
The Fatal Circle of NETs and NET-Associated DAMPs Contributing to Organ Dysfunction



Abstract
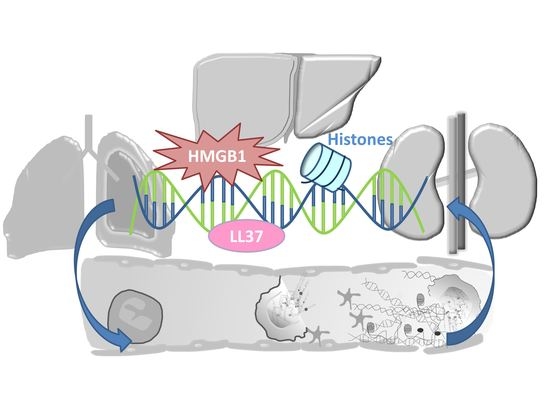
:
1. Introduction
2. Mechanisms of Extracellular Trap Formation and Clearance
2.1. Lytic or Suicidal NET Formation
2.2. Non-Lytic or Vital NET Formation
2.3. Other Forms of Extracellular Trap Formation
2.4. Degradation or Anti-Inflammatory Properties of NETs
3. DAMPs Associated with NETs or Capable of Inducing NETs
3.1. Histones
3.2. Cell Free DNA (cfDNA)
3.3. LL37—mCRAMP
3.4. High-Motility Group Box 1 (HMGB1)
3.5. Cold-Inducible RNA Binding Protein (CIRP)
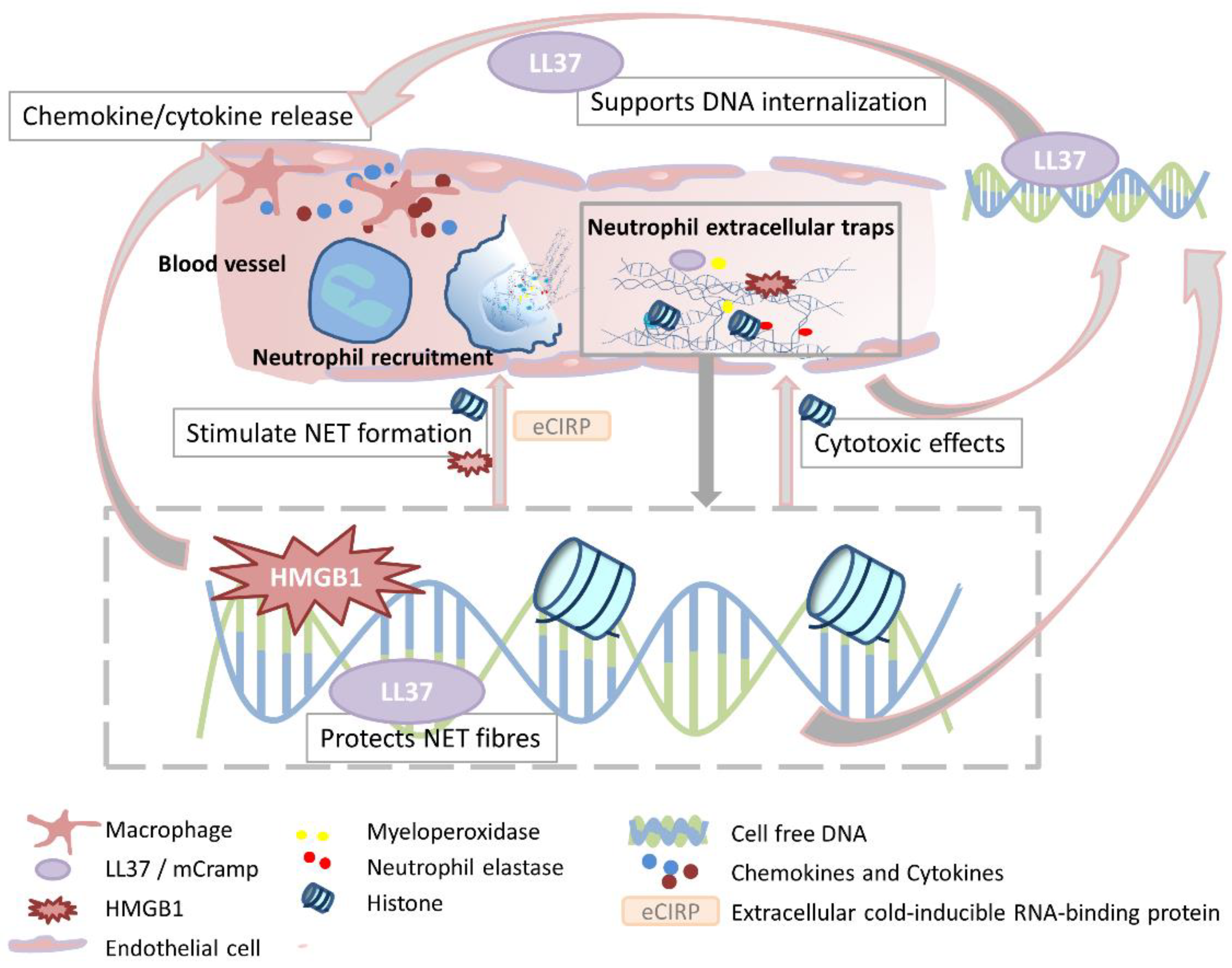
4. Organ Dysfunction and Remote Organ Injury
4.1. Kidney
4.1.1. Anti-Neutrophil Cytoplasmic Antibody (ANCA)-Associated Vasculitis
4.1.2. Acute Kidney Injury (AKI)
4.2. Lung
4.3. Liver
4.4. Immunothrombosis
5. NET-Targeting Therapies
5.1. Dnase1
5.2. Histones
5.3. HMGB1
5.4. Other Treatments
6. Summary
Author Contributions
Funding
Conflicts of Interest
References
- Li, D.; Wu, M. Pattern recognition receptors in health and diseases. Signal. Transduct. Target. Ther. 2021, 6, 291. [Google Scholar] [CrossRef] [PubMed]
- Amarante-Mendes, G.P.; Adjemian, S.; Branco, L.M.; Zanetti, L.C.; Weinlich, R.; Bortoluci, K.R. Pattern Recognition Receptors and the Host Cell Death Molecular Machinery. Front. Immunol. 2018, 9, 2379. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fukata, M.; Vamadevan, A.S.; Abreu, M.T. Toll-like receptors (TLRs) and Nod-like receptors (NLRs) in inflammatory disorders. Semin. Immunol. 2009, 21, 242–253. [Google Scholar] [CrossRef] [PubMed]
- Zindel, J.; Kubes, P. DAMPs, PAMPs, and LAMPs in Immunity and Sterile Inflammation. Annu. Rev. Pathol. 2020, 15, 493–518. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huber-Lang, M.; Lambris, J.D.; Ward, P.A. Innate immune responses to trauma. Nat. Immunol. 2018, 19, 327–341. [Google Scholar] [CrossRef]
- Andonegui, G.; Kerfoot, S.M.; McNagny, K.; Ebbert, K.V.; Patel, K.D.; Kubes, P. Platelets express functional Toll-like receptor-4. Blood 2005, 106, 2417–2423. [Google Scholar] [CrossRef]
- Wagner, D.D.; Burger, P.C. Platelets in inflammation and thrombosis. Arterioscler. Thromb. Vasc. Biol. 2003, 23, 2131–2137. [Google Scholar] [CrossRef]
- Zarbock, A.; Singbartl, K.; Ley, K. Complete reversal of acid-induced acute lung injury by blocking of platelet-neutrophil aggregation. J. Clin. Investig. 2006, 116, 3211–3219. [Google Scholar] [CrossRef] [Green Version]
- Auffray, C.; Sieweke, M.H.; Geissmann, F. Blood monocytes: Development, heterogeneity, and relationship with dendritic cells. Annu. Rev. Immunol. 2009, 27, 669–692. [Google Scholar] [CrossRef] [Green Version]
- Cappenberg, A.; Kardell, M.; Zarbock, A. Selectin-Mediated Signaling-Shedding Light on the Regulation of Integrin Activity in Neutrophils. Cells 2022, 11, 1310. [Google Scholar] [CrossRef]
- Brinkmann, V.; Reichard, U.; Goosmann, C.; Fauler, B.; Uhlemann, Y.; Weiss, D.S.; Weinrauch, Y.; Zychlinsky, A. Neutrophil extracellular traps kill bacteria. Science 2004, 303, 1532–1535. [Google Scholar] [CrossRef] [PubMed]
- Urban, C.F.; Reichard, U.; Brinkmann, V.; Zychlinsky, A. Neutrophil extracellular traps capture and kill Candida albicans yeast and hyphal forms. Cell. Microbiol. 2006, 8, 668–676. [Google Scholar] [CrossRef] [PubMed]
- Dwyer, M.; Shan, Q.; D’Ortona, S.; Maurer, R.; Mitchell, R.; Olesen, H.; Thiel, S.; Huebner, J.; Gadjeva, M. Cystic fibrosis sputum DNA has NETosis characteristics and neutrophil extracellular trap release is regulated by macrophage migration-inhibitory factor. J. Innate Immun. 2014, 6, 765–779. [Google Scholar] [CrossRef] [PubMed]
- Khandpur, R.; Carmona-Rivera, C.; Vivekanandan-Giri, A.; Gizinski, A.; Yalavarthi, S.; Knight, J.S.; Friday, S.; Li, S.; Patel, R.M.; Subramanian, V.; et al. NETs are a source of citrullinated autoantigens and stimulate inflammatory responses in rheumatoid arthritis. Sci. Transl. Med. 2013, 5, 178ra40. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, T.; Li, Y.; Sun, R.; Hu, H.; Liu, Y.; Herrmann, M.; Zhao, Y.; Munoz, L.E. Receptor-Mediated NETosis on Neutrophils. Front. Immunol. 2021, 12, 775267. [Google Scholar] [CrossRef]
- Gupta, A.K.; Giaglis, S.; Hasler, P.; Hahn, S. Efficient neutrophil extracellular trap induction requires mobilization of both intracellular and extracellular calcium pools and is modulated by cyclosporine A. PLoS ONE 2014, 9, e97088. [Google Scholar] [CrossRef] [Green Version]
- Hakkim, A.; Fuchs, T.A.; Martinez, N.E.; Hess, S.; Prinz, H.; Zychlinsky, A.; Waldmann, H. Activation of the Raf-MEK-ERK pathway is required for neutrophil extracellular trap formation. Nat. Chem. Biol. 2011, 7, 75–77. [Google Scholar] [CrossRef]
- Metzler, K.D.; Goosmann, C.; Lubojemska, A.; Zychlinsky, A.; Papayannopoulos, V. A myeloperoxidase-containing complex regulates neutrophil elastase release and actin dynamics during NETosis. Cell Rep. 2014, 8, 883–896. [Google Scholar] [CrossRef] [Green Version]
- Papayannopoulos, V.; Metzler, K.D.; Hakkim, A.; Zychlinsky, A. Neutrophil elastase and myeloperoxidase regulate the formation of neutrophil extracellular traps. J. Cell Biol. 2010, 191, 677–691. [Google Scholar] [CrossRef] [Green Version]
- Li, P.; Li, M.; Lindberg, M.R.; Kennett, M.J.; Xiong, N.; Wang, Y. PAD4 is essential for antibacterial innate immunity mediated by neutrophil extracellular traps. J. Exp. Med. 2010, 207, 1853–1862. [Google Scholar] [CrossRef]
- Parker, H.; Dragunow, M.; Hampton, M.B.; Kettle, A.J.; Winterbourn, C.C. Requirements for NADPH oxidase and myeloperoxidase in neutrophil extracellular trap formation differ depending on the stimulus. J. Leukoc. Biol. 2012, 92, 841–849. [Google Scholar] [CrossRef] [PubMed]
- Diaz-Godinez, C.; Fonseca, Z.; Nequiz, M.; Laclette, J.P.; Rosales, C.; Carrero, J.C. Entamoeba histolytica Trophozoites Induce a Rapid Non-classical NETosis Mechanism Independent of NOX2-Derived Reactive Oxygen Species and PAD4 Activity. Front. Cell. Infect. Microbiol. 2018, 8, 184. [Google Scholar] [CrossRef] [PubMed]
- Martinod, K.; Witsch, T.; Farley, K.; Gallant, M.; Remold-O’Donnell, E.; Wagner, D.D. Neutrophil elastase-deficient mice form neutrophil extracellular traps in an experimental model of deep vein thrombosis. J. Thromb. Haemost. 2016, 14, 551–558. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Amulic, B.; Knackstedt, S.L.; Abu Abed, U.; Deigendesch, N.; Harbort, C.J.; Caffrey, B.E.; Brinkmann, V.; Heppner, F.L.; Hinds, P.W.; Zychlinsky, A. Cell-Cycle Proteins Control Production of Neutrophil Extracellular Traps. Dev. Cell 2017, 43, 449–462.e5. [Google Scholar] [CrossRef] [PubMed]
- Fuchs, T.A.; Abed, U.; Goosmann, C.; Hurwitz, R.; Schulze, I.; Wahn, V.; Weinrauch, Y.; Brinkmann, V.; Zychlinsky, A. Novel cell death program leads to neutrophil extracellular traps. J. Cell Biol. 2007, 176, 231–241. [Google Scholar] [CrossRef] [PubMed]
- Chen, K.W.; Monteleone, M.; Boucher, D.; Sollberger, G.; Ramnath, D.; Condon, N.D.; von Pein, J.B.; Broz, P.; Sweet, M.J.; Schroder, K. Noncanonical inflammasome signaling elicits gasdermin D-dependent neutrophil extracellular traps. Sci. Immunol. 2018, 3, eaar6676. [Google Scholar] [CrossRef] [Green Version]
- Sollberger, G.; Choidas, A.; Burn, G.L.; Habenberger, P.; Di Lucrezia, R.; Kordes, S.; Menninger, S.; Eickhoff, J.; Nussbaumer, P.; Klebl, B.; et al. Gasdermin D plays a vital role in the generation of neutrophil extracellular traps. Sci. Immunol. 2018, 3, eaar6689. [Google Scholar] [CrossRef] [Green Version]
- Clark, S.R.; Ma, A.C.; Tavener, S.A.; McDonald, B.; Goodarzi, Z.; Kelly, M.M.; Patel, K.D.; Chakrabarti, S.; McAvoy, E.; Sinclair, G.D.; et al. Platelet TLR4 activates neutrophil extracellular traps to ensnare bacteria in septic blood. Nat. Med. 2007, 13, 463–469. [Google Scholar] [CrossRef]
- McDonald, B.; Urrutia, R.; Yipp, B.G.; Jenne, C.N.; Kubes, P. Intravascular neutrophil extracellular traps capture bacteria from the bloodstream during sepsis. Cell Host Microbe 2012, 12, 324–333. [Google Scholar] [CrossRef] [Green Version]
- Yipp, B.G.; Petri, B.; Salina, D.; Jenne, C.N.; Scott, B.N.; Zbytnuik, L.D.; Pittman, K.; Asaduzzaman, M.; Wu, K.; Meijndert, H.C.; et al. Infection-induced NETosis is a dynamic process involving neutrophil multitasking in vivo. Nat. Med. 2012, 18, 1386–1393. [Google Scholar] [CrossRef] [Green Version]
