
A Method for In Situ Reverse Genetic Analysis of Proteins Involved mtDNA Replication



Abstract
:
1. Introduction
2. Materials and Methods
2.1. Cell Growth and Treatment
2.2. Recombinant DNA
2.3. GeneSwap
2.4. mtDNA Diagnostics
2.5. Production of Retroviruses
2.6. Retroviral Transduction
2.7. PhiC31-Mediated Excision of Proviral Inserts
2.8. Genotyping of hTFAM Excision in 143B#6 Cells
2.9. Cellular Respiration
2.10. Western Blotting
2.11. Amino Acid Alignments
2.12. Testing oTFAM MTSs
2.13. Growth Rates
2.14. Quantitation of Mitochondrial Transcripts
2.15. Gene Inactivation by CRISPR-Cas9
2.16. TFAM Orthologs
2.17. MtDNA Copy Number (mtCN)
2.18. Statistical Analyses
3. Results
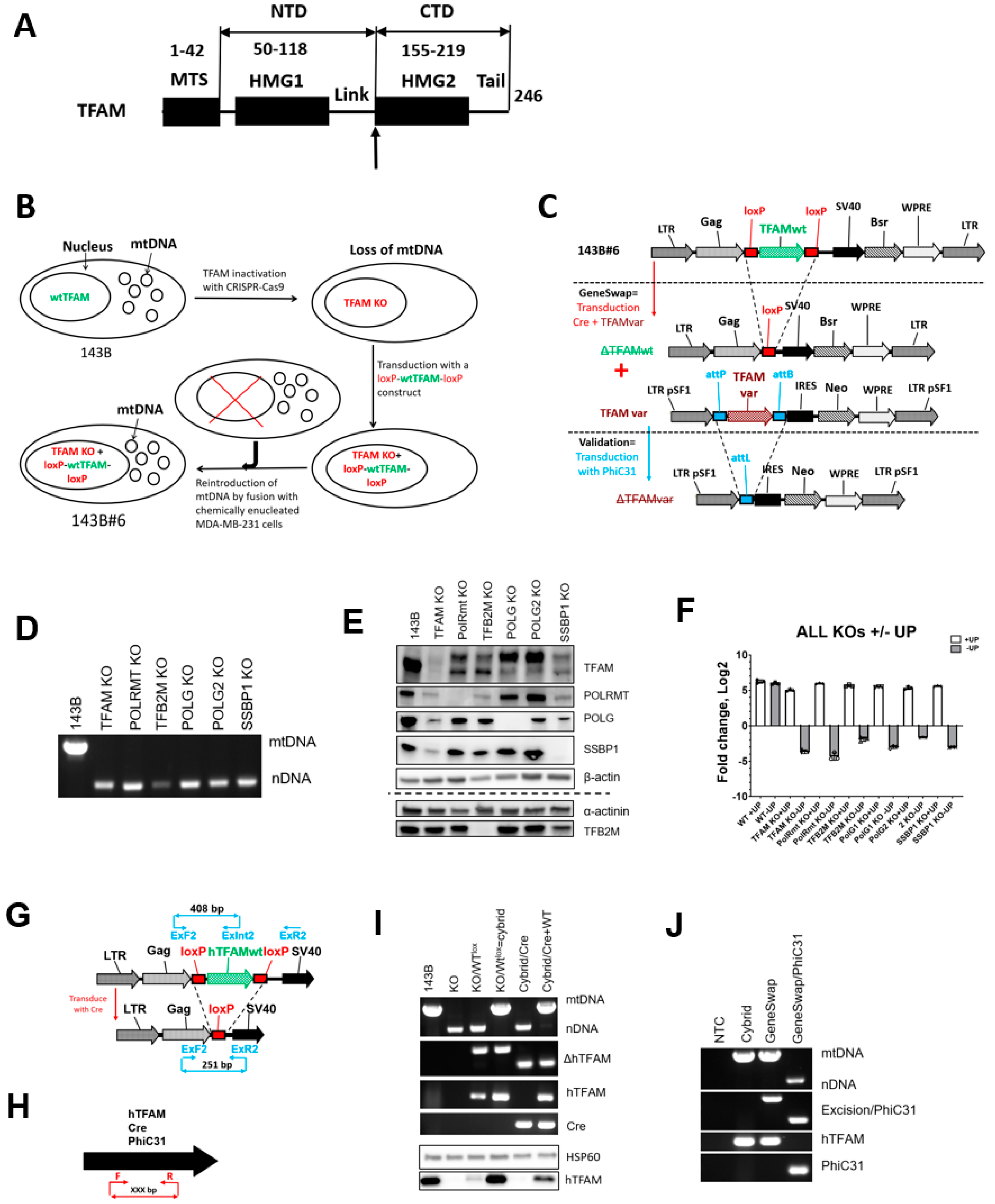
3.1. TFAM, POLRMT, TFB2M, POLG1, POLG2, and SSBP1 Are Dispensable for Viability in Cultured Cells
3.2. The GeneSwap Technique
3.3. GeneSwap of the hTFAM
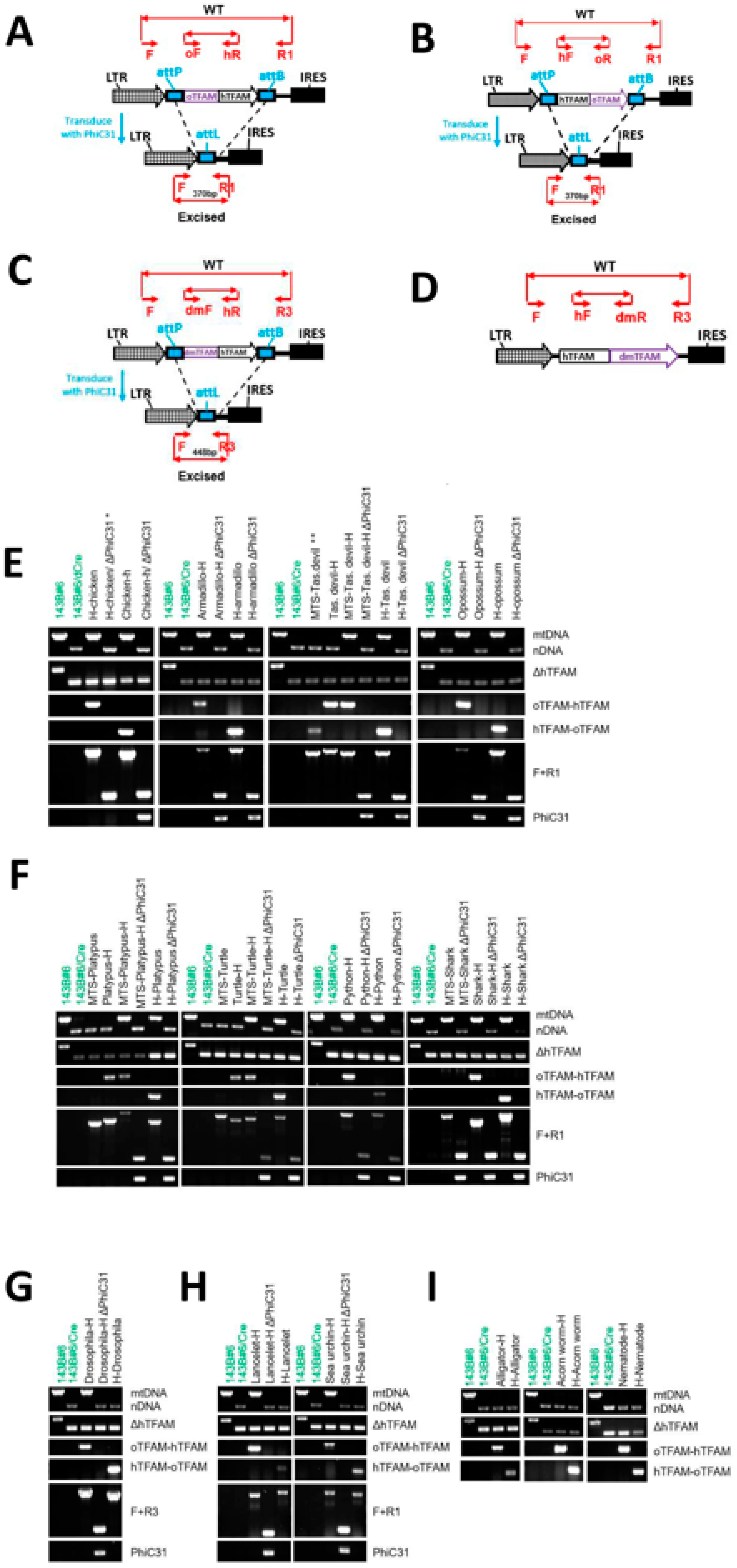
3.4. The Phylogenetic Relationships Do Not Determine the Ability of TFAM Orthologs to Support hmtDNA Replication
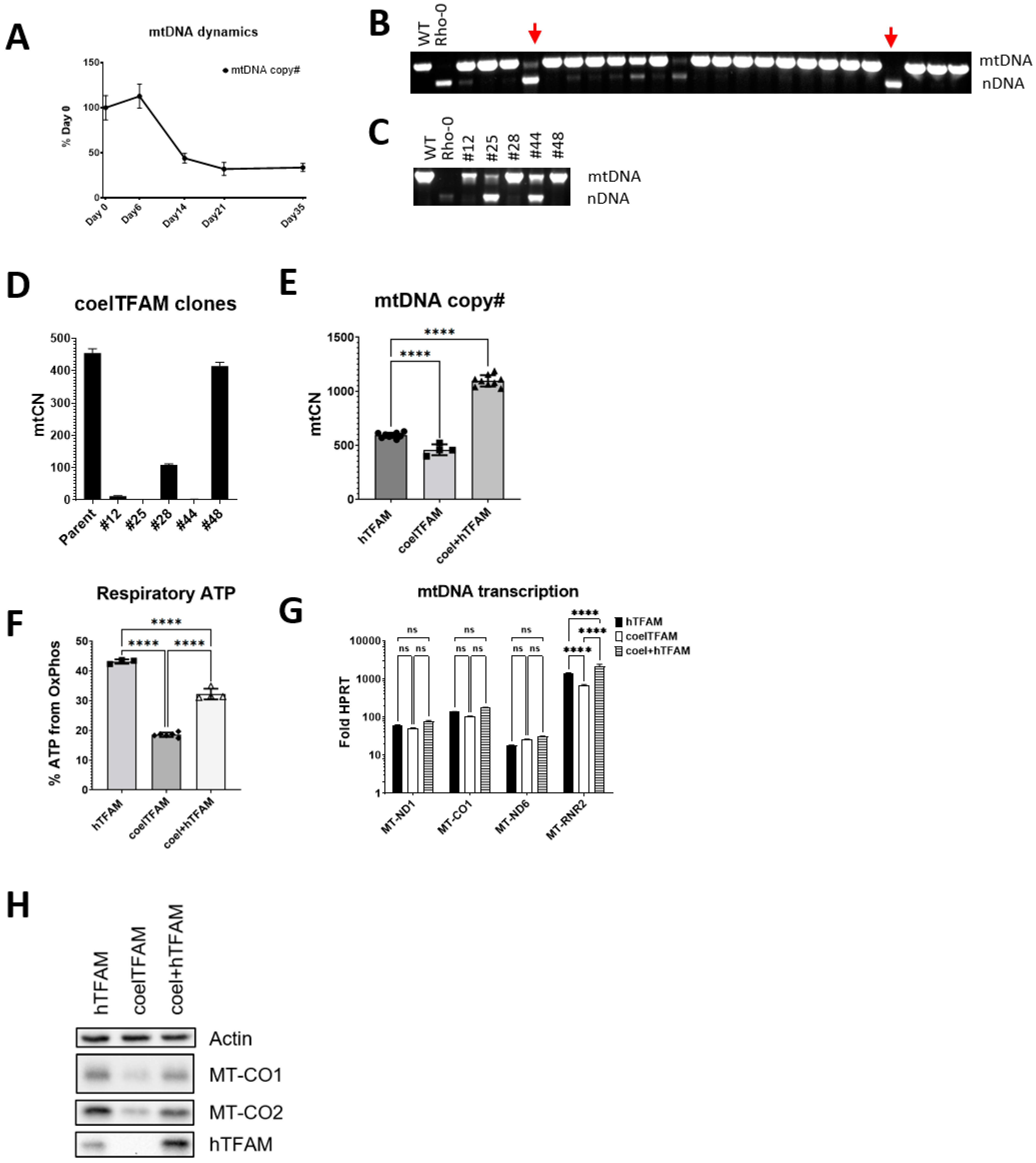
3.5. Coelacanth TFAM Promotes hmtDNA Instability
3.6. oTFAM MTSs Are Functional in Human Cells
3.7. TFAM C- and N-Terminal Domains Are Functionally Independent
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Clay Montier, L.L.; Deng, J.J.; Bai, Y. Number matters: Control of mammalian mitochondrial DNA copy number. J. Genet. Genom. 2009, 36, 125–131. [Google Scholar] [CrossRef] [Green Version]
- Rotig, A.; Poulton, J. Genetic causes of mitochondrial DNA depletion in humans. Biochim. Biophys. Acta 2009, 1792, 1103–1108. [Google Scholar] [CrossRef] [Green Version]
- Wallace, D.C. Mitochondria and cancer. Nat. Rev. Cancer 2012, 12, 685–698. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, H.Z.; Li, R.Y.; Li, M. A review of maternally inherited diabetes and deafness. Front. Biosci. (Landmark Ed.) 2014, 19, 777–782. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sobenin, I.A.; Zhelankin, A.V.; Mitrofanov, K.Y.; Sinyov, V.V.; Sazonova, M.A.; Postnov, A.Y.; Orekhov, A.N. Mutations of mitochondrial DNA in atherosclerosis and atherosclerosis-related diseases. Curr. Pharm. Des. 2015, 21, 1158–1163. [Google Scholar] [CrossRef]
- Cha, M.Y.; Kim, D.K.; Mook-Jung, I. The role of mitochondrial DNA mutation on neurodegenerative diseases. Exp. Mol. Med. 2015, 47, e150. [Google Scholar] [CrossRef] [Green Version]
