
LINCing Senescence and Nuclear Envelope Changes

 ,
,
Abstract
:1. Introduction
1.1. Senescence
1.2. The LINC Complex
2. LINC Proteins in Senescence
2.1. Nesprin-1
2.2. Nesprin-2
2.3. Nesprin-3
2.4. SUN1/SUN2
3. The LINC Complex, Mechanical Stress and Senescence
4. Concluding Remarks
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Rajagopalan, S.; Long, E.O. Cellular Senescence Induced by CD158d Reprograms Natural Killer Cells to Promote Vascular Remodeling. Proc. Natl. Acad. Sci. USA 2012, 109, 20596–20601. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Muñoz-Espín, D.; Cañamero, M.; Maraver, A.; Gómez-López, G.; Contreras, J.; Murillo-Cuesta, S.; Rodríguez-Baeza, A.; Varela-Nieto, I.; Ruberte, J.; Collado, M.; et al. Programmed Cell Senescence during Mammalian Embryonic Development. Cell 2013, 155, 1104–1118. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Storer, M.; Mas, A.; Robert-Moreno, A.; Pecoraro, M.; Ortells, M.C.; Di Giacomo, V.; Yosef, R.; Pilpel, N.; Krizhanovsky, V.; Sharpe, J.; et al. Senescence Is a Developmental Mechanism That Contributes to Embryonic Growth and Patterning. Cell 2013, 155, 1119–1130. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jun, J.-I.; Lau, L.F. Cellular Senescence Controls Fibrosis in Wound Healing. Aging 2010, 2, 627–631. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kong, X.; Feng, D.; Wang, H.; Hong, F.; Bertola, A.; Wang, F.-S.; Gao, B. Interleukin-22 Induces Hepatic Stellate Cell Senescence and Restricts Liver Fibrosis in Mice. Hepatology 2012, 56, 1150–1159. [Google Scholar] [CrossRef] [PubMed]
- De Cecco, M.; Criscione, S.W.; Peckham, E.J.; Hillenmeyer, S.; Hamm, E.A.; Manivannan, J.; Peterson, A.L.; Kreiling, J.A.; Neretti, N.; Sedivy, J.M. Genomes of Replicatively Senescent Cells Undergo Global Epigenetic Changes Leading to Gene Silencing and Activation of Transposable Elements. Aging Cell 2013, 12, 247–256. [Google Scholar] [CrossRef]
- Ivanov, A.; Pawlikowski, J.; Manoharan, I.; van Tuyn, J.; Nelson, D.M.; Rai, T.S.; Shah, P.P.; Hewitt, G.; Korolchuk, V.I.; Passos, J.F.; et al. Lysosome-Mediated Processing of Chromatin in Senescence. J. Cell Biol. 2013, 202, 129–143. [Google Scholar] [CrossRef]
- Wang, J.; Geesman, G.J.; Hostikka, S.L.; Atallah, M.; Blackwell, B.; Lee, E.; Cook, P.J.; Pasaniuc, B.; Shariat, G.; Halperin, E.; et al. Inhibition of Activated Pericentromeric SINE/Alu Repeat Transcription in Senescent Human Adult Stem Cells Reinstates Self-Renewal. Cell Cycle 2011, 10, 3016–3030. [Google Scholar] [CrossRef]
- Demaria, M.; Desprez, P.Y.; Campisi, J.; Velarde, M.C. Cell Autonomous and Non-Autonomous Effects of Senescent Cells in the Skin. J. Investig. Dermatol. 2015, 135, 1722–1726. [Google Scholar] [CrossRef] [Green Version]
- Shay, J.W.; Wright, W.E. Senescence and Immortalization: Role of Telomeres and Telomerase. Carcinogenesis 2005, 26, 867–874. [Google Scholar] [CrossRef]
- Fumagalli, M.; Rossiello, F.; Clerici, M.; Barozzi, S.; Cittaro, D.; Kaplunov, J.M.; Bucci, G.; Dobreva, M.; Matti, V.; Beausejour, C.M.; et al. Telomeric DNA Damage Is Irreparable and Causes Persistent DNA-Damage-Response Activation. Nat. Cell Biol. 2012, 14, 355–365. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Herbig, U.; Jobling, W.A.; Chen, B.P.; Chen, D.J.; Sedivy, J.M. Telomere Shortening Triggers Senescence of Human Cells through a Pathway Involving ATM, P53, and P21CIP1, but Not P16INK4a. Mol. Cell 2004, 14, 501–513. [Google Scholar] [CrossRef]
- Elmore, L.W.; Rehder, C.W.; Di, X.; McChesney, P.A.; Jackson-Cook, C.K.; Gewirtz, D.A.; Holt, S.E. Adriamycin-Induced Senescence in Breast Tumor Cells Involves Functional P53 and Telomere Dysfunction. J. Biol. Chem. 2002, 277, 35509–35515. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vasileiou, P.; Evangelou, K.; Vlasis, K.; Fildisis, G.; Panayiotidis, M.; Chronopoulos, E.; Passias, P.-G.; Kouloukoussa, M.; Gorgoulis, V.; Havaki, S. Mitochondrial Homeostasis and Cellular Senescence. Cells 2019, 8, 686. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Colavitti, R.; Finkel, T. Reactive Oxygen Species as Mediators of Cellular Senescence. IUBMB Life (Int. Union Biochem. Mol. Biol. Life) 2005, 57, 277–281. [Google Scholar] [CrossRef]
- Lee, A.C.; Fenster, B.E.; Ito, H.; Takeda, K.; Bae, N.S.; Hirai, T.; Yu, Z.-X.; Ferrans, V.J.; Howard, B.H.; Finkel, T. Ras Proteins Induce Senescence by Altering the Intracellular Levels of Reactive Oxygen Species. J. Biol. Chem. 1999, 274, 7936–7940. [Google Scholar] [CrossRef] [Green Version]
- Ogrunc, M.; Di Micco, R.; Liontos, M.; Bombardelli, L.; Mione, M.; Fumagalli, M.; Gorgoulis, V.G.; d’Adda di Fagagna, F. Oncogene-Induced Reactive Oxygen Species Fuel Hyperproliferation and DNA Damage Response Activation. Cell Death Differ. 2014, 21, 998–1012. [Google Scholar] [CrossRef] [Green Version]
