
Standardization of Cell Culture Conditions and Routine Genomic Screening under a Quality Management System Leads to Reduced Genomic Instability in hPSCs

 ,
,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Literature Search
2.2. Cell Lines
2.3. Initial Expansion of hPSC Lines
2.4. Human PSC Maintenance in mTeSR1 Media with ROCK Inhibitor
2.5. Sample Fixation for Karyotyping (G-Banding)
2.6. Sample Preparation for Microarray-Based Comparative Genome Hybridization (aCGH)
2.7. Alteration Probability Analysis
2.8. Statistical Analysis
2.9. Quality Management System (QMS)
3. Results
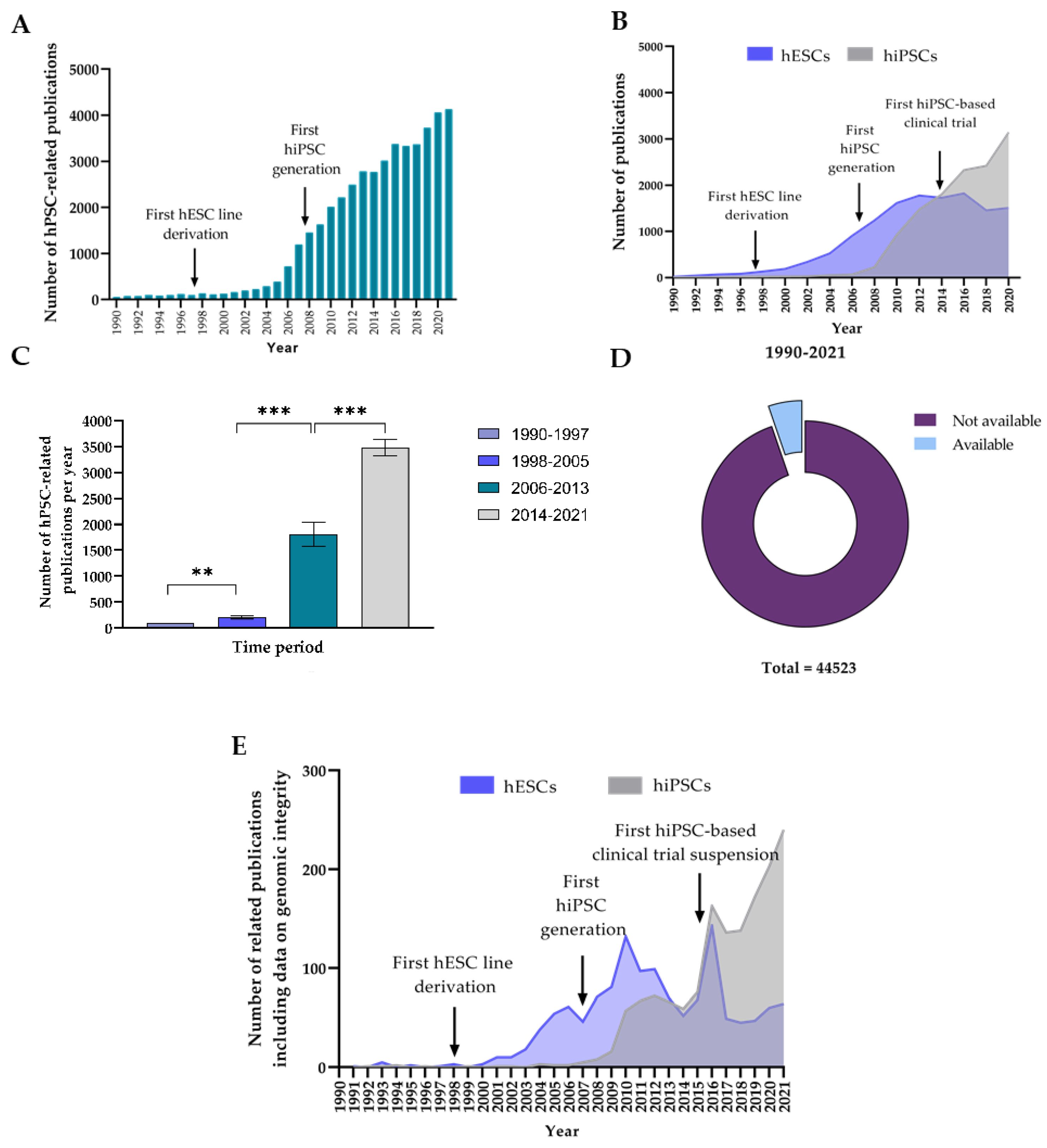
3.1. Analysis of the Evolution in Number of hPSC-Related Publications and Level of Reporting of Genomic Integrity Monitoring
3.1.1. The Number of Publications on hPSCs per Year Has Experienced a Large Increase during the Last 20 Years
3.1.2. Data on hPSC Genomic Integrity Remains Underreported despite the Fast Growth of hPSC Research
3.2. Analysis of the Types and Prevalence of Genomic Alterations Acquired by hPSCs In Vitro
3.2.1. Descriptive Statistics of the Analyzed hPSC Dataset
3.2.2. Recurrent Karyotypic Aberrations Are Detected by G-Banding with a Low Frequency
3.2.3. The hiPSCs Show an Increased Propensity to Acquire Chromosomic Aberrations
3.2.4. Comprehensive aCGH of hPSCs Reveals Hidden Recurrent Copy Number Variations (CNVs) and Pathological Copy Number Alterations (CNAs)
3.2.5. Polymorphic CNVs Are More Prevalent in hiPSCs Than in hESCs, while Potentially Pathogenic CNAs Are More Frequent in hESCs
3.3. Analysis on the Effect of QMS Implementation on the Acquisition Rate of Genomic Alterations by hPSCs
3.3.1. The Introduction of SOPs Results in a Reduction in the Prevalence and Types of De Novo Karyotypic Pathogenic Alterations and FPAs in hPSCs Cultured In Vitro
3.3.2. The Use of SOPs also Results in a Reduction in the Prevalence and Types of De Novo CNVs and CNAs in hPSCs Maintained In Vitro
3.3.3. The Standardization of hPSC Maintenance Procedures via QMS Implementation Leads to a Huge Reduction in the Probability of hPSCs Acquiring De Novo Karyotypic Aberrations and Subchromosomal CNAs during In Vitro Culture
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Evans, M.J.; Kaufman, M.H. Establishment in culture of pluripotential cells from mouse embryos. Nature 1981, 292, 154–156. [Google Scholar] [CrossRef] [PubMed]
- Thomson, J.A.; Itskovitz-Eldor, J.; Shapiro, S.S.; Waknitz, M.A.; Swiergiel, J.J.; Marshall, V.S.; Jones, J.M. Embryonic Stem Cell Lines Derived from Human Blastocysts. Science 1998, 282, 1145–1147. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Takahashi, K.; Yamanaka, S. Induction of Pluripotent Stem Cells from Mouse Embryonic and Adult Fibroblast Cultures by Defined Factors. Cell 2006, 126, 663–676. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Takahashi, K.; Tanabe, K.; Ohnuki, M.; Narita, M.; Ichisaka, T.; Tomoda, K.; Yamanaka, S. Induction of Pluripotent Stem Cells from Adult Human Fibroblasts by Defined Factors. Cell 2007, 131, 861–872. [Google Scholar] [CrossRef] [Green Version]