- Byrd, A.S.; O’Brien, X.M.; Johnson, C.M.; Lavigne, L.M.; Reichner, J.S. An extracellular matrix-based mechanism of rapid neutrophil extracellular trap formation in response to Candida albicans. J. Immunol. 2013, 190, 4136–4148. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pilsczek, F.H.; Salina, D.; Poon, K.K.; Fahey, C.; Yipp, B.G.; Sibley, C.D.; Robbins, S.M.; Green, F.H.; Surette, M.G.; Sugai, M.; et al. A novel mechanism of rapid nuclear neutrophil extracellular trap formation in response to Staphylococcus aureus. J. Immunol. 2010, 185, 7413–7425. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yipp, B.G.; Kubes, P. NETosis: How vital is it? Blood 2013, 122, 2784–2794. [Google Scholar] [CrossRef] [PubMed]
- Jorch, S.K.; Kubes, P. An emerging role for neutrophil extracellular traps in noninfectious disease. Nat. Med. 2017, 23, 279–287. [Google Scholar] [CrossRef] [PubMed]
- Roos, D.; Voetman, A.A.; Meerhof, L.J. Functional activity of enucleated human polymorphonuclear leukocytes. J. Cell Biol. 1983, 97, 368–377. [Google Scholar] [CrossRef]
- Krishnamoorthy, N.; Douda, D.N.; Bruggemann, T.R.; Ricklefs, I.; Duvall, M.G.; Abdulnour, R.E.; Martinod, K.; Tavares, L.; Wang, X.; Cernadas, M.; et al. Neutrophil cytoplasts induce TH17 differentiation and skew inflammation toward neutrophilia in severe asthma. Sci. Immunol. 2018, 3, eaao4747. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yousefi, S.; Mihalache, C.; Kozlowski, E.; Schmid, I.; Simon, H.U. Viable neutrophils release mitochondrial DNA to form neutrophil extracellular traps. Cell Death Differ. 2009, 16, 1438–1444. [Google Scholar] [CrossRef] [PubMed]
- Yousefi, S.; Gold, J.A.; Andina, N.; Lee, J.J.; Kelly, A.M.; Kozlowski, E.; Schmid, I.; Straumann, A.; Reichenbach, J.; Gleich, G.J.; et al. Catapult-like release of mitochondrial DNA by eosinophils contributes to antibacterial defense. Nat. Med. 2008, 14, 949–953. [Google Scholar] [CrossRef] [PubMed]
- Doster, R.S.; Rogers, L.M.; Gaddy, J.A.; Aronoff, D.M. Macrophage Extracellular Traps: A Scoping Review. J. Innate Immun. 2018, 10, 3–13. [Google Scholar] [CrossRef]
- Farrera, C.; Fadeel, B. Macrophage clearance of neutrophil extracellular traps is a silent process. J. Immunol. 2013, 191, 2647–2656. [Google Scholar] [CrossRef] [Green Version]
- Lazzaretto, B.; Fadeel, B. Intra- and Extracellular Degradation of Neutrophil Extracellular Traps by Macrophages and Dendritic Cells. J. Immunol. 2019, 203, 2276–2290. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Apel, F.; Andreeva, L.; Knackstedt, L.S.; Streeck, R.; Frese, C.K.; Goosmann, C.; Hopfner, K.P.; Zychlinsky, A. The cytosolic DNA sensor cGAS recognizes neutrophil extracellular traps. Sci. Signal. 2021, 14, eaax7942. [Google Scholar] [CrossRef] [PubMed]
- Nakazawa, D.; Shida, H.; Kusunoki, Y.; Miyoshi, A.; Nishio, S.; Tomaru, U.; Atsumi, T.; Ishizu, A. The responses of macrophages in interaction with neutrophils that undergo NETosis. J. Autoimmun. 2016, 67, 19–28. [Google Scholar] [CrossRef]
- Knopf, J.; Leppkes, M.; Schett, G.; Herrmann, M.; Munoz, L.E. Aggregated NETs Sequester and Detoxify Extracellular Histones. Front. Immunol. 2019, 10, 2176. [Google Scholar] [CrossRef] [PubMed]
- Schauer, C.; Janko, C.; Munoz, L.E.; Zhao, Y.; Kienhofer, D.; Frey, B.; Lell, M.; Manger, B.; Rech, J.; Naschberger, E.; et al. Aggregated neutrophil extracellular traps limit inflammation by degrading cytokines and chemokines. Nat. Med. 2014, 20, 511–517. [Google Scholar] [CrossRef]
- Hahn, J.; Schauer, C.; Czegley, C.; Kling, L.; Petru, L.; Schmid, B.; Weidner, D.; Reinwald, C.; Biermann, M.H.C.; Blunder, S.; et al. Aggregated neutrophil extracellular traps resolve inflammation by proteolysis of cytokines and chemokines and protection from antiproteases. FASEB J. 2019, 33, 1401–1414. [Google Scholar] [CrossRef] [PubMed]
- Xu, J.; Zhang, X.; Monestier, M.; Esmon, N.L.; Esmon, C.T. Extracellular histones are mediators of death through TLR2 and TLR4 in mouse fatal liver injury. J. Immunol. 2011, 187, 2626–2631. [Google Scholar] [CrossRef] [Green Version]
- Nakazawa, D.; Kumar, S.V.; Marschner, J.; Desai, J.; Holderied, A.; Rath, L.; Kraft, F.; Lei, Y.; Fukasawa, Y.; Moeckel, G.W.; et al. Histones and Neutrophil Extracellular Traps Enhance Tubular Necrosis and Remote Organ Injury in Ischemic AKI. J. Am. Soc. Nephrol. 2017, 28, 1753–1768. [Google Scholar] [CrossRef] [Green Version]
- Huang, H.; Tohme, S.; Al-Khafaji, A.B.; Tai, S.; Loughran, P.; Chen, L.; Wang, S.; Kim, J.; Billiar, T.; Wang, Y.; et al. Damage-associated molecular pattern-activated neutrophil extracellular trap exacerbates sterile inflammatory liver injury. Hepatology 2015, 62, 600–614. [Google Scholar] [CrossRef] [Green Version]
- Magna, M.; Pisetsky, D.S. The Alarmin Properties of DNA and DNA-associated Nuclear Proteins. Clin. Ther. 2016, 38, 1029–1041. [Google Scholar] [CrossRef] [Green Version]
- Ganguly, D.; Chamilos, G.; Lande, R.; Gregorio, J.; Meller, S.; Facchinetti, V.; Homey, B.; Barrat, F.J.; Zal, T.; Gilliet, M. Self-RNA-antimicrobial peptide complexes activate human dendritic cells through TLR7 and TLR8. J. Exp. Med. 2009, 206, 1983–1994. [Google Scholar] [CrossRef] [PubMed]
- Lande, R.; Gregorio, J.; Facchinetti, V.; Chatterjee, B.; Wang, Y.H.; Homey, B.; Cao, W.; Wang, Y.H.; Su, B.; Nestle, F.O.; et al. Plasmacytoid dendritic cells sense self-DNA coupled with antimicrobial peptide. Nature 2007, 449, 564–569. [Google Scholar] [CrossRef] [PubMed]
- Herster, F.; Bittner, Z.; Archer, N.K.; Dickhofer, S.; Eisel, D.; Eigenbrod, T.; Knorpp, T.; Schneiderhan-Marra, N.; Loffler, M.W.; Kalbacher, H.; et al. Neutrophil extracellular trap-associated RNA and LL37 enable self-amplifying inflammation in psoriasis. Nat. Commun. 2020, 11, 105. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Larrick, J.W.; Hirata, M.; Balint, R.F.; Lee, J.; Zhong, J.; Wright, S.C. Human CAP18: A novel antimicrobial lipopolysaccharide-binding protein. Infect. Immun. 1995, 63, 1291–1297. [Google Scholar] [CrossRef] [Green Version]
- Rosenfeld, Y.; Papo, N.; Shai, Y. Endotoxin (lipopolysaccharide) neutralization by innate immunity host-defense peptides. Peptide properties and plausible modes of action. J. Biol. Chem. 2006, 281, 1636–1643. [Google Scholar] [CrossRef] [Green Version]
- Di Nardo, A.; Braff, M.H.; Taylor, K.R.; Na, C.; Granstein, R.D.; McInturff, J.E.; Krutzik, S.; Modlin, R.L.; Gallo, R.L. Cathelicidin antimicrobial peptides block dendritic cell TLR4 activation and allergic contact sensitization. J. Immunol. 2007, 178, 1829–1834. [Google Scholar] [CrossRef] [Green Version]
- Peng, H.H.; Liu, Y.J.; Ojcius, D.M.; Lee, C.M.; Chen, R.H.; Huang, P.R.; Martel, J.; Young, J.D. Mineral particles stimulate innate immunity through neutrophil extracellular traps containing HMGB1. Sci. Rep. 2017, 7, 16628. [Google Scholar] [CrossRef] [Green Version]
- Schiraldi, M.; Raucci, A.; Munoz, L.M.; Livoti, E.; Celona, B.; Venereau, E.; Apuzzo, T.; De Marchis, F.; Pedotti, M.; Bachi, A.; et al. HMGB1 promotes recruitment of inflammatory cells to damaged tissues by forming a complex with CXCL12 and signaling via CXCR4. J. Exp. Med. 2012, 209, 551–563. [Google Scholar] [CrossRef] [Green Version]
- Wu, J.; Li, J.; Salcedo, R.; Mivechi, N.F.; Trinchieri, G.; Horuzsko, A. The proinflammatory myeloid cell receptor TREM-1 controls Kupffer cell activation and development of hepatocellular carcinoma. Cancer Res. 2012, 72, 3977–3986. [Google Scholar] [CrossRef] [Green Version]
- Vogel, S.; Bodenstein, R.; Chen, Q.; Feil, S.; Feil, R.; Rheinlaender, J.; Schaffer, T.E.; Bohn, E.; Frick, J.S.; Borst, O.; et al. Platelet-derived HMGB1 is a critical mediator of thrombosis. J. Clin. Investig. 2015, 125, 4638–4654. [Google Scholar] [CrossRef] [Green Version]
