
The Influence of PARP, ATR, CHK1 Inhibitors on Premature Mitotic Entry and Genomic Instability in High-Grade Serous BRCAMUT and BRCAWT Ovarian Cancer Cells

 , ,
, ,  , and
, and {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Reagents
2.2. Cell Culture and Drug Administration
2.3. MTT Assay
2.4. Metaphase Spread
2.5. Micronucleus Assay
2.6. Determination of Proliferation Rate
2.7. Cell-Cycle Analysis
2.8. Immunofluorescence and Immunocytochemistry Staining
2.9. Measurement of Chromatin Condensation
2.10. Detection of S-Phase Progression Using 5-Ethynyl-2′-deoxyuridine(EdU) Incorporation with Click-iT Chemistry
2.11. Measurement of Phosphatidylserine Externalization
2.12. Morphological Assessment of Apoptosis and Necrosis: Double Staining with Hoechst 33258 and Propidium Iodide (PI)
2.13. Western Blot Analysis
2.14. Statistical Analysis
3. Results
3.1. CHK1 or ATR Inhibition Is Cytotoxic to Ovarian Cancer Cells
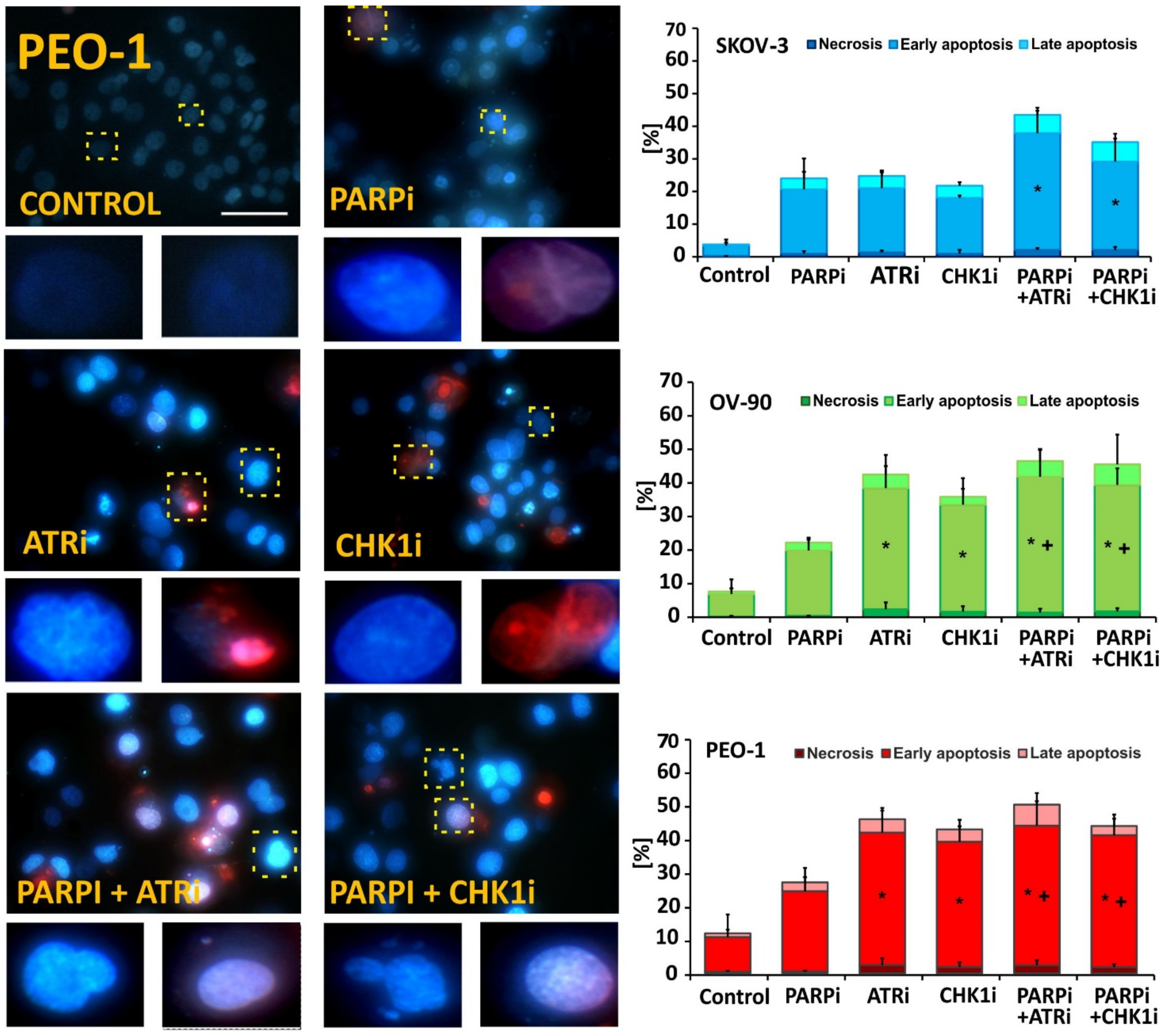
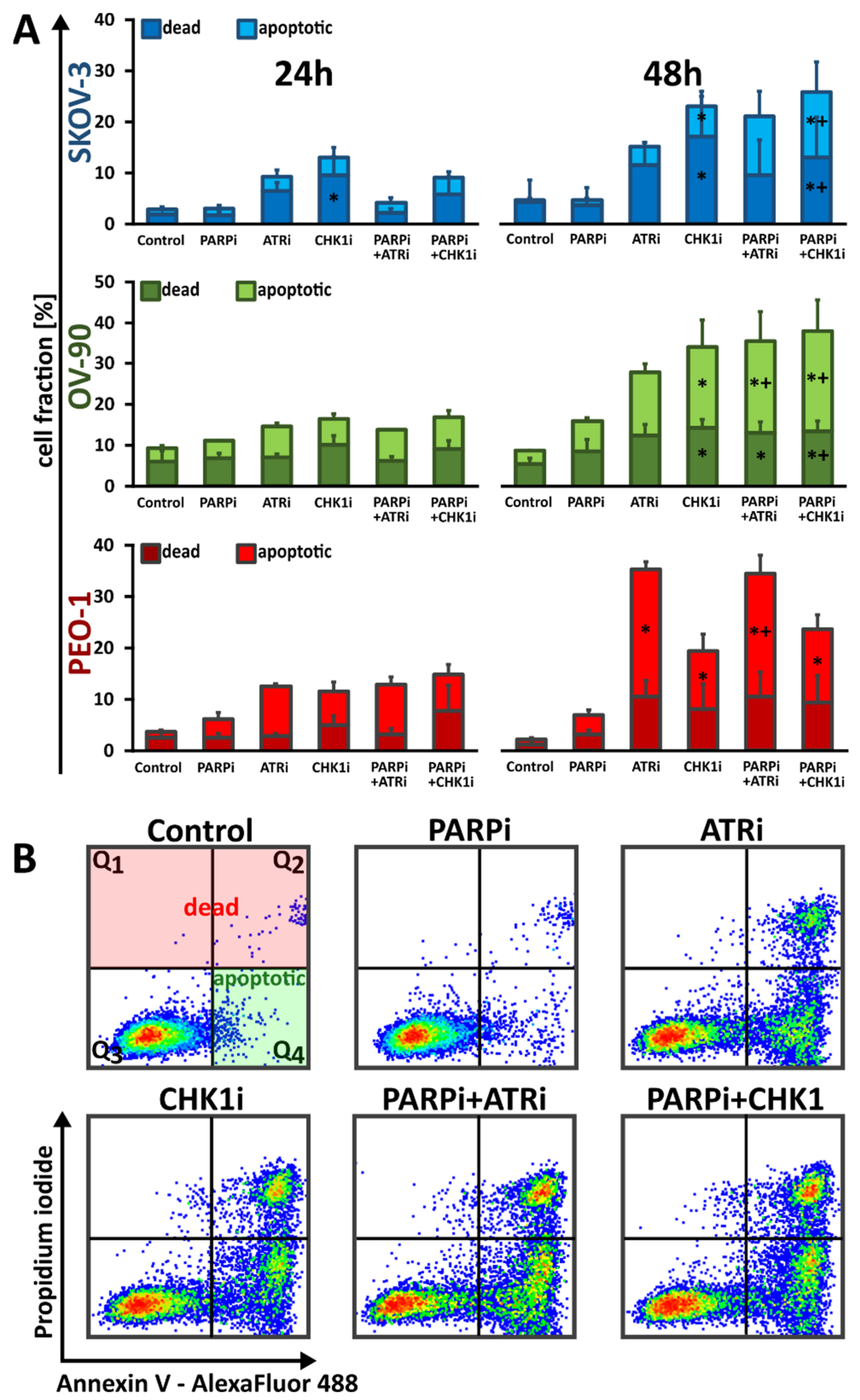
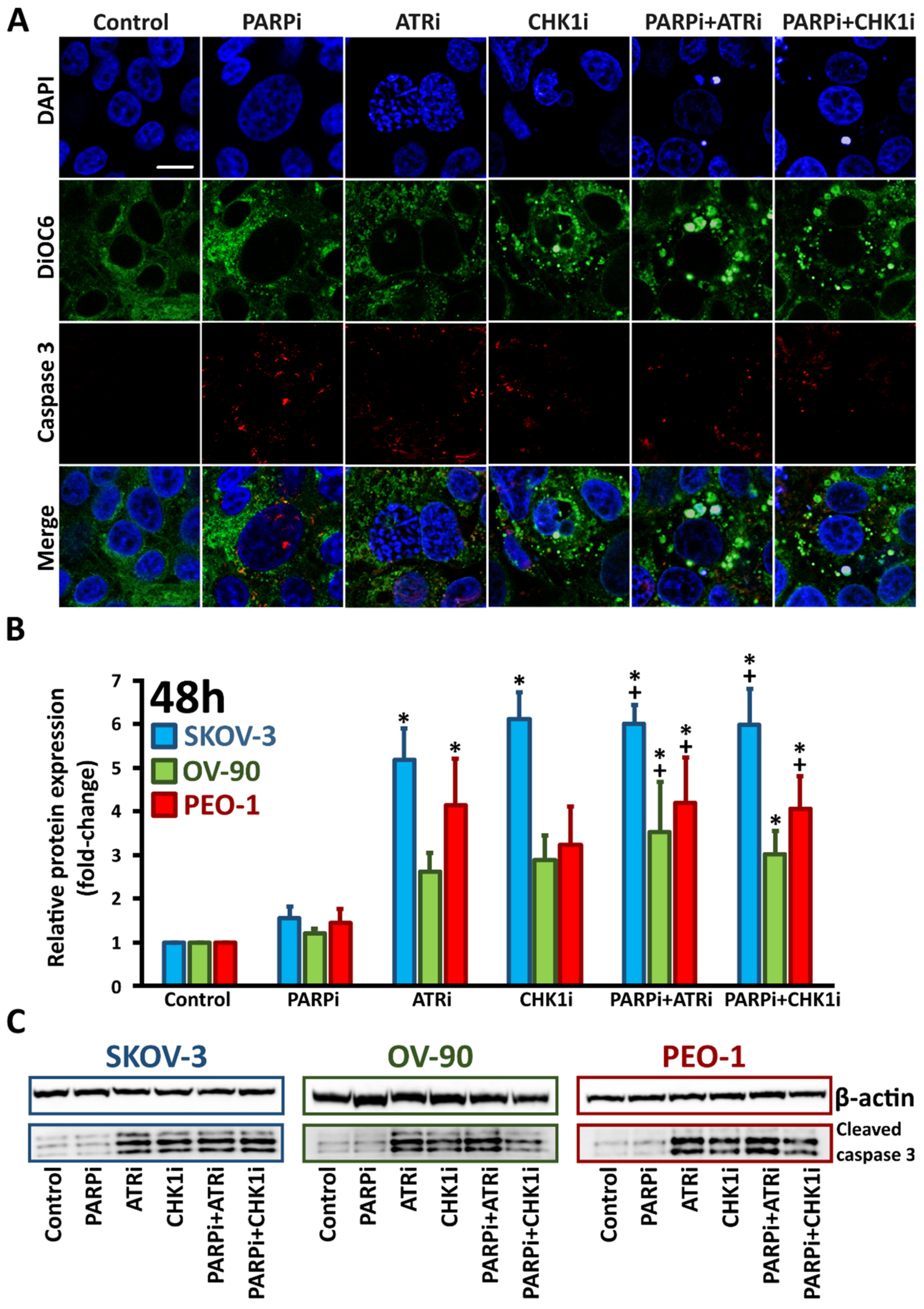
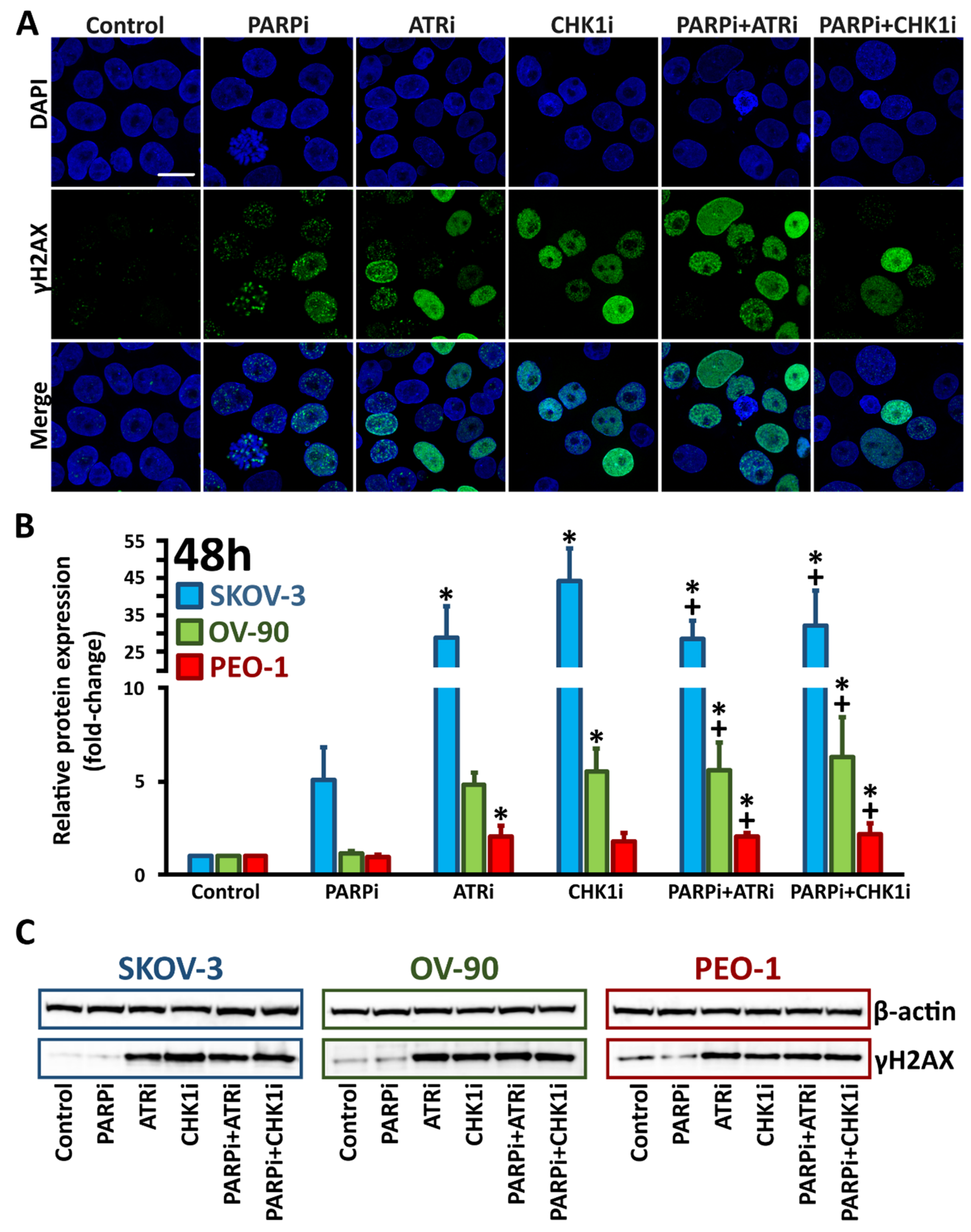
3.2. ATRi and CHK1imonotherapy Increases Apoptosis and DNA Damage
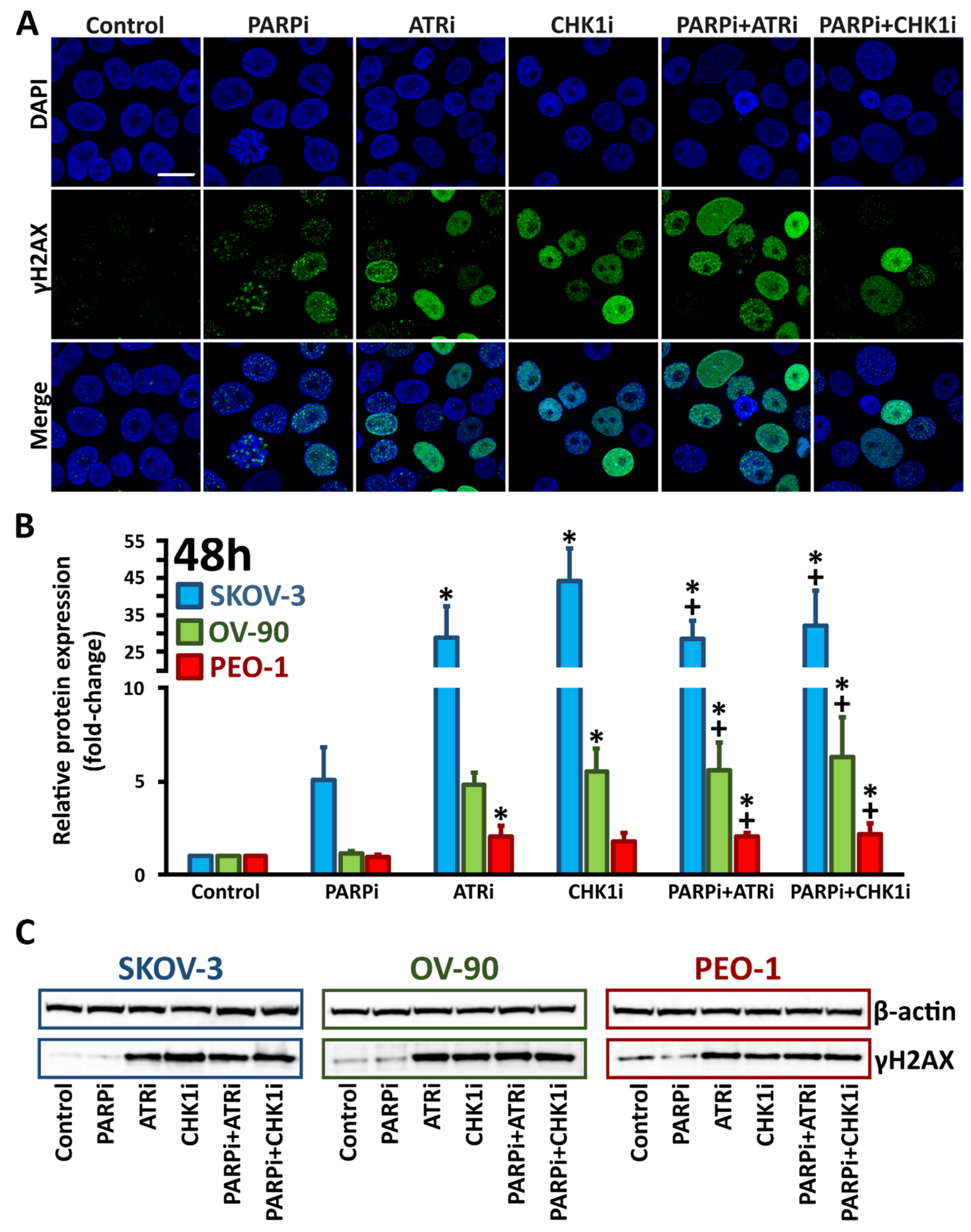
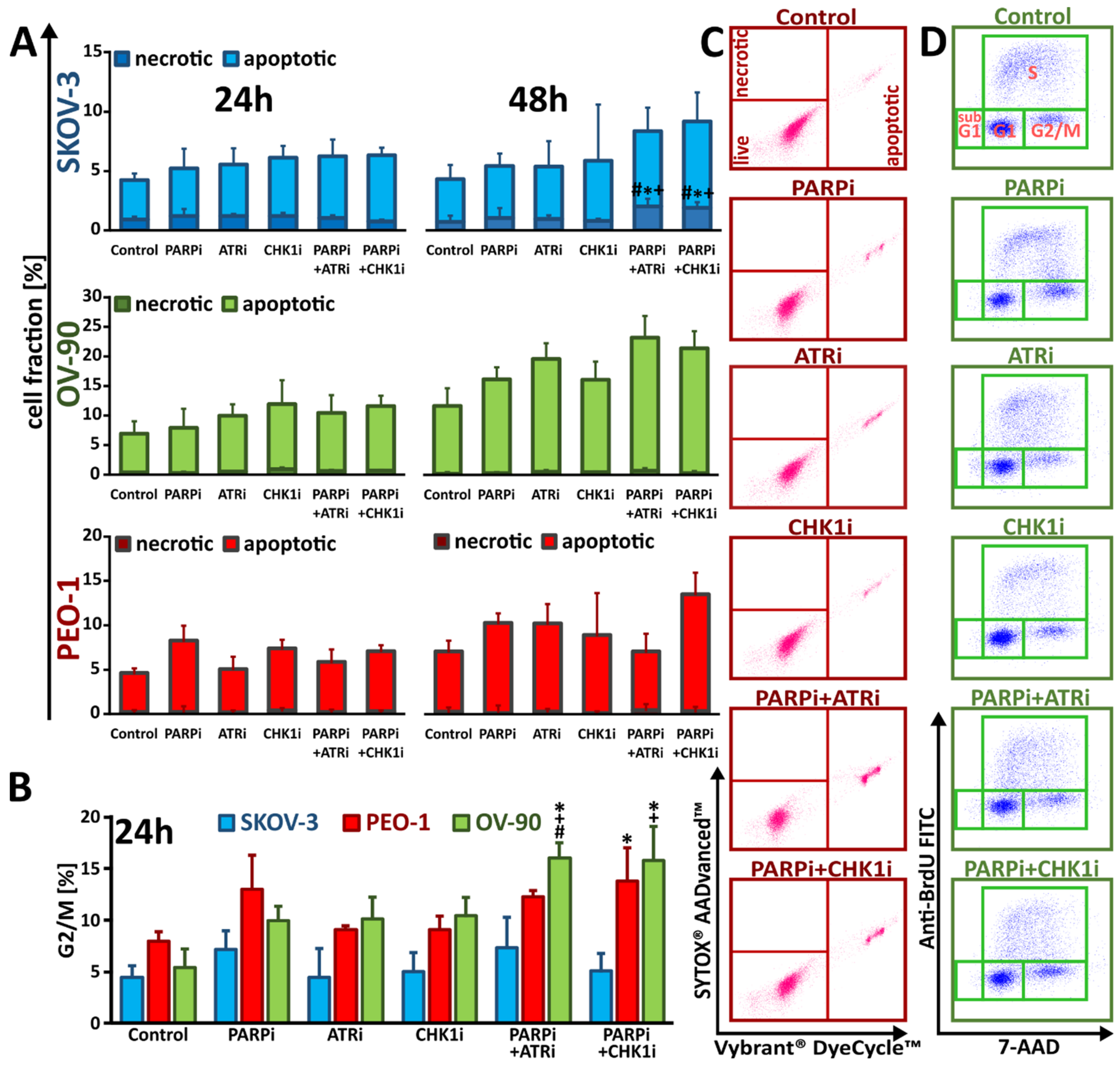
3.3. Monotherapy with ATRi/CHK1i and Combination Treatment with PARPi Activates DNA Damage Response Signaling through Chromatin Condensation and Progression through the G2/M Checkpoint
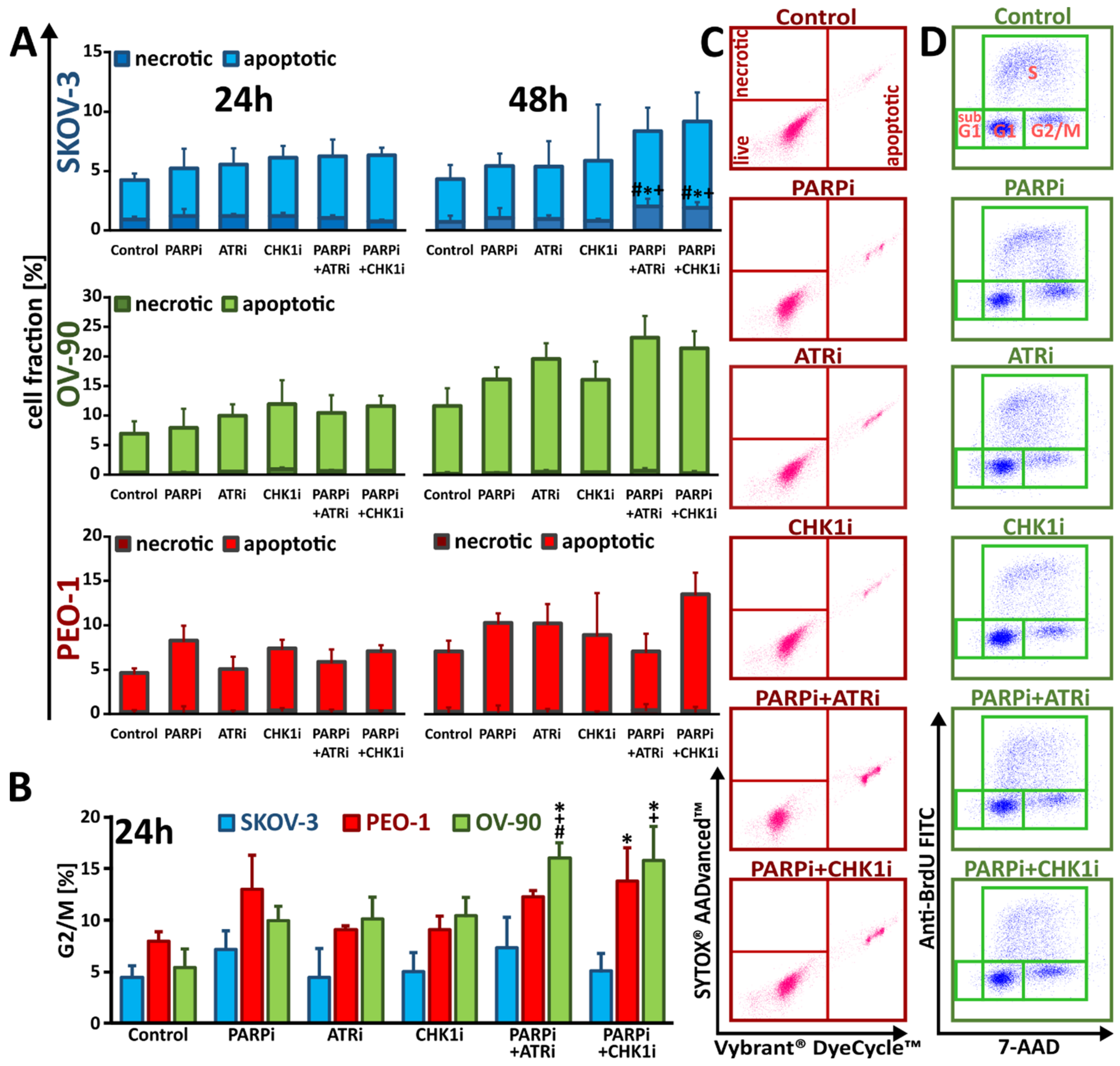
3.4. Treatment with ATRi/CHK1i Induces Aberrant Cell Cycle Progression and Increases RS Levels