- Jackson, J.G.; Pereira-Smith, O.M. Primary and Compensatory Roles for RB Family Members at Cell Cycle Gene Promoters That Are Deacetylated and Downregulated in Doxorubicin-Induced Senescence of Breast Cancer Cells. Mol. Cell. Biol. 2006, 26, 2501–2510. [Google Scholar] [CrossRef] [Green Version]
- Huun, J.; Lønning, P.E.; Knappskog, S. Effects of Concomitant Inactivation of P53 and PRb on Response to Doxorubicin Treatment in Breast Cancer Cell Lines. Cell Death Discov. 2017, 3, 17026. [Google Scholar] [CrossRef]
- Nemade, H.; Chaudhari, U.; Acharya, A.; Hescheler, J.; Hengstler, J.G.; Papadopoulos, S.; Sachinidis, A. Cell Death Mechanisms of the Anti-Cancer Drug Etoposide on Human Cardiomyocytes Isolated from Pluripotent Stem Cells. Arch. Toxicol. 2018, 92, 1507–1524. [Google Scholar] [CrossRef] [Green Version]
- Campisi, J. Aging, Cellular Senescence, and Cancer. Annu. Rev. Physiol. 2013, 75, 685–705. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- d’Adda di Fagagna, F.; Reaper, P.M.; Clay-Farrace, L.; Fiegler, H.; Carr, P.; von Zglinicki, T.; Saretzki, G.; Carter, N.P.; Jackson, S.P. A DNA Damage Checkpoint Response in Telomere-Initiated Senescence. Nature 2003, 426, 194–198. [Google Scholar] [CrossRef] [PubMed]
- Demaria, M.; Velarde, M.C. Senescence and Cellular Immortality. In Reference Module in Biomedical Sciences; Elsevier: Amsterdam, The Netherlands, 2017. [Google Scholar]
- Solana, R.; Tarazona, R.; Gayoso, I.; Lesur, O.; Dupuis, G.; Fulop, T. Innate Immunosenescence: Effect of Aging on Cells and Receptors of the Innate Immune System in Humans. Semin. Immunol. 2012, 24, 331–341. [Google Scholar] [CrossRef]
- Feng, Z.; Hu, W.; Teresky, A.K.; Hernando, E.; Cordon-Cardo, C.; Levine, A.J. Declining P53 Function in the Aging Process: A Possible Mechanism for the Increased Tumor Incidence in Older Populations. Proc. Natl. Acad. Sci. USA 2007, 104, 16633–16638. [Google Scholar] [CrossRef] [Green Version]
- Van Deursen, J.M. The Role of Senescent Cells in Ageing. Nature 2014, 509, 439–446. [Google Scholar] [CrossRef] [Green Version]
- Campisi, J. Senescent Cells, Tumor Suppression, and Organismal Aging: Good Citizens, Bad Neighbors. Cell 2005, 120, 513–522. [Google Scholar] [CrossRef]
- Krtolica, A.; Campisi, J. Cancer and Aging: A Model for the Cancer Promoting Effects of the Aging Stroma. Int. J. Biochem. Cell Biol. 2002, 34, 1401–1414. [Google Scholar] [CrossRef]
- Parrinello, S.; Coppe, J.-P.; Krtolica, A.; Campisi, J. Stromal-Epithelial Interactions in Aging and Cancer: Senescent Fibroblasts Alter Epithelial Cell Differentiation. J. Cell Sci. 2005, 118, 485–496. [Google Scholar] [CrossRef] [Green Version]
- Coppé, J.-P.; Desprez, P.-Y.; Krtolica, A.; Campisi, J. The Senescence-Associated Secretory Phenotype: The Dark Side of Tumor Suppression. Annu. Rev. Pathol. Mech. Dis. 2010, 5, 99–118. [Google Scholar] [CrossRef] [Green Version]
- Georgakopoulou, E.; Tsimaratou, K.; Evangelou, K.; Fernandez, M.-P.; Zoumpourlis, V.; Trougakos, I.; Kletsas, D.; Bartek, J.; Serrano, M.; Gorgoulis, V. Specific Lipofuscin Staining as a Novel Biomarker to Detect Replicative and Stress-Induced Senescence. A Method Applicable in Cryo-Preserved and Archival Tissues. Aging 2012, 5, 37–50. [Google Scholar] [CrossRef] [Green Version]
- Hernandez-Segura, A.; Nehme, J.; Demaria, M. Hallmarks of Cellular Senescence. Trends Cell Biol. 2018, 28, 436–453. [Google Scholar] [CrossRef] [PubMed]
- Dreesen, O.; Ong, P.F.; Chojnowski, A.; Colman, A. The Contrasting Roles of Lamin B1 in Cellular Aging and Human Disease. Nucleus 2013, 4, 283–290. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Freund, A.; Laberge, R.-M.; Demaria, M.; Campisi, J. Lamin B1 Loss Is a Senescence-Associated Biomarker. Mol. Biol. Cell 2012, 23, 2066–2075. [Google Scholar] [CrossRef] [PubMed]
- Shimi, T.; Butin-Israeli, V.; Adam, S.A.; Hamanaka, R.B.; Goldman, A.E.; Lucas, C.A.; Shumaker, D.K.; Kosak, S.T.; Chandel, N.S.; Goldman, R.D. The Role of Nuclear Lamin B1 in Cell Proliferation and Senescence. Genes Dev. 2011, 25, 2579–2593. [Google Scholar] [CrossRef] [Green Version]
- Dreesen, O.; Chojnowski, A.; Ong, P.F.; Zhao, T.Y.; Common, J.E.; Lunny, D.; Lane, E.B.; Lee, S.J.; Vardy, L.A.; Stewart, C.L.; et al. Lamin B1 Fluctuations Have Differential Effects on Cellular Proliferation and Senescence. J. Cell Biol. 2013, 200, 605–617. [Google Scholar] [CrossRef] [Green Version]
- Hernandez, L.; Roux, K.J.; Wong, E.S.M.; Mounkes, L.C.; Mutalif, R.; Navasankari, R.; Rai, B.; Cool, S.; Jeong, J.-W.; Wang, H.; et al. Functional Coupling between the Extracellular Matrix and Nuclear Lamina by Wnt Signaling in Progeria. Dev. Cell 2010, 19, 413–425. [Google Scholar] [CrossRef] [Green Version]
- Eriksson, M.; Brown, W.T.; Gordon, L.B.; Glynn, M.W.; Singer, J.; Scott, L.; Erdos, M.R.; Robbins, C.M.; Moses, T.Y.; Berglund, P.; et al. Recurrent de Novo Point Mutations in Lamin A Cause Hutchinson–Gilford Progeria Syndrome. Nature 2003, 423, 293–298. [Google Scholar] [CrossRef] [Green Version]