- Wallace, D.C. A mitochondrial paradigm of metabolic and degenerative diseases, aging, and cancer: A dawn for evolutionary medicine. Annu. Rev. Genet. 2005, 39, 359–407. [Google Scholar] [CrossRef] [Green Version]
- Matsushima, Y.; Matsumura, K.; Ishii, S.; Inagaki, H.; Suzuki, T.; Matsuda, Y.; Beck, K.; Kitagawa, Y. Functional domains of chicken mitochondrial transcription factor A for the maintenance of mitochondrial DNA copy number in lymphoma cell line DT40. J. Biol. Chem. 2003, 278, 31149–31158. [Google Scholar] [CrossRef] [Green Version]
- Blomen, V.A.; Majek, P.; Jae, L.T.; Bigenzahn, J.W.; Nieuwenhuis, J.; Staring, J.; Sacco, R.; van Diemen, F.R.; Olk, N.; Stukalov, A.; et al. Gene essentiality and synthetic lethality in haploid human cells. Science 2015, 350, 1092–1096. [Google Scholar] [CrossRef]
- Wang, T.; Birsoy, K.; Hughes, N.W.; Krupczak, K.M.; Post, Y.; Wei, J.J.; Lander, E.S.; Sabatini, D.M. Identification and characterization of essential genes in the human genome. Science 2015, 350, 1096–1101. [Google Scholar] [CrossRef] [Green Version]
- Morgens, D.W.; Deans, R.M.; Li, A.; Bassik, M.C. Systematic comparison of CRISPR/Cas9 and RNAi screens for essential genes. Nat. Biotechnol. 2016, 34, 634–636. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Inatomi, T.; Matsuda, S.; Ishiuchi, T.; Do, Y.; Nakayama, M.; Abe, S.; Kasho, K.; Wanrooij, S.; Nakada, K.; Ichiyanagi, K.; et al. TFB2M and POLRMT are essential for mammalian mitochondrial DNA replication. Biochim. Biophys. Acta Mol. Cell Res. 2022, 1869, 119167. [Google Scholar] [CrossRef] [PubMed]
- Do, Y.; Matsuda, S.; Inatomi, T.; Nakada, K.; Yasukawa, T.; Kang, D. The accessory subunit of human DNA polymerase gamma is required for mitochondrial DNA maintenance and is able to stabilize the catalytic subunit. Mitochondrion 2020, 53, 133–139. [Google Scholar] [CrossRef] [PubMed]
- Reeves, R. High mobility group (HMG) proteins: Modulators of chromatin structure and DNA repair in mammalian cells. DNA Repair. 2015, 36, 122–136. [Google Scholar] [CrossRef]
- Larsson, N.G.; Wang, J.; Wilhelmsson, H.; Oldfors, A.; Rustin, P.; Lewandoski, M.; Barsh, G.S.; Clayton, D.A. Mitochondrial transcription factor A is necessary for mtDNA maintenance and embryogenesis in mice. Nat. Genet. 1998, 18, 231–236. [Google Scholar] [CrossRef]