- Zhu, Z.; Huangfu, D. Human pluripotent stem cells: An emerging model in developmental biology. Development 2013, 140, 705–717. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Avior, Y.; Sagi, I.; Benvenisty, N. Pluripotent stem cells in disease modelling and drug discovery. Nat. Rev. Mol. Cell Biol. 2016, 17, 170–182. [Google Scholar] [CrossRef] [PubMed]
- Yamanaka, S. Pluripotent Stem Cell-Based Cell Therapy-Promise and Challenges. Cell Stem Cell 2020, 27, 3–531. [Google Scholar] [CrossRef] [PubMed]
- Keller, A.; Spits, C. The Impact of Acquired Genetic Abnormalities on the Clinical Translation of Human Pluripotent Stem Cells. Cells 2021, 10, 3246. [Google Scholar] [CrossRef]
- Mandai, M.; Kurimoto, Y.; Takahashi, M. Autologous Induced Stem-Cell-Derived Retinal Cells for Macular Degeneration. N. Engl. J. Med. 2017, 377, 792–793. [Google Scholar] [CrossRef]
- Schwartz, S.D.; Regillo, C.D.; Lam, B.L.; Eliott, D.; Rosenfeld, P.J.; Gregori, N.Z.; Hubschman, J.-P.; Davis, J.L.; Heilwell, G.; Spirn, M.; et al. Human embryonic stem cell-derived retinal pigment epithelium in patients with age-related macular degeneration and Stargardt’s macular dystrophy: Follow-up of two open-label phase 1/2 studies. Lancet 2015, 385, 509–516. [Google Scholar] [CrossRef]
- da Cruz, L.; Fynes, K.; Georgiadis, O.; Kerby, J.; Luo, Y.H.; Ahmado, A.; Vernon, A.; Daniels, J.T.; Nommiste, B.; Hasan, S.M.; et al. Phase 1 clinical study of an embryonic stem cell-derived retinal pigment epithelium patch in age-related macular degeneration. Nat. Biotechnol. 2018, 36, 328–337. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schwartz, S.D.; Hubschman, J.-P.; Heilwell, G.; Franco-Cardenas, V.; Pan, C.K.; Ostrick, R.M.; Mickunas, E.; Gay, R.; Klimanskaya, I.; Lanza, R. Embryonic stem cell trials for macular degeneration: A preliminary report. Lancet 2012, 379, 713–720. [Google Scholar] [CrossRef]
- Song, W.K.; Park, K.-M.; Kim, H.-J.; Lee, J.H.; Choi, J.; Chong, S.Y.; Shim, S.H.; Del Priore, L.V.; Lanza, R. Treatment of Macular Degeneration Using Embryonic Stem Cell-Derived Retinal Pigment Epithelium: Preliminary Results in Asian Patients. Stem Cell Rep. 2015, 4, 860–872. [Google Scholar] [CrossRef] [PubMed]
- Barker, R.A.; Parmar, M.; Studer, L.; Takahashi, J. Human Trials of Stem Cell-Derived Dopamine Neurons for Parkinson’s Disease: Dawn of a New Era. Cell Stem Cell 2017, 21, 569–573. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schweitzer, J.S.; Song, B.; Herrington, T.M.; Park, T.-Y.; Lee, N.; Ko, S.; Jeon, J.; Cha, Y.; Kim, K.; Li, Q.; et al. Personalized iPSC-Derived Dopamine Progenitor Cells for Parkinson’s Disease. N. Engl. J. Med. 2020, 382, 1926–1932. [Google Scholar] [CrossRef] [PubMed]
- Halliwell, J.; Barbaric, I.; Andrews, P.W. Acquired genetic changes in human pluripotent stem cells: Origins and consequences. Nat. Rev. Mol. Cell Biol. 2020, 21, 715–728. [Google Scholar] [CrossRef] [PubMed]