- Vahabikashi, A.; Adam, S.A.; Medalia, O.; Goldman, R.D. Nuclear Lamins: Structure and Function in Mechanobiology. APL Bioeng. 2022, 6, 011503. [Google Scholar] [CrossRef]
- Tran, J.R.; Chen, H.; Zheng, X.; Zheng, Y. Lamin in Inflammation and Aging. Curr. Opin. Cell Biol. 2016, 40, 124–130. [Google Scholar] [CrossRef] [Green Version]
- Jahed, Z.; Domkam, N.; Ornowski, J.; Yerima, G.; Mofrad, M.R.K. Molecular Models of LINC Complex Assembly at the Nuclear Envelope. J. Cell Sci. 2021, 134, jcs258194. [Google Scholar] [CrossRef]
- Horn, H.F. LINC Complex Proteins in Development and Disease, 1st ed.; Elsevier Inc.: Amsterdam, The Netherlands, 2014; Volume 109, ISBN 9780123979209. [Google Scholar]
- Cartwright, S.; Karakesisoglou, I. Nesprins in Health and Disease. Semin. Cell Dev. Biol. 2014, 29, 169–179. [Google Scholar] [CrossRef] [PubMed]
- Ketema, M.; Sonnenberg, A. Nesprin-3: A Versatile Connector between the Nucleus and the Cytoskeleton. Biochem. Soc. Trans. 2011, 39, 1719–1724. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Roux, K.J.; Crisp, M.L.; Liu, Q.; Kim, D.; Kozlov, S.; Stewart, C.L.; Burke, B. Nesprin 4 Is an Outer Nuclear Membrane Protein That Can Induce Kinesin-Mediated Cell Polarization. Proc. Natl. Acad. Sci. USA 2009, 106, 2194–2199. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Horn, H.F.; Kim, D.I.; Wright, G.D.; Wong, E.S.M.; Stewart, C.L.; Burke, B.; Roux, K.J. A Mammalian KASH Domain Protein Coupling Meiotic Chromosomes to the Cytoskeleton. J. Cell Biol. 2013, 202, 1023–1039. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Morimoto, A.; Shibuya, H.; Zhu, X.; Kim, J.; Ishiguro, K.I.; Han, M.; Watanabe, Y. A Conserved KASH Domain Protein Associates with Telomeres, SUN1, and Dynactin during Mammalian Meiosis. J. Cell Biol. 2012, 198, 165–172. [Google Scholar] [CrossRef] [PubMed]
- Sur-Erdem, I.; Hussain, M.S.; Asif, M.; Pınarbası, N.; Aksu, A.C.; Noegel, A.A. Nesprin-1 Impact on Tumorigenic Cell Phenotypes. Mol. Biol. Rep. 2020, 47, 921–934. [Google Scholar] [CrossRef]
- Sur, I.; Neumann, S.; Noegel, A.A. Nesprin-1 Role in DNA Damage Response. Nucleus 2014, 5, 173–191. [Google Scholar] [CrossRef] [Green Version]
- Warren, D.T.; Tajsic, T.; Porter, L.J.; Minaisah, R.M.; Cobb, A.; Jacob, A.; Rajgor, D.; Zhang, Q.P.; Shanahan, C.M. Nesprin-2-Dependent ERK1/2 Compartmentalisation Regulates the DNA Damage Response in Vascular Smooth Muscle Cell Ageing. Cell Death Differ. 2015, 22, 1540–1550. [Google Scholar] [CrossRef] [Green Version]
- Petrini, S.; Borghi, R.; D’Oria, V.; Restaldi, F.; Moreno, S.; Novelli, A.; Bertini, E.; Compagnucci, C. Aged Induced Pluripotent Stem Cell (IPSCs) as a New Cellular Model for Studying Premature Aging. Aging 2017, 9, 1453–1469. [Google Scholar] [CrossRef] [Green Version]
- Sola-Carvajal, A.; Revêchon, G.; Helgadottir, H.T.; Whisenant, D.; Hagblom, R.; Döhla, J.; Katajisto, P.; Brodin, D.; Fagerström-Billai, F.; Viceconte, N.; et al. Accumulation of Progerin Affects the Symmetry of Cell Division and Is Associated with Impaired Wnt Signaling and the Mislocalization of Nuclear Envelope Proteins. J. Investig. Dermatol. 2019, 139, 2272–2280. [Google Scholar] [CrossRef] [Green Version]
- Lei, K.; Zhu, X.; Xu, R.; Shao, C.; Xu, T.; Zhuang, Y.; Han, M. Inner Nuclear Envelope Proteins SUN1 and SUN2 Play a Prominent Role in the DNA Damage Response. Curr. Biol. 2012, 22, 1609–1615. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gutiérrez-Fernández, A.; Soria-Valles, C.; Osorio, F.G.; Gutiérrez-Abril, J.; Garabaya, C.; Aguirre, A.; Fueyo, A.; Fernández-García, M.S.; Puente, X.S.; López-Otín, C. Loss of MT 1-MMP Causes Cell Senescence and Nuclear Defects Which Can Be Reversed by Retinoic Acid. EMBO J. 2015, 34, 1875–1888. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, C.-Y.; Chi, Y.-H.; Mutalif, R.A.; Starost, M.F.; Myers, T.G.; Anderson, S.A.; Stewart, C.L.; Jeang, K.-T. Accumulation of the Inner Nuclear Envelope Protein Sun1 Is Pathogenic in Progeric and Dystrophic Laminopathies. Cell 2012, 149, 565–577. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, Z.-J.; Wang, W.-P.; Chen, Y.-C.; Wang, J.-Y.; Lin, W.-H.; Tai, L.-A.; Liou, G.-G.; Yang, C.-S.; Chi, Y.-H. Dysregulated Interactions between Lamin A and SUN1 Induce Abnormalities in the Nuclear Envelope and Endoplasmic Reticulum in Progeric Laminopathies. J. Cell Sci. 2014, 127, 1792–1804. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Burke, B.; Roux, K.J. Nuclei Take a Position: Managing Nuclear Location. Dev. Cell 2009, 17, 587–597. [Google Scholar] [CrossRef] [Green Version]
- Lüke, Y.; Zaim, H.; Karakesisoglou, I.; Jaeger, V.M.; Sellin, L.; Lu, W.; Schneider, M.; Neumann, S.; Beijer, A.; Munck, M.; et al. Nesprin-2 Giant (NUANCE) Maintains Nuclear Envelope Architecture and Composition in Skin. J. Cell Sci. 2008, 121, 1887–1898. [Google Scholar] [CrossRef] [Green Version]