- Baris, O.R.; Klose, A.; Kloepper, J.E.; Weiland, D.; Neuhaus, J.F.; Schauen, M.; Wille, A.; Muller, A.; Merkwirth, C.; Langer, T.; et al. The Mitochondrial Electron Transport Chain is Dispensable for Proliferation and Differentiation of Epidermal Progenitor Cells. Stem Cells 2011, 29, 1459–1468. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hansson, A.; Hance, N.; Dufour, E.; Rantanen, A.; Hultenby, K.; Clayton, D.A.; Wibom, R.; Larsson, N.G. A switch in metabolism precedes increased mitochondrial biogenesis in respiratory chain-deficient mouse hearts. Proc. Natl. Acad. Sci. USA 2004, 101, 3136–3141. [Google Scholar] [CrossRef] [Green Version]
- Desdin-Mico, G.; Soto-Heredero, G.; Aranda, J.F.; Oller, J.; Carrasco, E.; Gabande-Rodriguez, E.; Blanco, E.M.; Alfranca, A.; Cusso, L.; Desco, M.; et al. T cells with dysfunctional mitochondria induce multimorbidity and premature senescence. Science 2020, 368, 1371–1376. [Google Scholar] [CrossRef] [PubMed]
- Watanabe, A.; Arai, M.; Koitabashi, N.; Niwano, K.; Ohyama, Y.; Yamada, Y.; Kato, N.; Kurabayashi, M. Mitochondrial transcription factors TFAM and TFB2M regulate Serca2 gene transcription. Cardiovasc. Res. 2011, 90, 57–67. [Google Scholar] [CrossRef] [Green Version]
- Lee, E.J.; Kang, Y.C.; Park, W.H.; Jeong, J.H.; Pak, Y.K. Negative transcriptional regulation of mitochondrial transcription factor A (TFAM) by nuclear TFAM. Biochem. Biophys. Res. Commun. 2014, 450, 166–171. [Google Scholar] [CrossRef]
- De Oliveira, V.C.; Santos Roballo, K.C.; Mariano Junior, C.G.; Santos, S.I.P.; Bressan, F.F.; Chiaratti, M.R.; Tucker, E.J.; Davis, E.E.; Concordet, J.-P.; Ambrósio, C.E. HEK293T Cells with TFAM Disruption by CRISPR-Cas9 as a Model for Mitochondrial Regulation. Life 2021, 12, 22. [Google Scholar] [CrossRef] [PubMed]
- Wu, Z.; Oeck, S.; West, A.P.; Mangalhara, K.C.; Sainz, A.G.; Newman, L.E.; Zhang, X.O.; Wu, L.; Yan, Q.; Bosenberg, M.; et al. Mitochondrial DNA Stress Signalling Protects the Nuclear Genome. Nat. Metab. 2019, 1, 1209–1218. [Google Scholar] [CrossRef] [PubMed]
- De Oliveira, V.C.; Gomes Mariano Junior, C.; Belizario, J.E.; Krieger, J.E.; Fernandes Bressan, F.; Roballo, K.C.S.; Fantinato-Neto, P.; Meirelles, F.V.; Chiaratti, M.R.; Concordet, J.P.; et al. Characterization of post-edited cells modified in the TFAM gene by CRISPR/Cas9 technology in the bovine model. PLoS ONE 2020, 15, e0235856. [Google Scholar] [CrossRef] [PubMed]
