1. Introduction
Alzheimer’s disease (AD) is the most common and burdensome of the late-onset degenerative dementias: the World Alzheimer’s Report estimated a global prevalence of over 50 million worldwide, a number expected to triple by 2050 [
1,
2]. AD manifests as progressive memory impairment initially and a faster rate of cognitive decline and neurodegeneration in later stages, along with behavioral and physiological manifestations [
3]. Despite the convoluted etiology of AD, experimental and theoretical investigation suggests that synapses are the primary targets in the early stage of the disease [
4]. Histologically, the AD brain is marked by the extracellular deposition of senile beta-amyloid (Aβ) plaques—in early-onset FAD, this accumulation has been traced to abnormalities in the genes encoding amyloid precursor protein (APP) or intramembrane protease presenilin 1 and 2 (PS1, PS2). While rare (5% of AD cases), this form of Alzheimer’s disease holds an estimated heritability over 90% [
5]. These abnormalities are accompanied by the intracellular accumulation of neurofibrillary tangles (NFTs)—composed of hyperphosphorylated tau proteins (pTau)—that litter the cerebral and hippocampal cortices [
6,
7,
8,
9]. Although the exact mechanism is still being debated, numerous experimental studies implicate elevated intracellular Ca
2+ levels as one of the main mechanisms underlying Aβ toxicity [
10,
11]. These studies show that the AD brain is surfeit with dysregulation of Ca
2+ signaling pathways [
10,
11,
12,
13], motivating researchers to propose the Ca
2+ hypothesis of AD and aging [
14]. Indeed, it has been shown that both intra- and extracellular Aβ oligomers and FAD-causing mutations in presenilin result in enhanced Ca
2+ release from the ER through inositol (1,4,5)-triphosphate (IP3) receptors (IP
3Rs) and/or ryanodine receptors (RyRs) [
10,
11,
12,
13,
14,
15,
16,
17,
18,
19,
20,
21,
22]. This upregulated Ca
2+ release can contribute to aberrant plasticity and the functional disruption of neuronal networks [
11,
23].
Intracellular Ca
2+ is an important second messenger, regulating a multitude of neuronal functions, including neurotransmitter release. Synaptic function at nerve terminals is tightly coupled to the intracellular Ca
2+ concentration [Ca
2+], as Ca
2+ primarily regulates the biological machinery responsible for exocytosis and short-term plasticity [
24,
25]. The precise temporal control of synaptic transmission by Ca
2+ is achieved via local signal transduction mechanisms that aim to regulate Ca
2+ excitability at the axonal bouton. Voltage-gated Ca
2+ channels (VGCCs) are the primary mediators of the transduction of depolarization-induced Ca
2+ transients into neurotransmitter release. Furthermore, Ca
2+ influx through VGCCs leads to physiological events that alter plasma membrane functions underpinning synaptic plasticity, protein expression, spine maintenance, and the regulation of excitability in excitatory synapses [
26,
27]. Likewise, investigations of the Ca
2+ dependence of vesicular release have highlighted the role of intracellular stores in Ca
2+ handling and spontaneous exocytosis [
28,
29]. Thus, the close association between these Ca
2+ pathways, their effect on numerous neuronal processes, and their high sensitivity to pathological perturbations make it especially valuable to elucidate the exact nature of the coupling.
Extensive evidence supports the presence of the ER in the nerve terminal of CA3 pyramidal neurons [
23,
28,
29,
30,
31,
32]. In neurons, activation of Ca
2+-sensitive channels such as IP
3Rs and RyRs triggers the release of Ca
2+ from the ER. Opening of IP
3Rs primarily depends on Ca
2+ and IP
3. To achieve this, glutamate released into the synaptic cleft elicits the production of IP
3 by the activation of membrane-bound metabotropic glutamate receptors (mGLuRs). RyRs activation, on the other hand, is largely controlled by cytosolic [Ca
2+]. This specialized cascade underscores the importance of IP
3Rs and RyRs in the regulation of the Ca
2+-induced Ca
2+ release (CICR) mechanism of the ER. Previous works have shown that CICR is necessary for ER stores to adequately influence spontaneous vesicle release and homosynaptic plasticity [
28,
33]. Indeed, in vitro studies confirm that properly sensitized CICR is necessary for normal synaptic function, whereas aberrant CICR underlies the presynaptic impairment associated with AD [
34,
35,
36,
37]. Despite this evidence, the precise role of ER Ca
2+ handling in action potential (AP)-evoked presynaptic Ca
2+ dynamics and its downstream effect on presynaptic neuronal processes remain unclear [
28,
34,
35,
37].
Information encoding at the CA3 to CA1 synapses in the hippocampus, which is crucial for learning and memory storage, relies on the spatiotemporal organization of Ca
2+ events leading up to synaptic transmission [
38]. AP arrival at the nerve terminal activates VGCCs, leading to high-amplitude, short-lived Ca
2+ influx events into the AZ. Coupled with this specialized pathway, Ca
2+ sensors initiate a heterogeneous fusion of neurotransmitter vesicles with the plasma membrane that often culminates in either fast synchronous or slow asynchronous release. Neuronal communication primarily relies on the synchronous mode of exocytosis, which is regulated by synaptotagmin-1 (Syt1) sensors with low Ca
2+ affinity and rapid kinetics that are critical for the exquisite temporal precision of vesicle fusion that characterizes synaptic transmission at most CA3-CA1 terminals [
39,
40,
41]. Such a high degree of synchrony is in part achieved by the steep dose dependence of evoked release on the short-lived Ca
2+ transients constrained to micro- or nanodomains within the vicinity of VGCCs [
24,
42]. A global buildup of [Ca
2+], on the other hand, accelerates the recruitment of release-ready vesicles, controlling the degree of synaptic plasticity [
43]. Thus, changes in Ca
2+ signals at the local or global scale are expected to disrupt synaptic transmission and plasticity. Consistent with this assertion, experimental manipulations that perturb evoked Ca
2+ influx alter the contribution of the synchronous mode of release to overall exocytosis and compromise synaptic plasticity [
44,
45,
46]. These findings highlight the need for a thorough investigation of the potential link between impaired synaptic function and disrupted Ca
2+ homeostasis in AD.