- Lu, W.; Schneider, M.; Neumann, S.; Jaeger, V.-M.; Taranum, S.; Munck, M.; Cartwright, S.; Richardson, C.; Carthew, J.; Noh, K.; et al. Nesprin Interchain Associations Control Nuclear Size. Cell. Mol. Life Sci. 2012, 69, 3493–3509. [Google Scholar] [CrossRef] [Green Version]
- Rajgor, D.; Mellad, J.A.; Autore, F.; Zhang, Q.; Shanahan, C.M. Multiple Novel Nesprin-1 and Nesprin-2 Variants Act as Versatile Tissue-Specific Intracellular Scaffolds. PLoS ONE 2012, 7, e40098. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Q.; Skepper, J.N.; Yang, F.; Davies, J.D.; Hegyi, L.; Roberts, R.G.; Weissberg, P.L.; Ellis, J.A.; Shanahan, C.M. Nesprins: A Novel Family of Spectrin-Repeat-Containing Proteins That Localize to the Nuclear Membrane in Multiple Tissues. J. Cell Sci. 2001, 114, 4485–4498. [Google Scholar] [CrossRef]
- Padmakumar, V.; Abraham, S.; Braune, S.; Noegel, A.A.; Tunggal, B.; Karakesisoglou, I.; Korenbaum, E. Enaptin, a Giant Actin-Binding Protein, Is an Element of the Nuclear Membrane and the Actin Cytoskeleton. Exp. Cell Res. 2004, 295, 330–339. [Google Scholar] [CrossRef]
- Haque, F.; Mazzeo, D.; Patel, J.T.; Smallwood, D.T.; Ellis, J.A.; Shanahan, C.M.; Shackleton, S. Mammalian SUN Protein Interaction Networks at the Inner Nuclear Membrane and Their Role in Laminopathy Disease Processes. J. Biol. Chem. 2010, 285, 3487–3498. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Warren, D.T.; Tajsic, T.; Mellad, J.A.; Searles, R.; Zhang, Q.; Shanahan, C.M. Novel Nuclear Nesprin-2 Variants Tether Active Extracellular Signal-Regulated MAPK1 and MAPK2 at Promyelocytic Leukemia Protein Nuclear Bodies and Act to Regulate Smooth Muscle Cell Proliferation. J. Biol. Chem. 2010, 285, 1311–1320. [Google Scholar] [CrossRef] [Green Version]
- Zhang, J.; Felder, A.; Liu, Y.; Guo, L.T.; Lange, S.; Dalton, N.D.; Gu, Y.; Peterson, K.L.; Mizisin, A.P.; Shelton, G.D.; et al. Nesprin 1 Is Critical for Nuclear Positioning and Anchorage. Hum. Mol. Genet. 2010, 19, 329–341. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Broers, J.L.V.; Ramaekers, F.C.S.; Bonne, G.; Yaou, R.B.E.N.; Hutchison, C.J. Nuclear Lamins: Laminopathies and Their Role in Premature Ageing. Physiol. Rev. 2006, 86, 967–1008. [Google Scholar] [CrossRef] [PubMed]
- Li, M.; You, L.; Xue, J.; Lu, Y. Ionizing Radiation-Induced Cellular Senescence in Normal, Non-Transformed Cells and the Involved DNA Damage Response: A Mini Review. Front. Pharmacol. 2018, 9, 522. [Google Scholar] [CrossRef]
- Noda, A. Radiation-Induced Unrepairable DSBs: Their Role in the Late Effects of Radiation and Possible Applications to Biodosimetry. J. Radiat. Res. 2018, 59, ii114–ii120. [Google Scholar] [CrossRef] [Green Version]
- Alena, S.K.; Eva, B.; Aleš, K.; Emilie, L. Spatiotemporal Mislocalization of Nuclear Membrane-Associated Proteins in γ-Irradiation-Induced Senescent Cells. Cells 2020, 9, 999. [Google Scholar] [CrossRef] [Green Version]
- Rodier, F.; Coppé, J.-P.; Patil, C.K.; Hoeijmakers, W.A.M.; Muñoz, D.P.; Raza, S.R.; Freund, A.; Campeau, E.; Davalos, A.R.; Campisi, J. Persistent DNA Damage Signalling Triggers Senescence-Associated Inflammatory Cytokine Secretion. Nat. Cell Biol. 2009, 11, 973–979. [Google Scholar] [CrossRef]
- Crouch, J.; Shvedova, M.; Thanapaul, R.J.R.S.; Botchkarev, V.; Roh, D. Epigenetic Regulation of Cellular Senescence. Cells 2022, 11, 672. [Google Scholar] [CrossRef]
- Swartz, R.K.; Rodriguez, E.C.; King, M.C. A Role for Nuclear Envelope–Bridging Complexes in Homology-Directed Repair. Mol. Biol. Cell 2014, 25, 2461–2471. [Google Scholar] [CrossRef]
- Warren, J.J.; Pohlhaus, T.J.; Changela, A.; Iyer, R.R.; Modrich, P.L.; Beese, L.S. Structure of the Human MutSα DNA Lesion Recognition Complex. Mol. Cell 2007, 26, 579–592. [Google Scholar] [CrossRef] [PubMed]
- Li, F.; Mao, G.; Tong, D.; Huang, J.; Gu, L.; Yang, W.; Li, G.-M. The Histone Mark H3K36me3 Regulates Human DNA Mismatch Repair through Its Interaction with MutSα. Cell 2013, 153, 590–600. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, Q.; Bethmann, C.; Worth, N.F.; Davies, J.D.; Wasner, C.; Feuer, A.; Ragnauth, C.D.; Yi, Q.; Mellad, J.A.; Warren, D.T.; et al. Nesprin-1 and -2 Are Involved in the Pathogenesis of Emery–Dreifuss Muscular Dystrophy and Are Critical for Nuclear Envelope Integrity. Hum. Mol. Genet. 2007, 16, 2816–2833. [Google Scholar] [CrossRef] [PubMed]
- Janin, A.; Gache, V. Nesprins and Lamins in Health and Diseases of Cardiac and Skeletal Muscles. Front. Physiol. 2018, 9, 1277. [Google Scholar] [CrossRef] [PubMed]
- Kandert, S.; Lüke, Y.; Kleinhenz, T.; Neumann, S.; Lu, W.; Jaeger, V.M.; Munck, M.; Wehnert, M.; Müller, C.R.; Zhou, Z.; et al. Nesprin-2 Giant Safeguards Nuclear Envelope Architecture in LMNA S143F Progeria Cells. Hum. Mol. Genet. 2007, 16, 2944–2959. [Google Scholar] [CrossRef] [Green Version]
- Rana, A.; Rera, M.; Walker, D.W. Parkin Overexpression during Aging Reduces Proteotoxicity, Alters Mitochondrial Dynamics, and Extends Lifespan. Proc. Natl. Acad. Sci. USA 2013, 110, 8638–8643. [Google Scholar] [CrossRef] [Green Version]
