazee, D.; Xiao, C. The microRNA miR-148a functions as a critical regulator of B cell tolerance and autoimmunity. Nat. Immunol. 2016, 17, 433–440. [Google Scholar] [CrossRef]
- Benhamou, D.; Labi, V.; Novak, R.; Dai, I.; Shafir-Alon, S.; Weiss, A.; Gaujoux, R.; Arnold, R.; Shen-Orr, S.S.; Rajewsky, K.; et al. A c-Myc/miR17-92/Pten Axis Controls PI3K-Mediated Positive and Negative Selection in B Cell Development and Reconstitutes CD19 Deficiency. Cell Rep. 2016, 16, 419–431. [Google Scholar] [CrossRef] [Green Version]
- Lu, D.; Nakagawa, R.; Lazzaro, S.; Staudacher, P.; Abreu-Goodger, C.; Henley, T.; Boiani, S.; Leyland, R.; Galloway, A.; Andrews, S.; et al. The miR-155-PU.1 axis acts on Pax5 to enable efficient terminal B cell differentiation. J. Exp. Med. 2014, 211, 2183–2198. [Google Scholar] [CrossRef] [Green Version]
- Teng, G.; Hakimpour, P.; Landgraf, P.; Rice, A.; Tuschl, T.; Casellas, R.; Papavasiliou, F.N. MicroRNA-155 is a negative regulator of activation-induced cytidine deaminase. Immunity 2008, 28, 621–629. [Google Scholar] [CrossRef] [Green Version]
- Momen-Heravi, F.; Bala, S. miRNA regulation of innate immunity. J. Leukoc. Biol. 2018, 103, 1205–1217. [Google Scholar] [CrossRef]
- Berrien-Elliott, M.M.; Sun, Y.; Neal, C.; Ireland, A.; Trissal, M.C.; Sullivan, R.P.; Wagner, J.A.; Leong, J.W.; Wong, P.; Mah-Som, A.Y.; et al. MicroRNA-142 Is Critical for the Homeostasis and Function of Type 1 Innate Lymphoid Cells. Immunity 2019, 51, 479–490.e476. [Google Scholar] [CrossRef]
- Fionda, C.; Stabile, H.; Cerboni, C.; Soriani, A.; Gismondi, A.; Cippitelli, M.; Santoni, A. Hitting More Birds with a Stone: Impact of TGF-β on ILC Activity in Cancer. J. Clin. Med. 2020, 9, 143. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Roberts, L.B.; Jowett, G.M.; Read, E.; Zabinski, T.; Berkachy, R.; Selkirk, M.E.; Jackson, I.; Niazi, U.; Anandagoda, N.; Araki, M.; et al. MicroRNA-142 Critically Regulates Group 2 Innate Lymphoid Cell Homeostasis and Function. J. Immunol. 2021, 206, 2725–2739. [Google Scholar] [CrossRef]
- Singh, P.B.; Pua, H.H.; Happ, H.C.; Schneider, C.; von Moltke, J.; Locksley, R.M.; Baumjohann, D.; Ansel, K.M. MicroRNA regulation of type 2 innate lymphoid cell homeostasis and function in allergic inflammation. J. Exp. Med. 2017, 214, 3627–3643. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pelosi, A.; Alicata, C.; Tumino, N.; Ingegnere, T.; Loiacono, F.; Mingari, M.C.; Moretta, L.; Vacca, P. An Anti-inflammatory microRNA Signature Distinguishes Group 3 Innate Lymphoid Cells From Natural Killer Cells in Human Decidua. Front. Immunol. 2020, 11, 133. [Google Scholar] [CrossRef] [PubMed]
- Li, C.; Xu, M.M.; Wang, K.; Adler, A.J.; Vella, A.T.; Zhou, B. Macrophage polarization and meta-inflammation. Transl. Res. J. Lab. Clin. Med. 2018, 191, 29–44. [Google Scholar] [CrossRef]
- Essandoh, K.; Li, Y.; Huo, J.; Fan, G.C. MiRNA-Mediated Macrophage Polarization and its Potential Role in the Regulation of Inflammatory Response. Shock 2016, 46, 122–131. [Google Scholar] [CrossRef]
- Wang, Z.; Brandt, S.; Medeiros, A.; Wang, S.; Wu, H.; Dent, A.; Serezani, C.H. MicroRNA 21 is a homeostatic regulator of macrophage polarization and prevents prostaglandin E2-mediated M2 generation. PLoS ONE 2015, 10, e0115855. [Google Scholar] [CrossRef]
- Lu, T.X.; Munitz, A.; Rothenberg, M.E. MicroRNA-21 is up-regulated in allergic airway inflammation and regulates IL-12p35 expression. J. Immunol. 2009, 182, 4994–5002. [Google Scholar] [CrossRef] [Green Version]
- Sheedy, F.J. Turning 21: Induction of miR-21 as a Key Switch in the Inflammatory Response. Front. Immunol. 2015, 6, 19. [Google Scholar] [CrossRef] [Green Version]
- Caescu, C.I.; Guo, X.; Tesfa, L.; Bhagat, T.D.; Verma, A.; Zheng, D.; Stanley, E.R. Colony stimulating factor-1 receptor signaling networks inhibit mouse macrophage inflammatory responses by induction of microRNA-21. Blood 2015, 125, e1–e13. [Google Scholar] [CrossRef]
- Song, N.; Zhang, T.; Xu, X.; Lu, Z.; Yu, X.; Fang, Y.; Hu, J.; Jia, P.; Teng, J.; Ding, X. miR-21 Protects Against Ischemia/Reperfusion-Induced Acute Kidney Injury by Preventing Epithelial Cell Apoptosis and Inhibiting Dendritic Cell Maturation. Front. Physiol. 2018, 9, 790. [Google Scholar] [CrossRef] [PubMed]
- Christodoulou, M.I.; Kapsogeorgou, E.K.; Moutsopoulos, H.M. Characteristics of the minor salivary gland infiltrates in Sjogren’s syndrome. J. Autoimmun. 2010, 34, 400–407. [Google Scholar] [CrossRef] [PubMed]
- Rodrigues, A.R.; Soares, R. Inflammation in Sjogren’s syndrome: Cause or consequence? Autoimmunity 2017, 50, 141–150. [Google Scholar] [CrossRef] [PubMed]
- Mingueneau, M.; Boudaoud, S.; Haskett, S.; Reynolds, T.L.; Nocturne, G.; Norton, E.; Zhang, X.; Constant, M.; Park, D.; Wang, W.; et al. Cytometry by time-of-flight immunophenotyping identifies a blood Sjogren’s signature correlating with disease activity and glandular inflammation. J. Allergy Clin. Immunol. 2016, 137, 1809–1821.e1812. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, C.Q.; Hu, M.H.; Li, Y.; Stewart, C.; Peck, A.B. Salivary gland tissue expression of interleukin-23 and interleukin-17 in Sjögren’s syndrome: Findings in humans and mice. Arthritis Rheum. 2008, 58, 734–743. [Google Scholar] [CrossRef] [Green Version]
