
CLL-Derived Extracellular Vesicles Impair T-Cell Activation and Foster T-Cell Exhaustion via Multiple Immunological Checkpoints

 ,
,  ,
,  ,
, 
Abstract
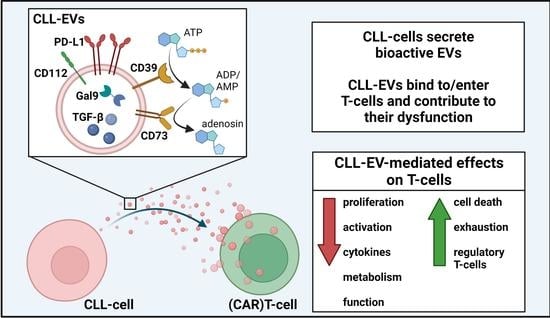
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
2.1. Cell Lines and Cells
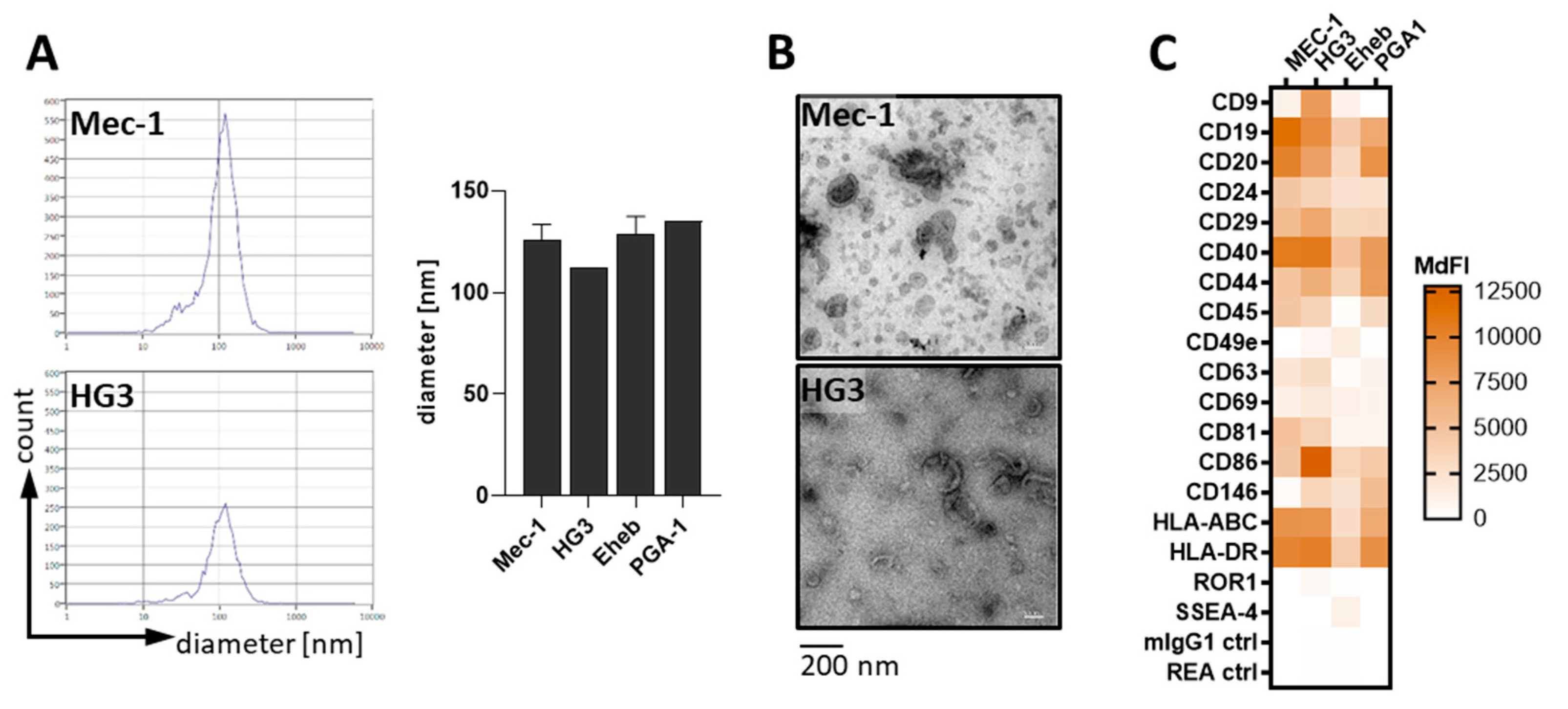
2.2. Extracellular Vesicles
2.3. Cell Culture
2.4. Flow Cytometry
2.5. Fluorescent Microscopy
2.6. Metabolic Analyses
2.7. Imaging Cytometry
2.8. Statistics
3. Results
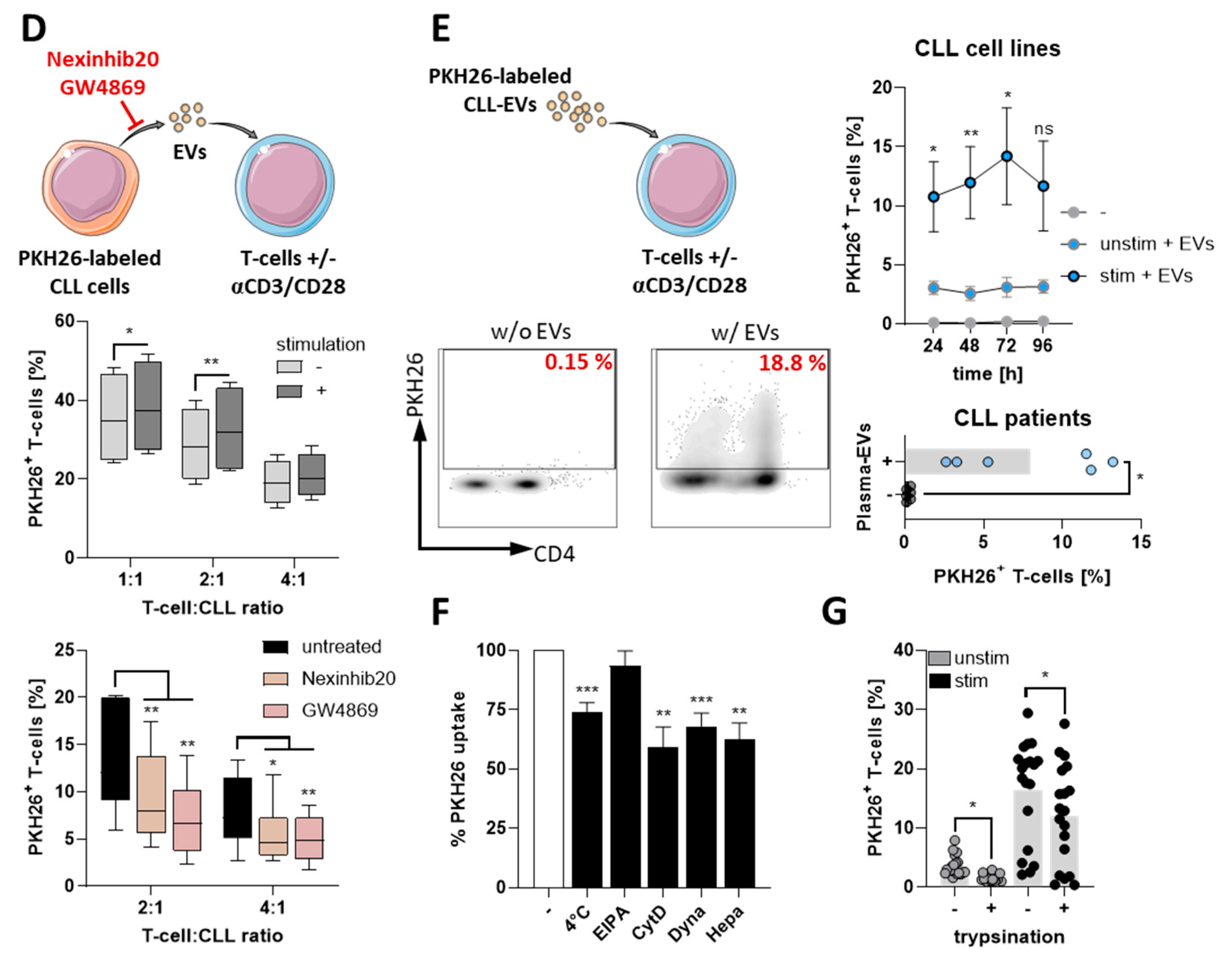
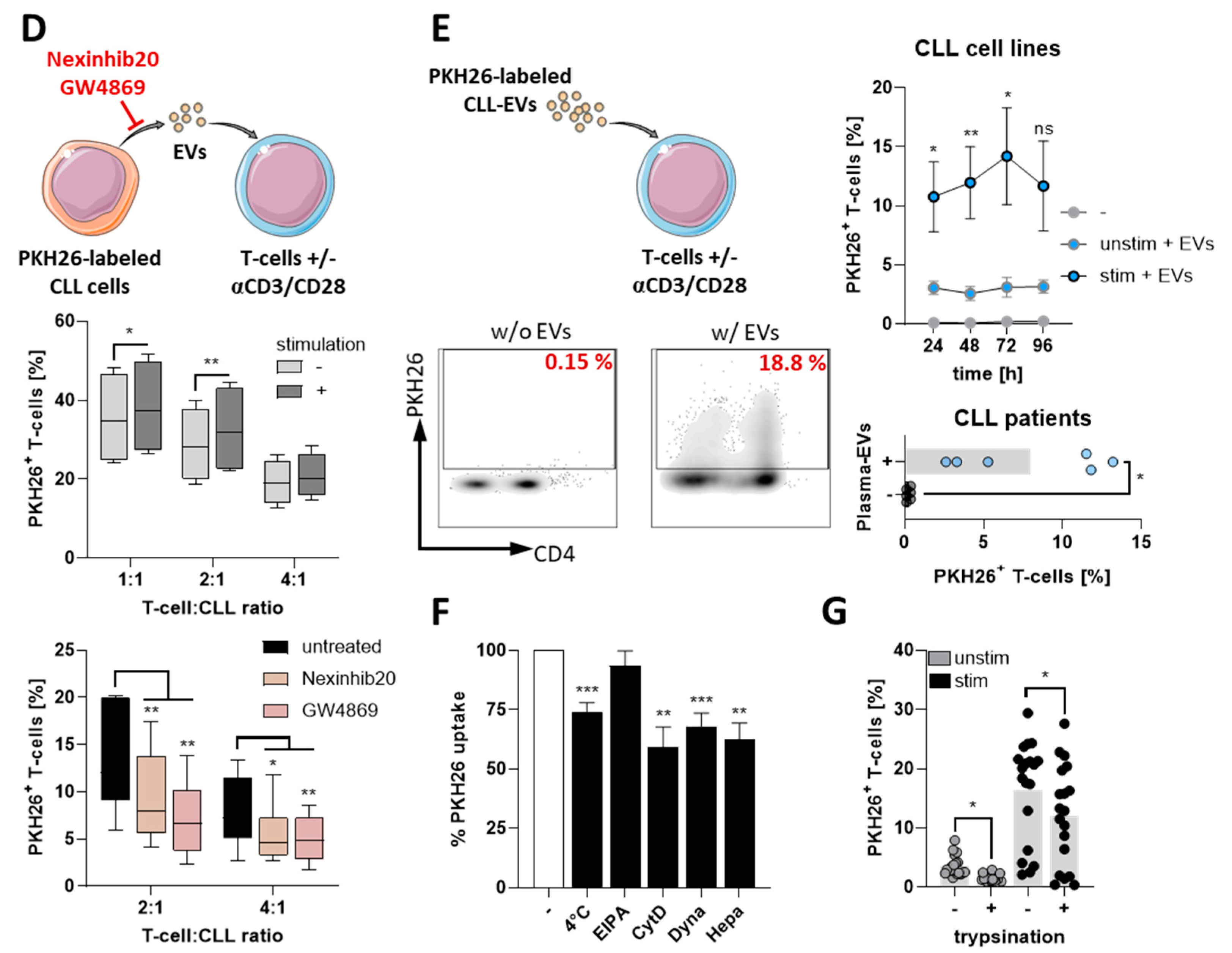
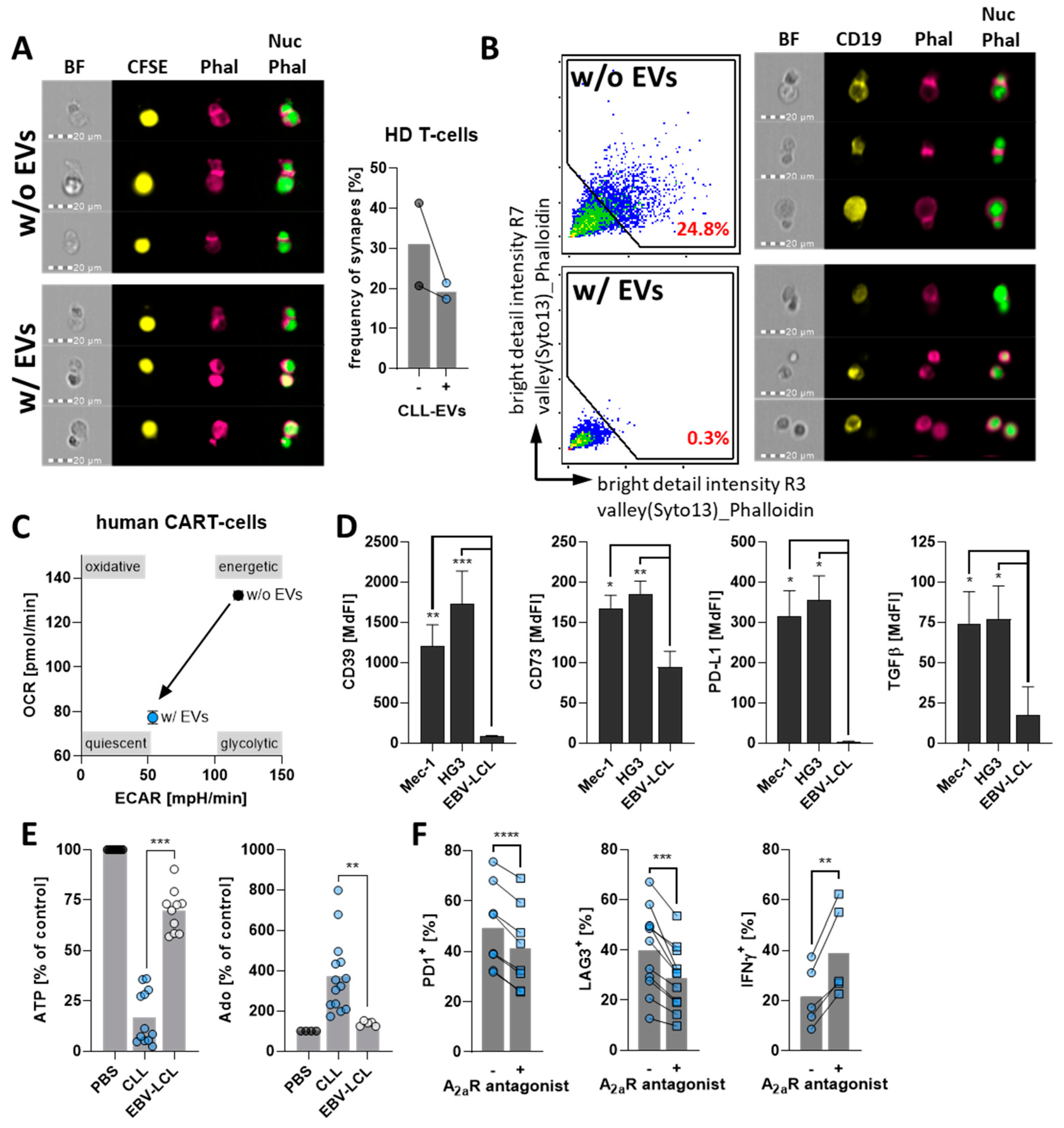
3.1. CLL-Cells Secrete EVs That Interact with T-Cells
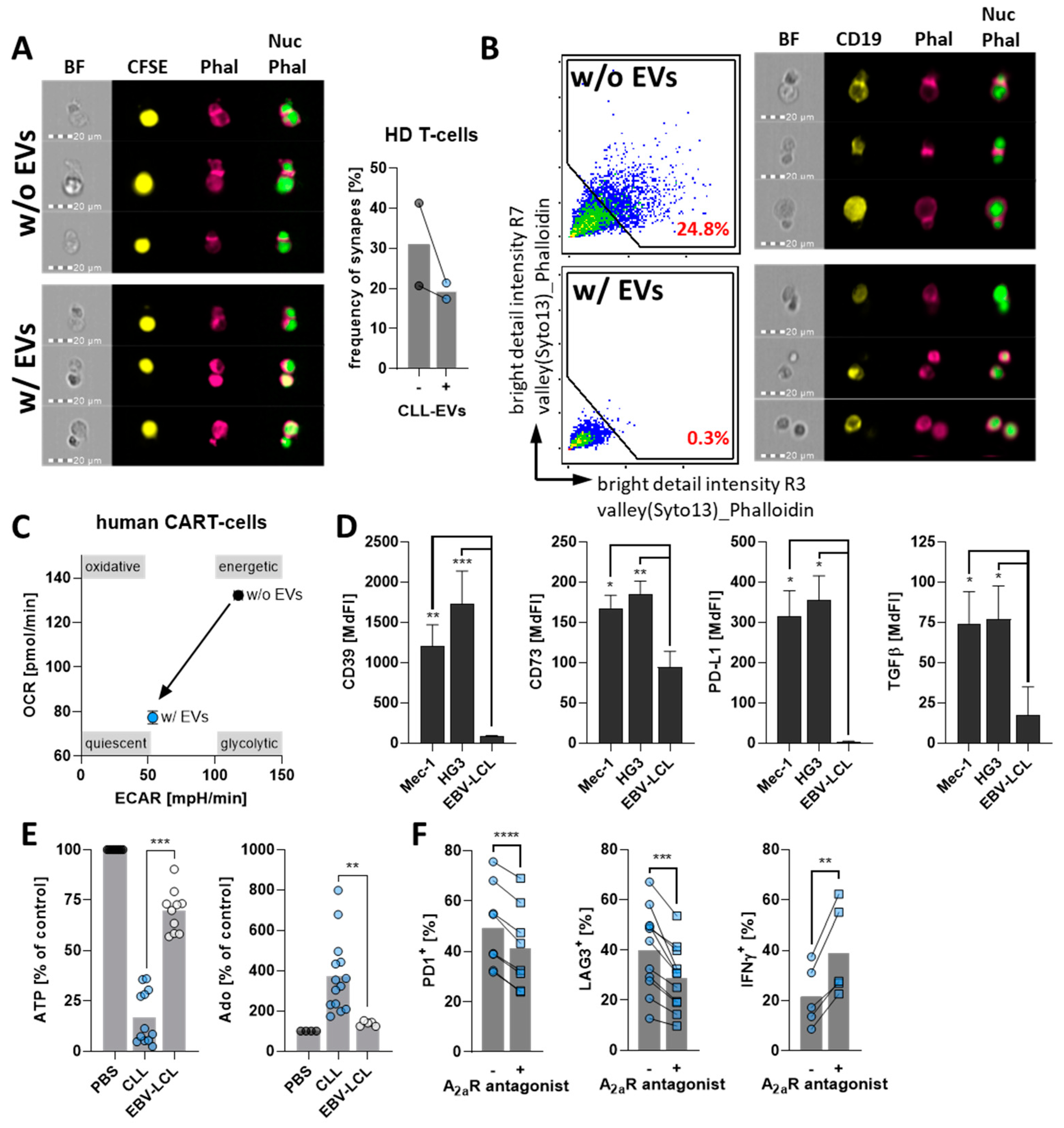
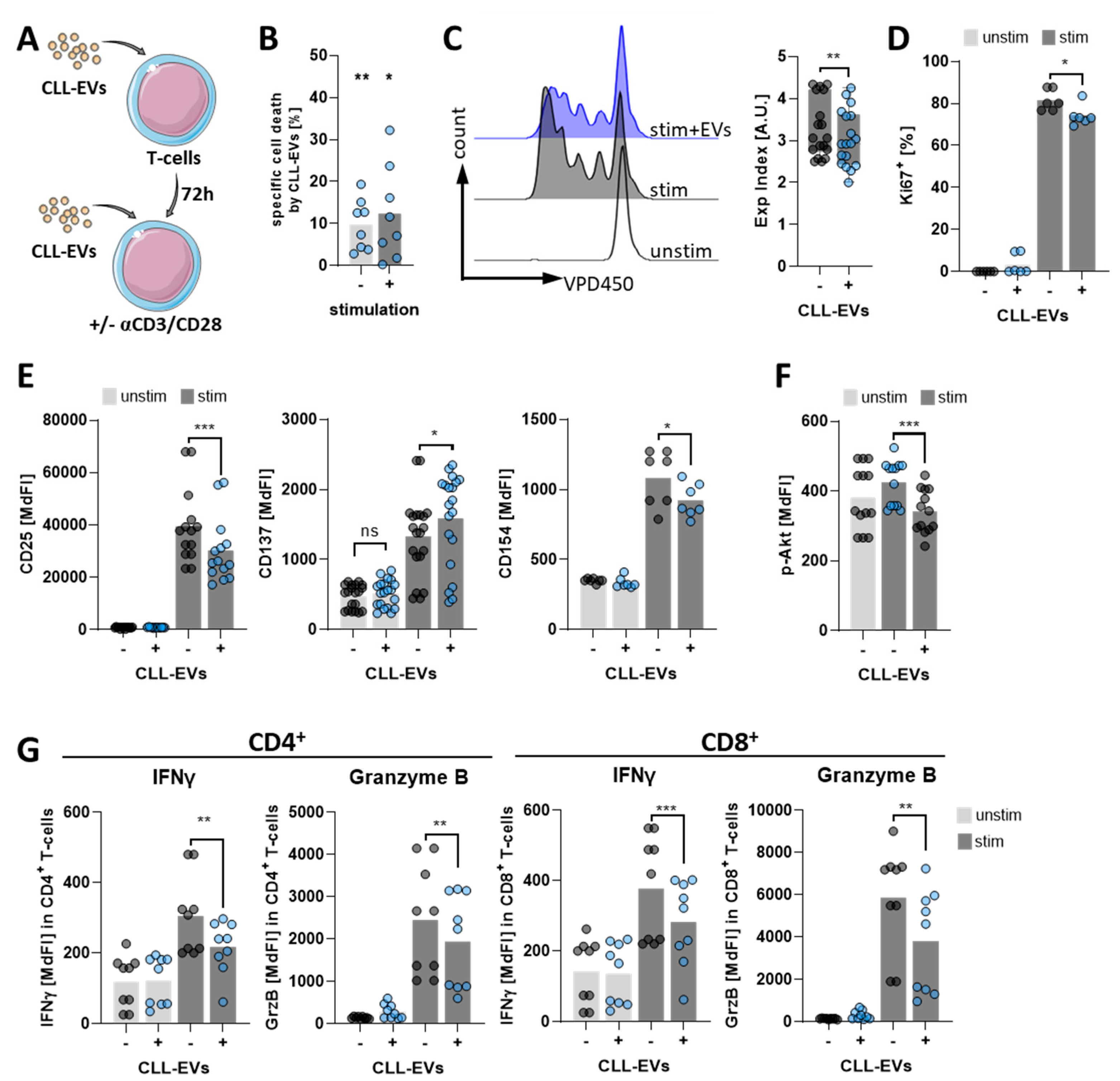
3.2. CLL-EVs Impair T-Cell Survival, Proliferation, and Activation
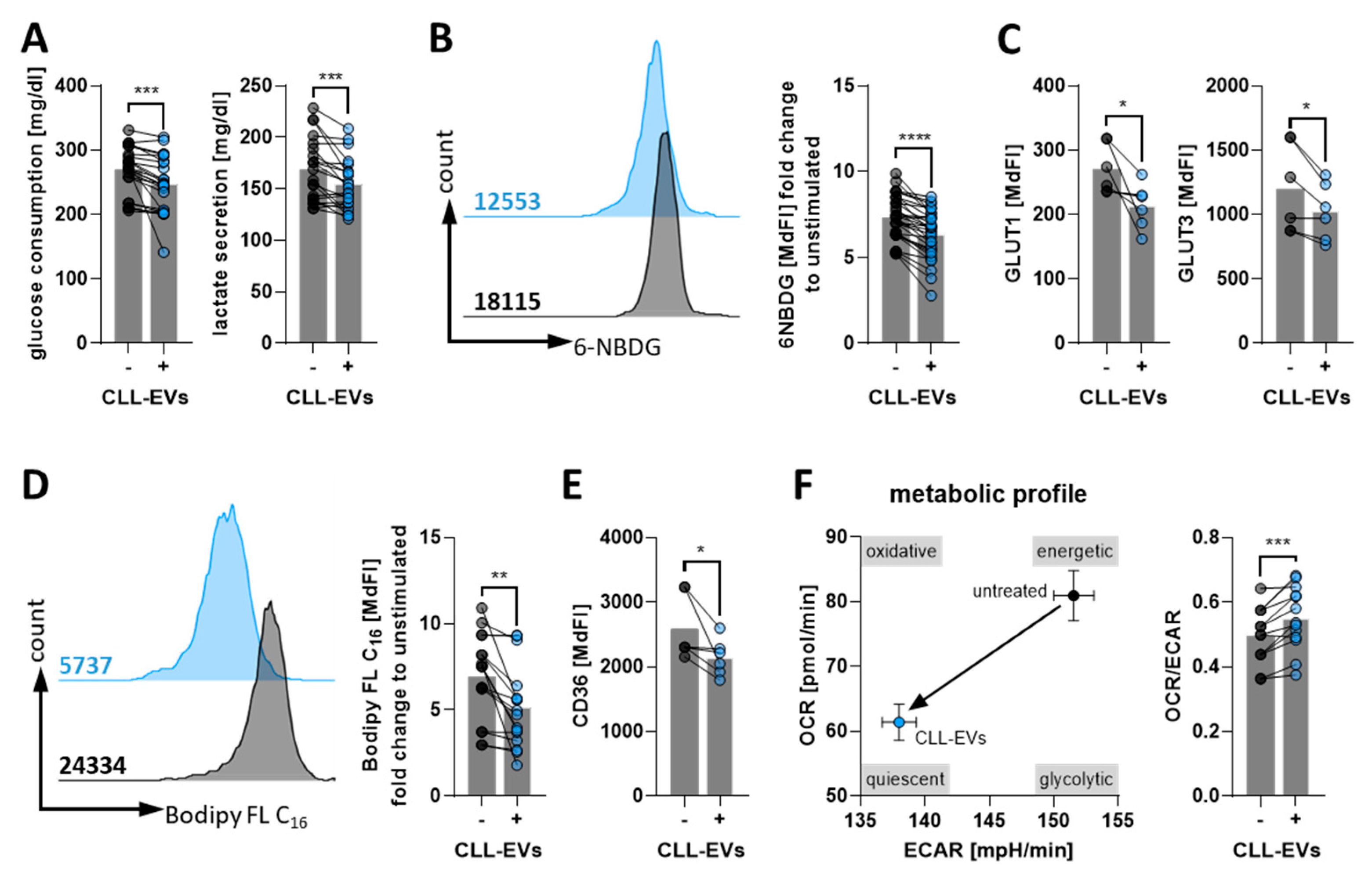
3.3. CLL-EVs Alter T-Cell Metabolism and Skew Their Subset Composition
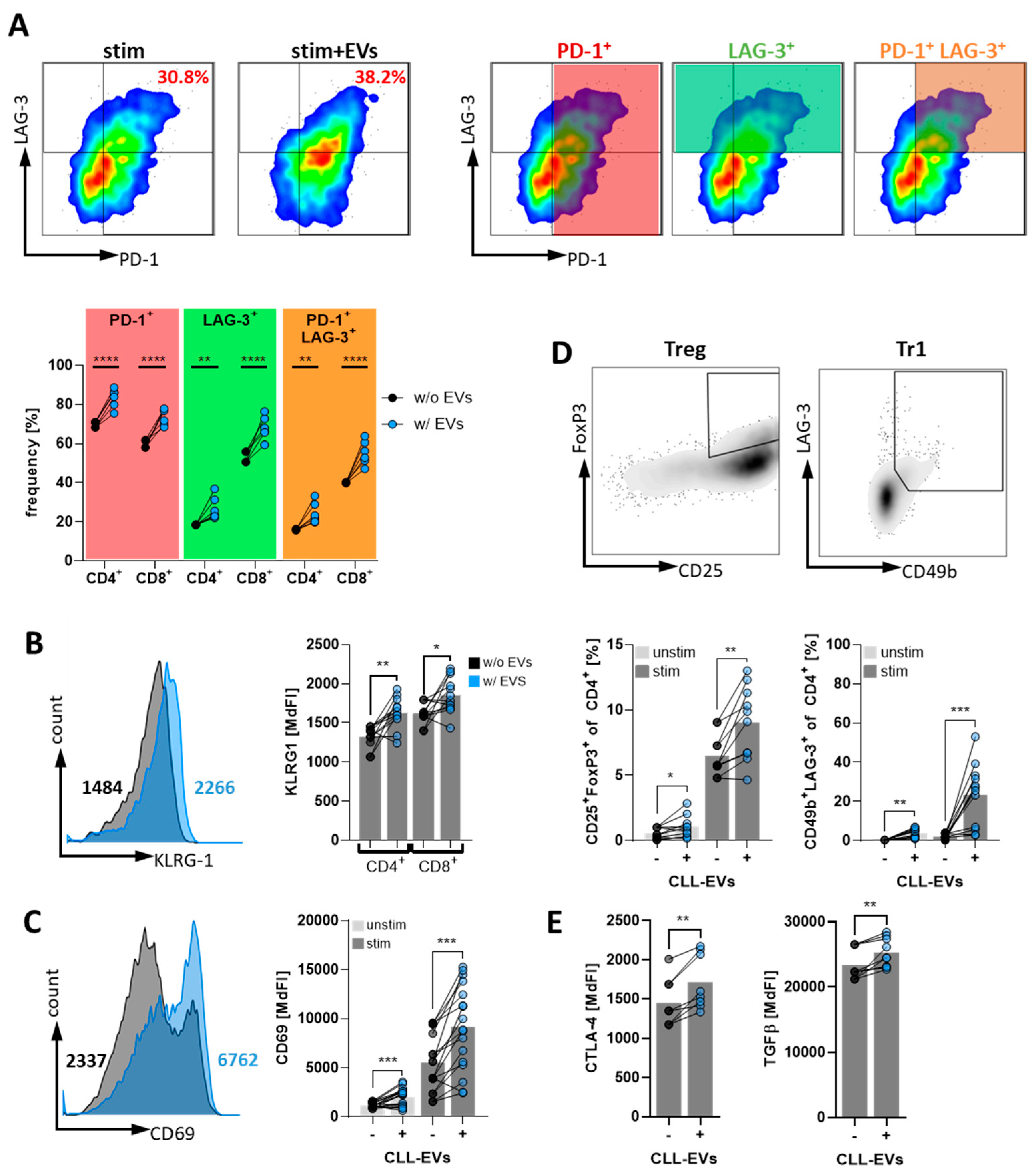
3.4. CLL-EVs Promote T-Cell Exhaustion and Regulatory T-Cell Formation