- Kang, H.T.; Park, J.T.; Choi, K.; Kim, Y.; Choi, H.J.C.; Jung, C.W.; Lee, Y.-S.; Park, S.C. Chemical Screening Identifies ATM as a Target for Alleviating Senescence. Nat. Chem. Biol. 2017, 13, 616–623. [Google Scholar] [CrossRef]
- Masotti, A.; Celluzzi, A.; Petrini, S.; Bertini, E.; Zanni, G.; Compagnucci, C. Aged IPSCs Display an Uncommon Mitochondrial Appearance and Fail to Undergo in Vitro Neurogenesis. Aging 2014, 6, 1094–1108. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Q. Nesprin-2 Is a Multi-Isomeric Protein That Binds Lamin and Emerin at the Nuclear Envelope and Forms a Subcellular Network in Skeletal Muscle. J. Cell Sci. 2005, 118, 673–687. [Google Scholar] [CrossRef] [Green Version]
- Aksenova, A.Y.; Mirkin, S.M. At the Beginning of the End and in the Middle of the Beginning: Structure and Maintenance of Telomeric DNA Repeats and Interstitial Telomeric Sequences. Genes 2019, 10, 118. [Google Scholar] [CrossRef] [Green Version]
- Bettin, N.; Oss Pegorar, C.; Cusanelli, E. The Emerging Roles of TERRA in Telomere Maintenance and Genome Stability. Cells 2019, 8, 246. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Srinivas, N.; Rachakonda, S.; Kumar, R. Telomeres and Telomere Length: A General Overview. Cancers 2020, 12, 558. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barnes, R.P.; Fouquerel, E.; Opresko, P.L. The Impact of Oxidative DNA Damage and Stress on Telomere Homeostasis. Mech. Ageing Dev. 2019, 177, 37–45. [Google Scholar] [CrossRef] [PubMed]
- Fausti, F.; Di Agostino, S.; Cioce, M.; Bielli, P.; Sette, C.; Pandolfi, P.P.; Oren, M.; Sudol, M.; Strano, S.; Blandino, G. ATM Kinase Enables the Functional Axis of YAP, PML and P53 to Ameliorate Loss of Werner Protein-Mediated Oncogenic Senescence. Cell Death Differ. 2013, 20, 1498–1509. [Google Scholar] [CrossRef] [Green Version]
- Adams, B.R.; Golding, S.E.; Rao, R.R.; Valerie, K. Dynamic Dependence on ATR and ATM for Double-Strand Break Repair in Human Embryonic Stem Cells and Neural Descendants. PLoS ONE 2010, 5, e10001. [Google Scholar] [CrossRef]
- Mombach, J.C.; Bugs, C.A.; Chaouiya, C. Modelling the Onset of Senescence at the G1/S Cell Cycle Checkpoint. BMC Genom. 2014, 15, S7. [Google Scholar] [CrossRef] [Green Version]
- Wei, F.; Xie, Y.; Tao, L.; Tang, D. Both ERK1 and ERK2 Kinases Promote G2/M Arrest in Etoposide-Treated MCF7 Cells by Facilitating ATM Activation. Cell. Signal. 2010, 22, 1783–1789. [Google Scholar] [CrossRef]
- De Sandre-Giovannoli, A. Lamin A Truncation in Hutchinson-Gilford Progeria. Science 2003, 300, 2055. [Google Scholar] [CrossRef]
- Kubben, N.; Misteli, T. Shared Molecular and Cellular Mechanisms of Premature Ageing and Ageing-Associated Diseases. Nat. Rev. Mol. Cell Biol. 2017, 18, 595–609. [Google Scholar] [CrossRef]
- Espada, J.; Varela, I.; Flores, I.; Ugalde, A.P.; Cadiñanos, J.; Pendás, A.M.; Stewart, C.L.; Tryggvason, K.; Blasco, M.A.; Freije, J.M.P.; et al. Nuclear Envelope Defects Cause Stem Cell Dysfunction in Premature-Aging Mice. J. Cell Biol. 2008, 181, 27–35. [Google Scholar] [CrossRef] [Green Version]
- Choi, J.Y.; Lai, J.K.; Xiong, Z.-M.; Ren, M.; Moorer, M.C.; Stains, J.P.; Cao, K. Diminished Canonical β-Catenin Signaling During Osteoblast Differentiation Contributes to Osteopenia in Progeria. J. Bone Miner. Res. 2018, 33, 2059–2070. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Markiewicz, E.; Tilgner, K.; Barker, N.; van de Wetering, M.; Clevers, H.; Dorobek, M.; Hausmanowa-Petrusewicz, I.; Ramaekers, F.C.S.; Broers, J.L.V.; Blankesteijn, W.M.; et al. The Inner Nuclear Membrane Protein Emerin Regulates β-Catenin Activity by Restricting Its Accumulation in the Nucleus. EMBO J. 2006, 25, 3275–3285. [Google Scholar] [CrossRef] [PubMed]
- Neumann, S.; Schneider, M.; Daugherty, R.L.; Gottardi, C.J.; Eming, S.A.; Beijer, A.; Noegel, A.A.; Karakesisoglou, I. Nesprin-2 Interacts with α-Catenin and Regulates Wnt Signaling at the Nuclear Envelope. J. Biol. Chem. 2010, 285, 34932–34938. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McKenna, T.; Rosengardten, Y.; Viceconte, N.; Baek, J.; Grochová, D.; Eriksson, M. Embryonic Expression of the Common Progeroid Lamin A Splice Mutation Arrests Postnatal Skin Development. Aging Cell 2014, 13, 292–302. [Google Scholar] [CrossRef]
- Zhang, Y.; Wang, X. Targeting the Wnt/β-Catenin Signaling Pathway in Cancer. J. Hematol. Oncol. 2020, 13, 165. [Google Scholar] [CrossRef]
- Adams, P.D.; Enders, G.H. Wnt Signaling and Senescence: A Tug of War in Early Neoplasia? Cancer Biol. Ther. 2008, 7, 1706–1711. [Google Scholar] [CrossRef] [Green Version]
- Fanjul-Fernández, M.; Folgueras, A.R.; Cabrera, S.; López-Otín, C. Matrix Metalloproteinases: Evolution, Gene Regulation and Functional Analysis in Mouse Models. Biochim. Biophys. Acta-Mol. Cell Res. 2010, 1803, 3–19. [Google Scholar] [CrossRef] [Green Version]