- van Woerkom, J.M.; Kruize, A.A.; Wenting-van Wijk, M.J.; Knol, E.; Bihari, I.C.; Jacobs, J.W.; Bijlsma, J.W.; Lafeber, F.P.; van Roon, J.A. Salivary gland and peripheral blood T helper 1 and 2 cell activity in Sjögren’s syndrome compared with non-Sjögren’s sicca syndrome. Ann. Rheum. Dis. 2005, 64, 1474–1479. [Google Scholar] [CrossRef] [Green Version]
- Ogawa, N.; Ping, L.; Zhenjun, L.; Takada, Y.; Sugai, S. Involvement of the interferon-gamma-induced T cell-attracting chemokines, interferon-gamma-inducible 10-kd protein (CXCL10) and monokine induced by interferon-gamma (CXCL9), in the salivary gland lesions of patients with Sjögren’s syndrome. Arthritis Rheum. 2002, 46, 2730–2741. [Google Scholar] [CrossRef]
- Crotty, S. T follicular helper cell differentiation, function, and roles in disease. Immunity 2014, 41, 529–542. [Google Scholar] [CrossRef] [Green Version]
- Yao, Y.; Ma, J.F.; Chang, C.; Xu, T.; Gao, C.Y.; Gershwin, M.E.; Lian, Z.X. Immunobiology of T Cells in Sjogren’s Syndrome. Clin. Rev. Allergy Immunol. 2021, 60, 111–131. [Google Scholar] [CrossRef]
- Singh, N.; Cohen, P.L. The T cell in Sjogren’s syndrome: Force majeure, not spectateur. J. Autoimmun. 2012, 39, 229–233. [Google Scholar] [CrossRef] [Green Version]
- Christodoulou, M.I.; Kapsogeorgou, E.K.; Moutsopoulos, N.M.; Moutsopoulos, H.M. Foxp3+ T-regulatory cells in Sjogren’s syndrome: Correlation with the grade of the autoimmune lesion and certain adverse prognostic factors. Am. J. Pathol. 2008, 173, 1389–1396. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kamali, A.N.; Noorbakhsh, S.M.; Hamedifar, H.; Jadidi-Niaragh, F.; Yazdani, R.; Bautista, J.M.; Azizi, G. A role for Th1-like Th17 cells in the pathogenesis of inflammatory and autoimmune disorders. Mol. Immunol. 2019, 105, 107–115. [Google Scholar] [CrossRef] [PubMed]
- Fallahi, P.; Ferrari, S.M.; Ragusa, F.; Ruffilli, I.; Elia, G.; Paparo, S.R.; Antonelli, A. Th1 Chemokines in Autoimmune Endocrine Disorders. J. Clin. Endocrinol. Metab. 2020, 105, 1046–1060. [Google Scholar] [CrossRef] [PubMed]
- Yasuda, K.; Takeuchi, Y.; Hirota, K. The pathogenicity of Th17 cells in autoimmune diseases. Semin. Immunopathol. 2019, 41, 283–297. [Google Scholar] [CrossRef]
- Lee, J.Y.; Hall, J.A.; Kroehling, L.; Wu, L.; Najar, T.; Nguyen, H.H.; Lin, W.Y.; Yeung, S.T.; Silva, H.M.; Li, D.; et al. Serum Amyloid A Proteins Induce Pathogenic Th17 Cells and Promote Inflammatory Disease. Cell 2020, 183, 2036–2039. [Google Scholar] [CrossRef]
- Ogawa, Y.; Shimizu, E.; Tsubota, K. Interferons and Dry Eye in Sjögren’s Syndrome. Int. J. Mol. Sci. 2018, 19, 3548. [Google Scholar] [CrossRef] [Green Version]
- Voigt, A.; Esfandiary, L.; Wanchoo, A.; Glenton, P.; Donate, A.; Craft, W.F.; Craft, S.L.; Nguyen, C.Q. Sexual dimorphic function of IL-17 in salivary gland dysfunction of the C57BL/6.NOD-Aec1Aec2 model of Sjogren’s syndrome. Sci. Rep. 2016, 6, 38717. [Google Scholar] [CrossRef] [Green Version]
- Verstappen, G.M.; Corneth, O.B.J.; Bootsma, H.; Kroese, F.G.M. Th17 cells in primary Sjogren’s syndrome: Pathogenicity and plasticity. J. Autoimmun. 2018, 87, 16–25. [Google Scholar] [CrossRef]
- Fasano, S.; Mauro, D.; Macaluso, F.; Xiao, F.; Zhao, Y.; Lu, L.; Guggino, G.; Ciccia, F. Pathogenesis of primary Sjögren’s syndrome beyond B lymphocytes. Clin. Exp. Rheumatol. 2020, 38 (Suppl. S126), 315–323. [Google Scholar]
- Crotty, S. T Follicular Helper Cell Biology: A Decade of Discovery and Diseases. Immunity 2019, 50, 1132–1148. [Google Scholar] [CrossRef]
- Jogdand, G.M.; Mohanty, S.; Devadas, S. Regulators of Tfh Cell Differentiation. Front. Immunol. 2016, 7, 520. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, W.; Yang, F.; Xu, G.; Ma, J.; Lin, J. Follicular helper T cells and follicular regulatory T cells in the immunopathology of primary Sjögren’s syndrome. J. Leukoc. Biol. 2021, 109, 437–447. [Google Scholar] [CrossRef] [PubMed]
- Szabo, K.; Papp, G.; Barath, S.; Gyimesi, E.; Szanto, A.; Zeher, M. Follicular helper T cells may play an important role in the severity of primary Sjogren’s syndrome. Clin. Immunol. 2013, 147, 95–104. [Google Scholar] [CrossRef] [Green Version]
- King, I.L.; Mohrs, M. IL-4-producing CD4+ T cells in reactive lymph nodes during helminth infection are T follicular helper cells. J. Exp. Med. 2009, 206, 1001–1007. [Google Scholar] [CrossRef] [PubMed]
- Katsifis, G.E.; Moutsopoulos, N.M.; Wahl, S.M. T lymphocytes in Sjögren’s syndrome: Contributors to and regulators of pathophysiology. Clin. Rev. Allergy Immunol. 2007, 32, 252–264. [Google Scholar] [CrossRef] [PubMed]