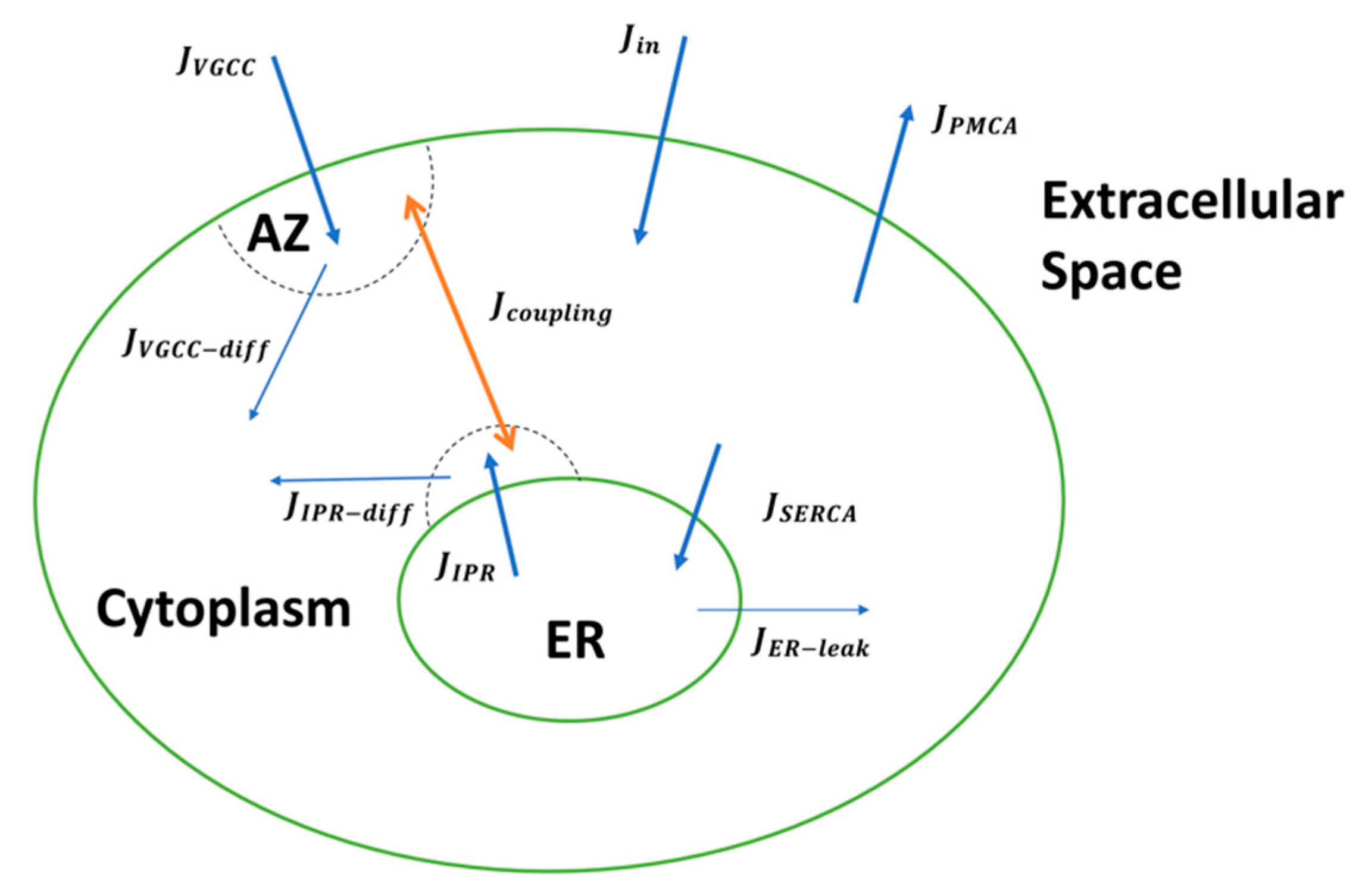
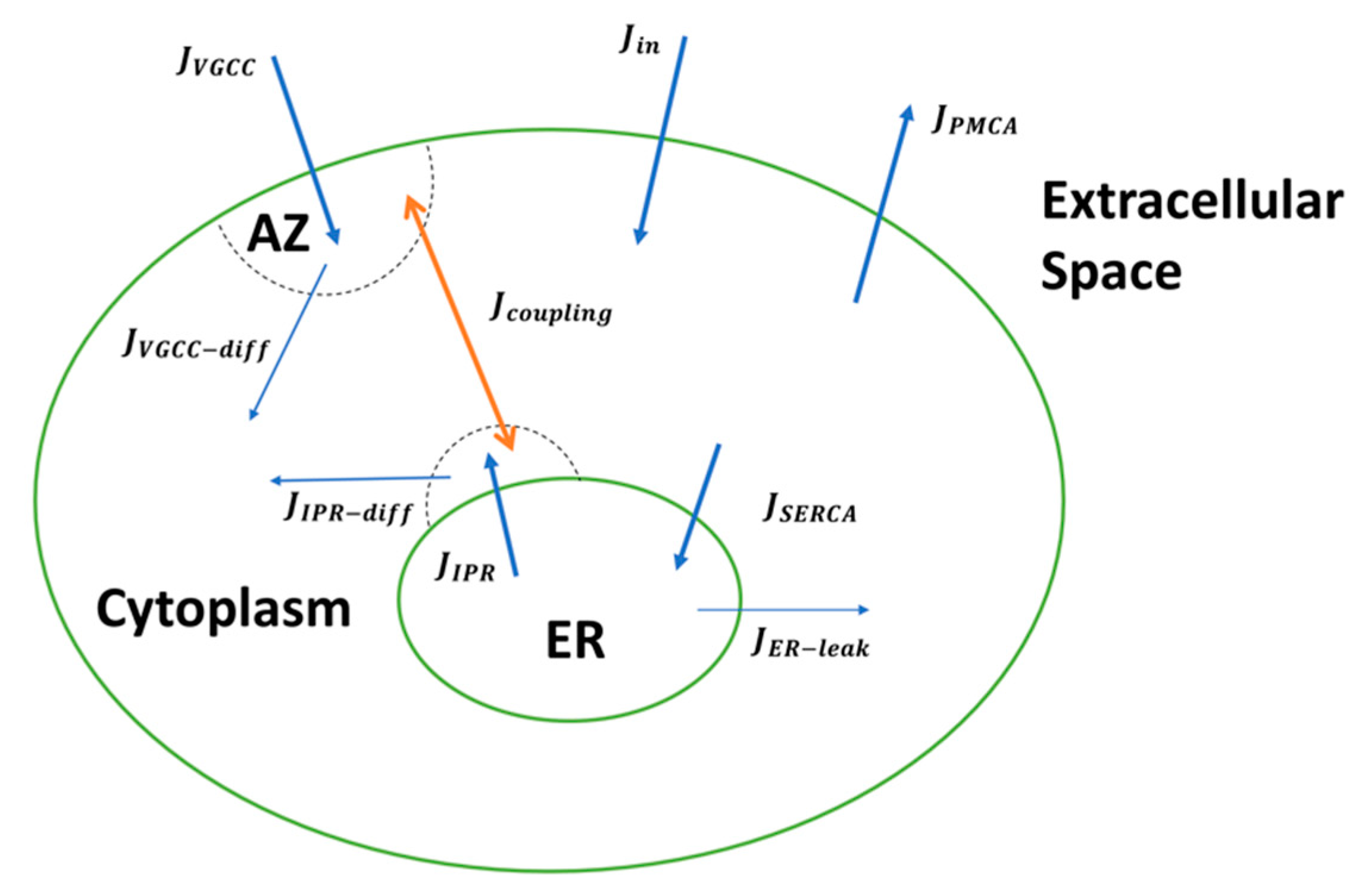
In this study, we incorporate findings from extensive experimental and computational studies to develop a detailed biophysical model of Ca2+-driven exocytosis at the CA3 presynaptic terminal. The model accounts for the observed Ca2+ and IP3 signaling pathways necessary for intracellular Ca2+ regulation and integrates the elaborate kinetics of neurotransmitter release—vesicle docking, mobilization, priming, and fusion—aided by distinct Ca2+ sensors. We reproduce crucial statistics of both Ca2+ and release events reported at small excitatory synapses, such as transient timescale, amplitude, and decay time. By developing a mathematical framework for coupling the Ca2+ domains surrounding the ER and AZ, we study how FAD-associated pathological Ca2+ release from the ER disrupts presynaptic neurotransmitter release rates and consequently alters synaptic plasticity and facilitation at affected synapses. Overall, our work provides novel insights on the pathologic role of aberrant neuronal Ca2+ handling on glutamate release and the downstream effects on synaptic dysfunction and cognitive decline observed in FAD. We address the limitations of this model, noting that the inclusion of the differential enrichment of VGCC subtypes, mitochondrial function, Ca2+ buffers, and RyRs activity in future extensions of the current work will provide a comprehensive computational framework that can be used to investigate key cellular mechanisms and processes, which in turn can be targeted for reversing presynaptic impairment in FAD.
4. Discussion
Despite extensive experimental studies reporting the ubiquitous presence of ER in both the axonal and dendritic compartments of neurons, little is known about its role in modifying the major components of synaptic transmission during AD-related pathologies [
23,
108,
109]. In this study, we fill this gap by building a detailed biophysical model that accurately captures the compartmentalized signaling of Ca
2+ at axonal terminals. In particular, our model incorporates ER-driven and AP-triggered presynaptic Ca
2+ signaling as well as the resulting release mechanism in WT and FAD-affected synapses. This is especially motivated by reports of distinct regulatory mechanisms for intracellular Ca
2+, which primarily includes a cluster of ligand-gated IP
3R Ca
2+ channels situated on the ER, as well as voltage-activated Ca