3.5. CLL-EVs Impair (CAR-)T-Cell Function and Express Multiple Immune Checkpoints
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Hallek, M. Chronic lymphocytic leukemia: 2020 update on diagnosis, risk stratification and treatment. Am. J. Hematol. 2019, 94, 1266–1287. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chiorazzi, N.; Rai, K.R.; Ferrarini, M. Chronic lymphocytic leukemia. N. Engl. J. Med. 2005, 352, 804–815. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ten Hacken, E.; Burger, J.A. Microenvironment interactions and B-cell receptor signaling in Chronic Lymphocytic Leukemia: Implications for disease pathogenesis and treatment. Biochim. Biophys. Acta 2016, 1863, 401–413. [Google Scholar] [CrossRef] [PubMed]
- Bewarder, M.; Stilgenbauer, S.; Thurner, L.; Kaddu-Mulindwa, D. Current treatment options in CLL. Cancers 2021, 13, 2468. [Google Scholar] [CrossRef] [PubMed]
- Wiedmeier-Nutor, J.; Leis, J. Chronic lymphocytic leukemia: Chemotherapy free and other novel therapies including CAR T. Curr. Treat. Options Oncol. 2022, 23, 904–919. [Google Scholar] [CrossRef]
- Os, A.; Burgler, S.; Ribes, A.P.; Funderud, A.; Wang, D.; Thompson, K.M.; Tjonnfjord, G.E.; Bogen, B.; Munthe, L.A. Chronic lymphocytic leukemia cells are activated and proliferate in response to specific T helper cells. Cell Rep. 2013, 4, 566–577. [Google Scholar] [CrossRef] [Green Version]
- Griggio, V.; Perutelli, F.; Salvetti, C.; Boccellato, E.; Boccadoro, M.; Vitale, C.; Coscia, M. Immune dysfunctions and immune-based therapeutic interventions in chronic lymphocytic leukemia. Front. Immunol. 2020, 11, 594556. [Google Scholar] [CrossRef]
- Abels, E.R.; Breakefield, X.O. Introduction to extracellular vesicles: Biogenesis, RNA cargo selection, content, release, and uptake. Cell Mol. Neurobiol. 2016, 36, 301–312. [Google Scholar] [CrossRef]
- Robbins, P.D.; Morelli, A.E. Regulation of immune responses by extracellular vesicles. Nat. Rev. Immunol. 2014, 14, 195–208. [Google Scholar] [CrossRef] [Green Version]