- Kessenbrock, K.; Plaks, V.; Werb, Z. Matrix Metalloproteinases: Regulators of the Tumor Microenvironment. Cell 2010, 141, 52–67. [Google Scholar] [CrossRef] [Green Version]
- Overall, C.M.; López-Otín, C. Strategies for MMP Inhibition in Cancer: Innovations for the Post-Trial Era. Nat. Rev. Cancer 2002, 2, 657–672. [Google Scholar] [CrossRef]
- Sabeh, F.; Ota, I.; Holmbeck, K.; Birkedal-Hansen, H.; Soloway, P.; Balbin, M.; Lopez-Otin, C.; Shapiro, S.; Inada, M.; Krane, S.; et al. Tumor Cell Traffic through the Extracellular Matrix Is Controlled by the Membrane-Anchored Collagenase MT1-MMP. J. Cell Biol. 2004, 167, 769–781. [Google Scholar] [CrossRef] [Green Version]
- Holmbeck, K.; Bianco, P.; Caterina, J.; Yamada, S.; Kromer, M.; Kuznetsov, S.A.; Mankani, M.; Gehron Robey, P.; Poole, A.R.; Pidoux, I.; et al. MT1-MMP-Deficient Mice Develop Dwarfism, Osteopenia, Arthritis, and Connective Tissue Disease Due to Inadequate Collagen Turnover. Cell 1999, 99, 81–92. [Google Scholar] [CrossRef] [Green Version]
- Zhou, Z.; Apte, S.S.; Soininen, R.; Cao, R.; Baaklini, G.Y.; Rauser, R.W.; Wang, J.; Cao, Y.; Tryggvason, K. Impaired Endochondral Ossification and Angiogenesis in Mice Deficient in Membrane-Type Matrix Metalloproteinase I. Proc. Natl. Acad. Sci. USA 2000, 97, 4052–4057. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Atkinson, J.J.; Holmbeck, K.; Yamada, S.; Birkedal-Hansen, H.; Parks, W.C.; Senior, R.M. Membrane-Type 1 Matrix Metalloproteinase Is Required for Normal Alveolar Development. Dev. Dyn. 2005, 232, 1079–1090. [Google Scholar] [CrossRef]
- Chun, T.-H.; Hotary, K.B.; Sabeh, F.; Saltiel, A.R.; Allen, E.D.; Weiss, S.J. A Pericellular Collagenase Directs the 3-Dimensional Development of White Adipose Tissue. Cell 2006, 125, 577–591. [Google Scholar] [CrossRef] [Green Version]
- Postel, R.; Ketema, M.; Kuikman, I.; de Pereda, J.M.; Sonnenberg, A. Nesprin-3 Augments Peripheral Nuclear Localization of Intermediate Filaments in Zebrafish. J. Cell Sci. 2011, 124, 755–764. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Swift, J.; Ivanovska, I.L.; Buxboim, A.; Harada, T.; Dingal, P.C.D.P.; Pinter, J.; Pajerowski, J.D.; Spinler, K.R.; Shin, J.-W.; Tewari, M.; et al. Nuclear Lamin-A Scales with Tissue Stiffness and Enhances Matrix-Directed Differentiation. Science 2013, 341. [Google Scholar] [CrossRef] [Green Version]
- Csoka, A.B.; English, S.B.; Simkevich, C.P.; Ginzinger, D.G.; Butte, A.J.; Schatten, G.P.; Rothman, F.G.; Sedivy, J.M. Genome-Scale Expression Profiling of Hutchinson-Gilford Progeria Syndrome Reveals Widespread Transcriptional Misregulation Leading to Mesodermal/Mesenchymal Defects and Accelerated Atherosclerosis. Aging Cell 2004, 3, 235–243. [Google Scholar] [CrossRef]
- De la Rosa, J.; Freije, J.M.P.; Cabanillas, R.; Osorio, F.G.; Fraga, M.F.; Fernández-García, M.S.; Rad, R.; Fanjul, V.; Ugalde, A.P.; Liang, Q.; et al. Prelamin A Causes Progeria through Cell-Extrinsic Mechanisms and Prevents Cancer Invasion. Nat. Commun. 2013, 4, 2268. [Google Scholar] [CrossRef] [Green Version]
- Rothballer, A.; Kutay, U. The Diverse Functional LINCs of the Nuclear Envelope to the Cytoskeleton and Chromatin. Chromosoma 2013, 122, 415–429. [Google Scholar] [CrossRef] [Green Version]
- Crisp, M.; Liu, Q.; Roux, K.; Rattner, J.B.B.; Shanahan, C.; Burke, B.; Stahl, P.D.; Hodzic, D. Coupling of the Nucleus and Cytoplasm: Role of the LINC Complex. J. Cell Biol. 2006, 172, 41–53. [Google Scholar] [CrossRef] [Green Version]
- Ding, X.; Xu, R.; Yu, J.; Xu, T.; Zhuang, Y.; Han, M. SUN1 Is Required for Telomere Attachment to Nuclear Envelope and Gametogenesis in Mice. Dev. Cell 2007, 12, 863–872. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schmitt, J.; Benavente, R.; Hodzic, D.; Höög, C.; Stewart, C.L.; Alsheimer, M. Transmembrane Protein Sun2 Is Involved in Tethering Mammalian Meiotic Telomeres to the Nuclear Envelope. Proc. Natl. Acad. Sci. USA 2007, 104, 7426–7431. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lei, K.; Zhang, X.; Ding, X.; Guo, X.; Chen, M.; Zhu, B.; Xu, T.; Zhuang, Y.; Xu, R.; Han, M. SUN1 and SUN2 Play Critical but Partially Redundant Roles in Anchoring Nuclei in Skeletal Muscle Cells in Mice. Proc. Natl. Acad. Sci. USA 2009, 106, 10207–10212. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, X.; Lei, K.; Yuan, X.; Wu, X.; Zhuang, Y.; Xu, T.; Xu, R.; Han, M. SUN1/2 and Syne/Nesprin-1/2 Complexes Connect Centrosome to the Nucleus during Neurogenesis and Neuronal Migration in Mice. Neuron 2009, 64, 173–187. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yu, J.; Lei, K.; Zhou, M.; Craft, C.M.; Xu, G.; Xu, T.; Zhuang, Y.; Xu, R.; Han, M. KASH Protein Syne-2/Nesprin-2 and SUN Proteins SUN1/2 Mediate Nuclear Migration during Mammalian Retinal Development. Hum. Mol. Genet. 2011, 20, 1061–1073. [Google Scholar] [CrossRef] [Green Version]