3.5. Treatment with ATRi/CHK1i in Combination with PARPi Increases Chromosomal Abnormalities
3.6. ATR Inhibition Intensifies PARPi-Induced Formation of MN in BRCAMUT Cells
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Ahmed, N.; Kadife, E.; Raza, A.; Short, M.; Jubinsky, P.T.; Kannourakis, G. Ovarian Cancer, Cancer Stem Cells and Current Treatment Strategies: A Potential Role of Magmas in the Current Treatment Methods. Cells 2020, 9, 719. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Beaufort, C.M.; Helmijr, J.C.; Piskorz, A.M.; Hoogstraat, M.; Ruigrok-Ritstier, K.; Besselink, N.; Murtaza, M.; Van, I.W.F.; Heine, A.A.; Smid, M.; et al. Ovarian cancer cell line panel (OCCP): Clinical importance of in vitro morphological subtypes. PLoS ONE 2014, 9, e103988. [Google Scholar] [CrossRef] [PubMed]
- Da Cunha Colombo Bonadio, R.R.; Fogace, R.N.; Miranda, V.C.; Diz, M. Homologous recombination deficiency in ovarian cancer: A review of its epidemiology and management. Clinics 2018, 73, e450s. [Google Scholar] [CrossRef] [PubMed]
- Konstantinopoulos, P.A.; Lheureux, S.; Moore, K.N. PARP Inhibitors for Ovarian Cancer: Current Indications, Future Combinations, and Novel Assets in Development to Target DNA Damage Repair. Am. Soc. Clin. Oncol. Educ. Book 2020, 40, e116–e131. [Google Scholar] [CrossRef]
- Barnes, D.E.; Lindahl, T. Repair and genetic consequences of endogenous DNA base damage in mammalian cells. Annu. Rev. Genet. 2004, 38, 445–476. [Google Scholar] [CrossRef] [Green Version]
- Ito, S.; Murphy, C.G.; Doubrovina, E.; Jasin, M.; Moynahan, M.E. PARP Inhibitors in Clinical Use Induce Genomic Instability in Normal Human Cells. PLoS ONE 2016, 11, e0159341. [Google Scholar] [CrossRef]