2+ channels constrained to the AZ, which result in tight microdomain signaling [
47,
53,
56,
71,
110]. In hippocampal synapses, the formation of evoked transient Ca
2+ microdomains in the AZ is predominantly mediated by the rapid kinetics of P/Q-type VGCCs, which open with minimal delay upon the arrival of AP, while subcellular domains in the vicinity of the ER occur via the stochastic gating of IP
3Rs that require IP
3 and Ca
2+ binding. In order to make meaningful quantitative predictions, we proceeded by developing a model that accurately captures the characteristic IP
3R’s gating in WT and FAD-affected neurons [
18,
19,
21]. We next incorporated a comprehensive description of presynaptic processes, including evoked Ca
2+ influx through VGCCs; Ca
2+ release and uptake by the ER; and synchronous, asynchronous, and spontaneous modes of synaptic transmission. Inspired by several lines of evidence elucidating the existence of a bidirectional interaction of intracellular Ca
2+ channels and presynaptic VGCCs mediated by stromal interaction molecules (STIM-1) and Orai channels, we have implemented a bidirectional coupling between the ER and AZ in our model that uncovers the unique biphasic dependence of the decay times of release and [Ca
2+] on the baseline probability [
91,
111]. Our model incorporates these critical components of presynaptic signaling and especially reproduces the observed spatiotemporal characteristics of intracellular Ca
2+—rise times, decay times, and amplitudes of corresponding events—and accounts for the stochasticity of presynaptic Ca
2+ dynamics driven by stochastic channel openings [
18,
53,
67,
112]. Release rate, facilitation, and depression vary dramatically among phasic and tonic synapses—phasic synapses are dominated by depression, whereas tonic synapses can be facilitated tremendously by vesicle recruitment [
24,
68]. Constrained by kinetics data of vesicular release observed in other phasic cell types, our model closely reproduces the essential determinants of neurotransmitter release, which consequently promoted the close estimation of PPR and facilitation requirements necessary to maintain normal plasma membrane function [
24,
73,
94]. Following these independent validations, it is reasonable to assume that the biophysical model and protocols developed in this work are physiologically realistic representations of neuronal processes in control and disease cases.