- Garber, K. RIKEN suspends first clinical trial involving induced pluripotent stem cells. Nat. Biotechnol. 2015, 33, 890–891. [Google Scholar] [CrossRef]
- Yasuda, S.; Kusakawa, S.; Kuroda, T.; Miura, T.; Tano, K.; Takada, N.; Matsuyama, S.; Matsuyama, A.; Nasu, M.; Umezawa, A.; et al. Tumorigenicity-associated characteristics of human iPS cell lines. PLoS ONE 2018, 13, e0205022. [Google Scholar] [CrossRef] [Green Version]
- Sato, Y.; Bando, H.; Di Piazza, M.; Gowing, G.; Herberts, C.; Jackman, S.; Leoni, G.; Libertini, S.; MacLachlan, T.; McBlane, J.W.; et al. Tumorigenicity assessment of cell therapy products: The need for global consensus and points to consider. Cytotherapy 2019, 21, 1095–1111. [Google Scholar] [CrossRef]
- Baker, D.E.C.; Harrison, N.J.; Maltby, E.; Smith, K.; Moore, H.D.; Shaw, P.J.; Heath, P.R.; Holden, H.; Andrews, P.W. Adaptation to culture of human embryonic stem cells and oncogenesis in vivo. Nat. Biotechnol. 2007, 25, 207–215. [Google Scholar] [CrossRef]
- Draper, J.S.; Smith, K.; Gokhale, P.; Moore, H.D.; Maltby, E.; Johnson, J.; Meisner, L.; Zwaka, T.P.; Thomson, J.A.; Andrews, P.W. Recurrent gain of chromosomes 17q and 12 in cultured human embryonic stem cells. Nat. Biotechnol. 2004, 22, 53–54. [Google Scholar] [CrossRef]
- Initiative, I.S.C.; Amps, K.; Andrews, P.W.; Anyfantis, G.; Armstrong, L.; Avery, S.; Baharvand, H.; Baker, J.; Baker, D.; Munoz, M.B.; et al. Screening ethnically diverse human embryonic stem cells identifies a chromosome 20 minimal amplicon conferring growth advantage. Nat. Biotechnol. 2011, 29, 1132–1144. [Google Scholar]
- Ronen, D.; Benvenisty, N. Genomic stability in reprogramming. Curr. Opin. Genet. Dev. 2012, 22, 444–449. [Google Scholar] [CrossRef] [PubMed]
- Martins-Taylor, K.; Xu, R.-H. Concise review: Genomic stability of human induced pluripotent stem cells. Stem Cells 2012, 30, 22–27. [Google Scholar] [CrossRef] [PubMed]
- Lund, R.J.; Närvä, E.; Lahesmaa, R. Genetic and epigenetic stability of human pluripotent stem cells. Nat. Rev. Genet. 2012, 13, 732–744. [Google Scholar] [CrossRef] [PubMed]
- Taapken, S.M.; Nisler, B.S.; Newton, M.A.; Sampsell-Barron, T.L.; Leonhard, K.A.; McIntire, E.M.; Montgomery, K.D. Karotypic abnormalities in human induced pluripotent stem cells and embryonic stem cells. Nat. Biotechnol. 2011, 29, 313–314. [Google Scholar] [CrossRef]
- Peterson, S.E.; Westra, J.W.; Rehen, S.K.; Young, H.; Bushman, D.M.; Paczkowski, C.M.; Yung, Y.C.; Lynch, C.L.; Tran, H.T.; Nickey, K.S.; et al. Normal human pluripotent stem cell lines exhibit pervasive mosaic aneuploidy. PLoS ONE 2011, 6, e23018. [Google Scholar] [CrossRef]
- Laurent, L.C.; Ulitsky, I.; Slavin, I.; Tran, H.; Schork, A.; Morey, R.; Lynch, C.; Harness, J.V.; Lee, S.; Barrero, M.J.; et al. Dynamic changes in the copy number of pluripotency and cell proliferation genes in human ESCs and iPSCs during reprogramming and time in culture. Cell Stem Cell 2011, 8, 106–118. [Google Scholar] [CrossRef] [Green Version]