- Lotvall, J.; Hill, A.F.; Hochberg, F.; Buzas, E.I.; Di Vizio, D.; Gardiner, C.; Gho, Y.S.; Kurochkin, I.V.; Mathivanan, S.; Quesenberry, P.; et al. Minimal experimental requirements for definition of extracellular vesicles and their functions: A position statement from the International Society for Extracellular Vesicles. J. Extracell Vesicles 2014, 3, 26913. [Google Scholar] [CrossRef]
- Yeh, Y.Y.; Ozer, H.G.; Lehman, A.M.; Maddocks, K.; Yu, L.; Johnson, A.J.; Byrd, J.C. Characterization of CLL exosomes reveals a distinct microRNA signature and enhanced secretion by activation of BCR signaling. Blood 2015, 125, 3297–3305. [Google Scholar] [CrossRef]
- Smallwood, D.T.; Apollonio, B.; Willimott, S.; Lezina, L.; Alharthi, A.; Ambrose, A.R.; De Rossi, G.; Ramsay, A.G.; Wagner, S.D. Extracellular vesicles released by CD40/IL-4-stimulated CLL cells confer altered functional properties to CD4+ T cells. Blood 2016, 128, 542–552. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Trino, S.; Lamorte, D.; Caivano, A.; De Luca, L.; Sgambato, A.; Laurenzana, I. Clinical relevance of extracellular vesicles in hematological neoplasms: From liquid biopsy to cell biopsy. Leukemia 2021, 35, 661–678. [Google Scholar] [CrossRef] [PubMed]