- Paviolo, N.S.; Vega, M.B.; Pansa, M.F.; Garcia, I.A.; Calzetta, N.L.; Soria, G.; Gottifredi, V. Persistent double strand break accumulation does not precede cell death in an Olaparib-sensitive BRCA-deficient colorectal cancer cell model. Genet. Mol. Biol. 2019, 43, e20190070. [Google Scholar] [CrossRef] [Green Version]
- Ray Chaudhuri, A.; Nussenzweig, A. The multifaceted roles of PARP1 in DNA repair and chromatin remodelling. Nat. Rev. Mol. Cell Biol 2017, 18, 610–621. [Google Scholar] [CrossRef]
- Huang, X.Z.; Jia, H.; Xiao, Q.; Li, R.Z.; Wang, X.S.; Yin, H.Y.; Zhou, X. Efficacy and Prognostic Factors for PARP Inhibitors in Patients With Ovarian Cancer. Front. Oncol. 2020, 10, 958. [Google Scholar] [CrossRef]
- Gralewska, P.; Gajek, A.; Marczak, A.; Mikula, M.; Ostrowski, J.; Sliwinska, A.; Rogalska, A. PARP Inhibition Increases the Reliance on ATR/CHK1 Checkpoint Signaling Leading to Synthetic Lethality-An Alternative Treatment Strategy for Epithelial Ovarian Cancer Cells Independent from HR Effectiveness. Int. J. Mol. Sci. 2020, 21, 9715. [Google Scholar] [CrossRef]
- Zhao, H.; Piwnica-Worms, H. ATR-mediated checkpoint pathways regulate phosphorylation and activation of human Chk1. Mol. Cell. Biol. 2001, 21, 4129–4139. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rybaczek, D.; Kowalewicz-Kulbat, M. Premature chromosome condensation induced by caffeine, 2-aminopurine, staurosporine and sodium metavanadate in S-phase arrested HeLa cells is associated with a decrease in Chk1 phosphorylation, formation of phospho-H2AX and minor cytoskeletal rearrangements. Histochem. Cell Biol. 2011, 135, 263–280. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De Gooijer, M.C.; Van den Top, A.; Bockaj, I.; Beijnen, J.H.; Wurdinger, T.; Van Tellingen, O. The G2 checkpoint-a node-based molecular switch. FEBS Open Bio 2017, 7, 439–455. [Google Scholar] [CrossRef] [PubMed]
- Schoonen, P.M.; Talens, F.; Stok, C.; Gogola, E.; Heijink, A.M.; Bouwman, P.; Foijer, F.; Tarsounas, M.; Blatter, S.; Jonkers, J.; et al. Progression through mitosis promotes PARP inhibitor-induced cytotoxicity in homologous recombination-deficient cancer cells. Nat. Commun. 2017, 8, 15981. [Google Scholar] [CrossRef]
- Helleday, T. Putting poly (ADP-ribose) polymerase and other DNA repair inhibitors into clinical practice. Curr. Opin. Oncol. 2013, 25, 609–614. [Google Scholar] [CrossRef]
- Carmichael, J.; Degraff, W.G.; Gazdar, A.F.; Minna, J.D.; Mitchell, J.B. Evaluation of a Tetrazolium-Based Semiautomated Colorimetric Assay—Assessment of Radiosensitivity. Cancer Res. 1987, 47, 943–946. [Google Scholar]
- Lopez-Acevedo, M.; Grace, L.; Teoh, D.; Whitaker, R.; Adams, D.J.; Jia, J.; Nixon, A.B.; Secord, A.A. Dasatinib (BMS-35482) potentiates the activity of gemcitabine and docetaxel in uterine leiomyosarcoma cell lines. Gynecol. Oncol. Res. Pract. 2014, 1, 2. [Google Scholar] [CrossRef] [Green Version]
- Kaltenbach, J.P.; Kaltenbach, M.H.; Lyons, W.B. Nigrosin as a dye for differentiating live and dead ascites cells. Exp. Cell Res. 1958, 15, 112–117. [Google Scholar] [CrossRef]