- Bai, Q.; Desprat, R.; Klein, B.; Lemaitre, J.-M.; De Vos, J. Embryonic Stem Cells or Induced Pluripotent Stem Cells? A DNA Integrity Perspective. Curr. Gene Ther. 2013, 13, 93–98. [Google Scholar] [CrossRef] [Green Version]
- Lefort, N.; Perrier, A.L.; Laâbi, Y.; Varela, C.; Peschanski, M. Human embryonic stem cells and genomic instability. Regen. Med. 2009, 4, 899–909. [Google Scholar] [CrossRef]
- Oliveira, P.H.; da Silva, C.L.; Cabral, J.M.S. Concise Review: Genomic Instability in Human Stem Cells: Current Status and Future Challenges. Stem Cells 2014, 32, 2824–2832. [Google Scholar] [CrossRef] [PubMed]
- Qi, S.; Fang, Z.; Wang, D.; Menendez, P.; Yao, K.; Ji, J. Concise Review: Induced Pluripotency by Defined Factors: Prey of Oxidative Stress. Stem Cells 2015, 33, 1371–1376. [Google Scholar] [CrossRef] [PubMed]
- Ben-David, U.; Benvenisty, N.; Mayshar, Y. Genetic instability in human induced pluripotent stem cells: Classification of causes and possible safeguards. Cell Cycle 2010, 9, 4603–4604. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ben-David, U.; Arad, G.; Weissbein, U.; Mandefro, B.; Maimon, A.; Golan-Lev, T.; Narwani, K.; Clark, A.T.; Andrews, P.W.; Benvenisty, N.; et al. Aneuploidy induces profound changes in gene expression, proliferation and tumorigenicity of human pluripotent stem cells. Nat. Commun. 2014, 5, 4825. [Google Scholar] [CrossRef] [PubMed]
- Thompson, O.; von Meyenn, F.; Hewitt, Z.; Alexander, J.; Wood, A.; Weightman, R.; Gregory, S.; Krueger, F.; Andrews, S.; Barbaric, I.; et al. Low rates of mutation in clinical grade human pluripotent stem cells under different culture conditions. Nat. Commun. 2020, 11, 1528. [Google Scholar] [CrossRef] [Green Version]
- Rouhani, F.J.; Nik-Zainal, S.; Wuster, A.; Li, Y.; Conte, N.; Koike-Yusa, H.; Kumasaka, N.; Vallier, L.; Yusa, K.; Bradley, A. Mutational History of a Human Cell Lineage from Somatic to Induced Pluripotent Stem Cells. PLoS Genet. 2016, 12, e1005932. [Google Scholar] [CrossRef] [Green Version]
- Jacobs, K.; Zambelli, F.; Mertzanidou, A.; Smolders, I.; Geens, M.; Nguyen, H.T.; Barbé, L.; Sermon, K.; Spits, C. Higher-Density Culture in Human Embryonic Stem Cells Results in DNA Damage and Genome Instability. Stem Cell Rep. 2016, 6, 330–341. [Google Scholar] [CrossRef] [Green Version]
- Forsyth, N.R.; Musio, A.; Vezzoni, P.; Simpson, A.H.R.W.; Noble, B.S.; McWhir, J. Physiologic oxygen enhances human embryonic stem cell clonal recovery and reduces chromosomal abnormalities. Cloning Stem Cells 2006, 8, 16–23. [Google Scholar] [CrossRef]
- Garitaonandia, I.; Amir, H.; Boscolo, F.S.; Wambua, G.K.; Schultheisz, H.L.; Sabatini, K.; Morey, R.; Waltz, S.; Wang, Y.-C.; Tran, H.; et al. Increased risk of genetic and epigenetic instability in human embryonic stem cells associated with specific culture conditions. PLoS ONE 2015, 10, e0118307. [Google Scholar] [CrossRef]
- Guidance Document on Good In Vitro Method Practices (GIVIMP); OECD Series on Testing and Assessment; OECD: Paris, France, 2018; ISBN 9789264311008.