- Ríos-Ríos, W.J.; Sosa-Luis, S.A.; Torres-Aguilar, H. T Cells Subsets in the Immunopathology and Treatment of Sjogren’s Syndrome. Biomolecules 2020, 10, 1539. [Google Scholar] [CrossRef]
- Verstappen, G.M.; Kroese, F.G.M.; Bootsma, H. T cells in primary Sjogren’s syndrome: Targets for early intervention. Rheumatology 2019, 60, 3088–3098. [Google Scholar] [CrossRef] [Green Version]
- Raimondi, G.; Shufesky, W.J.; Tokita, D.; Morelli, A.E.; Thomson, A.W. Regulated compartmentalization of programmed cell death-1 discriminates CD4+CD25+ resting regulatory T cells from activated T cells. J. Immunol. 2006, 176, 2808–2816. [Google Scholar] [CrossRef] [Green Version]
- Wang, Y.; Luo, J.; Gao, C.; Zhao, X.C. Sat0195 Abnormity Tfh Subsets Indicate Disease Activity While Sirolimus Therapy Restores the Pd-1+Icos+Tfh/Activated Tfr Balance in Primary Sjogren’s Syndrome Patients. BMJ 2020, 79, 1039–1040. [Google Scholar] [CrossRef]
- Verstappen, G.M.; Nakshbandi, U.; Mossel, E.; Haacke, E.A.; van der Vegt, B.; Vissink, A.; Bootsma, H.; Kroese, F.G.M. Is the T Follicular Regulatory:Follicular Helper T Cell Ratio in Blood a Biomarker for Ectopic Lymphoid Structure Formation in Sjögren’s Syndrome? Comment on the Article by Fonseca et al. Arthritis Rheumatol. 2018, 70, 1354–1355. [Google Scholar] [CrossRef] [Green Version]
- Sage, P.T.; Paterson, A.M.; Lovitch, S.B.; Sharpe, A.H. The coinhibitory receptor CTLA-4 controls B cell responses by modulating T follicular helper, T follicular regulatory, and T regulatory cells. Immunity 2014, 41, 1026–1039. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Borges da Silva, H. Navigating in Deep Waters: How Tissue Damage and Inflammation Shape Effector and Memory CD8(+) T Cell Responses. ImmunoHorizons 2021, 5, 338–348. [Google Scholar] [CrossRef] [PubMed]
- Blokland, S.L.M.; van den Hoogen, L.L.; Leijten, E.F.A.; Hartgring, S.A.Y.; Fritsch, R.; Kruize, A.A.; van Roon, J.A.G.; Radstake, T. Increased expression of Fas on group 2 and 3 innate lymphoid cells is associated with an interferon signature in systemic lupus erythematosus and Sjögren’s syndrome. Rheumatology 2019, 58, 1740–1745. [Google Scholar] [CrossRef] [PubMed]
- Nocturne, G.; Mariette, X. B cells in the pathogenesis of primary Sjögren syndrome. Nat. Rev. Rheumatol. 2018, 14, 133–145. [Google Scholar] [CrossRef] [PubMed]
- Bohnhorst, J.; Bjørgan, M.B.; Thoen, J.E.; Natvig, J.B.; Thompson, K.M. Bm1-Bm5 classification of peripheral blood B cells reveals circulating germinal center founder cells in healthy individuals and disturbance in the B cell subpopulations in patients with primary Sjögren’s syndrome. J. Immunol. 2001, 167, 3610–3618. [Google Scholar] [CrossRef]
- Carbone, J.; Perez-Fernandez, R.; Muñoz, A.; Sabin, P.; Carreño, L.; Fernandez-Cruz, E. Combined therapy with rituximab plus cyclophosphamide/vincristine/prednisone for Sjogren’s syndrome-associated B-cell non-Hodgkin’s lymphoma. Clin. Rev. Allergy Immunol. 2008, 34, 80–84. [Google Scholar] [CrossRef]
- Xian, Z.; Fu, D.; Liu, S.; Yao, Y.; Gao, C. Association between B Cell Growth Factors and Primary Sjögren’s Syndrome-Related Autoantibodies in Patients with Non-Hodgkin’s Lymphoma. J. Immunol. Res. 2019, 2019, 7627384. [Google Scholar] [CrossRef] [Green Version]
- Hurdayal, R.; Nieuwenhuizen, N.E.; Khutlang, R.; Brombacher, F. Inflammatory Dendritic Cells, Regulated by IL-4 Receptor Alpha Signaling, Control Replication, and Dissemination of Leishmania major in Mice. Front. Cell. Infect. Microbiol. 2020, 9, 479. [Google Scholar] [CrossRef]
- Youinou, P.; Pers, J.O. Disturbance of cytokine networks in Sjögren’s syndrome. Arthritis Res. Ther. 2011, 13, 227. [Google Scholar] [CrossRef] [Green Version]
- Shen, L.; Suresh, L.; Lindemann, M.; Xuan, J.; Kowal, P.; Malyavantham, K.; Ambrus, J.L., Jr. Novel autoantibodies in Sjogren’s syndrome. Clin. Immunol. 2012, 145, 251–255. [Google Scholar] [CrossRef]
- Vivier, E.; Artis, D.; Colonna, M.; Diefenbach, A.; Di Santo, J.P.; Eberl, G.; Koyasu, S.; Locksley, R.M.; McKenzie, A.N.J.; Mebius, R.E.; et al. Innate Lymphoid Cells: 10 Years On. Cell 2018, 174, 1054–1066. [Google Scholar] [CrossRef] [Green Version]
- Panda, S.K.; Colonna, M. Innate Lymphoid Cells in Mucosal Immunity. Front. Immunol. 2019, 10, 861. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ciccia, F.; Guggino, G.; Giardina, A.; Ferrante, A.; Carrubbi, F.; Giacomelli, R.; Triolo, G. The role of innate and lymphoid IL-22-producing cells in the immunopathology of primary Sjögren’s syndrome. Expert Rev. Clin. Immunol. 2014, 10, 533–541. [Google Scholar] [CrossRef] [PubMed]
- Wildenberg, M.E.; Welzen-Coppens, J.M.; van Helden-Meeuwsen, C.G.; Bootsma, H.; Vissink, A.; van Rooijen, N.; van de Merwe, J.P.; Drexhage, H.A.; Versnel, M.A. Increased frequency of CD16+ monocytes and the presence of activated dendritic cells in salivary glands in primary Sjögren syndrome. Ann. Rheum. Dis. 2009, 68, 420–426. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kapsogeorgou, E.K.; Abu-Helu, R.F.; Moutsopoulos, H.M.; Manoussakis, M.N. Salivary gland epithelial cell exosomes: A source of autoantigenic ribonucleoproteins. Arthritis Rheum. 2005, 52, 1517–1521. [Google Scholar] [CrossRef] [PubMed]