- De Oliveira, V.C.; Moreira, G.S.A.; Bressan, F.F.; Gomes Mariano Junior, C.; Roballo, K.C.S.; Charpentier, M.; Concordet, J.P.; Meirelles, F.V.; Ambrosio, C.E. Edition of TFAM gene by CRISPR/Cas9 technology in bovine model. PLoS ONE 2019, 14, e0213376. [Google Scholar] [CrossRef]
- Kanki, T.; Ohgaki, K.; Gaspari, M.; Gustafsson, C.M.; Fukuoh, A.; Sasaki, N.; Hamasaki, N.; Kang, D. Architectural role of mitochondrial transcription factor A in maintenance of human mitochondrial DNA. Mol. Cell. Biol. 2004, 24, 9823–9834. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ullah, F.; Rauf, W.; Khan, K.; Khan, S.; Bell, K.M.; de Oliveira, V.C.; Tariq, M.; Bakhshalizadeh, S.; Touraine, P.; Katsanis, N.; et al. A recessive variant in TFAM causes mtDNA depletion associated with primary ovarian insufficiency, seizures, intellectual disability and hearing loss. Hum. Genet. 2021, 140, 1733–1751. [Google Scholar] [CrossRef]
- Stiles, A.R.; Simon, M.T.; Stover, A.; Eftekharian, S.; Khanlou, N.; Wang, H.L.; Magaki, S.; Lee, H.; Partynski, K.; Dorrani, N.; et al. Mutations in TFAM, encoding mitochondrial transcription factor A, cause neonatal liver failure associated with mtDNA depletion. Mol. Genet. Metab. 2016, 119, 91–99. [Google Scholar] [CrossRef]
- Sambrook, J.; Russel, D.W. Molecular Cloning. A Laboratory Manual; Cold Spring Harbor Laboratory Press: New York, NY, USA, 2001. [Google Scholar]
- Ho, S.N.; Hunt, H.D.; Horton, R.M.; Pullen, J.K.; Pease, L.R. Site-directed mutagenesis by overlap extension using the polymerase chain reaction. Gene 1989, 77, 51–59. [Google Scholar] [CrossRef]
- Shokolenko, I.N.; Fayzulin, R.Z.; Katyal, S.; McKinnon, P.J.; Wilson, G.L.; Alexeyev, M.F. Mitochondrial DNA ligase is dispensable for the viability of cultured cells but essential for mtDNA maintenance. J. Biol. Chem. 2013, 288, 26594–26605. [Google Scholar] [CrossRef] [Green Version]
- Khozhukhar, N.; Spadafora, D.; Rodriguez, Y.; Alexeyev, M. Elimination of Mitochondrial DNA from Mammalian Cells. Curr. Protoc. Cell Biol. 2018, 78. [Google Scholar] [CrossRef]
- Stemmer, M.; Thumberger, T.; Del Sol Keyer, M.; Wittbrodt, J.; Mateo, J.L. CCTop: An Intuitive, Flexible and Reliable CRISPR/Cas9 Target Prediction Tool. PLoS ONE 2015, 10, e0124633. [Google Scholar] [CrossRef] [Green Version]