- Meiser, I.; Majer, J.; Katsen-Globa, A.; Schulz, A.; Schmidt, K.; Stracke, F.; Koutsouraki, E.; Witt, G.; Keminer, O.; Pless, O.; et al. Droplet-based vitrification of adherent human induced pluripotent stem cells on alginate microcarrier influenced by adhesion time and matrix elasticity. Cryobiology 2021, 103, 57–69. [Google Scholar] [CrossRef]
- Bradley, C.K.; Scott, H.A.; Chami, O.; Peura, T.T.; Dumevska, B.; Schmidt, U.; Stojanov, T. Derivation of Huntington’s disease-affected human embryonic stem cell lines. Stem Cells Dev. 2011, 20, 495–502. [Google Scholar] [CrossRef] [PubMed]
- Consortium, H.D. iPSC Induced pluripotent stem cells from patients with Huntington’s disease show CAG-repeat-expansion-associated phenotypes. Cell Stem Cell 2012, 11, 264–278. [Google Scholar]
- Ooi, J.; Langley, S.R.; Xu, X.; Utami, K.H.; Sim, B.; Huang, Y.; Harmston, N.P.; Tay, Y.L.; Ziaei, A.; Zeng, R.; et al. Unbiased Profiling of Isogenic Huntington Disease hPSC-Derived CNS and Peripheral Cells Reveals Strong Cell-Type Specificity of CAG Length Effects. Cell Rep. 2019, 26, 2494–2508.e7. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Warmflash, A.; Sorre, B.; Etoc, F.; Siggia, E.D.; Brivanlou, A.H. A method to recapitulate early embryonic spatial patterning in human embryonic stem cells. Nat. Methods 2014, 11, 847–854. [Google Scholar] [CrossRef] [Green Version]
- Cyranoski, D. Japanese woman is first recipient of next-generation stem cells. Nature 2014. Available online: https://www.nature.com/articles/nature.2014.15915 (accessed on 12 September 2014).
- Henry, M.P.; Hawkins, J.R.; Boyle, J.; Bridger, M. The Genomic Health of Human Pluripotent Stem Cells: Genomic Instability and the Consequences on Nuclear Organization. Front. Genet. 2019, 9, 623. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mitalipova, M.M.; Rao, R.R.; Hoyer, D.M.; Johnson, J.A.; Meisner, L.F.; Jones, K.L.; Dalton, S.; Stice, S.L. Preserving the genetic integrity of human embryonic stem cells. Nat. Biotechnol. 2005, 23, 19–20. [Google Scholar] [CrossRef] [PubMed]
- Olariu, V.; Harrison, N.J.; Coca, D.; Gokhale, P.J.; Baker, D.; Billings, S.; Kadirkamanathan, V.; Andrews, P.W. Modeling the evolution of culture-adapted human embryonic stem cells. Stem Cell Res. 2010, 4, 50–56. [Google Scholar] [CrossRef] [Green Version]
- Rossi, A.; Lickfett, S.; Martins, S.; Prigione, A. A call for consensus guidelines on monitoring the integrity of nuclear and mitochondrial genomes in human pluripotent stem cells. Stem Cell Reports 2022, 17, 707–710. [Google Scholar] [CrossRef]
- Steinemann, D.; Göhring, G.; Schlegelberger, B. Genetic instability of modified stem cells-a first step towards malignant transformation? Am. J. Stem Cells 2013, 2, 39. [Google Scholar]
- Valli, R.; Marletta, C.; Pressato, B.; Montalbano, G.; Lo Curto, F.; Pasquali, F.; Maserati, E. Comparative genomic hybridization on microarray (a-CGH) in constitutional and acquired mosaicism may detect as low as 8% abnormal cells. Mol. Cytogenet. 2011, 4, 13. [Google Scholar] [CrossRef] [Green Version]