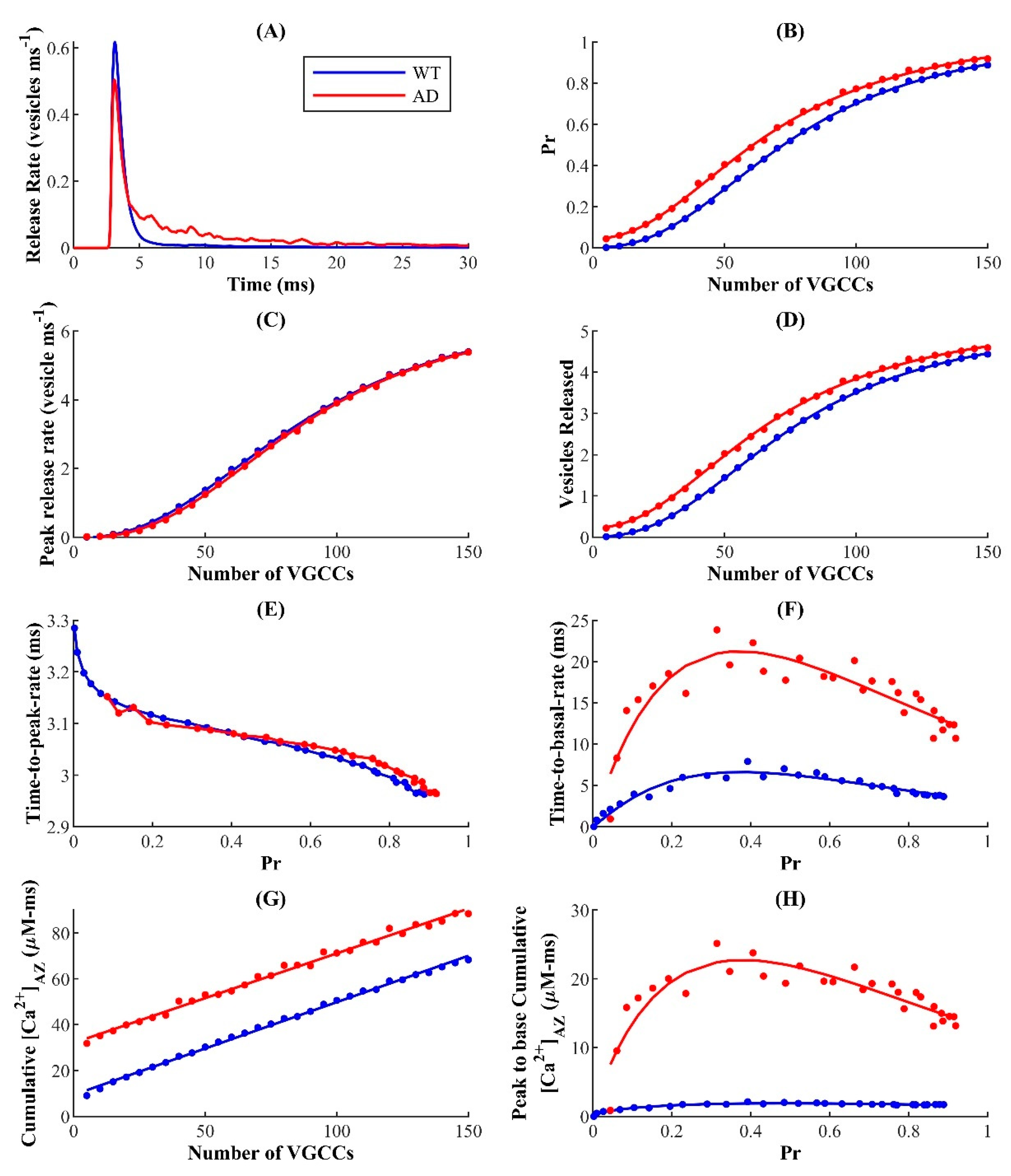
Broadening of Ca
2+ waveforms and response profiles affects the reliability of synaptic information transfer at affected terminals. Our results suggest that the FAD-associated increase in Ca
2+ release from the ER affects nearly all aspects of SVR. Despite the high stochasticity at the hippocampal CA3 terminal, the ER allows individual synapses operating in the low- and intermediate-probability regime to maintain relatively higher reliability of information-rate coding [
93]. Here, our results show that the FAD-associated enhanced [Ca
2+] selectively diminishes the reliability of intermediate-P
r synapses, suggesting that low and intermediate P
r synapses are more susceptible to FAD-associated Ca
2+ disruptions. Importantly, the model predicts that the aberrant Ca
2+ rise in AD-affected neurons may trigger hyperactivity over very short timescales (~30–60 ms) and lowers facilitation during prolonged (~450 ms) stimulation. We also report enhanced excitability in pathological synapses when simulated with higher coupling, which corresponds to a tighter feedback loop between the ER and AZ. Thus, our findings here provide a plausible explanation for why alterations in ER Ca
2+ handling, which result in excessive efflux, induce a severe perturbation of neuronal processes that can in turn decrease the reliability of information encoded in the firing rate of neurons affected by FAD pathology [
93,
113]. Overall, our findings provide novel insights into the role of aberrant ER Ca
2+ release in altering the release profile of a synapse in AD and other neurodegenerative diseases where such Ca
2+ impairments are observed [
114].