- Cong, L.; Ran, F.A.; Cox, D.; Lin, S.; Barretto, R.; Habib, N.; Hsu, P.D.; Wu, X.; Jiang, W.; Marraffini, L.A.; et al. Multiplex genome engineering using CRISPR/Cas systems. Science 2013, 339, 819–823. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Certo, M.T.; Gwiazda, K.S.; Kuhar, R.; Sather, B.; Curinga, G.; Mandt, T.; Brault, M.; Lambert, A.R.; Baxter, S.K.; Jacoby, K.; et al. Coupling endonucleases with DNA end-processing enzymes to drive gene disruption. Nat. Methods 2012, 9, 973–975. [Google Scholar] [CrossRef] [PubMed]
- Kozhukhar, N.; Fant, A.; Alexeyev, M.F. Quantification of mtDNA content in cultured cells by direct droplet digital PCR. Mitochondrion 2021, 61, 102–113. [Google Scholar] [CrossRef] [PubMed]
- Kuhl, I.; Miranda, M.; Posse, V.; Milenkovic, D.; Mourier, A.; Siira, S.J.; Bonekamp, N.A.; Neumann, U.; Filipovska, A.; Polosa, P.L.; et al. POLRMT regulates the switch between replication primer formation and gene expression of mammalian mtDNA. Sci. Adv. 2016, 2, e1600963. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hance, N.; Ekstrand, M.I.; Trifunovic, A. Mitochondrial DNA polymerase gamma is essential for mammalian embryogenesis. Hum. Mol. Genet. 2005, 14, 1775–1783. [Google Scholar] [CrossRef] [PubMed]
- Humble, M.M.; Young, M.J.; Foley, J.F.; Pandiri, A.R.; Travlos, G.S.; Copeland, W.C. Polg2 is essential for mammalian embryogenesis and is required for mtDNA maintenance. Hum. Mol. Genet. 2012, 22, 1017–1025. [Google Scholar] [CrossRef] [Green Version]
- Maier, D.; Farr, C.L.; Poeck, B.; Alahari, A.; Vogel, M.; Fischer, S.; Kaguni, L.S.; Schneuwly, S. Mitochondrial single-stranded DNA-binding protein is required for mitochondrial DNA replication and development in Drosophila melanogaster. Mol. Biol. Cell 2001, 12, 821–830. [Google Scholar] [CrossRef] [Green Version]
- Kenyon, L.; Moraes, C.T. Expanding the functional human mitochondrial DNA database by the establishment of primate xenomitochondrial cybrids. Proc. Natl. Acad. Sci. USA 1997, 94, 9131–9135. [Google Scholar] [CrossRef] [Green Version]
- Fisher, R.P.; Clayton, D.A. Purification and characterization of human mitochondrial transcription factor 1. Mol. Cell. Biol. 1988, 8, 3496–3509. [Google Scholar]
- Fisher, R.P.; Topper, J.N.; Clayton, D.A. Promoter selection in human mitochondria involves binding of a transcription factor to orientation-independent upstream regulatory elements. Cell 1987, 50, 247–258. [Google Scholar] [CrossRef]