- Lu, J.; Liu, H.; Huang, C.T.-L.; Chen, H.; Du, Z.; Liu, Y.; Sherafat, M.A.; Zhang, S.-C. Generation of integration-free and region-specific neural progenitors from primate fibroblasts. Cell Rep. 2013, 3, 1580–1591. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hussein, S.M.; Batada, N.N.; Vuoristo, S.; Ching, R.W.; Autio, R.; Närvä, E.; Ng, S.; Sourour, M.; Hämäläinen, R.; Olsson, C.; et al. Copy number variation and selection during reprogramming to pluripotency. Nature 2011, 471, 58–62. [Google Scholar] [CrossRef] [PubMed]
- Avery, S.; Hirst, A.J.; Baker, D.; Lim, C.Y.; Alagaratnam, S.; Skotheim, R.I.; Lothe, R.A.; Pera, M.F.; Colman, A.; Robson, P.; et al. BCL-XL mediates the strong selective advantage of a 20q11.21 amplification commonly found in human embryonic stem cell cultures. Stem Cell Rep. 2013, 1, 379–386. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nguyen, H.T.; Geens, M.; Mertzanidou, A.; Jacobs, K.; Heirman, C.; Breckpot, K.; Spits, C. Gain of 20q11.21 in human embryonic stem cells improves cell survival by increased expression of Bcl-xL. Mol. Hum. Reprod. 2014, 20, 168–177. [Google Scholar] [CrossRef]
- Baker, D.; Hirst, A.J.; Gokhale, P.J.; Juarez, M.A.; Williams, S.; Wheeler, M.; Bean, K.; Allison, T.F.; Moore, H.D.; Andrews, P.W.; et al. Detecting Genetic Mosaicism in Cultures of Human Pluripotent Stem Cells. Stem Cell Rep. 2016, 7, 998–1012. [Google Scholar] [CrossRef] [Green Version]
- Werbowetski-Ogilvie, T.E.; Bossé, M.; Stewart, M.; Schnerch, A.; Ramos-Mejia, V.; Rouleau, A.; Wynder, T.; Smith, M.-J.; Dingwall, S.; Carter, T.; et al. Characterization of human embryonic stem cells with features of neoplastic progression. Nat. Biotechnol. 2009, 27, 91–97. [Google Scholar] [CrossRef]
- Markouli, C.; Couvreu De Deckersberg, E.; Regin, M.; Nguyen, H.T.; Zambelli, F.; Keller, A.; Dziedzicka, D.; De Kock, J.; Tilleman, L.; Van Nieuwerburgh, F.; et al. Gain of 20q11.21 in Human Pluripotent Stem Cells Impairs TGF-β-Dependent Neuroectodermal Commitment. Stem Cell Rep. 2019, 13, 163–176. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Andrews, P.W. From teratocarcinomas to embryonic stem cells. Philos. Trans. R. Soc. London. Ser. B Biol. Sci. 2002, 357, 405–417. [Google Scholar] [CrossRef]
- Atkin, N.B.; Baker, M.C. Specific chromosome change, i(12p), in testicular tumours? Lancet 1982, 2, 1349. [Google Scholar] [CrossRef]
- Nguyen Thi, H.; Duong, H. The molecular characteristics of colorectal cancer: Implications for diagnosis and therapy (Review). Oncol. Lett. 2018, 16, 9–18. [Google Scholar] [CrossRef] [Green Version]
- Scotto, L.; Narayan, G.; Nandula, S.V.; Arias-Pulido, H.; Subramaniyam, S.; Schneider, A.; Kaufmann, A.M.; Wright, J.D.; Pothuri, B.; Mansukhani, M.; et al. Identification of copy number gain and overexpressed genes on chromosome arm 20q by an integrative genomic approach in cervical cancer: Potential role in progression. Genes. Chromosomes Cancer 2008, 47, 755–765. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Beroukhim, R.; Mermel, C.H.; Porter, D.; Wei, G.; Raychaudhuri, S.; Donovan, J.; Barretina, J.; Boehm, J.S.; Dobson, J.; Urashima, M.; et al. The landscape of somatic copy-number alteration across human cancers. Nature 2010, 463, 899–905. [Google Scholar] [CrossRef] [PubMed]