- Maehara, T.; Moriyama, M.; Hayashida, J.N.; Tanaka, A.; Shinozaki, S.; Kubo, Y.; Matsumura, K.; Nakamura, S. Selective localization of T helper subsets in labial salivary glands from primary Sjogren’s syndrome patients. Clin. Exp. Immunol. 2012, 169, 89–99. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Psianou, K.; Panagoulias, I.; Papanastasiou, A.D.; de Lastic, A.L.; Rodi, M.; Spantidea, P.I.; Degn, S.E.; Georgiou, P.; Mouzaki, A. Clinical and immunological parameters of Sjogren’s syndrome. Autoimmun. Rev. 2018, 17, 1053–1064. [Google Scholar] [CrossRef] [PubMed]
- Jin, J.O.; Kawai, T.; Cha, S.; Yu, Q. Interleukin-7 enhances the Th1 response to promote the development of Sjogren’s syndrome-like autoimmune exocrinopathy in mice. Arthritis Rheum. 2013, 65, 2132–2142. [Google Scholar] [CrossRef] [Green Version]
- Gong, Y.Z.; Nititham, J.; Taylor, K.; Miceli-Richard, C.; Sordet, C.; Wachsmann, D.; Bahram, S.; Georgel, P.; Criswell, L.A.; Sibilia, J.; et al. Differentiation of follicular helper T cells by salivary gland epithelial cells in primary Sjogren’s syndrome. J. Autoimmun. 2014, 51, 57–66. [Google Scholar] [CrossRef]
- Verstappen, G.M.; Meiners, P.M.; Corneth, O.B.J.; Visser, A.; Arends, S.; Abdulahad, W.H.; Hendriks, R.W.; Vissink, A.; Kroese, F.G.M.; Bootsma, H. Attenuation of Follicular Helper T Cell-Dependent B Cell Hyperactivity by Abatacept Treatment in Primary Sjogren’s Syndrome. Arthritis Rheumatol. 2017, 69, 1850–1861. [Google Scholar] [CrossRef]
- Thompson, N.; Isenberg, D.A.; Jury, E.C.; Ciurtin, C. Exploring BAFF: Its expression, receptors and contribution to the immunopathogenesis of Sjogren’s syndrome. Rheumatology 2016, 55, 1548–1555. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pestka, S.; Krause, C.D.; Walter, M.R. Interferons, interferon-like cytokines, and their receptors. Immunol. Rev. 2004, 202, 8–32. [Google Scholar] [CrossRef] [PubMed]
- Lazear, H.M.; Schoggins, J.W.; Diamond, M.S. Shared and Distinct Functions of Type I and Type III Interferons. Immunity 2019, 50, 907–923. [Google Scholar] [CrossRef]
- Lovgren, T.; Eloranta, M.L.; Kastner, B.; Wahren-Herlenius, M.; Alm, G.V.; Ronnblom, L. Induction of interferon-alpha by immune complexes or liposomes containing systemic lupus erythematosus autoantigen- and Sjogren’s syndrome autoantigen-associated RNA. Arthritis Rheum. 2006, 54, 1917–1927. [Google Scholar] [CrossRef] [PubMed]
- Ali, S.; Mann-Nuttel, R.; Schulze, A.; Richter, L.; Alferink, J.; Scheu, S. Sources of Type I Interferons in Infectious Immunity: Plasmacytoid Dendritic Cells Not Always in the Driver’s Seat. Front. Immunol. 2019, 10, 778. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ittah, M.; Miceli-Richard, C.; Gottenberg, J.E.; Sellam, J.; Eid, P.; Lebon, P.; Pallier, C.; Lepajolec, C.; Mariette, X. Viruses induce high expression of BAFF by salivary gland epithelial cells through TLR- and type-I IFN-dependent and -independent pathways. Eur. J. Immunol. 2008, 38, 1058–1064. [Google Scholar] [CrossRef] [PubMed]
- Kyriakidis, N.C.; Kapsogeorgou, E.K.; Gourzi, V.C.; Konsta, O.D.; Baltatzis, G.E.; Tzioufas, A.G. Toll-like receptor 3 stimulation promotes Ro52/TRIM21 synthesis and nuclear redistribution in salivary gland epithelial cells, partially via type I interferon pathway. Clin. Exp. Immunol. 2014, 178, 548–560. [Google Scholar] [CrossRef] [Green Version]
- Schreiber, G. The molecular basis for differential type I interferon signaling. J. Biol. Chem. 2017, 292, 7285–7294. [Google Scholar] [CrossRef] [Green Version]
- Negishi, H.; Taniguchi, T.; Yanai, H. The Interferon (IFN) Class of Cytokines and the IFN Regulatory Factor (IRF) Transcription Factor Family. Cold Spring Harb. Perspect. Biol. 2018, 10, a028423. [Google Scholar] [CrossRef]
- Li, S.F.; Gong, M.J.; Zhao, F.R.; Shao, J.J.; Xie, Y.L.; Zhang, Y.G.; Chang, H.Y. Type I Interferons: Distinct Biological Activities and Current Applications for Viral Infection. Cell Physiol. Biochem. 2018, 51, 2377–2396. [Google Scholar] [CrossRef]
- Le Bon, A.; Schiavoni, G.; D’Agostino, G.; Gresser, I.; Belardelli, F.; Tough, D.F. Type i interferons potently enhance humoral immunity and can promote isotype switching by stimulating dendritic cells in vivo. Immunity 2001, 14, 461–470. [Google Scholar] [CrossRef] [Green Version]
- Hervas-Stubbs, S.; Perez-Gracia, J.L.; Rouzaut, A.; Sanmamed, M.F.; Le Bon, A.; Melero, I. Direct effects of type I interferons on cells of the immune system. Clin. Cancer Res. 2011, 17, 2619–2627. [Google Scholar] [CrossRef] [Green Version]