- Reiners, K.S.; Shatnyeva, O.; Vasyutina, E.; Bosl, T.; Hansen, H.P.; Hallek, M.; Herling, M.; von Strandmann, E.P. Extracellular vesicles released from chronic lymphocytic leukemia cells exhibit a disease relevant mRNA signature and transfer mRNA to bystander cells. Haematologica 2017, 102, e100–e103. [Google Scholar] [CrossRef] [PubMed]
- Bruns, H.; Bottcher, M.; Qorraj, M.; Fabri, M.; Jitschin, S.; Dindorf, J.; Busch, L.; Jitschin, R.; Mackensen, A.; Mougiakakos, D. CLL-cell-mediated MDSC induction by exosomal miR-155 transfer is disrupted by vitamin D. Leukemia 2017, 31, 985–988. [Google Scholar] [CrossRef]
- Dubois, N.; Crompot, E.; Meuleman, N.; Bron, D.; Lagneaux, L.; Stamatopoulos, B. Importance of crosstalk between chronic lymphocytic leukemia cells and the stromal microenvironment: Direct contact, soluble factors, and extracellular vesicles. Front. Oncol. 2020, 10, 1422. [Google Scholar] [CrossRef]
- Cox, M.J.; Lucien, F.; Sakemura, R.; Boysen, J.C.; Kim, Y.; Horvei, P.; Manriquez Roman, C.; Hansen, M.J.; Tapper, E.E.; Siegler, E.L.; et al. Leukemic extracellular vesicles induce chimeric antigen receptor T cell dysfunction in chronic lymphocytic leukemia. Mol. Ther. 2021, 29, 1529–1540. [Google Scholar] [CrossRef]
- Thery, C.; Amigorena, S.; Raposo, G.; Clayton, A. Isolation and characterization of exosomes from cell culture supernatants and biological fluids. Curr. Protoc. Cell Biol. 2006, 30, 3.22.1–3.22.29. [Google Scholar] [CrossRef]
- Bottcher, M.; Renner, K.; Berger, R.; Mentz, K.; Thomas, S.; Cardenas-Conejo, Z.E.; Dettmer, K.; Oefner, P.J.; Mackensen, A.; Kreutz, M.; et al. D-2-hydroxyglutarate interferes with HIF-1alpha stability skewing T-cell metabolism towards oxidative phosphorylation and impairing Th17 polarization. Oncoimmunology 2018, 7, e1445454. [Google Scholar] [CrossRef] [Green Version]
- Arguello, R.J.; Combes, A.J.; Char, R.; Gigan, J.P.; Baaziz, A.I.; Bousiquot, E.; Camosseto, V.; Samad, B.; Tsui, J.; Yan, P.; et al. SCENITH: A flow cytometry-based method to functionally profile energy metabolism with single-cell resolution. Cell Metab. 2020, 32, 1063–1075.e7. [Google Scholar] [CrossRef]