- Uzbekov, R.E. Analysis of the cell cycle and a method employing synchronized cells for study of protein expression at various stages of the cell cycle. Biochemistry 2004, 69, 485–496. [Google Scholar] [CrossRef]
- Korzyńska, A.; Zychowicz, M. A method of estimation of the cell doubling time on basis of the cell culture monitoring data. Biocybern. Biomed. Eng. 2008, 28, 75–82. [Google Scholar]
- Rogalska, A.; Gajek, A.; Marczak, A. Suppression of autophagy enhances preferential toxicity of epothilone A and epothilone B in ovarian cancer cells. Phytomedicine 2019, 61, 152847. [Google Scholar] [CrossRef]
- Gajek, A.; Poczta, A.; Łukawska, M.; Cecuda- Adamczewska, V.; Tobiasz, J.; Marczak, A. Chemical modification of melphalan as a key to improving treatment of haematological malignancies. Sci. Rep. 2020, 10, 4479. [Google Scholar] [CrossRef] [Green Version]
- Rogalska, A.; Marczak, A.; Gajek, A.; Szwed, M.; Śliwińska, A.; Drzewoski, J.; Jóźwiak, Z. Induction of apoptosis in human ovarian cancer cells by new anticancer compounds, epothilone A and B. Toxicol. In Vitr. 2013, 27, 239–249. [Google Scholar] [CrossRef]
- Gralewska, P.; Gajek, A.; Marczak, A.; Rogalska, A. Metformin Affects Olaparib Sensitivity through Induction of Apoptosis in Epithelial Ovarian Cancer Cell Lines. Int. J. Mol. Sci. 2021, 22, 10557. [Google Scholar] [CrossRef]
- Olins, A.L.; Olins, D.E. Spheroid chromatin units (v bodies). Science 1974, 183, 330–332. [Google Scholar] [CrossRef]
- Thoma, F.; Koller, T.; Klug, A. Involvement of histone H1 in the organization of the nucleosome and of the salt-dependent superstructures of chromatin. J. Cell Biol. 1979, 83, 403–427. [Google Scholar] [CrossRef]