- Tabach, Y.; Kogan-Sakin, I.; Buganim, Y.; Solomon, H.; Goldfinger, N.; Hovland, R.; Ke, X.-S.; Oyan, A.M.; Kalland, K.-H.; Rotter, V.; et al. Amplification of the 20q Chromosomal Arm Occurs Early in Tumorigenic Transformation and May Initiate Cancer. PLoS ONE 2011, 6, e14632. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Riggs, M.J.; Sheridan, S.D.; Rao, R.R. ARHGDIA Confers Selective Advantage to Dissociated Human Pluripotent Stem Cells. Stem Cells Dev. 2021, 30, 705–713. [Google Scholar] [CrossRef]
- Tackett, J.L.; Miller, J.D. Introduction to the special section on increasing replicability, transparency, and openness in clinical psychology. J. Abnorm. Psychol. 2019, 128, 487–492. [Google Scholar] [CrossRef]
- Miyakawa, T. No raw data, no science: Another possible source of the reproducibility crisis. Mol. Brain 2020, 13, 24. [Google Scholar] [CrossRef] [Green Version]
- Baker, M. Is there a reproducibility crisis? Nature 2016, 533, 452–454. [Google Scholar] [CrossRef] [Green Version]
- Pegliasco, J.; Hirsch, P.; Marzac, C.; Isnard, F.; Meniane, J.-C.; Deswarte, C.; Pellet, P.; Lemaitre, C.; Leroy, G.; Rabadan Moraes, G.; et al. Germline ATG2B/GSKIP-containing 14q32 duplication predisposes to early clonal hematopoiesis leading to myeloid neoplasms. Leukemia 2022, 36, 126–137. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Cell Line | hPSCreg®/NIH ID | Disease Status | hESC/hiPSC | Institution * |
|---|---|---|---|---|
| GENEA-019 | GENEAe020-A | Unaffected | hESC | GENEA |
| H9 | WAe009-A | Unaffected | hESC | WCRI |
| RUES2 | RUESe002-A | Unaffected | hESC | RU |
| RUES2 2123 | RUESe002-A-1 | Unaffected | hESC | RU |
| CTR2161 | CHDIi013-A | Unaffected | hiPSC | CHDI |
| CTR2162 | CHDIi014-A | Unaffected | hiPSC | CHDI |
| CTR2164 | CHDIi016-A | Unaffected | hiPSC | CHDI |
| CTR2175 | CHDIi027-A | Unaffected | hiPSC | CHDI |
| CTR2190 | CHDIi042-A | Unaffected | hiPSC | CHDI |
| CTR33n1 | CS83iCTR33n1 | Unaffected | hiPSC | CSRMI |
| GENEA-018 | NIHhESC-12-0169 | Affected HD | hESC | GENEA |
| GENEA-020 | GENEAe015-A | Affected HD | hESC | GENEA |
| HD65Q | NA | Affected HD | hESC | TLGM |
| RUES2 1811 | RUESe002-A-2 | Affected HD | hESC | RU |
| HD60n5 | CS21iHD60n5 | Affected HD | hiPSC | CSRMI |
| HD2158 | CHDIi010-A | Affected HD | hiPSC | CHDI |
| HD2170 | CHDIi022-A | Affected HD | hiPSC | CHDI |
| HD2174 | CHDIi026-A | Affected HD | hiPSC | CHDI |
| HD2197 | CHDIi049-A | Affected HD | hiPSC | CHDI |
| Alteration | % of Affected hPSCs | % of Affected hESCs | % of Affected hiPSCs | |
|---|---|---|---|---|
| Chromosomal pathogenic alterations | 47,XX,+8 | 2.78 | NA | 6.12 |
| 47,XX,+12 | 1.85 | NA | 4.08 | |
| 46,XX,i(20) (q10) | 11.11 | 10.17 | 12.24 | |
| 47,XX,+20 | 0.93 | 1.69 | NA | |
| Frequent Polymorphic Alterations (FPAs) | 46,XX, inv(9) (p11q13) | 3.70 | 1.69 | 6.12 |
| 46,XX,21qs+ | 3.70 | NA | 8.16 | |
| Alteration | Region | hESC/hiPSC | % Cells Affected | % of Total CNVs |
|---|---|---|---|---|
| Deletion | 1p11.2 | hiPSC | 11.54 | 23.08 |
| Deletion | 3q26.1 | hiPSC | 11.54 | 23.08 |
| Insertion | 4q35.2 | hiPSC | 7.69 | 15.38 |
| Insertion | 5p15.33 | hiPSC | 7.69 | 15.38 |
| Deletion | 10q21.3 | hiPSC | 7.69 | 15.38 |
| Deletion | 22q11.21 | hiPSC | 3.85 | 7.69 |