- Ittah, M.; Miceli-Richard, C.; Eric Gottenberg, J.; Lavie, F.; Lazure, T.; Ba, N.; Sellam, J.; Lepajolec, C.; Mariette, X. B cell-activating factor of the tumor necrosis factor family (BAFF) is expressed under stimulation by interferon in salivary gland epithelial cells in primary Sjögren’s syndrome. Arthritis Res. Ther. 2006, 8, R51. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cha, S.; van Blockland, S.C.; Versnel, M.A.; Homo-Delarche, F.; Nagashima, H.; Brayer, J.; Peck, A.B.; Humphreys-Beher, M.G. Abnormal organogenesis in salivary gland development may initiate adult onset of autoimmune exocrinopathy. Exp. Clin. Immunogenet. 2001, 18, 143–160. [Google Scholar] [CrossRef]
- Szczerba, B.M.; Rybakowska, P.D.; Dey, P.; Payerhin, K.M.; Peck, A.B.; Bagavant, H.; Deshmukh, U.S. Type I interferon receptor deficiency prevents murine Sjogren’s syndrome. J. Dent. Res. 2013, 92, 444–449. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hall, J.C.; Casciola-Rosen, L.; Berger, A.E.; Kapsogeorgou, E.K.; Cheadle, C.; Tzioufas, A.G.; Baer, A.N.; Rosen, A. Precise probes of type II interferon activity define the origin of interferon signatures in target tissues in rheumatic diseases. Proc. Natl. Acad. Sci. USA 2012, 109, 17609–17614. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Peck, A.B.; Nguyen, C.Q. What can Sjogren’s syndrome-like disease in mice contribute to human Sjogren’s syndrome? Clin. Immunol. 2017, 182, 14–23. [Google Scholar] [CrossRef] [Green Version]
- Pontarini, E.; Murray-Brown, W.J.; Croia, C.; Lucchesi, D.; Conway, J.; Rivellese, F.; Fossati-Jimack, L.; Astorri, E.; Prediletto, E.; Corsiero, E.; et al. Unique expansion of IL-21+ Tfh and Tph cells under control of ICOS identifies Sjogren’s syndrome with ectopic germinal centres and MALT lymphoma. Ann. Rheum. Dis. 2020, 79, 1588–1599. [Google Scholar] [CrossRef]
- Reksten, T.R.; Lessard, C.J.; Sivils, K.L. Genetics in Sjogren Syndrome. Rheum. Dis. Clin. N. Am. 2016, 42, 435–447. [Google Scholar] [CrossRef]
- Wang, F.; Zhang, S.; Jeon, R.; Vuckovic, I.; Jiang, X.; Lerman, A.; Folmes, C.D.; Dzeja, P.D.; Herrmann, J. Interferon Gamma Induces Reversible Metabolic Reprogramming of M1 Macrophages to Sustain Cell Viability and Pro-Inflammatory Activity. EBioMedicine 2018, 30, 303–316. [Google Scholar] [CrossRef] [Green Version]
- Manoussakis, M.N.; Dimitriou, I.D.; Kapsogeorgou, E.K.; Xanthou, G.; Paikos, S.; Polihronis, M.; Moutsopoulos, H.M. Expression of B7 costimulatory molecules by salivary gland epithelial cells in patients with Sjogren’s syndrome. Arthritis Rheum. 1999, 42, 229–239. [Google Scholar] [CrossRef]
- Dimitriou, I.D.; Kapsogeorgou, E.K.; Moutsopoulos, H.M.; Manoussakis, M.N. CD40 on salivary gland epithelial cells: High constitutive expression by cultured cells from Sjogren’s syndrome patients indicating their intrinsic activation. Clin. Exp. Immunol. 2002, 127, 386–392. [Google Scholar] [CrossRef] [PubMed]
- Tsunawaki, S.; Nakamura, S.; Ohyama, Y.; Sasaki, M.; Ikebe-Hiroki, A.; Hiraki, A.; Kadena, T.; Kawamura, E.; Kumamaru, W.; Shinohara, M.; et al. Possible function of salivary gland epithelial cells as nonprofessional antigen-presenting cells in the development of Sjogren’s syndrome. J. Rheumatol. 2002, 29, 1884–1896. [Google Scholar] [PubMed]
- Abu-Helu, R.F.; Dimitriou, I.D.; Kapsogeorgou, E.K.; Moutsopoulos, H.M.; Manoussakis, M.N. Induction of Salivary Gland Epithelial Cell Injury in Sjogren’s Syndrome: In Vitro Assessment of T Cell-derived Cytokines and Fas Protein Expression. J. Autoimmun. 2001, 17, 141–153. [Google Scholar] [CrossRef] [PubMed]
- Ewert, P.; Aguilera, S.; Alliende, C.; Kwon, Y.J.; Albornoz, A.; Molina, C.; Urzúa, U.; Quest, A.; Olea, N.; Rheum, P.P.J.A. Disruption of tight junction structure in salivary glands from Sjgren’s syndrome patients is linked to proinflammatory cytokine exposure. Arthritis Rheum. 2010, 62, 1280–1289. [Google Scholar] [CrossRef]
- Apostolou, E.; Kapsogeorgou, E.K.; Konsta, O.D.; Giotakis, I.; Saridaki, M.I.; Andreakos, E.; Tzioufas, A.G. Expression of type III interferons (IFNlambdas) and their receptor in Sjogren’s syndrome. Clin. Exp. Immunol. 2016, 186, 304–312. [Google Scholar] [CrossRef] [Green Version]
- Roescher, N.; Tak, P.P.; Illei, G.G. Cytokines in Sjogren’s syndrome. Oral Dis. 2009, 15, 519–526. [Google Scholar] [CrossRef] [Green Version]
- Katsifis, G.E.; Rekka, S.; Moutsopoulos, N.M.; Pillemer, S.; Wahl, S.M. Systemic and local interleukin-17 and linked cytokines associated with Sjogren’s syndrome immunopathogenesis. Am. J. Pathol. 2009, 175, 1167–1177. [Google Scholar] [CrossRef] [Green Version]
- Fei, Y.; Zhang, W.; Lin, D.; Wu, C.; Li, M.; Zhao, Y.; Zeng, X.; Zhang, F. Clinical parameter and Th17 related to lymphocytes infiltrating degree of labial salivary gland in primary Sjogren’s syndrome. Clin. Rheumatol. 2014, 33, 523–529. [Google Scholar] [CrossRef]
- Sakai, A.; Sugawara, Y.; Kuroishi, T.; Sasano, T.; Sugawara, S. Identification of IL-18 and Th17 cells in salivary glands of patients with Sjogren’s syndrome, and amplification of IL-17-mediated secretion of inflammatory cytokines from salivary gland cells by IL-18. J. Immunol. 2008, 181, 2898–2906. [Google Scholar] [CrossRef]