- Gotoh, E. Visualizing the dynamics of chromosome structure formation coupled with DNA replication. Chromosoma 2007, 116, 453–462. [Google Scholar] [CrossRef]
- Balcerczyk, A.; Rybaczek, D.; Wojtala, M.; Pirola, L.; Okabe, J.; El-Osta, A. Pharmacological inhibition of arginine and lysine methyltransferases induces nuclear abnormalities and suppresses angiogenesis in human endothelial cells. Biochem. Pharmacol. 2016, 121, 18–32. [Google Scholar] [CrossRef]
- Luzhna, L.; Kathiria, P.; Kovalchuk, O. Micronuclei in genotoxicity assessment: From genetics to epigenetics and beyond. Front. Genet. 2013, 4, 131. [Google Scholar] [CrossRef] [Green Version]
- Fenech, M. Cytokinesis-block micronucleus cytome assay. Nat. Protoc. 2007, 2, 1084–1104. [Google Scholar] [CrossRef] [Green Version]
- Xu, Q.; Li, Z. Update on Poly ADP-Ribose Polymerase Inhibitors in Ovarian Cancer with Non-BRCA Mutations. Front. Pharmacol. 2021, 12, 743073. [Google Scholar] [CrossRef]
- FDA. FDA Approved Olaparib (LYNPARZA, AstraZeneca Pharmaceuticals LP) for the Maintenance Treatment of Adult Patients with Deleterious or Suspected Deleterious Germline or Somatic BRCA-Mutated (gBRCAm or sBRCAm) Advanced Epithelial Ovarian, Fallopian Tube or Primary Peritoneal Cancer Who Are in Complete or Partial Response to First-Line Platinum-Based. Available online: https://www.fda.gov/drugs/fda-approved-olaparib-lynparza-astrazeneca-pharmaceuticals-lp-maintenance-treatment-adult-patients (accessed on 2 June 2022).