- Vogel, S.; Chatterjee, M.; Metzger, K.; Borst, O.; Geisler, T.; Seizer, P.; Muller, I.; Mack, A.; Schumann, S.; Buhring, H.J.; et al. Activated platelets interfere with recruitment of mesenchymal stem cells to apoptotic cardiac cells via high mobility group box 1/Toll-like receptor 4-mediated down-regulation of hepatocyte growth factor receptor MET. J. Biol. Chem. 2014, 289, 11068–11082. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xiang, M.; Shi, X.; Li, Y.; Xu, J.; Yin, L.; Xiao, G.; Scott, M.J.; Billiar, T.R.; Wilson, M.A.; Fan, J. Hemorrhagic shock activation of NLRP3 inflammasome in lung endothelial cells. J. Immunol. 2011, 187, 4809–4817. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ha, T.; Xia, Y.; Liu, X.; Lu, C.; Liu, L.; Kelley, J.; Kalbfleisch, J.; Kao, R.L.; Williams, D.L.; Li, C. Glucan phosphate attenuates myocardial HMGB1 translocation in severe sepsis through inhibiting NF-kappaB activation. Am. J. Physiol. Heart Circ. Physiol. 2011, 301, H848–H855. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yu, M.; Wang, H.; Ding, A.; Golenbock, D.T.; Latz, E.; Czura, C.J.; Fenton, M.J.; Tracey, K.J.; Yang, H. HMGB1 signals through toll-like receptor (TLR) 4 and TLR2. Shock 2006, 26, 174–179. [Google Scholar] [CrossRef] [PubMed]
- Ivanov, S.; Dragoi, A.M.; Wang, X.; Dallacosta, C.; Louten, J.; Musco, G.; Sitia, G.; Yap, G.S.; Wan, Y.; Biron, C.A.; et al. A novel role for HMGB1 in TLR9-mediated inflammatory responses to CpG-DNA. Blood 2007, 110, 1970–1981. [Google Scholar] [CrossRef]
- Tian, J.; Avalos, A.M.; Mao, S.Y.; Chen, B.; Senthil, K.; Wu, H.; Parroche, P.; Drabic, S.; Golenbock, D.; Sirois, C.; et al. Toll-like receptor 9-dependent activation by DNA-containing immune complexes is mediated by HMGB1 and RAGE. Nat. Immunol. 2007, 8, 487–496. [Google Scholar] [CrossRef] [PubMed]
- Maugeri, N.; Campana, L.; Gavina, M.; Covino, C.; De Metrio, M.; Panciroli, C.; Maiuri, L.; Maseri, A.; D’Angelo, A.; Bianchi, M.E.; et al. Activated platelets present high mobility group box 1 to neutrophils, inducing autophagy and promoting the extrusion of neutrophil extracellular traps. J. Thromb. Haemost. 2014, 12, 2074–2088. [Google Scholar] [CrossRef]
- Qiang, X.; Yang, W.L.; Wu, R.; Zhou, M.; Jacob, A.; Dong, W.; Kuncewitch, M.; Ji, Y.; Yang, H.; Wang, H.; et al. Cold-inducible RNA-binding protein (CIRP) triggers inflammatory responses in hemorrhagic shock and sepsis. Nat. Med. 2013, 19, 1489–1495. [Google Scholar] [CrossRef] [Green Version]
- Ode, Y.; Aziz, M.; Jin, H.; Arif, A.; Nicastro, J.G.; Wang, P. Cold-inducible RNA-binding Protein Induces Neutrophil Extracellular Traps in the Lungs during Sepsis. Sci. Rep. 2019, 9, 6252. [Google Scholar] [CrossRef]
- Ode, Y.; Aziz, M.; Wang, P. CIRP increases ICAM-1(+) phenotype of neutrophils exhibiting elevated iNOS and NETs in sepsis. J. Leukoc. Biol. 2018, 103, 693–707. [Google Scholar] [CrossRef]
- Venkatesh, S.; Workman, J.L. Histone exchange, chromatin structure and the regulation of transcription. Nat. Rev. Mol. Cell Biol. 2015, 16, 178–189. [Google Scholar] [CrossRef] [PubMed]
- Szatmary, P.; Huang, W.; Criddle, D.; Tepikin, A.; Sutton, R. Biology, role and therapeutic potential of circulating histones in acute inflammatory disorders. J. Cell. Mol. Med. 2018, 22, 4617–4629. [Google Scholar] [CrossRef] [PubMed]
- Nair, R.R.; Mazza, D.; Brambilla, F.; Gorzanelli, A.; Agresti, A.; Bianchi, M.E. LPS-Challenged Macrophages Release Microvesicles Coated With Histones. Front. Immunol. 2018, 9, 1463. [Google Scholar] [CrossRef] [PubMed]
- Xu, J.; Zhang, X.; Pelayo, R.; Monestier, M.; Ammollo, C.T.; Semeraro, F.; Taylor, F.B.; Esmon, N.L.; Lupu, F.; Esmon, C.T. Extracellular histones are major mediators of death in sepsis. Nat. Med. 2009, 15, 1318–1321. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Saffarzadeh, M.; Juenemann, C.; Queisser, M.A.; Lochnit, G.; Barreto, G.; Galuska, S.P.; Lohmeyer, J.; Preissner, K.T. Neutrophil extracellular traps directly induce epithelial and endothelial cell death: A predominant role of histones. PLoS ONE 2012, 7, e32366. [Google Scholar] [CrossRef] [PubMed]
- Alhamdi, Y.; Abrams, S.T.; Cheng, Z.; Jing, S.; Su, D.; Liu, Z.; Lane, S.; Welters, I.; Wang, G.; Toh, C.H. Circulating Histones Are Major Mediators of Cardiac Injury in Patients With Sepsis. Crit. Care Med. 2015, 43, 2094–2103. [Google Scholar] [CrossRef] [PubMed]
- Locke, M.; Francis, R.J.; Tsaousi, E.; Longstaff, C. Fibrinogen protects neutrophils from the cytotoxic effects of histones and delays neutrophil extracellular trap formation induced by ionomycin. Sci. Rep. 2020, 10, 11694. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Q.; Itagaki, K.; Hauser, C.J. Mitochondrial DNA is released by shock and activates neutrophils via p38 map kinase. Shock 2010, 34, 55–59. [Google Scholar] [CrossRef]
- Bhagirath, V.C.; Dwivedi, D.J.; Liaw, P.C. Comparison of the Proinflammatory and Procoagulant Properties of Nuclear, Mitochondrial, and Bacterial DNA. Shock 2015, 44, 265–271. [Google Scholar] [CrossRef]
- Mondelo-Macia, P.; Castro-Santos, P.; Castillo-Garcia, A.; Muinelo-Romay, L.; Diaz-Pena, R. Circulating Free DNA and Its Emerging Role in Autoimmune Diseases. J. Pers. Med. 2021, 11, 151. [Google Scholar] [CrossRef]
- Zhang, X.; Oglecka, K.; Sandgren, S.; Belting, M.; Esbjorner, E.K.; Norden, B.; Graslund, A. Dual functions of the human antimicrobial peptide LL-37-target membrane perturbation and host cell cargo delivery. Biochim. Biophys. Acta 2010, 1798, 2201–2208. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yamasaki, K.; Schauber, J.; Coda, A.; Lin, H.; Dorschner, R.A.; Schechter, N.M.; Bonnart, C.; Descargues, P.; Hovnanian, A.; Gallo, R.L. Kallikrein-mediated proteolysis regulates the antimicrobial effects of cathelicidins in skin. FASEB J. 2006, 20, 2068–2080. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sorensen, O.E.; Follin, P.; Johnsen, A.H.; Calafat, J.; Tjabringa, G.S.; Hiemstra, P.S.; Borregaard, N. Human cathelicidin, hCAP-18, is processed to the antimicrobial peptide LL-37 by extracellular cleavage with proteinase 3. Blood 2001, 97, 3951–3959. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Oren, Z.; Lerman, J.C.; Gudmundsson, G.H.; Agerberth, B.; Shai, Y. Structure and organization of the human antimicrobial peptide LL-37 in phospholipid membranes: Relevance to the molecular basis for its non-cell-selective activity. Biochem. J. 1999, 341 Pt 3, 501–513. [Google Scholar] [CrossRef] [PubMed]
- Bucki, R.; Janmey, P.A. Interaction of the gelsolin-derived antibacterial PBP 10 peptide with lipid bilayers and cell membranes. Antimicrob. Agents Chemother. 2006, 50, 2932–2940. [Google Scholar] [CrossRef] [Green Version]
- Lee, C.C.; Sun, Y.; Qian, S.; Huang, H.W. Transmembrane pores formed by human antimicrobial peptide LL-37. Biophys. J. 2011, 100, 1688–1696. [Google Scholar] [CrossRef] [Green Version]
- Ding, B.; Soblosky, L.; Nguyen, K.; Geng, J.; Yu, X.; Ramamoorthy, A.; Chen, Z. Physiologically-relevant modes of membrane interactions by the human antimicrobial peptide, LL-37, revealed by SFG experiments. Sci. Rep. 2013, 3, 1854. [Google Scholar] [CrossRef] [Green Version]
- Vandamme, D.; Landuyt, B.; Luyten, W.; Schoofs, L. A comprehensive summary of LL-37, the factotum human cathelicidin peptide. Cell. Immunol. 2012, 280, 22–35. [Google Scholar] [CrossRef]
- Neumann, A.; Vollger, L.; Berends, E.T.; Molhoek, E.M.; Stapels, D.A.; Midon, M.; Friaes, A.; Pingoud, A.; Rooijakkers, S.H.; Gallo, R.L.; et al. Novel role of the antimicrobial peptide LL-37 in the protection of neutrophil extracellular traps against degradation by bacterial nucleases. J. Innate Immun. 2014, 6, 860–868. [Google Scholar] [CrossRef]
- Nizet, V.; Ohtake, T.; Lauth, X.; Trowbridge, J.; Rudisill, J.; Dorschner, R.A.; Pestonjamasp, V.; Piraino, J.; Huttner, K.; Gallo, R.L. Innate antimicrobial peptide protects the skin from invasive bacterial infection. Nature 2001, 414, 454–457. [Google Scholar] [CrossRef]
- Iimura, M.; Gallo, R.L.; Hase, K.; Miyamoto, Y.; Eckmann, L.; Kagnoff, M.F. Cathelicidin mediates innate intestinal defense against colonization with epithelial adherent bacterial pathogens. J. Immunol. 2005, 174, 4901–4907. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chromek, M.; Slamova, Z.; Bergman, P.; Kovacs, L.; Podracka, L.; Ehren, I.; Hokfelt, T.; Gudmundsson, G.H.; Gallo, R.L.; Agerberth, B.; et al. The antimicrobial peptide cathelicidin protects the urinary tract against invasive bacterial infection. Nat. Med. 2006, 12, 636–641. [Google Scholar] [CrossRef] [PubMed]