- Iwakura, Y.; Ishigame, H.; Saijo, S.; Nakae, S. Functional specialization of interleukin-17 family members. Immunity 2011, 34, 149–162. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, L.W.; Cong, X.; Zhang, Y.; Wei, T.; Wu, L.L. Interleukin-17 Impairs Salivary Tight Junction Integrity in Sjgren’s Syndrome. J. Dent. Res. 2016, 95, 784. [Google Scholar] [CrossRef] [PubMed]
- Fogli, L.K.; Sundrud, M.S.; Goel, S.; Bajwa, S.; Jensen, K.; Derudder, E.; Sun, A.; Coffre, M.; Uyttenhove, C.; Van Snick, J.; et al. T Cell-Derived IL-17 Mediates Epithelial Changes in the Airway and Drives Pulmonary Neutrophilia. J. Immunol. 2013, 191, 3100–3111. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pérez, P.; Kwon, Y.J.; Alliende, C.; Leyton, L.; Aguilera, S.; Molina, C.; Labra, C.; Julio, M.; Leyton, C.; Arthritis, M.-J.G.J.; et al. Increased acinar damage of salivary glands of patients with Sjögren’s syndrome is paralleled by simultaneous imbalance of matrix metalloproteinase 3/tissue inhibitor of metalloproteinases 1 and matrix metalloproteinase 9/tissue inhibitor of metalloproteinases 1 ratios. Arthritis Rheum. 2005, 59, 2751–2760. [Google Scholar]
- Nguyen, C.Q.; Yin, H.; Lee, B.H.; Carcamo, W.C.; Chiorini, J.A.; Peck, A.B. Pathogenic effect of interleukin-17A in induction of Sjögren’s syndrome-like disease using adenovirus-mediated gene transfer. Arthritis Res. Ther. 2010, 12, R220. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liang, S.C.; Tan, X.Y.; Luxenberg, D.P.; Karim, R.; Dunussi-Joannopoulos, K.; Collins, M.; Fouser, L.A. Interleukin (IL)-22 and IL-17 are coexpressed by Th17 cells and cooperatively enhance expression of antimicrobial peptides. J. Exp. Med. 2006, 203, 2271–2279. [Google Scholar] [CrossRef]
- Ciccia, F.; Guggino, G.; Rizzo, A.; Ferrante, A.; Raimondo, S.; Giardina, A.; Dieli, F.; Campisi, G.; Alessandro, R.; Triolo, G. Potential involvement of IL-22 and IL-22-producing cells in the inflamed salivary glands of patients with Sjogren’s syndrome. Ann. Rheum. Dis. 2012, 71, 295–301. [Google Scholar] [CrossRef]
- Barone, F.; Nayar, S.; Campos, J.; Cloake, T.; Withers, D.R.; Toellner, K.M.; Zhang, Y.; Fouser, L.; Fisher, B.; Bowman, S.; et al. IL-22 regulates lymphoid chemokine production and assembly of tertiary lymphoid organs. Proc. Natl. Acad. Sci. USA 2015, 112, 11024–11029. [Google Scholar] [CrossRef] [Green Version]
- Kang, K.Y.; Kim, H.O.; Kwok, S.K.; Ju, J.H.; Park, K.S.; Sun, D.I.; Jhun, J.Y.; Oh, H.J.; Park, S.H.; Kim, H.Y. Impact of interleukin-21 in the pathogenesis of primary Sjögren’s syndrome: Increased serum levels of interleukin-21 and its expression in the labial salivary glands. Arthritis Res. Ther. 2011, 13, R179. [Google Scholar] [CrossRef] [Green Version]
- Kwok, S.K.; Lee, J.; Yu, D.; Kang, K.Y.; Cho, M.L.; Kim, H.R.; Ju, J.H.; Lee, S.H.; Park, S.H.; Kim, H.Y. A pathogenetic role for IL-21 in primary Sjogren syndrome. Nat. Rev. Rheumatol. 2015, 11, 368–374. [Google Scholar] [CrossRef]
- McGuire, H.M.; Vogelzang, A.; Ma, C.S.; Hughes, W.E.; Silveira, P.A.; Tangye, S.G.; Christ, D.; Fulcher, D.; Falcone, M.; King, C. A subset of interleukin-21+ chemokine receptor CCR9+ T helper cells target accessory organs of the digestive system in autoimmunity. Immunity 2011, 34, 602–615. [Google Scholar] [CrossRef] [Green Version]
- Wei, L.; Laurence, A.; Elias, K.M.; O’Shea, J.J. IL-21 is produced by Th17 cells and drives IL-17 production in a STAT3-dependent manner. J. Biol. Chem. 2007, 282, 34605–34610. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zeng, R.; Spolski, R.; Finkelstein, S.E.; Oh, S.; Kovanen, P.E.; Hinrichs, C.S.; Pise-Masison, C.A.; Radonovich, M.F.; Brady, J.N.; Restifo, N.P.; et al. Synergy of IL-21 and IL-15 in regulating CD8+ T cell expansion and function. J. Exp. Med. 2005, 201, 139–148. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, H.; Liu, G.; Gong, L.; Zhang, Y.; Jiang, G. Local suppression of IL-21 in submandibular glands retards the development of Sjögren’s syndrome in non-obese diabetic mice. J. Oral Pathol. Med. 2012, 41, 728–735. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, C.Q.; Gao, J.-H.; Kim, H.; Saban, D.; Immunology, R.J. IL-4-STAT6 Signal Transduction-Dependent Induction of the Clinical Phase of Sjogren’s Syndrome-Like Disease of the Nonobese Diabetic Mouse. J. Immunol. 2007, 179, 382–390. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pandey, S.; Mourcin, F.; Marchand, T.; Nayar, S.; Guirriec, M.; Pangault, C.; Monvoisin, C.; Ame-Thomas, P.; Guilloton, F.; Dulong, J.; et al. IL-4/CXCL12 loop is a key regulator of lymphoid stroma function in follicular lymphoma. Blood 2017, 129, 2507–2518. [Google Scholar] [CrossRef] [Green Version]
- Barone, F.; Bombardieri, M.; Rosado, M.M.; Morgan, P.R.; Challacombe, S.J.; De Vita, S.; Carsetti, R.; Spencer, J.; Valesini, G.; Pitzalis, C. CXCL13, CCL21, and CXCL12 expression in salivary glands of patients with Sjogren’s syndrome and MALT lymphoma: Association with reactive and malignant areas of lymphoid organization. J. Immunol. 2008, 180, 5130–5140. [Google Scholar] [CrossRef]