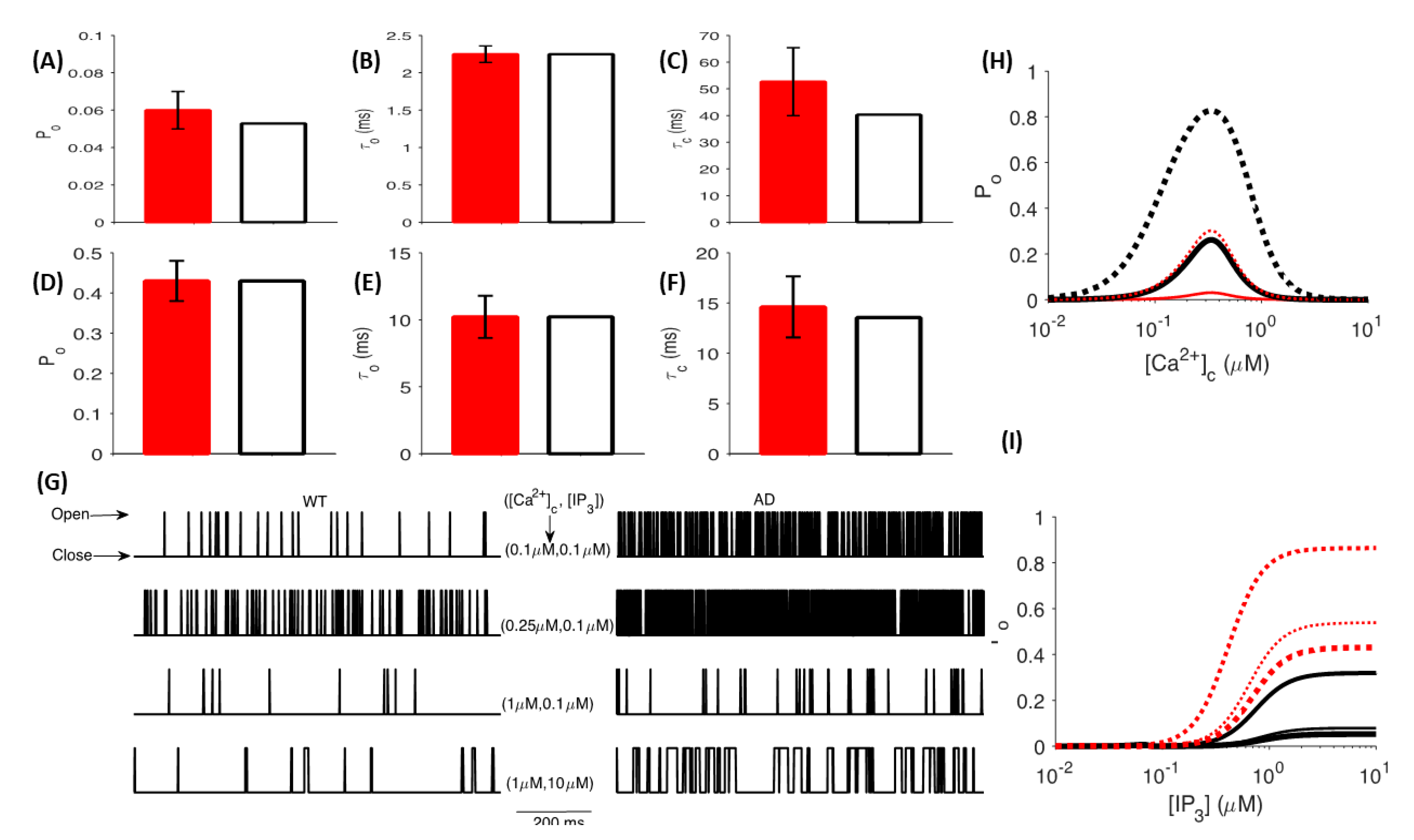
Many explorations into the Ca
2+ dependence of release have proposed that periodic Ca
2+ release from the ER, which occurs predominantly via the rapid gating kinetics of IP
3R, could trigger a series of physiological events—such as activating Ca
2+-sensing G-proteins—that eventually manifest as changes in the global [Ca
2+] and, in turn, influence spontaneous release: a miniature form of exocytosis [
28,
29]. Although IP
3Rs are differentially expressed according to cell type, several studies report the functional involvement of IP
3Rs in sculpting long-term potentiation (LTP) or depression (LTD) profiles in the CA1 region of the hippocampus [
115,
116]. In particular, immunocytochemical evidence reveals the expression of IP
3Rs in presynaptic terminals of the rat CA3-CA1 hippocampal synapses and elucidates their modulatory role in presynaptic neurotransmitter release and synaptic plasticity [
109]. Shorter opening times and longer closing times are characteristic features of the gating kinetics of IP
3Rs in cortical neurons and are essential for the homeostasis of local and global Ca
2+ in the norm. Consistent with these requirements, studies on AD patients have implicated exaggerated Ca
2+ release mediated by altered IP
3R activity in the dysregulation of bulk Ca
2+, which consequently triggers the progressive loss of synaptic function [
117,
118,
119,
120]. From our model, it is clear that these intrinsic biophysical properties of IP
3R make it highly effective at maintaining physiological bulk Ca
2+ and P
r, whereas upregulated IP
3R orchestrates exaggerated [Ca
2+] from the ER and in turn increases the P
r.
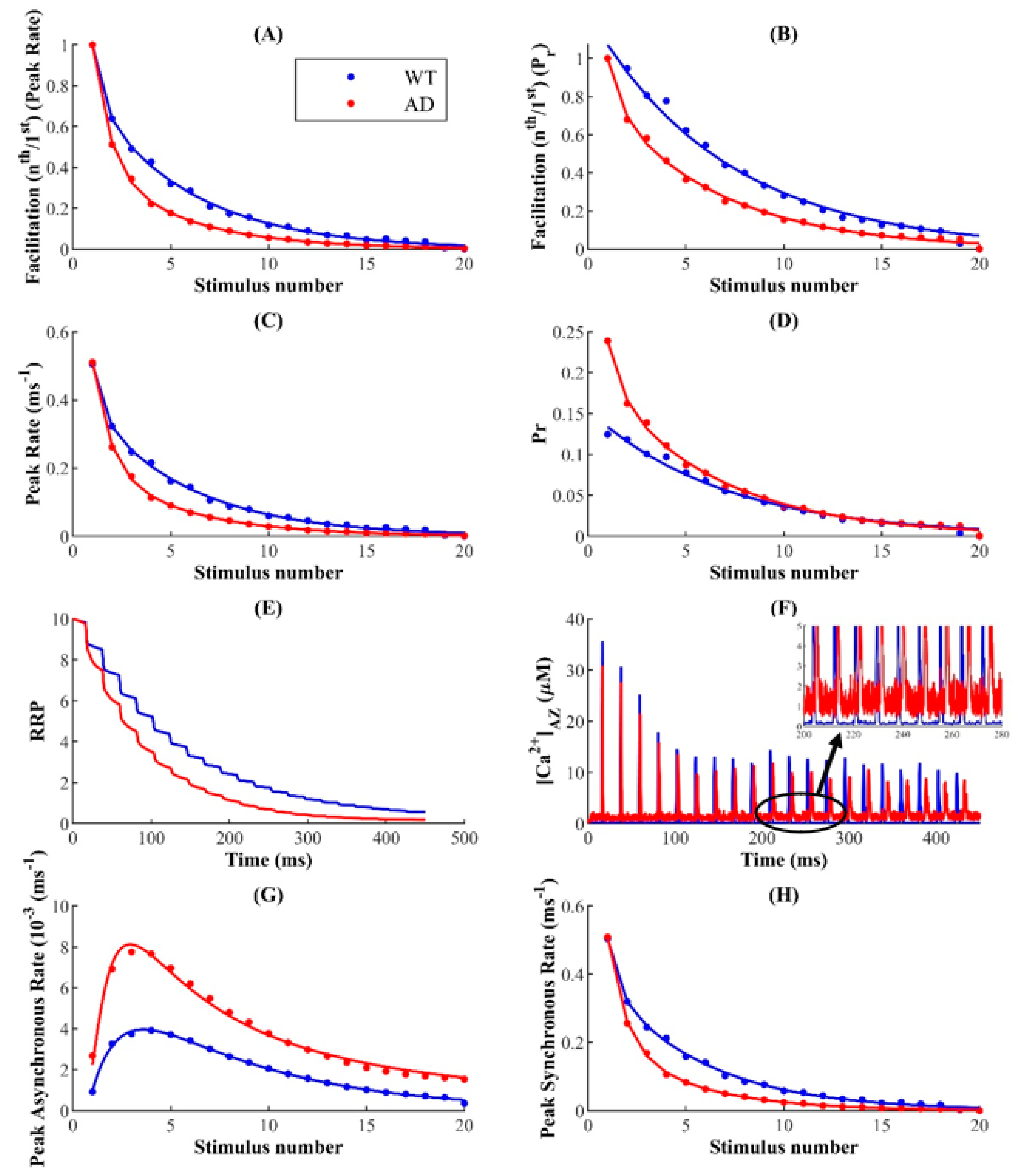
Consistent with experimental evidence, our model reproduces the well-established inverse relationship between the P
r and PPR but suggests that the enhanced STP over rapid timescales observed in FAD merely indicates hyperactivity rather than increased synaptic reliability [
73,
121]. The most crucial insight from this finding is that pathological synapses retain a longer history of Ca