- Freyer, C.; Park, C.B.; Ekstrand, M.I.; Shi, Y.; Khvorostova, J.; Wibom, R.; Falkenberg, M.; Gustafsson, C.M.; Larsson, N.G. Maintenance of respiratory chain function in mouse hearts with severely impaired mtDNA transcription. Nucleic Acids Res. 2010, 38, 6577–6588. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Szczesny, R.J.; Kowalska, K.; Klosowska-Kosicka, K.; Chlebowski, A.; Owczarek, E.P.; Warkocki, Z.; Kulinski, T.M.; Adamska, D.; Affek, K.; Jedroszkowiak, A.; et al. Versatile approach for functional analysis of human proteins and efficient stable cell line generation using FLP-mediated recombination system. PLoS ONE 2018, 13, e0194887. [Google Scholar] [CrossRef]
- Shutt, T.E.; Bestwick, M.; Shadel, G.S. The core human mitochondrial transcription initiation complex: It only takes two to tango. Transcription 2011, 2, 55–59. [Google Scholar] [CrossRef] [Green Version]
- Shutt, T.E.; Lodeiro, M.F.; Cotney, J.; Cameron, C.E.; Shadel, G.S. Core human mitochondrial transcription apparatus is a regulated two-component system in vitro. Proc. Natl. Acad. Sci. USA 2010, 107, 12133–12138. [Google Scholar] [CrossRef] [Green Version]
- Zollo, O.; Tiranti, V.; Sondheimer, N. Transcriptional requirements of the distal heavy-strand promoter of mtDNA. Proc. Natl. Acad. Sci. USA 2012, 109, 6508–6512. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Baixauli, F.; Acin-Perez, R.; Villarroya-Beltri, C.; Mazzeo, C.; Nunez-Andrade, N.; Gabande-Rodriguez, E.; Ledesma, M.D.; Blazquez, A.; Martin, M.A.; Falcon-Perez, J.M.; et al. Mitochondrial Respiration Controls Lysosomal Function during Inflammatory T Cell Responses. Cell Metab. 2015, 22, 485–498. [Google Scholar] [CrossRef] [Green Version]
- Castellani, C.A.; Longchamps, R.J.; Sumpter, J.A.; Newcomb, C.E.; Lane, J.A.; Grove, M.L.; Bressler, J.; Brody, J.A.; Floyd, J.S.; Bartz, T.M.; et al. Mitochondrial DNA copy number can influence mortality and cardiovascular disease via methylation of nuclear DNA CpGs. Genome Med. 2020, 12, 84. [Google Scholar] [CrossRef]
- Shi, Y.; Dierckx, A.; Wanrooij, P.H.; Wanrooij, S.; Larsson, N.G.; Wilhelmsson, L.M.; Falkenberg, M.; Gustafsson, C.M. Mammalian transcription factor A is a core component of the mitochondrial transcription machinery. Proc. Natl. Acad. Sci. USA 2012, 109, 16510–16515. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shokolenko, I.N.; Alexeyev, M.F. Mitochondrial transcription in mammalian cells. Front. Biosci. (Landmark Ed.) 2017, 22, 835–853. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lodeiro, M.F.; Uchida, A.; Bestwick, M.; Moustafa, I.M.; Arnold, J.J.; Shadel, G.S.; Cameron, C.E. Transcription from the second heavy-strand promoter of human mtDNA is repressed by transcription factor A in vitro. Proc. Natl. Acad. Sci. USA 2012, 109, 6513–6518. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Montoya, J.; Christianson, T.; Levens, D.; Rabinowitz, M.; Attardi, G. Identification of initiation sites for heavy-strand and light-strand transcription in human mitochondrial DNA. Proc. Natl. Acad. Sci. USA 1982, 79, 7195–7199. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Montoya, J.; Gaines, G.L.; Attardi, G. The pattern of transcription of the human mitochondrial rRNA genes reveals two overlapping transcription units. Cell 1983, 34, 151–159. [Google Scholar] [CrossRef]
- Martin, M.; Cho, J.; Cesare, A.J.; Griffith, J.D.; Attardi, G. Termination factor-mediated DNA loop between termination and initiation sites drives mitochondrial rRNA synthesis. Cell 2005, 123, 1227–1240. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gustafsson, C.M.; Falkenberg, M.; Larsson, N.G. Maintenance and Expression of Mammalian Mitochondrial DNA. Annu. Rev. Biochem. 2016, 85, 133–160. [Google Scholar] [CrossRef] [PubMed]
- Terzioglu, M.; Ruzzenente, B.; Harmel, J.; Mourier, A.; Jemt, E.; Lopez, M.D.; Kukat, C.; Stewart, J.B.; Wibom, R.; Meharg, C.; et al. MTERF1 binds mtDNA to prevent transcriptional interference at the light-strand promoter but is dispensable for rRNA gene transcription regulation. Cell Metab. 2013, 17, 618–626. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pastukh, V.; Shokolenko, I.; Wang, B.; Wilson, G.; Alexeyev, M. Human mitochondrial transcription factor A possesses multiple subcellular targeting signals. FEBS J. 2007, 274, 6488–6499. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Species, Trivial | Species Latin/GenPept | Consensus/Identity, % 1 | Comp 2 | Species, Trivial | Species Latin/GenPept | Consensus/Identity, % 1 | Comp 2 |
|---|---|---|---|---|---|---|---|
| Human | Homo sapiens/NP_003192.1 | 100/100 | + | Chicken | Gallus gallus/NP_989431 | 61/42.2 | − |
| Cat | Felis catus/XP_003993997 | 84.3/74.1 | + | Green Sea Turtle | Chelonia mydas/XP_007060730 | 58.5/43.2 | − |
| Pig | Sus scrofa/NP_001123683 | 84.3/71.6 | + | Bald eagle | Haliaeetus leucocephalus/XP_010578303 | 58.4/41.6 | − |
| Manatee | Trichechus manatus latirostris/XP_004369930 | 82.3/73.6 | + | Python | Python bivittatus/XP_007424936 | 56.2/40.4 | − |