- Mounkes, L.C.; Kozlov, S.; Hernandez, L.; Sullivan, T.; Stewart, C.L. A Progeroid Syndrome in Mice Is Caused by Defects in A-Type Lamins. Nature 2003, 423, 298–301. [Google Scholar] [CrossRef]
- Sullivan, T.; Escalante-Alcalde, D.; Bhatt, H.; Anver, M.; Bhat, N.; Nagashima, K.; Stewart, C.L.; Burke, B. Loss of A-Type Lamin Expression Compromises Nuclear Envelope Integrity Leading to Muscular Dystrophy. J. Cell Biol. 1999, 147, 913–920. [Google Scholar] [CrossRef] [Green Version]
- Pendás, A.M.; Zhou, Z.; Cadiñanos, J.; Freije, J.M.P.; Wang, J.; Hultenby, K.; Astudillo, A.; Wernerson, A.; Rodríguez, F.; Tryggvason, K.; et al. Defective Prelamin A Processing and Muscular and Adipocyte Alterations in Zmpste24 Metalloproteinase–Deficient Mice. Nat. Genet. 2002, 31, 94–99. [Google Scholar] [CrossRef]
- Bergo, M.O.; Gavino, B.; Ross, J.; Schmidt, W.K.; Hong, C.; Kendall, L.V.; Mohr, A.; Meta, M.; Genant, H.; Jiang, Y.; et al. Zmpste24 Deficiency in Mice Causes Spontaneous Bone Fractures, Muscle Weakness, and a Prelamin A Processing Defect. Proc. Natl. Acad. Sci. USA 2002, 99, 13049–13054. [Google Scholar] [CrossRef] [Green Version]
- Varela, I.; Cadiñanos, J.; Pendás, A.M.; Gutiérrez-Fernández, A.; Folgueras, A.R.; Sánchez, L.M.; Zhou, Z.; Rodríguez, F.J.; Stewart, C.L.; Vega, J.A.; et al. Accelerated Ageing in Mice Deficient in Zmpste24 Protease Is Linked to P53 Signalling Activation. Nature 2005, 437, 564–568. [Google Scholar] [CrossRef]
- Liu, B.; Wang, J.; Chan, K.M.; Tjia, W.M.; Deng, W.; Guan, X.; Huang, J.; Li, K.M.; Chau, P.Y.; Chen, D.J.; et al. Genomic Instability in Laminopathy-Based Premature Aging. Nat. Med. 2005, 11, 780–785. [Google Scholar] [CrossRef] [PubMed]
- Denoyelle, C.; Abou-Rjaily, G.; Bezrookove, V.; Verhaegen, M.; Johnson, T.M.; Fullen, D.R.; Pointer, J.N.; Gruber, S.B.; Su, L.D.; Nikiforov, M.A.; et al. Anti-Oncogenic Role of the Endoplasmic Reticulum Differentially Activated by Mutations in the MAPK Pathway. Nat. Cell Biol. 2006, 8, 1053–1063. [Google Scholar] [CrossRef] [PubMed]
- Cho, J.-H.; Saini, D.K.; Karunarathne, W.K.A.; Kalyanaraman, V.; Gautam, N. Alteration of Golgi Structure in Senescent Cells and Its Regulation by a G Protein γ Subunit. Cell. Signal. 2011, 23, 785–793. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sobol, R.W.; Horton, J.K.; Kühn, R.; Gu, H.; Singhal, R.K.; Prasad, R.; Rajewsky, K.; Wilson, S.H. Requirement of Mammalian DNA Polymerase-β in Base-Excision Repair. Nature 1996, 379, 183–186. [Google Scholar] [CrossRef] [PubMed]
- Davidson, D.; Amrein, L.; Panasci, L.; Aloyz, R. Small Molecules, Inhibitors of DNA-PK, Targeting DNA Repair, and Beyond. Front. Pharmacol. 2013, 4, 5. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yue, X.; Bai, C.; Xie, D.; Ma, T.; Zhou, P.-K. DNA-PKcs: A Multi-Faceted Player in DNA Damage Response. Front. Genet. 2020, 11, 1692. [Google Scholar] [CrossRef]
- Valon, L.; Levayer, R. Dying under Pressure: Cellular Characterisation and in Vivo Functions of Cell Death Induced by Compaction. Biol. Cell 2019, 111, 51–66. [Google Scholar] [CrossRef] [Green Version]
- Hu, M.; Hong, L.; Hong, S.; Min, J.; Zhao, Y.; Yang, Q.; Zhang, Q.; Tang, J.; Li, Y. Mechanical Stress Influences the Viability and Morphology of Human Parametrial Ligament Fibroblasts. Mol. Med. Rep. 2017, 15, 853–858. [Google Scholar] [CrossRef]
- Feng, C.; Yang, M.; Zhang, Y.; Lan, M.; Huang, B.; Liu, H.; Zhou, Y. Cyclic Mechanical Tension Reinforces DNA Damage and Activates the P53-P21-Rb Pathway to Induce Premature Senescence of Nucleus Pulposus Cells. Int. J. Mol. Med. 2018, 41, 3316–3326. [Google Scholar] [CrossRef]
- Li, P.; Hou, G.; Zhang, R.; Gan, Y.; Xu, Y.; Song, L.; Zhou, Q. High-Magnitude Compression Accelerates the Premature Senescence of Nucleus Pulposus Cells via the P38 MAPK-ROS Pathway. Arthritis Res. Ther. 2017, 19, 209. [Google Scholar] [CrossRef] [Green Version]
- Isermann, P.; Lammerding, J. Nuclear Mechanics and Mechanotransduction in Health and Disease. Curr. Biol. 2013, 23, R1113–R1121. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Donnaloja, F.; Jacchetti, E.; Soncini, M.; Raimondi, M.T. Mechanosensing at the Nuclear Envelope by Nuclear Pore Complex Stretch Activation and Its Effect in Physiology and Pathology. Front. Physiol. 2019, 10, 896. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wernig, F.; Xu, Q. Mechanical Stress-Induced Apoptosis in the Cardiovascular System. Prog. Biophys. Mol. Biol. 2002, 78, 105–137. [Google Scholar] [CrossRef]
- Maurer, M.; Lammerding, J. The Driving Force: Nuclear Mechanotransduction in Cellular Function, Fate, and Disease. Annu. Rev. Biomed. Eng. 2019, 21, 443–468. [Google Scholar] [CrossRef]
- Hennekam, R.C.M. Hutchinson–Gilford Progeria Syndrome: Review of the Phenotype. Am. J. Med. Genet. Part A 2006, 140A, 2603–2624. [Google Scholar] [CrossRef] [Green Version]
- Stehbens, W.E.; Delahunt, B.; Shozawa, T.; Gilbert-Barness, E. Smooth Muscle Cell Depletion and Collagen Types in Progeric Arteries. Cardiovasc. Pathol. 2001, 10, 133–136. [Google Scholar] [CrossRef]