- Hing, T.C.; Ho, S.; Shih, D.Q.; Ichikawa, R.; Cheng, M.; Chen, J.; Chen, X.; Law, I.; Najarian, R.; Kelly, C.P.; et al. The antimicrobial peptide cathelicidin modulates Clostridium difficile-associated colitis and toxin A-mediated enteritis in mice. Gut 2013, 62, 1295–1305. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barlow, P.G.; Svoboda, P.; Mackellar, A.; Nash, A.A.; York, I.A.; Pohl, J.; Davidson, D.J.; Donis, R.O. Antiviral activity and increased host defense against influenza infection elicited by the human cathelicidin LL-37. PLoS ONE 2011, 6, e25333. [Google Scholar] [CrossRef] [PubMed]
- Yu, J.; Mookherjee, N.; Wee, K.; Bowdish, D.M.; Pistolic, J.; Li, Y.; Rehaume, L.; Hancock, R.E. Host defense peptide LL-37, in synergy with inflammatory mediator IL-1beta, augments immune responses by multiple pathways. J. Immunol. 2007, 179, 7684–7691. [Google Scholar] [CrossRef] [Green Version]
- Barlow, P.G.; Li, Y.; Wilkinson, T.S.; Bowdish, D.M.; Lau, Y.E.; Cosseau, C.; Haslett, C.; Simpson, A.J.; Hancock, R.E.; Davidson, D.J. The human cationic host defense peptide LL-37 mediates contrasting effects on apoptotic pathways in different primary cells of the innate immune system. J. Leukoc. Biol. 2006, 80, 509–520. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, X.; Takai, T.; Xie, Y.; Niyonsaba, F.; Okumura, K.; Ogawa, H. Human antimicrobial peptide LL-37 modulates proinflammatory responses induced by cytokine milieus and double-stranded RNA in human keratinocytes. Biochem. Biophys. Res. Commun. 2013, 433, 532–537. [Google Scholar] [CrossRef]
- Brown, K.L.; Poon, G.F.; Birkenhead, D.; Pena, O.M.; Falsafi, R.; Dahlgren, C.; Karlsson, A.; Bylund, J.; Hancock, R.E.; Johnson, P. Host defense peptide LL-37 selectively reduces proinflammatory macrophage responses. J. Immunol. 2011, 186, 5497–5505. [Google Scholar] [CrossRef] [Green Version]
- Scott, M.G.; Davidson, D.J.; Gold, M.R.; Bowdish, D.; Hancock, R.E. The human antimicrobial peptide LL-37 is a multifunctional modulator of innate immune responses. J. Immunol. 2002, 169, 3883–3891. [Google Scholar] [CrossRef] [Green Version]
- Lai, Y.; Adhikarakunnathu, S.; Bhardwaj, K.; Ranjith-Kumar, C.T.; Wen, Y.; Jordan, J.L.; Wu, L.H.; Dragnea, B.; San Mateo, L.; Kao, C.C. LL37 and cationic peptides enhance TLR3 signaling by viral double-stranded RNAs. PLoS ONE 2011, 6, e26632. [Google Scholar] [CrossRef] [Green Version]
- Sessa, L.; Bianchi, M.E. The evolution of High Mobility Group Box (HMGB) chromatin proteins in multicellular animals. Gene 2007, 387, 133–140. [Google Scholar] [CrossRef] [PubMed]
- Rouhiainen, A.; Imai, S.; Rauvala, H.; Parkkinen, J. Occurrence of amphoterin (HMG1) as an endogenous protein of human platelets that is exported to the cell surface upon platelet activation. Thromb. Haemost. 2000, 84, 1087–1094. [Google Scholar] [PubMed]
- Ito, T.; Kawahara, K.; Nakamura, T.; Yamada, S.; Nakamura, T.; Abeyama, K.; Hashiguchi, T.; Maruyama, I. High-mobility group box 1 protein promotes development of microvascular thrombosis in rats. J. Thromb. Haemost. 2007, 5, 109–116. [Google Scholar] [CrossRef] [PubMed]
- Andersson, U.; Yang, H.; Harris, H. High-mobility group box 1 protein (HMGB1) operates as an alarmin outside as well as inside cells. Semin. Immunol. 2018, 38, 40–48. [Google Scholar] [CrossRef] [PubMed]
- Lu, B.; Wang, C.; Wang, M.; Li, W.; Chen, F.; Tracey, K.J.; Wang, H. Molecular mechanism and therapeutic modulation of high mobility group box 1 release and action: An updated review. Expert Rev. Clin. Immunol. 2014, 10, 713–727. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Whittall-Garcia, L.P.; Torres-Ruiz, J.; Zentella-Dehesa, A.; Tapia-Rodriguez, M.; Alcocer-Varela, J.; Mendez-Huerta, N.; Gomez-Martin, D. Neutrophil extracellular traps are a source of extracellular HMGB1 in lupus nephritis: Associations with clinical and histopathological features. Lupus 2019, 28, 1549–1557. [Google Scholar] [CrossRef]
- Janko, C.; Filipovic, M.; Munoz, L.E.; Schorn, C.; Schett, G.; Ivanovic-Burmazovic, I.; Herrmann, M. Redox modulation of HMGB1-related signaling. Antioxid. Redox Signal. 2014, 20, 1075–1085. [Google Scholar] [CrossRef] [Green Version]
- Venereau, E.; Casalgrandi, M.; Schiraldi, M.; Antoine, D.J.; Cattaneo, A.; De Marchis, F.; Liu, J.; Antonelli, A.; Preti, A.; Raeli, L.; et al. Mutually exclusive redox forms of HMGB1 promote cell recruitment or proinflammatory cytokine release. J. Exp. Med. 2012, 209, 1519–1528. [Google Scholar] [CrossRef] [Green Version]
- Venereau, E.; Schiraldi, M.; Uguccioni, M.; Bianchi, M.E. HMGB1 and leukocyte migration during trauma and sterile inflammation. Mol. Immunol. 2013, 55, 76–82. [Google Scholar] [CrossRef]
- Yang, H.; Antoine, D.J.; Andersson, U.; Tracey, K.J. The many faces of HMGB1: Molecular structure-functional activity in inflammation, apoptosis, and chemotaxis. J. Leukoc. Biol. 2013, 93, 865–873. [Google Scholar] [CrossRef] [Green Version]
- Tang, Y.; Zhao, X.; Antoine, D.; Xiao, X.; Wang, H.; Andersson, U.; Billiar, T.R.; Tracey, K.J.; Lu, B. Regulation of Posttranslational Modifications of HMGB1 During Immune Responses. Antioxid. Redox Signal. 2016, 24, 620–634. [Google Scholar] [CrossRef] [PubMed]
- Chen, R.; Kang, R.; Tang, D. The mechanism of HMGB1 secretion and release. Exp. Mol Med. 2022, 54, 91–102. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Ward, M.F.; Sama, A.E. Targeting HMGB1 in the treatment of sepsis. Expert Opin. Ther. Targets 2014, 18, 257–268. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Abraham, E.; Arcaroli, J.; Carmody, A.; Wang, H.; Tracey, K.J. HMG-1 as a mediator of acute lung inflammation. J. Immunol. 2000, 165, 2950–2954. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Youn, J.H.; Oh, Y.J.; Kim, E.S.; Choi, J.E.; Shin, J.S. High mobility group box 1 protein binding to lipopolysaccharide facilitates transfer of lipopolysaccharide to CD14 and enhances lipopolysaccharide-mediated TNF-alpha production in human monocytes. J. Immunol. 2008, 180, 5067–5074. [Google Scholar] [CrossRef] [Green Version]
- Anggayasti, W.L.; Mancera, R.L.; Bottomley, S.; Helmerhorst, E. The self-association of HMGB1 and its possible role in the binding to DNA and cell membrane receptors. FEBS Lett. 2017, 591, 282–294. [Google Scholar] [CrossRef] [Green Version]
- Kwak, M.S.; Lim, M.; Lee, Y.J.; Lee, H.S.; Kim, Y.H.; Youn, J.H.; Choi, J.E.; Shin, J.S. HMGB1 Binds to Lipoteichoic Acid and Enhances TNF-alpha and IL-6 Production through HMGB1-Mediated Transfer of Lipoteichoic Acid to CD14 and TLR2. J. Innate Immun. 2015, 7, 405–416. [Google Scholar] [CrossRef]
- Dyer, M.R.; Chen, Q.; Haldeman, S.; Yazdani, H.; Hoffman, R.; Loughran, P.; Tsung, A.; Zuckerbraun, B.S.; Simmons, R.L.; Neal, M.D. Deep vein thrombosis in mice is regulated by platelet HMGB1 through release of neutrophil-extracellular traps and DNA. Sci. Rep. 2018, 8, 2068. [Google Scholar] [CrossRef] [Green Version]
- Ward, P.A. An endogenous factor mediates shock-induced injury. Nat. Med. 2013, 19, 1368–1369. [Google Scholar] [CrossRef]
- Zhu, X.; Buhrer, C.; Wellmann, S. Cold-inducible proteins CIRP and RBM3, a unique couple with activities far beyond the cold. Cell. Mol. Life Sci. 2016, 73, 3839–3859. [Google Scholar] [CrossRef] [Green Version]
- Zhu, X.; Zelmer, A.; Kapfhammer, J.P.; Wellmann, S. Cold-inducible RBM3 inhibits PERK phosphorylation through cooperation with NF90 to protect cells from endoplasmic reticulum stress. FASEB J. 2016, 30, 624–634. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, K.; Murao, A.; Arif, A.; Takizawa, S.; Jin, H.; Jiang, J.; Aziz, M.; Wang, P. Inhibition of Efferocytosis by Extracellular CIRP-Induced Neutrophil Extracellular Traps. J. Immunol. 2021, 206, 797–806. [Google Scholar] [CrossRef] [PubMed]
- Block, H.; Zarbock, A. A Fragile Balance: Does Neutrophil Extracellular Trap Formation Drive Pulmonary Disease Progression? Cells 2021, 10, 1932. [Google Scholar] [CrossRef] [PubMed]
- Nakazawa, D.; Marschner, J.A.; Platen, L.; Anders, H.J. Extracellular traps in kidney disease. Kidney Int. 2018, 94, 1087–1098. [Google Scholar] [CrossRef]
- Jennette, J.C.; Falk, R.J. Small-vessel vasculitis. N. Engl. J. Med. 1997, 337, 1512–1523. [Google Scholar] [CrossRef]
- Falk, R.J.; Gross, W.L.; Guillevin, L.; Hoffman, G.S.; Jayne, D.R.; Jennette, J.C.; Kallenberg, C.G.; Luqmani, R.; Mahr, A.D.; Matteson, E.L.; et al. Granulomatosis with polyangiitis (Wegener’s): An alternative name for Wegener’s granulomatosis. Arthritis Rheum. 2011, 63, 863–864. [Google Scholar] [CrossRef]