- Mulcahy, L.A.; Pink, R.C.; Carter, D.R. Routes and mechanisms of extracellular vesicle uptake. J. Extracell Vesicles 2014, 3, 24641. [Google Scholar] [CrossRef] [Green Version]
- Buck, M.D.; O’Sullivan, D.; Pearce, E.L. T cell metabolism drives immunity. J. Exp. Med. 2015, 212, 1345–1360. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Geltink, R.I.K.; Kyle, R.L.; Pearce, E.L. Unraveling the complex interplay between T cell metabolism and function. Annu. Rev. Immunol. 2018, 36, 461–488. [Google Scholar] [CrossRef]
- Saravia, J.; Raynor, J.L.; Chapman, N.M.; Lim, S.A.; Chi, H. Signaling networks in immunometabolism. Cell Res. 2020, 30, 328–342. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- O’Sullivan, D. The metabolic spectrum of memory T cells. Immunol. Cell Biol. 2019, 97, 636–646. [Google Scholar] [CrossRef] [PubMed]
- Mahnke, Y.D.; Brodie, T.M.; Sallusto, F.; Roederer, M.; Lugli, E. The who’s who of T-cell differentiation: Human memory T-cell subsets. Eur. J. Immunol. 2013, 43, 2797–2809. [Google Scholar] [CrossRef]