- Balasubramaniam, S.; Beaver, J.A.; Horton, S.; Fernandes, L.L.; Tang, S.; Horne, H.N.; Liu, J.; Liu, C.; Schrieber, S.J.; Yu, J.; et al. FDA Approval Summary: Rucaparib for the Treatment of Patients with Deleterious BRCA Mutation–Associated Advanced Ovarian Cancer. Clin. Cancer Res. 2017, 23, 7165–7170. [Google Scholar] [CrossRef] [Green Version]
- Do, K.T.; Hill, S.J.; Kochupurakkal, B.; Supko, J.G.; Gannon, C.; Anderson, A.; Muzikansky, A.; Wolanski, A.; Hedglin, J.; Parmar, K.; et al. Abstract CT232: Phase I combination study of the CHK1 inhibitor prexasertib (LY2606368) and olaparib in patients with high-grade serous ovarian cancer and other advanced solid tumors. Cancer Res. 2019, 79, CT232. [Google Scholar] [CrossRef]
- Gomez, M.K.; Illuzzi, G.; Colomer, C.; Churchman, M.; Hollis, R.L.; O’Connor, M.J.; Gourley, C.; Leo, E.; Melton, D.W. Identifying and Overcoming Mechanisms of PARP Inhibitor Resistance in Homologous Recombination Repair-Deficient and Repair-Proficient High Grade Serous Ovarian Cancer Cells. Cancers 2020, 12, 1503. [Google Scholar] [CrossRef]
- Paik, J. Olaparib: A Review as First-Line Maintenance Therapy in Advanced Ovarian Cancer. Target Oncol. 2021, 16, 847–856. [Google Scholar] [CrossRef]
- Vakifahmetoglu, H.; Olsson, M.; Zhivotovsky, B. Death through a tragedy: Mitotic catastrophe. Cell Death Differ. 2008, 15, 1153–1162. [Google Scholar] [CrossRef] [Green Version]
- Lloyd, R.L.; Wijnhoven, P.W.G.; Ramos-Montoya, A.; Wilson, Z.; Illuzzi, G.; Falenta, K.; Jones, G.N.; James, N.; Chabbert, C.D.; Stott, J.; et al. Combined PARP and ATR inhibition potentiates genome instability and cell death in ATM-deficient cancer cells. Oncogene 2020, 39, 4869–4883. [Google Scholar] [CrossRef]
- Miyasaka, A.; Oda, K.; Ikeda, Y.; Wada-Hiraike, O.; Kashiyama, T.; Enomoto, A.; Hosoya, N.; Koso, T.; Fukuda, T.; Inaba, K.; et al. Anti-tumor activity of olaparib, a poly (ADP-ribose) polymerase (PARP) inhibitor, in cultured endometrial carcinoma cells. BMC Cancer 2014, 14, 179. [Google Scholar] [CrossRef] [Green Version]
- Dubois, C.; Martin, F.; Hassel, C.; Magnier, F.; Daumar, P.; Aubel, C.; Guerder, S.; Mounetou, E.; Penault-Lorca, F.; Bamdad, M. Low-Dose and Long-Term Olaparib Treatment Sensitizes MDA-MB-231 and SUM1315 Triple-Negative Breast Cancers Spheroids to Fractioned Radiotherapy. J. Clin. Med. 2019, 9, 64. [Google Scholar] [CrossRef] [Green Version]
- Kummar, S.; Gutierrez, M.; Doroshow, J.H.; Murgo, A.J. Drug development in oncology: Classical cytotoxics and molecularly targeted agents. Br. J. Clin. Pharmacol. 2006, 62, 15–26. [Google Scholar] [CrossRef] [Green Version]
- Poti, A.; Gyergyak, H.; Nemeth, E.; Rusz, O.; Toth, S.; Kovacshazi, C.; Chen, D.; Szikriszt, B.; Spisak, S.; Takeda, S.; et al. Correlation of homologous recombination deficiency induced mutational signatures with sensitivity to PARP inhibitors and cytotoxic agents. Genome Biol. 2019, 20, 240. [Google Scholar] [CrossRef] [PubMed]
- Burgess, R.C.; Burman, B.; Kruhlak, M.J.; Misteli, T. Activation of DNA damage response signaling by condensed chromatin. Cell Rep. 2014, 9, 1703–1717. [Google Scholar] [CrossRef] [Green Version]
- Soutoglou, E.; Misteli, T. Activation of the cellular DNA damage response in the absence of DNA lesions. Science 2008, 320, 1507–1510. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nalabothula, N.; Al-jumaily, T.; Eteleeb, A.M.; Flight, R.M.; Xiaorong, S.; Moseley, H.; Rouchka, E.C.; Fondufe-Mittendorf, Y.N. Genome-Wide Profiling of PARP1 Reveals an Interplay with Gene Regulatory Regions and DNA Methylation. PLoS ONE 2015, 10, e0135410. [Google Scholar] [CrossRef]
- Andronikou, C.; Rottenberg, S. Studying PAR-Dependent Chromatin Remodeling to Tackle PARPi Resistance. Trends Mol. Med. 2021, 27, 630–642. [Google Scholar] [CrossRef] [PubMed]
- Ji, Y.; Tulin, A.V. The roles of PARP1 in gene control and cell differentiation. Curr. Opin. Genet. Dev. 2010, 20, 512–518. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Khurana, S.; Kruhlak, M.J.; Kim, J.; Tran, A.D.; Liu, J.; Nyswaner, K.; Shi, L.; Jailwala, P.; Sung, M.H.; Hakim, O.; et al. A macrohistone variant links dynamic chromatin compaction to BRCA1-dependent genome maintenance. Cell Rep. 2014, 8, 1049–1062. [Google Scholar] [CrossRef] [Green Version]
- Smeenk, G.; Wiegant, W.W.; Vrolijk, H.; Solari, A.P.; Pastink, A.; Van Attikum, H. The NuRD chromatin-remodeling complex regulates signaling and repair of DNA damage. J. Cell Biol. 2010, 190, 741–749. [Google Scholar] [CrossRef]
- Colicchia, V.; Petroni, M.; Guarguaglini, G.; Sardina, F.; Sahún-Roncero, M.; Carbonari, M.; Ricci, B.; Heil, C.; Capalbo, C.; Belardinilli, F.; et al. PARP inhibitors enhance replication stress and cause mitotic catastrophe in MYCN-dependent neuroblastoma. Oncogene 2017, 36, 4682–4691. [Google Scholar] [CrossRef]
- Dillon, M.T.; Barker, H.E.; Pedersen, M.; Hafsi, H.; Bhide, S.A.; Newbold, K.L.; Nutting, C.M.; McLaughlin, M.; Harrington, K.J. Radiosensitization by the ATR Inhibitor AZD6738 through Generation of Acentric Micronuclei. Mol. Cancer Ther. 2017, 16, 25–34. [Google Scholar] [CrossRef] [Green Version]
- Michelena, J.; Lezaja, A.; Teloni, F.; Schmid, T.; Imhof, R.; Altmeyer, M. Analysis of PARP inhibitor toxicity by multidimensional fluorescence microscopy reveals mechanisms of sensitivity and resistance. Nat. Commun. 2018, 9, 2678. [Google Scholar] [CrossRef]