- Gadola, S.D.; Gross, W.L. Vasculitis in 2011: The renaissance of granulomatous inflammation in AAV. Nat. Rev. Rheumatol. 2012, 8, 74–76. [Google Scholar] [CrossRef]
- Kessenbrock, K.; Krumbholz, M.; Schonermarck, U.; Back, W.; Gross, W.L.; Werb, Z.; Grone, H.J.; Brinkmann, V.; Jenne, D.E. Netting neutrophils in autoimmune small-vessel vasculitis. Nat. Med. 2009, 15, 623–625. [Google Scholar] [CrossRef]
- Yoshida, M.; Sasaki, M.; Sugisaki, K.; Yamaguchi, Y.; Yamada, M. Neutrophil extracellular trap components in fibrinoid necrosis of the kidney with myeloperoxidase-ANCA-associated vasculitis. Clin. Kidney J. 2013, 6, 308–312. [Google Scholar] [CrossRef] [Green Version]
- Abreu-Velez, A.M.; Smith, J.G., Jr.; Howard, M.S. Presence of neutrophil extracellular traps and antineutrophil cytoplasmic antibodies associated with vasculitides. N. Am. J. Med. Sci. 2009, 1, 309–313. [Google Scholar]
- Sangaletti, S.; Tripodo, C.; Chiodoni, C.; Guarnotta, C.; Cappetti, B.; Casalini, P.; Piconese, S.; Parenza, M.; Guiducci, C.; Vitali, C.; et al. Neutrophil extracellular traps mediate transfer of cytoplasmic neutrophil antigens to myeloid dendritic cells toward ANCA induction and associated autoimmunity. Blood 2012, 120, 3007–3018. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nakazawa, D.; Tomaru, U.; Yamamoto, C.; Jodo, S.; Ishizu, A. Abundant neutrophil extracellular traps in thrombus of patient with microscopic polyangiitis. Front. Immunol. 2012, 3, 333. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Imamoto, T.; Nakazawa, D.; Shida, H.; Suzuki, A.; Otsuka, N.; Tomaru, U.; Ishizu, A. Possible linkage between microscopic polyangiitis and thrombosis via neutrophil extracellular traps. Clin. Exp. Rheumatol. 2014, 32, 149–150. [Google Scholar] [PubMed]
- Kambas, K.; Chrysanthopoulou, A.; Vassilopoulos, D.; Apostolidou, E.; Skendros, P.; Girod, A.; Arelaki, S.; Froudarakis, M.; Nakopoulou, L.; Giatromanolaki, A.; et al. Tissue factor expression in neutrophil extracellular traps and neutrophil derived microparticles in antineutrophil cytoplasmic antibody associated vasculitis may promote thromboinflammation and the thrombophilic state associated with the disease. Ann. Rheum. Dis. 2014, 73, 1854–1863. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Allenbach, Y.; Seror, R.; Pagnoux, C.; Teixeira, L.; Guilpain, P.; Guillevin, L.; French Vasculitis Study, G. High frequency of venous thromboembolic events in Churg-Strauss syndrome, Wegener’s granulomatosis and microscopic polyangiitis but not polyarteritis nodosa: A systematic retrospective study on 1130 patients. Ann. Rheum. Dis. 2009, 68, 564–567. [Google Scholar] [CrossRef] [PubMed]
- Stassen, P.M.; Derks, R.P.; Kallenberg, C.G.; Stegeman, C.A. Venous thromboembolism in ANCA-associated vasculitis--incidence and risk factors. Rheumatology 2008, 47, 530–534. [Google Scholar] [CrossRef] [Green Version]
- Kallenberg, C.G. Pathogenesis and treatment of ANCA-associated vasculitides. Clin. Exp. Rheumatol. 2015, 33, S11–S14. [Google Scholar]
- Xiao, H.; Heeringa, P.; Hu, P.; Liu, Z.; Zhao, M.; Aratani, Y.; Maeda, N.; Falk, R.J.; Jennette, J.C. Antineutrophil cytoplasmic autoantibodies specific for myeloperoxidase cause glomerulonephritis and vasculitis in mice. J. Clin. Investig. 2002, 110, 955–963. [Google Scholar] [CrossRef]
- Little, M.A.; Al-Ani, B.; Ren, S.; Al-Nuaimi, H.; Leite, M., Jr.; Alpers, C.E.; Savage, C.O.; Duffield, J.S. Anti-proteinase 3 anti-neutrophil cytoplasm autoantibodies recapitulate systemic vasculitis in mice with a humanized immune system. PLoS ONE 2012, 7, e28626. [Google Scholar] [CrossRef]
- Falk, R.J.; Terrell, R.S.; Charles, L.A.; Jennette, J.C. Anti-neutrophil cytoplasmic autoantibodies induce neutrophils to degranulate and produce oxygen radicals in vitro. Proc. Natl. Acad. Sci. USA 1990, 87, 4115–4119. [Google Scholar] [CrossRef] [Green Version]
- Radford, D.J.; Savage, C.O.; Nash, G.B. Treatment of rolling neutrophils with antineutrophil cytoplasmic antibodies causes conversion to firm integrin-mediated adhesion. Arthritis Rheum. 2000, 43, 1337–1345. [Google Scholar] [CrossRef]
- Xiao, H.; Schreiber, A.; Heeringa, P.; Falk, R.J.; Jennette, J.C. Alternative complement pathway in the pathogenesis of disease mediated by anti-neutrophil cytoplasmic autoantibodies. Am. J. Pathol. 2007, 170, 52–64. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schreiber, A.; Xiao, H.; Jennette, J.C.; Schneider, W.; Luft, F.C.; Kettritz, R. C5a receptor mediates neutrophil activation and ANCA-induced glomerulonephritis. J. Am. Soc. Nephrol. 2009, 20, 289–298. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kettritz, R. How anti-neutrophil cytoplasmic autoantibodies activate neutrophils. Clin. Exp. Immunol. 2012, 169, 220–228. [Google Scholar] [CrossRef] [PubMed]
- Thadhani, R.; Pascual, M.; Bonventre, J.V. Acute renal failure. N. Engl. J. Med. 1996, 334, 1448–1460. [Google Scholar] [CrossRef]
- Devarajan, P. Update on mechanisms of ischemic acute kidney injury. J. Am. Soc. Nephrol. 2006, 17, 1503–1520. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Raup-Konsavage, W.M.; Wang, Y.; Wang, W.W.; Feliers, D.; Ruan, H.; Reeves, W.B. Neutrophil peptidyl arginine deiminase-4 has a pivotal role in ischemia/reperfusion-induced acute kidney injury. Kidney Int. 2018, 93, 365–374. [Google Scholar] [CrossRef]
- Jansen, M.P.; Emal, D.; Teske, G.J.; Dessing, M.C.; Florquin, S.; Roelofs, J.J. Release of extracellular DNA influences renal ischemia reperfusion injury by platelet activation and formation of neutrophil extracellular traps. Kidney Int. 2017, 91, 352–364. [Google Scholar] [CrossRef] [Green Version]
- Ham, A.; Rabadi, M.; Kim, M.; Brown, K.M.; Ma, Z.; D’Agati, V.; Lee, H.T. Peptidyl arginine deiminase-4 activation exacerbates kidney ischemia-reperfusion injury. Am. J. Physiol. Renal. Physiol. 2014, 307, F1052–F1062. [Google Scholar] [CrossRef] [Green Version]
- Rabadi, M.; Kim, M.; D’Agati, V.; Lee, H.T. Peptidyl arginine deiminase-4-deficient mice are protected against kidney and liver injury after renal ischemia and reperfusion. Am. J. Physiol. Renal Physiol. 2016, 311, F437–F449. [Google Scholar] [CrossRef] [Green Version]
- Sonneveld, M.A.; de Maat, M.P.; Portegies, M.L.; Kavousi, M.; Hofman, A.; Turecek, P.L.; Rottensteiner, H.; Scheiflinger, F.; Koudstaal, P.J.; Ikram, M.A.; et al. Low ADAMTS13 activity is associated with an increased risk of ischemic stroke. Blood 2015, 126, 2739–2746. [Google Scholar] [CrossRef] [PubMed]
- Chang, X.; Yamada, R.; Suzuki, A.; Kochi, Y.; Sawada, T.; Yamamoto, K. Citrullination of fibronectin in rheumatoid arthritis synovial tissue. Rheumatology 2005, 44, 1374–1382. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chang, X.; Han, J.; Pang, L.; Zhao, Y.; Yang, Y.; Shen, Z. Increased PADI4 expression in blood and tissues of patients with malignant tumors. BMC Cancer 2009, 9, 40. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gosswein, S.; Lindemann, A.; Mahajan, A.; Maueroder, C.; Martini, E.; Patankar, J.; Schett, G.; Becker, C.; Wirtz, S.; Naumann-Bartsch, N.; et al. Citrullination Licenses Calpain to Decondense Nuclei in Neutrophil Extracellular Trap Formation. Front. Immunol. 2019, 10, 2481. [Google Scholar] [CrossRef] [PubMed]
- Armutcu, F. Organ crosstalk: The potent roles of inflammation and fibrotic changes in the course of organ interactions. Inflamm. Res. 2019, 68, 825–839. [Google Scholar] [CrossRef]
- Mehta, R.L.; Pascual, M.T.; Gruta, C.G.; Zhuang, S.; Chertow, G.M. Refining predictive models in critically ill patients with acute renal failure. J. Am. Soc. Nephrol. 2002, 13, 1350–1357. [Google Scholar] [CrossRef] [Green Version]
- Steiger, S.; Rossaint, J.; Zarbock, A.; Anders, H.J. Secondary Immunodeficiency Related to Kidney Disease (SIDKD)-Definition, Unmet Need, and Mechanisms. J. Am. Soc. Nephrol. 2022, 33, 259–278. [Google Scholar] [CrossRef]
- Apostolakis, E.; Filos, K.S.; Koletsis, E.; Dougenis, D. Lung dysfunction following cardiopulmonary bypass. J. Card. Surg. 2010, 25, 47–55. [Google Scholar] [CrossRef]
- Du, M.; Yang, L.; Gu, J.; Wu, J.; Ma, Y.; Wang, T. Inhibition of Peptidyl Arginine Deiminase-4 Prevents Renal Ischemia-Reperfusion-Induced Remote Lung Injury. Mediat. Inflamm. 2020, 2020, 1724206. [Google Scholar] [CrossRef]