- Wurster, A.L.; Rodgers, V.L.; White, M.F.; Rothstein, T.L.; Grusby, M.J. Interleukin-4-mediated protection of primary B cells from apoptosis through Stat6-dependent up-regulation of Bcl-xL. J. Biol. Chem. 2002, 277, 27169–27175. [Google Scholar] [CrossRef] [Green Version]
- Brayer, J.B.; Cha, S.; Nagashima, H.; Yasunari, U.; Lindberg, A.; Diggs, S.; Martinez, J.; Goa, J.; Humphreys-Beher, M.G.; Peck, A.B. IL-4-dependent effector phase in autoimmune exocrinopathy as defined by the NOD.IL-4-gene knockout mouse model of Sjögren’s syndrome. Scand. J. Immunol. 2001, 54, 133–140. [Google Scholar] [CrossRef]
- Xu, T. Leptin/OB-R pathway promotes IL-4 secretion from B lymphocytes and induces salivary gland epithelial cell apoptosis in Sjögren’s syndrome. In Proceedings of the 17th International Congress of Immunology, Beijing, China, 19–23 October 2019. [Google Scholar]
- Vanamee, E.S.; Faustman, D.L. Structural principles of tumor necrosis factor superfamily signaling. Sci. Signal. 2018, 11, aao4910. [Google Scholar] [CrossRef] [Green Version]
- Dostert, C.; Grusdat, M.; Letellier, E.; Brenner, D. The TNF Family of Ligands and Receptors: Communication Modules in the Immune System and Beyond. Physiol. Rev. 2019, 99, 115–160. [Google Scholar] [CrossRef]
- Holbrook, J.; Lara-Reyna, S.; Jarosz-Griffiths, H.; McDermott, M. Tumour necrosis factor signalling in health and disease. F1000Res 2019, 8, 111. [Google Scholar] [CrossRef] [PubMed]
- Morimoto, S.; Nakano, S.; Watanabe, T.; Tamayama, Y.; Mitsuo, A.; Nakiri, Y.; Suzuki, J.; Nozawa, K.; Amano, H.; Tokano, Y.; et al. Expression of B-cell activating factor of the tumour necrosis factor family (BAFF) in T cells in active systemic lupus erythematosus: The role of BAFF in T cell-dependent B cell pathogenic autoantibody production. Rheumatology 2007, 46, 1083–1086. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Szodoray, P.; Alex, P.; Jonsson, M.V.; Knowlton, N.; Dozmorov, I.; Nakken, B.; Delaleu, N.; Jonsson, R.; Centola, M. Distinct profiles of Sjogren’s syndrome patients with ectopic salivary gland germinal centers revealed by serum cytokines and BAFF. Clin. Immunol. 2005, 117, 168–176. [Google Scholar] [CrossRef]
- Kang, E.H.; Lee, Y.J.; Hyon, J.Y.; Yun, P.Y.; Song, Y.W. Salivary cytokine profiles in primary Sjogren’s syndrome differ from those in non-Sjogren sicca in terms of TNF-alpha levels and Th-1/Th-2 ratios. Clin. Exp. Rheumatol. 2011, 29, 970–976. [Google Scholar] [PubMed]
- Fox, R.I.; Kang, H.I.; Ando, D.; Abrams, J.; Pisa, E. Cytokine mRNA expression in salivary gland biopsies of Sjögren’s syndrome. J. Immunol. 1994, 152, 5532–5539. [Google Scholar] [PubMed]
- Chatzantoni, K.; Mouzaki, A. Anti-TNF-alpha antibody therapies in autoimmune diseases. Curr. Top. Med. Chem. 2006, 6, 1707–1714. [Google Scholar] [CrossRef]
- Jin, J.O.; Yu, Q. T Cell-Associated Cytokines in the Pathogenesis of Sjogren’s Syndrome. J. Clin. Cell. Immunol. 2013, 9, 1–7. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kamachi, M.; Kawakami, A.; Yamasaki, S.; Hida, A.; Nakashima, T.; Nakamura, H.; Ida, H.; Furuyama, M.; Nakashima, K.; Shibatomi, K.; et al. Regulation of apoptotic cell death by cytokines in a human salivary gland cell line: Distinct and synergistic mechanisms in apoptosis induced by tumor necrosis factor alpha and interferon gamma. J. Lab. Clin. Med. 2002, 139, 13–19. [Google Scholar] [CrossRef]
- Bossen, C.; Schneider, P. BAFF, APRIL and their receptors: Structure, function and signaling. Semin. Immunol. 2006, 18, 263–275. [Google Scholar] [CrossRef] [Green Version]
- Mackay, F.; Browning, J.L. BAFF: A fundamental survival factor for B cells. Nat. Rev. Immunol. 2002, 2, 465–475. [Google Scholar] [CrossRef] [PubMed]
- Mackay, F.; Schneider, P. Cracking the BAFF code. Nat. Rev. Immunol. 2009, 9, 491–502. [Google Scholar] [CrossRef] [Green Version]
- Litinskiy, M.B.; Nardelli, B.; Hilbert, D.M.; He, B.; Schaffer, A.; Casali, P.; Cerutti, A. DCs induce CD40-independent immunoglobulin class switching through BLyS and APRIL. Nat. Immunol. 2002, 3, 822–829. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Groom, J.; Kalled, S.L.; Cutler, A.H.; Olson, C.; Woodcock, S.A.; Schneider, P.; Tschopp, J.; Cachero, T.G.; Batten, M.; Wheway, J.; et al. Association of BAFF/BLyS overexpression and altered B cell differentiation with Sjogren’s syndrome. J. Clin. Investig. 2002, 109, 59–68. [Google Scholar] [CrossRef] [PubMed]
- Miceli-Richard, C.; Wang-Renault, S.F.; Boudaoud, S.; Busato, F.; Lallemand, C.; Bethune, K.; Belkhir, R.; Nocturne, G.; Mariette, X.; Tost, J. Overlap between differentially methylated DNA regions in blood B lymphocytes and genetic at-risk loci in primary Sjögren’s syndrome. Ann. Rheum. Dis. 2016, 75, 933–940. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Imgenberg-Kreuz, J.; Sandling, J.K.; Almlöf, J.C.; Nordlund, J.; Signér, L.; Norheim, K.B.; Omdal, R.; Rönnblom, L.; Eloranta, M.L.; Syvänen, A.C.; et al. Genome-wide DNA methylation analysis in multiple tissues in primary Sjögren’s syndrome reveals regulatory effects at interferon-induced genes. Ann. Rheum. Dis. 2016, 75, 2029–2036. [Google Scholar] [CrossRef] [Green Version]