- Schoonen, P.M.; Kok, Y.P.; Wierenga, E.; Bakker, B.; Foijer, F.; Spierings, D.C.J.; Van Vugt, M. Premature mitotic entry induced by ATR inhibition potentiates olaparib inhibition-mediated genomic instability, inflammatory signaling, and cytotoxicity in BRCA2-deficient cancer cells. Mol. Oncol. 2019, 13, 2422–2440. [Google Scholar] [CrossRef] [Green Version]
- Wang, J.Y.J. Cell Death Response to DNA Damage. Yale J. Biol. Med. 2019, 92, 771–779. [Google Scholar]
- Gravells, P.; Neale, J.; Grant, E.; Nathubhai, A.; Smith, K.M.; James, D.I.; Bryant, H.E. Radiosensitization with an inhibitor of poly(ADP-ribose) glycohydrolase: A comparison with the PARP1/2/3 inhibitor olaparib. DNA Repair 2018, 61, 25–36. [Google Scholar] [CrossRef]
- Saldivar, J.C.; Hamperl, S.; Bocek, M.J.; Chung, M.; Bass, T.E.; Cisneros-Soberanis, F.; Samejima, K.; Xie, L.; Paulson, J.R.; Earnshaw, W.C.; et al. An intrinsic S/G2 checkpoint enforced by ATR. Science 2018, 361, 806–810. [Google Scholar] [CrossRef] [Green Version]
- Patil, M.; Pabla, N.; Dong, Z. Checkpoint kinase 1 in DNA damage response and cell cycle regulation. Cell Mol. Life Sci. 2013, 70, 4009–4021. [Google Scholar] [CrossRef] [Green Version]
- Mikhailov, A.; Cole, R.W.; Rieder, C.L. DNA damage during mitosis in human cells delays the metaphase/anaphase transition via the spindle-assembly checkpoint. Curr. Biol. 2002, 12, 1797–1806. [Google Scholar] [CrossRef] [Green Version]
- Slade, D. Mitotic functions of poly(ADP-ribose) polymerases. Biochem. Pharmacol. 2019, 167, 33–43. [Google Scholar] [CrossRef]
- Young, L.A.; O’Connor, L.O.; De Renty, C.; Veldman-Jones, M.H.; Dorval, T.; Wilson, Z.; Jones, D.R.; Lawson, D.; Odedra, R.; Maya-Mendoza, A.; et al. Differential Activity of ATR and WEE1 Inhibitors in a Highly Sensitive Subpopulation of DLBCL Linked to Replication Stress. Cancer Res. 2019, 79, 3762–3775. [Google Scholar] [CrossRef] [Green Version]
- Vescarelli, E.; Gerini, G.; Megiorni, F.; Anastasiadou, E.; Pontecorvi, P.; Solito, L.; De Vitis, C.; Camero, S.; Marchetti, C.; Mancini, R.; et al. MiR-200c sensitizes Olaparib-resistant ovarian cancer cells by targeting Neuropilin 1. J. Exp. Clin. Cancer Res. 2020, 39, 3. [Google Scholar] [CrossRef]
- Fleury, H.; Malaquin, N.; Tu, V.; Gilbert, S.; Martinez, A.; Olivier, M.-A.; Sauriol, A.; Communal, L.; Leclerc-Desaulniers, K.; Carmona, E.; et al. Exploiting interconnected synthetic lethal interactions between PARP inhibition and cancer cell reversible senescence. Nat. Commun. 2019, 10, 2556. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sule, A.; Van Doorn, J.; Sundaram, R.K.; Ganesa, S.; Vasquez, J.C.; Bindra, R.S. Targeting IDH1/2 mutant cancers with combinations of ATR and PARP inhibitors. NAR Cancer 2021, 3, zcab018. [Google Scholar] [CrossRef]
- Huntoon, C.J.; Flatten, K.S.; Wahner Hendrickson, A.E.; Huehls, A.M.; Sutor, S.L.; Kaufmann, S.H.; Karnitz, L.M. ATR Inhibition Broadly Sensitizes Ovarian Cancer Cells to Chemotherapy Independent of BRCA Status. Cancer Res. 2013, 73, 3683–3691. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fong, P.C.; Boss, D.S.; Yap, T.A.; Tutt, A.; Wu, P.; Mergui-Roelvink, M.; Mortimer, P.; Swaisland, H.; Lau, A.; O’Connor, M.J.; et al. Inhibition of poly(ADP-ribose) polymerase in tumors from BRCA mutation carriers. N. Engl. J. Med. 2009, 361, 123–134. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kukolj, E.; Kaufmann, T.; Dick, A.E.; Zeillinger, R.; Gerlich, D.W.; Slade, D. PARP inhibition causes premature loss of cohesion in cancer cells. Oncotarget 2017, 8, 103931–103951. [Google Scholar] [CrossRef] [PubMed]
- Southgate, H.E.D.; Chen, L.; Tweddle, D.A.; Curtin, N.J. ATR Inhibition Potentiates PARP Inhibitor Cytotoxicity in High Risk Neuroblastoma Cell Lines by Multiple Mechanisms. Cancers 2020, 12, 1095. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.; George, E.; Ragland, R.L.; Rafail, S.; Zhang, R.; Krepler, C.; Morgan, M.A.; Herlyn, M.; Brown, E.J.; Simpkins, F. Targeting the ATR/CHK1 Axis with PARP Inhibition Results in Tumor Regression in BRCA-Mutant Ovarian Cancer Models. Clin. Cancer Res. 2017, 23, 3097–3108. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Engelke, C.G.; Parsels, L.A.; Qian, Y.; Zhang, Q.; Karnak, D.; Robertson, J.R.; Tanska, D.M.; Wei, D.; Davis, M.A.; Parsels, J.D.; et al. Sensitization of pancreatic cancer to chemoradiation by the Chk1 inhibitor MK8776. Clin. Cancer Res. 2013, 19, 4412–4421. [Google Scholar] [CrossRef] [Green Version]




Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gralewska, P.; Gajek, A.; Rybaczek, D.; Marczak, A.; Rogalska, A. The Influence of PARP, ATR, CHK1 Inhibitors on Premature Mitotic Entry and Genomic Instability in High-Grade Serous BRCAMUT and BRCAWT Ovarian Cancer Cells. Cells 2022, 11, 1889. https://doi.org/10.3390/cells11121889
Gralewska P, Gajek A, Rybaczek D, Marczak A, Rogalska A. The Influence of PARP, ATR, CHK1 Inhibitors on Premature Mitotic Entry and Genomic Instability in High-Grade Serous BRCAMUT and BRCAWT Ovarian Cancer Cells. Cells. 2022; 11(12):1889. https://doi.org/10.3390/cells11121889
Chicago/Turabian StyleGralewska, Patrycja, Arkadiusz Gajek, Dorota Rybaczek, Agnieszka Marczak, and Aneta Rogalska. 2022. "The Influence of PARP, ATR, CHK1 Inhibitors on Premature Mitotic Entry and Genomic Instability in High-Grade Serous BRCAMUT and BRCAWT Ovarian Cancer Cells" Cells 11, no. 12: 1889. https://doi.org/10.3390/cells11121889
APA StyleGralewska, P., Gajek, A., Rybaczek, D., Marczak, A., & Rogalska, A. (2022). The Influence of PARP, ATR, CHK1 Inhibitors on Premature Mitotic Entry and Genomic Instability in High-Grade Serous BRCAMUT and BRCAWT Ovarian Cancer Cells. Cells, 11(12), 1889. https://doi.org/10.3390/cells11121889