- Doi, K.; Ishizu, T.; Tsukamoto-Sumida, M.; Hiruma, T.; Yamashita, T.; Ogasawara, E.; Hamasaki, Y.; Yahagi, N.; Nangaku, M.; Noiri, E. The high-mobility group protein B1-Toll-like receptor 4 pathway contributes to the acute lung injury induced by bilateral nephrectomy. Kidney Int. 2014, 86, 316–326. [Google Scholar] [CrossRef] [Green Version]
- Zhan, Y.; Ling, Y.; Deng, Q.; Qiu, Y.; Shen, J.; Lai, H.; Chen, Z.; Huang, C.; Liang, L.; Li, X.; et al. HMGB1-Mediated Neutrophil Extracellular Trap Formation Exacerbates Intestinal Ischemia/Reperfusion-Induced Acute Lung Injury. J. Immunol. 2022, 208, 968–978. [Google Scholar] [CrossRef] [PubMed]
- Caudrillier, A.; Kessenbrock, K.; Gilliss, B.M.; Nguyen, J.X.; Marques, M.B.; Monestier, M.; Toy, P.; Werb, Z.; Looney, M.R. Platelets induce neutrophil extracellular traps in transfusion-related acute lung injury. J. Clin. Investig. 2012, 122, 2661–2671. [Google Scholar] [CrossRef] [PubMed]
- Bosmann, M.; Grailer, J.J.; Ruemmler, R.; Russkamp, N.F.; Zetoune, F.S.; Sarma, J.V.; Standiford, T.J.; Ward, P.A. Extracellular histones are essential effectors of C5aR- and C5L2-mediated tissue damage and inflammation in acute lung injury. FASEB J. 2013, 27, 5010–5021. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rossaint, J.; Herter, J.M.; Van Aken, H.; Napirei, M.; Döring, Y.; Weber, C.; Soehnlein, O.; Zarbock, A. Synchronized integrin engagement and chemokine activation is crucial in neutrophil extracellular trap-mediated sterile inflammation. Blood 2014, 123, 2573–2584. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Pan, P.; Su, X.; Liu, S.; Zhang, L.; Wu, D.; Li, H.; Dai, M.; Li, Y.; Hu, C.; et al. Neutrophil Extracellular Traps Are Pathogenic in Ventilator-Induced Lung Injury and Partially Dependent on TLR4. BioMed Res. Int. 2017, 2017, 8272504. [Google Scholar] [CrossRef] [PubMed]
- Mikacenic, C.; Moore, R.; Dmyterko, V.; West, T.E.; Altemeier, W.A.; Liles, W.C.; Lood, C. Neutrophil extracellular traps (NETs) are increased in the alveolar spaces of patients with ventilator-associated pneumonia. Crit. Care 2018, 22, 358. [Google Scholar] [CrossRef] [Green Version]
- Ackermann, M.; Verleden, S.E.; Kuehnel, M.; Haverich, A.; Welte, T.; Laenger, F.; Vanstapel, A.; Werlein, C.; Stark, H.; Tzankov, A.; et al. Pulmonary Vascular Endothelialitis, Thrombosis, and Angiogenesis in COVID-19. N. Engl. J. Med. 2020, 383, 120–128. [Google Scholar] [CrossRef]
- Barnes, B.J.; Adrover, J.M.; Baxter-Stoltzfus, A.; Borczuk, A.; Cools-Lartigue, J.; Crawford, J.M.; Dassler-Plenker, J.; Guerci, P.; Huynh, C.; Knight, J.S.; et al. Targeting potential drivers of COVID-19: Neutrophil extracellular traps. J. Exp. Med. 2020, 217, e20200652. [Google Scholar] [CrossRef]
- Barton, L.M.; Duval, E.J.; Stroberg, E.; Ghosh, S.; Mukhopadhyay, S. COVID-19 Autopsies, Oklahoma, USA. Am. J. Clin. Pathol. 2020, 153, 725–733. [Google Scholar] [CrossRef] [Green Version]
- Zhang, H.; Wang, C.Y.; Zhou, P.; Yue, H.; Du, R. Histopathologic Changes and SARS-CoV-2 Immunostaining in the Lung of a Patient With COVID-19. Ann. Intern. Med. 2020, 173, 324. [Google Scholar] [CrossRef]
- Wu, C.; Chen, X.; Cai, Y.; Xia, J.; Zhou, X.; Xu, S.; Huang, H.; Zhang, L.; Zhou, X.; Du, C.; et al. Risk Factors Associated With Acute Respiratory Distress Syndrome and Death in Patients With Coronavirus Disease 2019 Pneumonia in Wuhan, China. JAMA Intern. Med. 2020, 180, 934–943. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zuo, Y.; Yalavarthi, S.; Shi, H.; Gockman, K.; Zuo, M.; Madison, J.A.; Blair, C.; Weber, A.; Barnes, B.J.; Egeblad, M.; et al. Neutrophil extracellular traps (NETs) as markers of disease severity in COVID-19. medRxiv 2020. [Google Scholar] [CrossRef] [Green Version]
- van den Akker, J.P.; Egal, M.; Groeneveld, A.B. Invasive mechanical ventilation as a risk factor for acute kidney injury in the critically ill: A systematic review and meta-analysis. Crit. Care 2013, 17, R98. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kuiper, J.W.; Plotz, F.B.; Groeneveld, A.J.; Haitsma, J.J.; Jothy, S.; Vaschetto, R.; Zhang, H.; Slutsky, A.S. High tidal volume mechanical ventilation-induced lung injury in rats is greater after acid instillation than after sepsis-induced acute lung injury, but does not increase systemic inflammation: An experimental study. BMC Anesthesiol. 2011, 11, 26. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Slutsky, A.S.; Tremblay, L.N. Multiple system organ failure. Is mechanical ventilation a contributing factor? Am. J. Respir. Crit Care Med. 1998, 157, 1721–1725. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hepokoski, M.; Englert, J.A.; Baron, R.M.; Crotty-Alexander, L.E.; Fuster, M.M.; Beitler, J.R.; Malhotra, A.; Singh, P. Ventilator-induced lung injury increases expression of endothelial inflammatory mediators in the kidney. Am. J. Physiol. Renal. Physiol. 2017, 312, F654–F660. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Patterson, E.K.; Yao, L.J.; Ramic, N.; Lewis, J.F.; Cepinskas, G.; McCaig, L.; Veldhuizen, R.A.; Yamashita, C.M. Lung-derived mediators induce cytokine production in downstream organs via an NF-kappaB-dependent mechanism. Mediat. Inflamm. 2013, 2013, 586895. [Google Scholar] [CrossRef] [Green Version]
- Gonnert, F.A.; Kunisch, E.; Gajda, M.; Lambeck, S.; Weber, M.; Claus, R.A.; Bauer, M.; Kinne, R.W. Hepatic Fibrosis in a Long-term Murine Model of Sepsis. Shock 2012, 37, 399–407. [Google Scholar] [CrossRef]
- Protzer, U.; Maini, M.K.; Knolle, P.A. Living in the liver: Hepatic infections. Nat. Rev. Immunol. 2012, 12, 201–213. [Google Scholar] [CrossRef]
- Wong, C.H.; Jenne, C.N.; Petri, B.; Chrobok, N.L.; Kubes, P. Nucleation of platelets with blood-borne pathogens on Kupffer cells precedes other innate immunity and contributes to bacterial clearance. Nat. Immunol. 2013, 14, 785–792. [Google Scholar] [CrossRef] [Green Version]
- Kolaczkowska, E.; Jenne, C.N.; Surewaard, B.G.; Thanabalasuriar, A.; Lee, W.Y.; Sanz, M.J.; Mowen, K.; Opdenakker, G.; Kubes, P. Molecular mechanisms of NET formation and degradation revealed by intravital imaging in the liver vasculature. Nat. Commun. 2015, 6, 6673. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Klune, J.R.; Tsung, A. Molecular biology of liver ischemia/reperfusion injury: Established mechanisms and recent advancements. Surg. Clin. Am. 2010, 90, 665–677. [Google Scholar] [CrossRef] [PubMed]
- Kubes, P.; Mehal, W.Z. Sterile inflammation in the liver. Gastroenterology 2012, 143, 1158–1172. [Google Scholar] [CrossRef] [PubMed]
- Evankovich, J.; Cho, S.W.; Zhang, R.; Cardinal, J.; Dhupar, R.; Zhang, L.; Klune, J.R.; Zlotnicki, J.; Billiar, T.; Tsung, A. High mobility group box 1 release from hepatocytes during ischemia and reperfusion injury is mediated by decreased histone deacetylase activity. J. Biol. Chem. 2010, 285, 39888–39897. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, H.; Evankovich, J.; Yan, W.; Nace, G.; Zhang, L.; Ross, M.; Liao, X.; Billiar, T.; Xu, J.; Esmon, C.T.; et al. Endogenous histones function as alarmins in sterile inflammatory liver injury through Toll-like receptor 9 in mice. Hepatology 2011, 54, 999–1008. [Google Scholar] [CrossRef]
- Tsung, A.; Sahai, R.; Tanaka, H.; Nakao, A.; Fink, M.P.; Lotze, M.T.; Yang, H.; Li, J.; Tracey, K.J.; Geller, D.A.; et al. The nuclear factor HMGB1 mediates hepatic injury after murine liver ischemia-reperfusion. J. Exp. Med. 2005, 201, 1135–1143. [Google Scholar] [CrossRef]
- Al-Khafaji, A.B.; Tohme, S.; Yazdani, H.O.; Miller, D.; Huang, H.; Tsung, A. Superoxide induces Neutrophil Extracellular Trap Formation in a TLR-4 and NOX-dependent mechanism. Mol. Med. 2016, 22, 621–631. [Google Scholar] [CrossRef]
- Hathcock, J.J. Flow effects on coagulation and thrombosis. Arterioscler. Thromb. Vasc. Biol. 2006, 26, 1729–1737. [Google Scholar] [CrossRef]
- Ammollo, C.T.; Semeraro, F.; Xu, J.; Esmon, N.L.; Esmon, C.T. Extracellular histones increase plasma thrombin generation by impairing thrombomodulin-dependent protein C activation. J. Thromb. Haemost. 2011, 9, 1795–1803. [Google Scholar] [CrossRef]
- Liaw, P.C.; Ito, T.; Iba, T.; Thachil, J.; Zeerleder, S. DAMP and DIC: The role of extracellular DNA and DNA-binding proteins in the pathogenesis of DIC. Blood Rev. 2016, 30, 257–261. [Google Scholar] [CrossRef]