2+ dynamics, which consequently causes them to exhibit enhanced excitation in a paired-pulse protocol. The traditional view claiming the massive reduction in the efficacy of excitatory synaptic transmission in early stages of AD has been challenged recently by several studies reporting aberrant Ca
2+ homeostasis and hyperactivity in AD-affected neuronal networks [
84,
100,
101]. A key finding of these studies is that hyperactivity is an early dysfunction in hippocampal synapses, whereas neuronal silencing emerges during later stages of the disease. Our model predicts that synaptic facilitation and depression depend on the timescale and frequency of stimulation. Relatively short low-frequency stimuli cause facilitation, which supports the idea that by enhancing potentiation over rapid timescales, aberrant Ca
2+ release from intracellular stores plays an important role in the history-dependent neuronal hyperactivity observed during early stages of FAD.
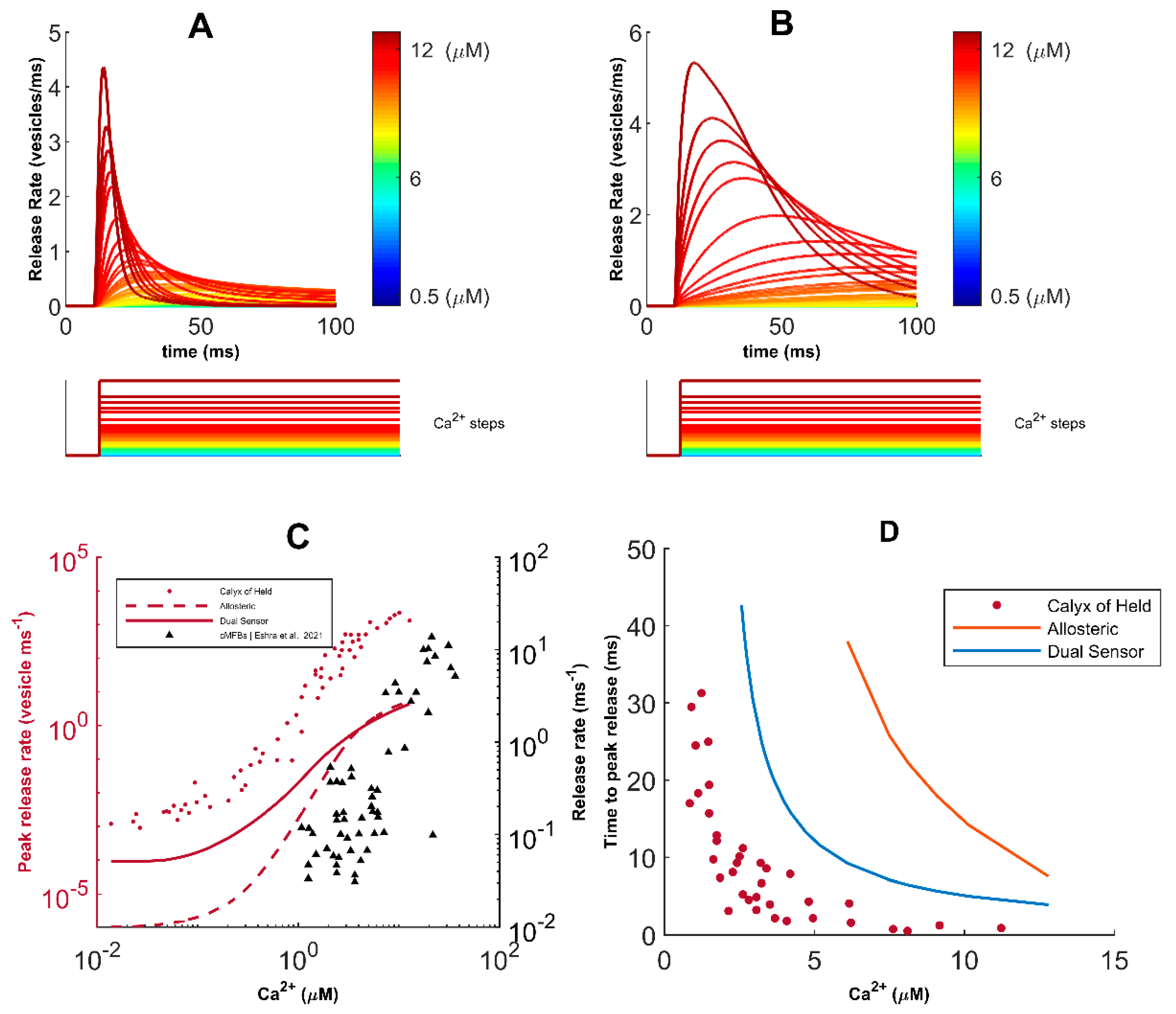
Our model shows that long high-frequency pulses trigger depression in FAD-affected synapses, which is governed by the depletion of RRP of vesicles. Experimental evidence reports three distinct molecular pathways for exocytosis [
39]. Indeed, synchronous, asynchronous, and spontaneous modes of vesicular release, characterized by distinct release timescales, have been reported in cultured hippocampal synaptic terminals [
28,
44]. Synaptotagmin-1 (Syt1) and 7 (Syt7) are the Ca
2+ sensors controlling the timescales of synchronous and asynchronous release in CA3 pyramidal neurons, respectively [
39,
122]. The rapid kinetics and low Ca
2+ affinity of Syt1 enable the remarkable temporal precision of synchronous release, where most vesicles immediately fuse with the membrane following stimulation. On the other hand, the slow kinetics of Syt7 promotes the progressive desynchronization of release later in the spike train [
123]. Studies on Syt1 knockout mice reported the selective abolishment of synchronous release and an increase in the magnitude of asynchronous release. Likewise, knockdown of Syt7 enhanced synchronous release in zebrafish’s neuromuscular junction, suggesting that the primary modes of exocytosis draw from the same vesicle resources in the RRP, which causes their activity patterns to be negatively correlated [