- Wang-Renault, S.F.; Boudaoud, S.; Nocturne, G.; Roche, E.; Sigrist, N.; Daviaud, C.; Bugge Tinggaard, A.; Renault, V.; Deleuze, J.F.; Mariette, X.; et al. Deregulation of microRNA expression in purified T and B lymphocytes from patients with primary Sjögren’s syndrome. Ann. Rheum. Dis. 2018, 77, 133–140. [Google Scholar] [CrossRef] [Green Version]
- Alevizos, I.; Alexander, S.; Turner, R.J.; Illei, G.G. MicroRNA expression profiles as biomarkers of minor salivary gland inflammation and dysfunction in Sjögren’s syndrome. Arthritis Rheum. 2011, 63, 535–544. [Google Scholar] [CrossRef] [Green Version]
- Marketos, N.; Cinoku, I.; Rapti, A.; Mavragani, C.P. Type I interferon signature in Sjögren’s syndrome: Pathophysiological and clinical implications. Clin. Exp. Rheumatol. 2019, 37 (Suppl. S118), 185–191. [Google Scholar]
- Toker, A.; Engelbert, D.; Garg, G.; Polansky, J.K.; Floess, S.; Miyao, T.; Baron, U.; Düber, S.; Geffers, R.; Giehr, P.; et al. Active demethylation of the Foxp3 locus leads to the generation of stable regulatory T cells within the thymus. J. Immunol. 2013, 190, 3180–3188. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yu, X.; Liang, G.; Yin, H.; Ngalamika, O.; Li, F.; Zhao, M.; Lu, Q. DNA hypermethylation leads to lower FOXP3 expression in CD4+ T cells of patients with primary Sjögren’s syndrome. Clin. Immunol. 2013, 148, 254–257. [Google Scholar] [CrossRef] [PubMed]
- Taylor, K.E.; Wong, Q.; Levine, D.M.; McHugh, C.; Laurie, C.; Doheny, K.; Lam, M.Y.; Baer, A.N.; Challacombe, S.; Lanfranchi, H.; et al. Genome-Wide Association Analysis Reveals Genetic Heterogeneity of Sjögren’s Syndrome According to Ancestry. Arthritis Rheumatol. 2017, 69, 1294–1305. [Google Scholar] [CrossRef] [Green Version]
- Nordmark, G.; Kristjansdottir, G.; Theander, E.; Eriksson, P.; Brun, J.G.; Wang, C.; Padyukov, L.; Truedsson, L.; Alm, G.; Eloranta, M.L.; et al. Additive effects of the major risk alleles of IRF5 and STAT4 in primary Sjögren’s syndrome. Genes Immun. 2009, 10, 68–76. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Reksten, T.R.; Johnsen, S.J.; Jonsson, M.V.; Omdal, R.; Brun, J.G.; Theander, E.; Eriksson, P.; Wahren-Herlenius, M.; Jonsson, R.; Nordmark, G. Genetic associations to germinal centre formation in primary Sjogren’s syndrome. Ann. Rheum. Dis. 2014, 73, 1253–1258. [Google Scholar] [CrossRef]
- Lindén, M.; Ramírez Sepúlveda, J.I.; James, T.; Thorlacius, G.E.; Brauner, S.; Gómez-Cabrero, D.; Olsson, T.; Kockum, I.; Wahren-Herlenius, M. Sex influences eQTL effects of SLE and Sjögren’s syndrome-associated genetic polymorphisms. Biol. Sex Differ. 2017, 8, 34. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cobb, B.L.; Fei, Y.; Jonsson, R.; Bolstad, A.I.; Brun, J.G.; Rischmueller, M.; Lester, S.E.; Witte, T.; Illei, G.; Brennan, M.; et al. Genetic association between methyl-CpG binding protein 2 (MECP2) and primary Sjogren’s syndrome. Ann. Rheum. Dis. 2010, 69, 1731–1732. [Google Scholar] [CrossRef] [Green Version]
- Webb, R.; Wren, J.D.; Jeffries, M.; Kelly, J.A.; Kaufman, K.M.; Tang, Y.; Frank, M.B.; Merrill, J.; Kimberly, R.P.; Edberg, J.C.; et al. Variants within MECP2, a key transcription regulator, are associated with increased susceptibility to lupus and differential gene expression in patients with systemic lupus erythematosus. Arthritis Rheum. 2009, 60, 1076–1084. [Google Scholar] [CrossRef] [Green Version]
- Zhao, J.; Wei, K.; Chang, C.; Xu, L.; Jiang, P.; Guo, S.; Schrodi, S.J.; He, D. DNA Methylation of T Lymphocytes as a Therapeutic Target: Implications for Rheumatoid Arthritis Etiology. Front. Immunol. 2022, 13, 863703. [Google Scholar] [CrossRef]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Li, P.; Han, M.; Zhao, X.; Ren, G.; Mei, S.; Zhong, C. Abnormal Epigenetic Regulations in the Immunocytes of Sjögren’s Syndrome Patients and Therapeutic Potentials. Cells 2022, 11, 1767. https://doi.org/10.3390/cells11111767
Li P, Han M, Zhao X, Ren G, Mei S, Zhong C. Abnormal Epigenetic Regulations in the Immunocytes of Sjögren’s Syndrome Patients and Therapeutic Potentials. Cells. 2022; 11(11):1767. https://doi.org/10.3390/cells11111767
Chicago/Turabian StyleLi, Peng, Mengwei Han, Xingyu Zhao, Guanqun Ren, Si Mei, and Chao Zhong. 2022. "Abnormal Epigenetic Regulations in the Immunocytes of Sjögren’s Syndrome Patients and Therapeutic Potentials" Cells 11, no. 11: 1767. https://doi.org/10.3390/cells11111767
APA StyleLi, P., Han, M., Zhao, X., Ren, G., Mei, S., & Zhong, C. (2022). Abnormal Epigenetic Regulations in the Immunocytes of Sjögren’s Syndrome Patients and Therapeutic Potentials. Cells, 11(11), 1767. https://doi.org/10.3390/cells11111767