- Fuchs, T.A.; Brill, A.; Duerschmied, D.; Schatzberg, D.; Monestier, M.; Myers, D.D., Jr.; Wrobleski, S.K.; Wakefield, T.W.; Hartwig, J.H.; Wagner, D.D. Extracellular DNA traps promote thrombosis. Proc. Natl. Acad. Sci. USA 2010, 107, 15880–15885. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rossaint, J.; Kuhne, K.; Skupski, J.; Van Aken, H.; Looney, M.R.; Hidalgo, A.; Zarbock, A. Directed transport of neutrophil-derived extracellular vesicles enables platelet-mediated innate immune response. Nat. Commun. 2016, 7, 13464. [Google Scholar] [CrossRef] [PubMed]
- Carestia, A.; Kaufman, T.; Rivadeneyra, L.; Landoni, V.I.; Pozner, R.G.; Negrotto, S.; D’Atri, L.P.; Gomez, R.M.; Schattner, M. Mediators and molecular pathways involved in the regulation of neutrophil extracellular trap formation mediated by activated platelets. J. Leukoc. Biol. 2016, 99, 153–162. [Google Scholar] [CrossRef] [PubMed]
- Longstaff, C.; Varju, I.; Sotonyi, P.; Szabo, L.; Krumrey, M.; Hoell, A.; Bota, A.; Varga, Z.; Komorowicz, E.; Kolev, K. Mechanical stability and fibrinolytic resistance of clots containing fibrin, DNA, and histones. J. Biol. Chem. 2013, 288, 6946–6956. [Google Scholar] [CrossRef] [Green Version]
- Shi, C.; Yang, L.; Braun, A.; Anders, H.J. Extracellular DNA-A Danger Signal Triggering Immunothrombosis. Front. Immunol. 2020, 11, 568513. [Google Scholar] [CrossRef]
- Al-Ani, F.; Chehade, S.; Lazo-Langner, A. Thrombosis risk associated with COVID-19 infection. A scoping review. Thromb. Res. 2020, 192, 152–160. [Google Scholar] [CrossRef]
- Ranucci, M.; Ballotta, A.; Di Dedda, U.; Baryshnikova, E.; Dei Poli, M.; Resta, M.; Falco, M.; Albano, G.; Menicanti, L. The procoagulant pattern of patients with COVID-19 acute respiratory distress syndrome. J. Thromb. Haemost. 2020, 18, 1747–1751. [Google Scholar] [CrossRef]
- Connors, J.M.; Levy, J.H. COVID-19 and its implications for thrombosis and anticoagulation. Blood 2020, 135, 2033–2040. [Google Scholar] [CrossRef]
- Manne, B.K.; Denorme, F.; Middleton, E.A.; Portier, I.; Rowley, J.W.; Stubben, C.; Petrey, A.C.; Tolley, N.D.; Guo, L.; Cody, M.; et al. Platelet gene expression and function in patients with COVID-19. Blood 2020, 136, 1317–1329. [Google Scholar] [CrossRef]
- Middleton, E.A.; He, X.Y.; Denorme, F.; Campbell, R.A.; Ng, D.; Salvatore, S.P.; Mostyka, M.; Baxter-Stoltzfus, A.; Borczuk, A.C.; Loda, M.; et al. Neutrophil extracellular traps contribute to immunothrombosis in COVID-19 acute respiratory distress syndrome. Blood 2020, 136, 1169–1179. [Google Scholar] [CrossRef]
- Nicolai, L.; Leunig, A.; Brambs, S.; Kaiser, R.; Weinberger, T.; Weigand, M.; Muenchhoff, M.; Hellmuth, J.C.; Ledderose, S.; Schulz, H.; et al. Immunothrombotic Dysregulation in COVID-19 Pneumonia Is Associated With Respiratory Failure and Coagulopathy. Circulation 2020, 142, 1176–1189. [Google Scholar] [CrossRef] [PubMed]
- Schurink, B.; Roos, E.; Radonic, T.; Barbe, E.; Bouman, C.S.C.; de Boer, H.H.; de Bree, G.J.; Bulle, E.B.; Aronica, E.M.; Florquin, S.; et al. Viral presence and immunopathology in patients with lethal COVID-19: A prospective autopsy cohort study. Lancet Microbe 2020, 1, e290–e299. [Google Scholar] [CrossRef]
- Radermecker, C.; Detrembleur, N.; Guiot, J.; Cavalier, E.; Henket, M.; d’Emal, C.; Vanwinge, C.; Cataldo, D.; Oury, C.; Delvenne, P.; et al. Neutrophil extracellular traps infiltrate the lung airway, interstitial, and vascular compartments in severe COVID-19. J. Exp. Med. 2020, 217, e20201012. [Google Scholar] [CrossRef] [PubMed]
- McDonald, B.; Davis, R.P.; Kim, S.J.; Tse, M.; Esmon, C.T.; Kolaczkowska, E.; Jenne, C.N. Platelets and neutrophil extracellular traps collaborate to promote intravascular coagulation during sepsis in mice. Blood 2017, 129, 1357–1367. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Medeiros, S.K.; Emery, B.; Bhagirath, V.; Parpia, S.; Dwivedi, D.J.; Dwivedi, N.J.; Kearon, C.; Liaw, P.C. Does cell-free DNA promote coagulation and inhibit fibrinolysis in patients with unprovoked venous thromboembolism? Thromb. Res. 2020, 186, 13–19. [Google Scholar] [CrossRef]
- Nasr, S.Z.; Strouse, P.J.; Soskolne, E.; Maxvold, N.J.; Garver, K.A.; Rubin, B.K.; Moler, F.W. Efficacy of recombinant human deoxyribonuclease I in the hospital management of respiratory syncytial virus bronchiolitis. Chest 2001, 120, 203–208. [Google Scholar] [CrossRef]
- Hodson, M.E.; McKenzie, S.; Harms, H.K.; Koch, C.; Mastella, G.; Navarro, J.; Strandvik, B.; Investigators of the Epidemiologic Registry of Cystic Fibrosis. Dornase alfa in the treatment of cystic fibrosis in Europe: A report from the Epidemiologic Registry of Cystic Fibrosis. Pediatr. Pulmonol. 2003, 36, 427–432. [Google Scholar] [CrossRef]
- Frederiksen, B.; Pressler, T.; Hansen, A.; Koch, C.; Hoiby, N. Effect of aerosolized rhDNase (Pulmozyme) on pulmonary colonization in patients with cystic fibrosis. Acta Paediatr. 2006, 95, 1070–1074. [Google Scholar] [CrossRef]
- Holliday, Z.M.; Earhart, A.P.; Alnijoumi, M.M.; Krvavac, A.; Allen, L.H.; Schrum, A.G. Non-Randomized Trial of Dornase Alfa for Acute Respiratory Distress Syndrome Secondary to COVID-19. Front. Immunol. 2021, 12, 714833. [Google Scholar] [CrossRef]
- Wygrecka, M.; Kosanovic, D.; Wujak, L.; Reppe, K.; Henneke, I.; Frey, H.; Didiasova, M.; Kwapiszewska, G.; Marsh, L.M.; Baal, N.; et al. Antihistone Properties of C1 Esterase Inhibitor Protect against Lung Injury. Am. J. Respir. Crit. Care Med. 2017, 196, 186–199. [Google Scholar] [CrossRef]
- Chirivi, R.G.S.; van Rosmalen, J.W.G.; van der Linden, M.; Euler, M.; Schmets, G.; Bogatkevich, G.; Kambas, K.; Hahn, J.; Braster, Q.; Soehnlein, O.; et al. Therapeutic ACPA inhibits NET formation: A potential therapy for neutrophil-mediated inflammatory diseases. Cell. Mol. Immunol. 2021, 18, 1528–1544. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Caliezi, C.; Wuillemin, W.A.; Zeerleder, S.; Redondo, M.; Eisele, B.; Hack, C.E. C1-Esterase inhibitor: An anti-inflammatory agent and its potential use in the treatment of diseases other than hereditary angioedema. Pharmacol. Rev. 2000, 52, 91–112. [Google Scholar] [PubMed]
- Liu, D.; Cai, S.; Gu, X.; Scafidi, J.; Wu, X.; Davis, A.E., 3rd. C1 inhibitor prevents endotoxin shock via a direct interaction with lipopolysaccharide. J. Immunol. 2003, 171, 2594–2601. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zeerleder, S.; Caliezi, C.; van Mierlo, G.; Eerenberg-Belmer, A.; Sulzer, I.; Hack, C.E.; Wuillemin, W.A. Administration of C1 inhibitor reduces neutrophil activation in patients with sepsis. Clin. Vaccine Immunol. 2003, 10, 529–535. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Igonin, A.A.; Protsenko, D.N.; Galstyan, G.M.; Vlasenko, A.V.; Khachatryan, N.N.; Nekhaev, I.V.; Shlyapnikov, S.A.; Lazareva, N.B.; Herscu, P. C1-esterase inhibitor infusion increases survival rates for patients with sepsis*. Crit. Care Med. 2012, 40, 770–777. [Google Scholar] [CrossRef] [PubMed]
- Deng, Q.; Pan, B.; Alam, H.B.; Liang, Y.; Wu, Z.; Liu, B.; Mor-Vaknin, N.; Duan, X.; Williams, A.M.; Tian, Y.; et al. Citrullinated Histone H3 as a Therapeutic Target for Endotoxic Shock in Mice. Front. Immunol. 2019, 10, 2957. [Google Scholar] [CrossRef] [PubMed]
- Wardrop, D.; Keeling, D. The story of the discovery of heparin and warfarin. Br. J. Haematol. 2008, 141, 757–763. [Google Scholar] [CrossRef]
- Wildhagen, K.C.; Garcia de Frutos, P.; Reutelingsperger, C.P.; Schrijver, R.; Areste, C.; Ortega-Gomez, A.; Deckers, N.M.; Hemker, H.C.; Soehnlein, O.; Nicolaes, G.A. Nonanticoagulant heparin prevents histone-mediated cytotoxicity in vitro and improves survival in sepsis. Blood 2014, 123, 1098–1101. [Google Scholar] [CrossRef] [Green Version]
- Iba, T.; Hashiguchi, N.; Nagaoka, I.; Tabe, Y.; Kadota, K.; Sato, K. Heparins attenuated histone-mediated cytotoxicity in vitro and improved the survival in a rat model of histone-induced organ dysfunction. Intensive Care Med. Exp. 2015, 3, 36. [Google Scholar] [CrossRef] [Green Version]
- Sanchez, J. Low Molecular Weight Heparins-A new tool to disetangle from the NETs. Pharmacol. Res. 2017, 123, 157. [Google Scholar] [CrossRef]
- Manfredi, A.A.; Rovere-Querini, P.; D’Angelo, A.; Maugeri, N. Low molecular weight heparins prevent the induction of autophagy of activated neutrophils and the formation of neutrophil extracellular traps. Pharmacol. Res. 2017, 123, 146–156. [Google Scholar] [CrossRef] [PubMed]