- Stehbens, W.E.; Wakefield, S.J.; Gilbert-Barness, E.; Olson, R.E.; Ackerman, J. Histological and Ultrastructural Features of Atherosclerosis in Progeria. Cardiovasc. Pathol. 1999, 8, 29–39. [Google Scholar] [CrossRef]
- Olive, M.; Harten, I.; Mitchell, R.; Beers, J.K.; Djabali, K.; Cao, K.; Erdos, M.R.; Blair, C.; Funke, B.; Smoot, L.; et al. Cardiovascular Pathology in Hutchinson-Gilford Progeria: Correlation With the Vascular Pathology of Aging. Arterioscler. Thromb. Vasc. Biol. 2010, 30, 2301–2309. [Google Scholar] [CrossRef] [Green Version]
- Kim, P.H.; Luu, J.; Heizer, P.; Tu, Y.; Weston, T.A.; Chen, N.; Lim, C.; Li, R.L.; Lin, P.-Y.; Dunn, J.C.Y.; et al. Disrupting the LINC Complex in Smooth Muscle Cells Reduces Aortic Disease in a Mouse Model of Hutchinson-Gilford Progeria Syndrome. Sci. Transl. Med. 2018, 10, eaat7163. [Google Scholar] [CrossRef] [Green Version]
- Varga, R.; Eriksson, M.; Erdos, M.R.; Olive, M.; Harten, I.; Kolodgie, F.; Capell, B.C.; Cheng, J.; Faddah, D.; Perkins, S.; et al. Progressive Vascular Smooth Muscle Cell Defects in a Mouse Model of Hutchinson–Gilford Progeria Syndrome. Proc. Natl. Acad. Sci. USA 2006, 103, 3250–3255. [Google Scholar] [CrossRef] [Green Version]
- Dahl, K.N.; Scaffidi, P.; Islam, M.F.; Yodh, A.G.; Wilson, K.L.; Misteli, T. Distinct Structural and Mechanical Properties of the Nuclear Lamina in Hutchinson–Gilford Progeria Syndrome. Proc. Natl. Acad. Sci. USA 2006, 103, 10271–10276. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Verstraeten, V.L.R.M.; Ji, J.Y.; Cummings, K.S.; Lee, R.T.; Lammerding, J. Increased Mechanosensitivity and Nuclear Stiffness in Hutchinson–Gilford Progeria Cells: Effects of Farnesyltransferase Inhibitors. Aging Cell 2008, 7, 383–393. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stewart-Hutchinson, P.J.; Hale, C.M.; Wirtz, D.; Hodzic, D. Structural Requirements for the Assembly of LINC Complexes and Their Function in Cellular Mechanical Stiffness. Exp. Cell Res. 2008, 314, 1892–1905. [Google Scholar] [CrossRef] [PubMed] [Green Version]

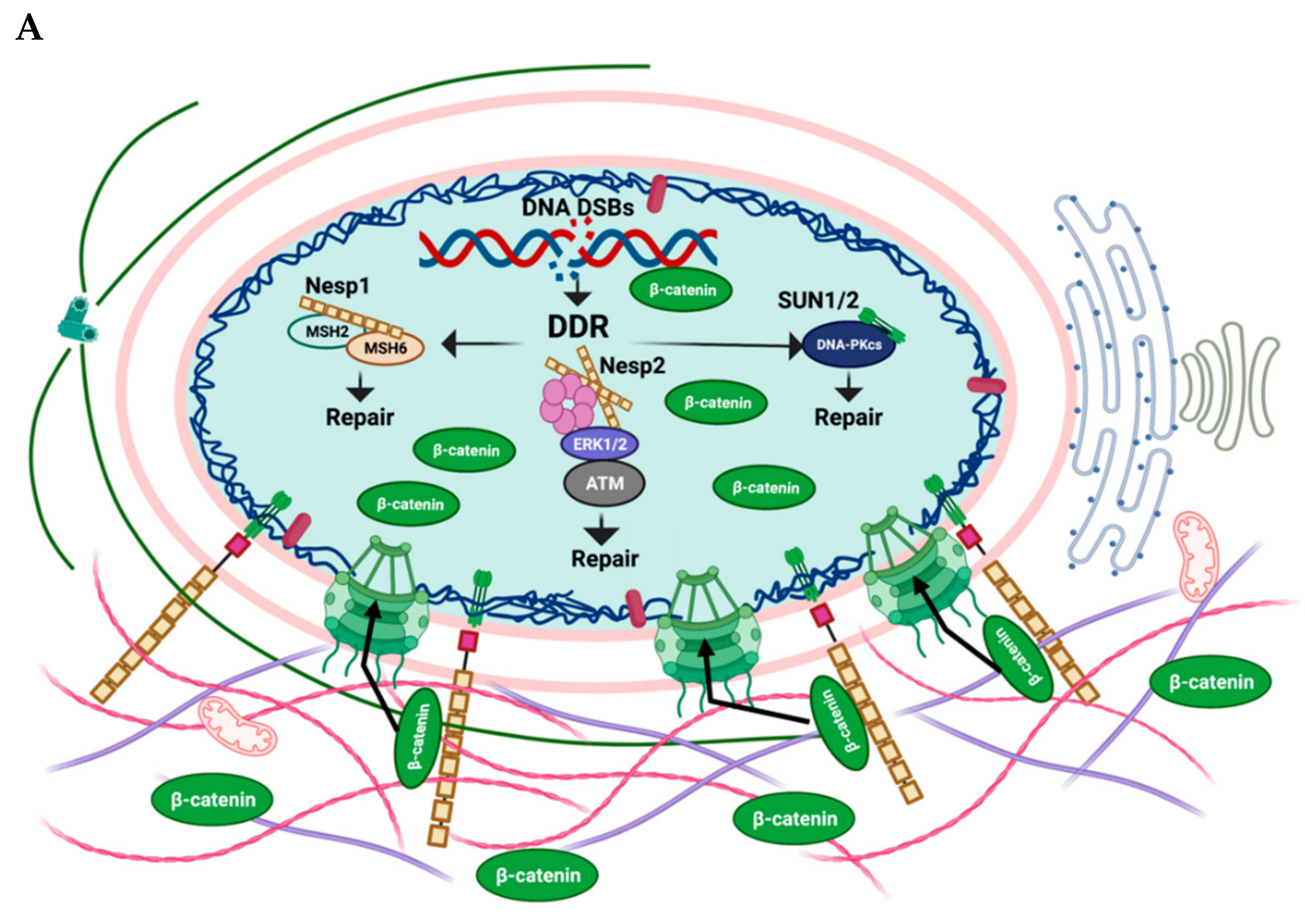


{kind=link}
{kind=link}
{kind=link}
| LINC Protein | Model/Cell Line | Mode of Senescence Induction | Cellular Mechanisms/Structures Affected | Ref’s |
|---|---|---|---|---|
| Nesprin-1 | Embryonic mouse mesenchymal stem cell line (CH310T1/2) |
| Increase (↑) DNA damage (DSBs) due to DNA mismatch repair (MMR) impairment
| [48,49] |
| Human dermal fibroblasts (HFs) |
|
Knockdown
:
Decrease (↓) MMR (etoposide)
| ||
| Human colorectal carcinoma cell line (DLD-1). These cells are Nesprin-1 deficient. | N/A |
Compared to normal human fibroblast
:
| ||
| Human hepatocellular carcinoma cell lines (Hep3B, Huh7) These cells have low endogenous Nesprin-1 expression. |
| Non-Treated (comparing Hep3B and Huh7 to normal liver cells)
| ||
| Nesprin-2 variant (β∆KASH1) | Human aortic vascular smooth muscle cells (VSMCs) | Population doublings (PDs) | Presenescence:
| [50] |
|
| |||
|
| |||
| Proliferative:
| |||
| Presenescent:
Proliferative: G2/M checkpoint failure:
| |||
| Nesprin-2 | Aged-Pluripotent stem cells (a-iPSCs) | iPSCs cultured for more than one year | mRNA expression levels:
| [51] |
| HGPS dermal fibroblasts HGPS mice interfollicular epidermis (IFE) | N/A |
Postnatal day 4 (mice):
Human HGPS fibroblasts:
| [52] | |
| SUN1/2 | SUN1/2-/- MEFs | N/A | ↓ Perinuclear heterochromatin compared to wild-type MEFs | [53] |
| Compared to wild type MEFs:
| |||
|
| |||
| Mmp14-/- mouse embryonic fibroblasts (MEFs) | N/A |
Perinuclear and nuclear region:
| [54] | |
| LMNA-/- mouse embryonic fibroblasts (MEFs) | N/A |
| [55] | |
| Lmna∆9 mutant MEFs | N/A | Irregularly shaped nucleus with frequent herniations and blebs that were reduced in LMNA/SUN1 DKO MEFs | ||
| Hutchinson-Gilford progeria syndrome (HGPS) human skin fibroblasts | N/A | Compared to normal human fibroblast:
| ||
| Hutchinson-Gilford progeria syndrome (HGPS) human skin fibroblasts | N/A |
Structural changes:
| [56] | |
| HeLa cells | Progerin overexpression |
| ||
| Progerin overexpression & SUN1 KD |
|
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Meqbel, B.R.M.; Gomes, M.; Omer, A.; Gallouzi, I.E.; Horn, H.F. LINCing Senescence and Nuclear Envelope Changes. Cells 2022, 11, 1787. https://doi.org/10.3390/cells11111787
Meqbel BRM, Gomes M, Omer A, Gallouzi IE, Horn HF. LINCing Senescence and Nuclear Envelope Changes. Cells. 2022; 11(11):1787. https://doi.org/10.3390/cells11111787
Chicago/Turabian StyleMeqbel, Bakhita R. M., Matilde Gomes, Amr Omer, Imed E. Gallouzi, and Henning F. Horn. 2022. "LINCing Senescence and Nuclear Envelope Changes" Cells 11, no. 11: 1787. https://doi.org/10.3390/cells11111787
APA StyleMeqbel, B. R. M., Gomes, M., Omer, A., Gallouzi, I. E., & Horn, H. F. (2022). LINCing Senescence and Nuclear Envelope Changes. Cells, 11(11), 1787. https://doi.org/10.3390/cells11111787