| Alteration | Region | hESC/ hiPSC | % Cells Affected (All hPSCs) | % of Cells Affected (hPSC Type) | % of Total CNAs (All hPSCs) | % of Total CNAs (hPSC Type) |
|---|---|---|---|---|---|---|
| Deletion | 1q21.2 | hiPSC | 3.85 | 5.88 | 2.70 | 4 |
| Deletion | 2q23.3 | hiPSC | 7.69 | 11.76 | 5.41 | 8 |
| Deletion | 2q37.3 | hiPSC | 7.69 | 11.76 | 5.41 | 8 |
| Deletion | 3q13.31 | hiPSC | 7.69 | 11.76 | 5.41 | 8 |
| Deletion | 4q28.3 | hiPSC | 7.69 | 11.76 | 5.41 | 8 |
| Insertion | 6q26 | hiPSC | 7.69 | 11.76 | 5.41 | 8 |
| Deletion | 7q11.23 | hiPSC | 3.85 | 5.88 | 2.70 | 4 |
| Deletion | 7p14.3 | hiPSC | 3.85 | 5.88 | 2.70 | 4 |
| Duplication | 8q24.3 | hiPSC | 7.69 | 11.76 | 5.41 | 8 |
| Insertion | 10q23.32 | hESC | 15.38 | 44.44 | 10.81 | 16 |
| Deletion | 11q11 | hiPSC | 7.69 | 11.76 | 5.41 | 8 |
| Insertion | 12p13.31 | hiPSC | 3.85 | 5.88 | 2.70 | 4 |
| Insertion | 14q23.3 | hESC | 7.69 | 22.22 | 5.41 | 8 |
| Insertion | 14q32 | hiPSC | 3.85 | 5.88 | 2.70 | 4 |
| Deletion | 15q11.1–11.2 | hiPSC | 3.85 | 5.88 | 2.70 | 4 |
| Deletion | 15q26.3 | hiPSC | 7.69 | 11.76 | 5.41 | 8 |
| Duplication | 20q11.21 | hESC and hiPSC | 26.92 | 66.67 (hESCs), 5.88 (hiPSCs) | 16.22 | 24 (hESCs), 4 (hiPSCs) |
| Insertion | 22q11.2 | hiPSC | 7.69 | 11.76 | 2.70 | 8 |
| Culture Conditions | Chromosomal Alterations (G-Banding) | Subchromosomal Alterations (aCGH) | ||
|---|---|---|---|---|
| FPA | Pathogenic | CNV | CNA | |
| Pre-adaptation (PrA) | 11.76% | 46.67% | 46.67% | 11.76% |
| Post-adaptation (PoA) | 5.49% | 18.18% | 18.18% | 5.49% |
| −53.31% | −61.05% | −61.05% | −53.31% | |
| Fold-change | −2.14 | −2.57 | −2.57 | −2.14 |
| Alteration | Region | Size | % of Affected hPSCs | |
|---|---|---|---|---|
| Pathogenic chromosomal alterations | Trisomy | +8 | NA | 3.30 |
| Trisomy | +12 | NA | 2.20 | |
| Isochromosome | i(20) (q10) | NA | 3.30 | |
| Chromosomal FPAs | Pericentric inversion | inv(9) (p11q13) | NA | 1.10 |
| Microsatellite | 21qs+ | NA | 4.40 | |
| Subchromosomal CNVs | Deletion | 3q26.21 | 62 kb | 9.09 |
| Insertion | 4q35.2 | 219 kb | 9.09 | |
| Subchromosomal CNAs | Insertion | 14q23.3 | 370 kb | 18.18 |
| Insertion | 14q32 | 136 kb | 9.09 | |
| Duplication | 20q11.21 | 4.7 Mb | 9.09 | |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Molina-Ruiz, F.J.; Introna, C.; Bombau, G.; Galofre, M.; Canals, J.M. Standardization of Cell Culture Conditions and Routine Genomic Screening under a Quality Management System Leads to Reduced Genomic Instability in hPSCs. Cells 2022, 11, 1984. https://doi.org/10.3390/cells11131984
Molina-Ruiz FJ, Introna C, Bombau G, Galofre M, Canals JM. Standardization of Cell Culture Conditions and Routine Genomic Screening under a Quality Management System Leads to Reduced Genomic Instability in hPSCs. Cells. 2022; 11(13):1984. https://doi.org/10.3390/cells11131984
Chicago/Turabian StyleMolina-Ruiz, Francisco J., Clelia Introna, Georgina Bombau, Mireia Galofre, and Josep M. Canals. 2022. "Standardization of Cell Culture Conditions and Routine Genomic Screening under a Quality Management System Leads to Reduced Genomic Instability in hPSCs" Cells 11, no. 13: 1984. https://doi.org/10.3390/cells11131984