- Tadie, J.M.; Bae, H.B.; Jiang, S.; Park, D.W.; Bell, C.P.; Yang, H.; Pittet, J.F.; Tracey, K.; Thannickal, V.J.; Abraham, E.; et al. HMGB1 promotes neutrophil extracellular trap formation through interactions with Toll-like receptor 4. Am. J. Physiol.-Lung Cell. Mol. Physiol. 2013, 304, L342–L349. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kang, R.; Zhang, Q.; Hou, W.; Yan, Z.; Chen, R.; Bonaroti, J.; Bansal, P.; Billiar, T.R.; Tsung, A.; Wang, Q.; et al. Intracellular Hmgb1 inhibits inflammatory nucleosome release and limits acute pancreatitis in mice. Gastroenterology 2014, 146, 1097–1107. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Menegazzo, L.; Scattolini, V.; Cappellari, R.; Bonora, B.M.; Albiero, M.; Bortolozzi, M.; Romanato, F.; Ceolotto, G.; Vigili de Kreutzeberg, S.; Avogaro, A.; et al. The antidiabetic drug metformin blunts NETosis in vitro and reduces circulating NETosis biomarkers in vivo. Acta Diabetol. 2018, 55, 593–601. [Google Scholar] [CrossRef] [PubMed]
- Gregoire, M.; Uhel, F.; Lesouhaitier, M.; Gacouin, A.; Guirriec, M.; Mourcin, F.; Dumontet, E.; Chalin, A.; Samson, M.; Berthelot, L.L.; et al. Impaired efferocytosis and neutrophil extracellular trap clearance by macrophages in ARDS. Eur. Respir. J. 2018, 52, 1702590. [Google Scholar] [CrossRef] [PubMed]
- Lapponi, M.J.; Carestia, A.; Landoni, V.I.; Rivadeneyra, L.; Etulain, J.; Negrotto, S.; Pozner, R.G.; Schattner, M. Regulation of neutrophil extracellular trap formation by anti-inflammatory drugs. J. Pharmacol. Exp. Ther. 2013, 345, 430–437. [Google Scholar] [CrossRef] [PubMed]
- Hirose, T.; Hamaguchi, S.; Matsumoto, N.; Irisawa, T.; Seki, M.; Tasaki, O.; Hosotsubo, H.; Yamamoto, N.; Yamamoto, K.; Akeda, Y.; et al. Presence of neutrophil extracellular traps and citrullinated histone H3 in the bloodstream of critically ill patients. PLoS ONE 2014, 9, e111755. [Google Scholar] [CrossRef] [Green Version]
- Eisen, D.P.; Leder, K.; Woods, R.L.; Lockery, J.E.; McGuinness, S.L.; Wolfe, R.; Pilcher, D.; Moore, E.M.; Shastry, A.; Nelson, M.R.; et al. Effect of aspirin on deaths associated with sepsis in healthy older people (ANTISEPSIS): A randomised, double-blind, placebo-controlled primary prevention trial. Lancet Respir. Med. 2021, 9, 186–195. [Google Scholar] [CrossRef]
- Gaidt, M.M.; Ebert, T.S.; Chauhan, D.; Ramshorn, K.; Pinci, F.; Zuber, S.; O’Duill, F.; Schmid-Burgk, J.L.; Hoss, F.; Buhmann, R.; et al. The DNA Inflammasome in Human Myeloid Cells Is Initiated by a STING-Cell Death Program Upstream of NLRP3. Cell 2017, 171, 1110–1124.e8. [Google Scholar] [CrossRef]
- An, J.; Woodward, J.J.; Sasaki, T.; Minie, M.; Elkon, K.B. Cutting edge: Antimalarial drugs inhibit IFN-beta production through blockade of cyclic GMP-AMP synthase-DNA interaction. J. Immunol. 2015, 194, 4089–4093. [Google Scholar] [CrossRef] [Green Version]
- Kuznik, A.; Bencina, M.; Svajger, U.; Jeras, M.; Rozman, B.; Jerala, R. Mechanism of endosomal TLR inhibition by antimalarial drugs and imidazoquinolines. J. Immunol. 2011, 186, 4794–4804. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Boone, B.A.; Murthy, P.; Miller-Ocuin, J.; Doerfler, W.R.; Ellis, J.T.; Liang, X.; Ross, M.A.; Wallace, C.T.; Sperry, J.L.; Lotze, M.T.; et al. Chloroquine reduces hypercoagulability in pancreatic cancer through inhibition of neutrophil extracellular traps. BMC Cancer 2018, 18, 678. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Murthy, P.; Singhi, A.D.; Ross, M.A.; Loughran, P.; Paragomi, P.; Papachristou, G.I.; Whitcomb, D.C.; Zureikat, A.H.; Lotze, M.T.; Zeh Iii, H.J.; et al. Enhanced Neutrophil Extracellular Trap Formation in Acute Pancreatitis Contributes to Disease Severity and Is Reduced by Chloroquine. Front. Immunol. 2019, 10, 28. [Google Scholar] [CrossRef] [PubMed]
- Gies, V.; Bekaddour, N.; Dieudonne, Y.; Guffroy, A.; Frenger, Q.; Gros, F.; Rodero, M.P.; Herbeuval, J.P.; Korganow, A.S. Beyond Anti-viral Effects of Chloroquine/Hydroxychloroquine. Front. Immunol. 2020, 11, 1409. [Google Scholar] [CrossRef]
- Lamphier, M.; Zheng, W.; Latz, E.; Spyvee, M.; Hansen, H.; Rose, J.; Genest, M.; Yang, H.; Shaffer, C.; Zhao, Y.; et al. Novel small molecule inhibitors of TLR7 and TLR9: Mechanism of action and efficacy in vivo. Mol. Pharmacol. 2014, 85, 429–440. [Google Scholar] [CrossRef] [Green Version]
- Vargas, A.; Boivin, R.; Cano, P.; Murcia, Y.; Bazin, I.; Lavoie, J.P. Neutrophil extracellular traps are downregulated by glucocorticosteroids in lungs in an equine model of asthma. Respir. Res. 2017, 18, 207. [Google Scholar] [CrossRef]
- Yost, C.C.; Schwertz, H.; Cody, M.J.; Wallace, J.A.; Campbell, R.A.; Vieira-de-Abreu, A.; Araujo, C.V.; Schubert, S.; Harris, E.S.; Rowley, J.W.; et al. Neonatal NET-inhibitory factor and related peptides inhibit neutrophil extracellular trap formation. J. Clin. Investig. 2016, 126, 3783–3798. [Google Scholar] [CrossRef] [Green Version]
- Leaker, B.R.; Barnes, P.J.; O’Connor, B. Inhibition of LPS-induced airway neutrophilic inflammation in healthy volunteers with an oral CXCR2 antagonist. Respir. Res. 2013, 14, 137. [Google Scholar] [CrossRef] [Green Version]
- Wolf, D.; Anto-Michel, N.; Blankenbach, H.; Wiedemann, A.; Buscher, K.; Hohmann, J.D.; Lim, B.; Bauml, M.; Marki, A.; Mauler, M.; et al. A ligand-specific blockade of the integrin Mac-1 selectively targets pathologic inflammation while maintaining protective host-defense. Nat. Commun. 2018, 9, 525. [Google Scholar] [CrossRef]
- Rossaint, J.; Thomas, K.; Mersmann, S.; Skupski, J.; Margraf, A.; Tekath, T.; Jouvene, C.C.; Dalli, J.; Hidalgo, A.; Meuth, S.G.; et al. Platelets orchestrate the resolution of pulmonary inflammation in mice by T reg cell repositioning and macrophage education. J. Exp. Med. 2021, 218, e20201353. [Google Scholar] [CrossRef]
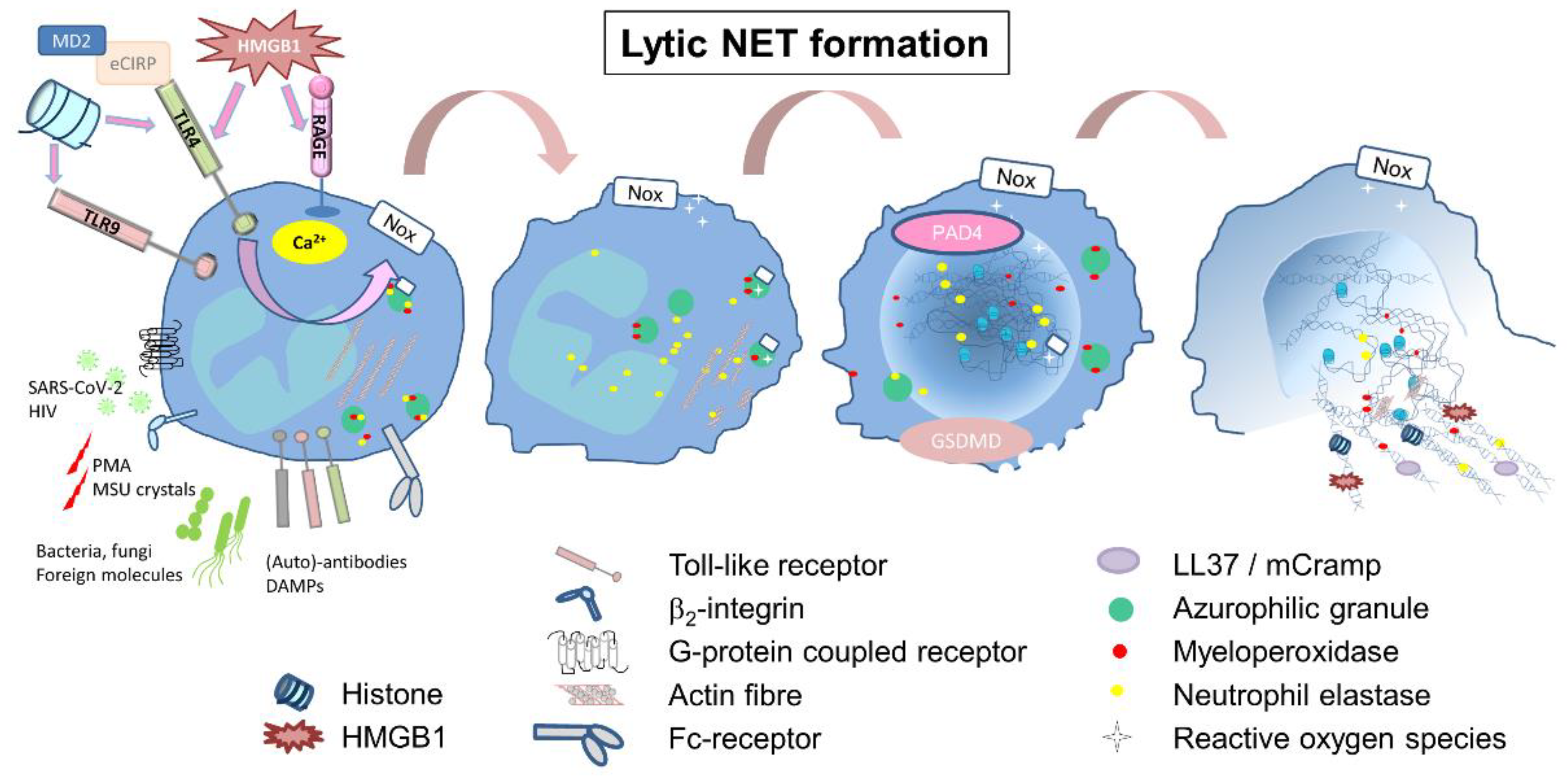


{kind=link}
{kind=link}
{kind=link}
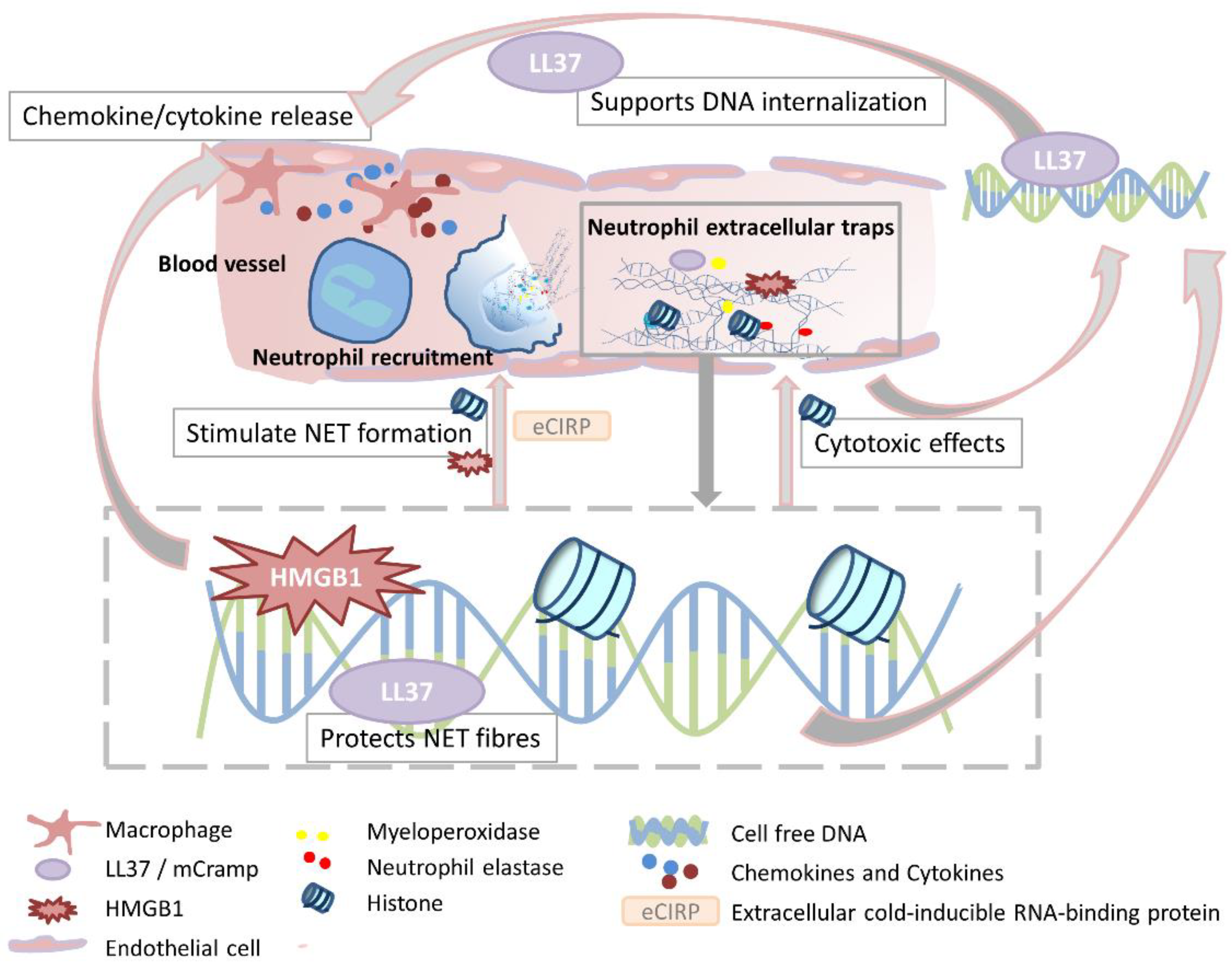{kind=link}
| DAMP | NET-Association | PRRs Involved | References |
|---|---|---|---|
| Histones | Part of NETs Induce NET formation | TLR2, TLR3, TLR4, TLR9 | [47,48,49] |
| cfDNA | Part of NETs | TLR9, cGAS, IFI16, AIM2, STING | [50] |
| LL37 | Protects NET-DNA Induction of NET formation through LL37 autoantibodies | TLR4, TLR7, TLR8, TLR9, TLR13 | [51,52,53,54,55,56] |
| HMGB1 | Induce NET formation Part of NETs | TLR2, TLR4, TLR9, CXCR4, RAGE, TREM | [57,58,59,60,61,62,63,64,65,66,67] |
| CIRP | Induce NET formation | TLR4 | [68,69,70] |
| Compound | Target | Model | Reference |
|---|---|---|---|
| Dornase Alfa/Dnase | DNA | Bronchiolitis Cystic fibrosis | [206,207,208] |
| COVID-19 | [209] | ||
| C1 esterase inhibitor | Histones | Sepsis patients | [210,214,215] |
| tACPA α-H3-cit | Citrullinated Histones | Inflammatory murine disease models | [211,216] |
| Heparin | Histones | Inflammatory murine disease models | [217,218,219,220,221] |
| HMGB-antibodies | HMGB1 blockade | LPS-treated mice | [222,223] |
| Metformin | HMGB/NET clearance | Diabetes patients | [224,225] |
| Aspirin (Hydroxy)chloroquine αCLEC Glucocorticoids NET-inhibiting factors | Inhibition of NET formation | COVID-19 Critically ill patients Inflammatory murine disease models | [226,227,228] [232,233,234] [235] [236] [237] |
| CXCR2 antagonist | Neutrophil recruitment | LPS-challenged humans | [238] |
| CD40L-M7 | Mac1 | Inflammatory murine disease models | [239] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Block, H.; Rossaint, J.; Zarbock, A. The Fatal Circle of NETs and NET-Associated DAMPs Contributing to Organ Dysfunction. Cells 2022, 11, 1919. https://doi.org/10.3390/cells11121919
Block H, Rossaint J, Zarbock A. The Fatal Circle of NETs and NET-Associated DAMPs Contributing to Organ Dysfunction. Cells. 2022; 11(12):1919. https://doi.org/10.3390/cells11121919
Chicago/Turabian StyleBlock, Helena, Jan Rossaint, and Alexander Zarbock. 2022. "The Fatal Circle of NETs and NET-Associated DAMPs Contributing to Organ Dysfunction" Cells 11, no. 12: 1919. https://doi.org/10.3390/cells11121919
APA StyleBlock, H., Rossaint, J., & Zarbock, A. (2022). The Fatal Circle of NETs and NET-Associated DAMPs Contributing to Organ Dysfunction. Cells, 11(12), 1919. https://doi.org/10.3390/cells11121919