40,
44,
123,
124,
125,
126]. The most critical insight of these findings is that manipulations that suppress synchronous release increase the vesicle resources available for asynchronous release and indirectly enhance its magnitude. In agreement with the above, during ongoing activity, we observed a shift from synchronous to asynchronous exocytosis, leading to the increased depression of synaptic transmission during FAD pathology.
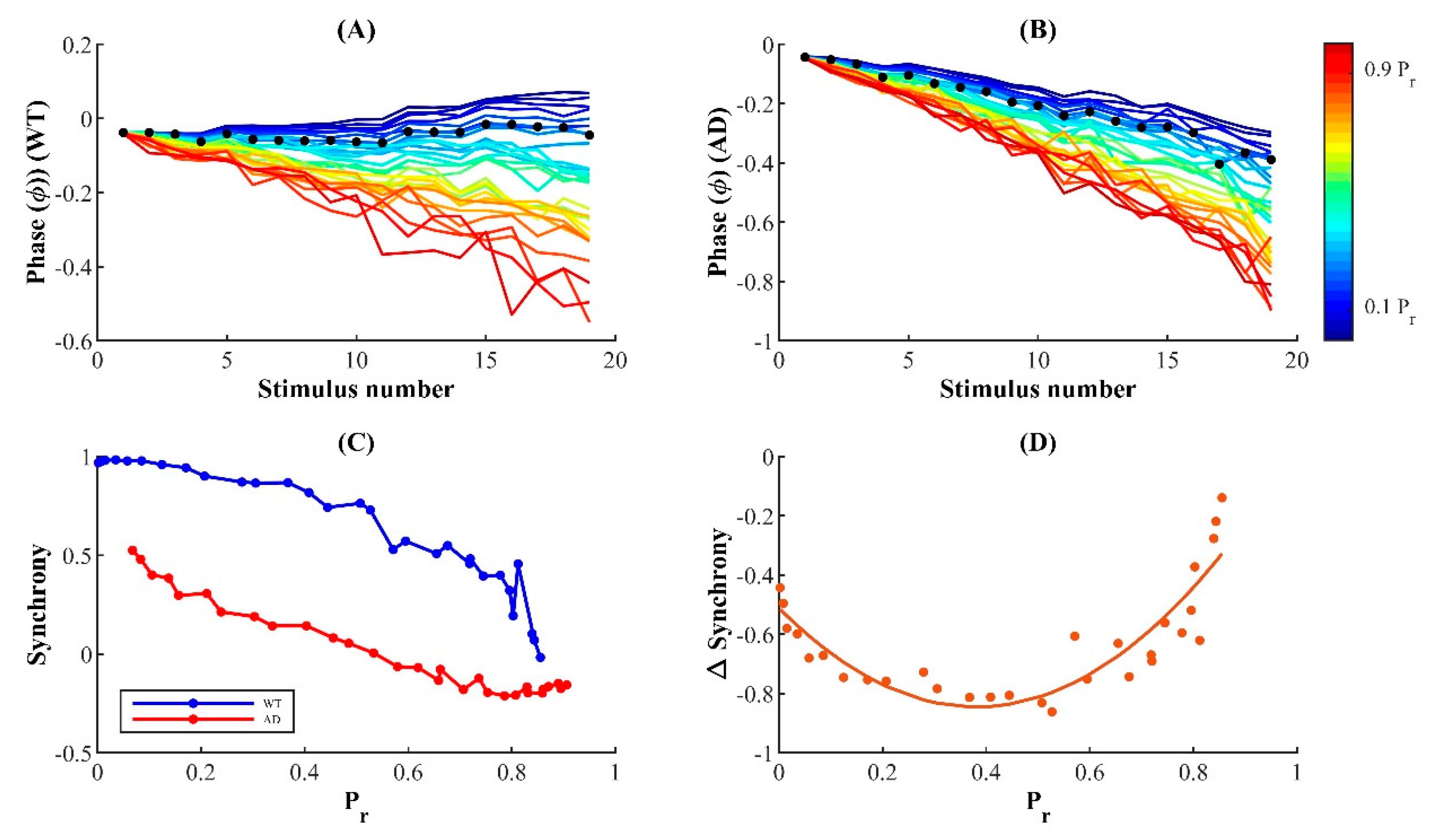
CA3-CA1 presynaptic terminals are equipped with a conspicuously low release probability that allows them to maintain a delicate balance between facilitation and reliability, giving rise to finely tuned rate codes with remarkable temporal precision. Thus, perturbations of release mechanisms are expected to alter the fidelity of neural rhythms in AD. Indeed, cortical neurons with Aβ peptide expressed reduced N-methyl D-aspartate (NMDA) receptor density, resulting in the rapid and persistent depression of NMDA-evoked currents [
106]. Moreover, the severe impairment of evoked synaptic response latency observed in Tg2576 mice overexpressing APP provides direct evidence for the reduced temporal coincidence of response in AD [
107]. These findings suggest that impaired response precision is an acute effect of perturbations due to AD that leads to overt cortical deficits. In agreement with these observations, our results reveal the increased latency of release events in FAD and uncover the unique dependence of the synchrony change—from control to FAD—on intrinsic Pr. The loss in temporal coordination of release in FAD is more severely expressed at a physiologically plausible P
r range for hippocampal synapses, although lower-P
r synapses exhibit relatively elevated temporal precision in both WT and FAD-affected synapses. Thus, despite the high fidelity of hippocampal synapses, their conspicuously low response success may make them more susceptible to AD pathology. We hope that future experiments will uncover the molecular mechanisms underlying the pathological enhancement of susceptibility in low-P
r hippocampal synapses.