| Pika | Ochotona princeps/XP_004583634 | 81.2/70.1 | + | Coelacanth | Latimeria chalumnae/XP_006004778 | 55.9/41.5 | + |
| Armadillo-like | Dasypus novemcinctus/XP_004473261 | 81.2/69.5 | + | Zebrafish | Danio rerio/NP_001070857 | 53.7/39 | + |
| Hedgehog | Echinops telfairi/XP_004701428 | 80.9/66.8 | + | Frog | Xenopus leavis/NP_001081106 | 53.7/37.1 | + |
| Elephant | Loxodonta Africana/XP_003409078 | 80.4/68.8 | + | Elephant shark | Callorhinchus milii/XP_007895218 | 50.7/37.0 | − |
| Elephant shrew | Elephantulus edwardii/XP_006895656 | 80.2/69.5 | + | Alligator | Alligator sinensis/XP_006032464 | 50.6/38.2 | − |
| Bat | Myotis lucifugus; XP_006098959 | 79.7/63.5 | + | Fruit fly | Drosophila melanogaster/NP_524415 | 47.3/30.9 | − |
| Armadillo * | Dasypus novemcinctus; XP_004473258 | 77.7/64 | - | Lancelet | Branchiostoma floridae/XP_035670496 | 39.1/25.3 | − |
| Mouse | Mus musculus/NP_033386.1 | 76.1/62.9 | + | Sea urchin | Strongylocentrotus purpuratus/XP_030834910.1 | 39.1/24.8 | − |
| Tasmanian devil | Sarcophilus harrisii/XP_003755126 | 67.8/48.7 | − | Acorn worm | Saccoglossus kowalevskii/XP_006813645 | 38.4/25.5 | − |
| Opossum | Monodelphis domestica/XP_007478397 | 65.8/48.7 | − | Nematode | Caenarhabditis elegans/NP_501245.1 | 38.4/21.2 | − |
| Platypus | Ornithorhynchus anatinus XP_001507982 | 65/44.7 | − | Yeast | Saccharomyces cerevisiae/NP_013788.1 | 27.2/17.4 | − |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kozhukhar, N.; Spadafora, D.; Rodriguez, Y.A.R.; Alexeyev, M.F. A Method for In Situ Reverse Genetic Analysis of Proteins Involved mtDNA Replication. Cells 2022, 11, 2168. https://doi.org/10.3390/cells11142168
Kozhukhar N, Spadafora D, Rodriguez YAR, Alexeyev MF. A Method for In Situ Reverse Genetic Analysis of Proteins Involved mtDNA Replication. Cells. 2022; 11(14):2168. https://doi.org/10.3390/cells11142168
Chicago/Turabian StyleKozhukhar, Natalya, Domenico Spadafora, Yelitza A. R. Rodriguez, and Mikhail F. Alexeyev. 2022. "A Method for In Situ Reverse Genetic Analysis of Proteins Involved mtDNA Replication" Cells 11, no. 14: 2168. https://doi.org/10.3390/cells11142168
APA StyleKozhukhar, N., Spadafora, D., Rodriguez, Y. A. R., & Alexeyev, M. F. (2022). A Method for In Situ Reverse Genetic Analysis of Proteins Involved mtDNA Replication. Cells, 11(14), 2168. https://doi.org/10.3390/cells11142168