We remark that while our model is developed to be consistent with most observations in the hippocampal CA3-CA1 synapses, it does not incorporate the uneven distribution of P/Q-type, N-type, and R-type VGCCs specifically found in the AZ of central synapses [
66,
127]. At hippocampal glutamatergic synapses, Cav2.1—P/Q-type—channels are thought to be most enriched at the presynaptic AZ and predominantly govern Ca
2+ influx at the axon terminal [
52]. Thus, similar to others, we value parsimony and use a formulation with only P/Q VGCCs [
67]. Furthermore, results from cultured hippocampal and superior cervical ganglion neurons provide evidence for direct interactions between the release machinery and VGCCs, implying that channel distribution is important for accurately predicting the spatiotemporal profiles of evoked release [
128,
129]. Our model does not capture the effect of the spatial distribution of VGCCs on synaptic transmission; neither does it incorporate other mechanisms for paired-pulse modulation expressed at putative single hippocampal synapses—lateral inhibition and release inactivation [
73,
94]. Thus, despite reproducing the observed inverse relationship between paired-pulse facilitation and initial P
r, our model falls short of the measured values, suggesting that we cannot eliminate additional mechanisms when investigating the interplay between residual Ca
2+, facilitation, and depression. Although IP
3R-dependent modulation of cytosolic Ca
2+ is usually adequate for explaining the critical aspects of ER Ca
2+ release and regulation of neurotransmission, the upregulation of RyR expression and modulation of IP
3Rs’ gating due to Ca
2+ release through RyRs (and vice versa) are also thought to play a key role in the aberrant Ca
2+ release from the ER, as well as the propagation of presynaptic signals [
10,
11,
12,
13,
14,
15,
16,
17,
18,
19,
20,
21,
22,
130]. It has been suggested that at the hippocampal Schaffer-collateral pathway, presynaptic presenilin inactivation perturbs STP and facilitation via impaired RyR function [
36,
131]. Furthermore, in 3xTg-AD mice, deviant RyR activity triggers Ca
2+ signaling alterations that promote synaptic depression [
37]. However, our model does not describe the contribution of presynaptic RyRs to vesicular release, as the biophysical properties of these receptors yield a distinct temporal range of Ca
2+ transients that can modulate LTP/LTD [
132,
133]. Presenilin mutations account for a significant portion of FAD cases; however, our current model does not incorporate ER-resident Ca
2+ permeable leak channels formed by Presenilins that are disrupted in FAD-affected neurons [
134]. Another key factor missing from our model is synaptic mitochondria. Mitochondria play a key role in shaping Ca
2+ gradients in synaptic terminals. Synaptic mitochondria are among the earliest targets in AD. Among other aspects, the ATP production and Ca
2+ buffering capacities of mitochondria are severely disrupted [
135,
136]. All these issues will be the subjects of our future research.
In summary, we have leveraged diverse experimental data to model Ca2+ homeostasis in the axonal terminal and explore how exocytosis is affected in FAD. Motivated by the difficulty in probing signaling cascades at the AZ of small hippocampal synapses, our main goal was to build a comprehensive but simple framework for unravelling the role of enhanced Ca2+ release from the ER in SVR during FAD pathology. In addition to agreeing closely with several observations about the kinetics of IP3Rs, SVR, and synaptic plasticity in both WT and diseased synapses, our modelling work provides key insights into impaired presynaptic function in FAD. Specifically, we make five key predictions:
The overall Pr in response to a single AP is upregulated in FAD-affected synapses;
Short-lived low-frequency stimuli promote potentiation in FAD-affected synapses;
During sustained high-frequency stimulation, FAD-affected terminals exhibit enhanced depression;
FAD-affected synapses operate less reliably, and
The effect of FAD pathology is exacerbated in synapses with low to intermediate Pr.
Although extensive experimental evidence corroborates our findings and demonstrates the significance of ER Ca
2+ dysregulation and loss of synaptic integrity in FAD, SVR therapeutics remain underdeveloped. For example, the current lack of validated in vivo biomarkers that can be used as proxies of synaptic dysfunction has directed most translational efforts towards indirect measures of synaptic integrity in AD [
137]. Our findings suggest that targeting the essential components of neurotransmission as well as related pathways of the ER could potentially aid biomarker identification and drug discovery efforts in FAD. In future research, we expect to incorporate a better and comprehensive understanding of the mechanisms underlying neuronal Ca
2+ dysregulation to provide better insights into the disease pathogenesis and therapeutic directions. Taken together with the aforementioned limitations, our analysis highlights the need for further studies investigating the role of perturbed Ca
2+ signaling due to intracellular organelles such as the ER and mitochondria in cognitive deficits associated with AD and other neurodegenerative diseases.





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}