- Franco, F.; Jaccard, A.; Romero, P.; Yu, Y.R.; Ho, P.C. Metabolic and epigenetic regulation of T-cell exhaustion. Nat. Metab. 2020, 2, 1001–1012. [Google Scholar] [CrossRef]
- Coe, D.J.; Kishore, M.; Marelli-Berg, F. Metabolic regulation of regulatory T cell development and function. Front. Immunol. 2014, 5, 590. [Google Scholar] [CrossRef] [Green Version]
- Roessner, P.M.; Seiffert, M. T-cells in chronic lymphocytic leukemia: Guardians or drivers of disease? Leukemia 2020, 34, 2012–2024. [Google Scholar] [CrossRef]
- Beltra, J.C.; Manne, S.; Abdel-Hakeem, M.S.; Kurachi, M.; Giles, J.R.; Chen, Z.; Casella, V.; Ngiow, S.F.; Khan, O.; Huang, Y.J.; et al. Developmental relationships of four exhausted CD8(+) T cell subsets reveals underlying transcriptional and epigenetic landscape control mechanisms. Immunity 2020, 52, 825–841. [Google Scholar] [CrossRef]
- Gao, R.; Shi, G.P.; Wang, J. Functional diversities of regulatory T cells in the context of cancer immunotherapy. Front. Immunol. 2022, 13, 833667. [Google Scholar] [CrossRef] [PubMed]
- Gagliani, N.; Magnani, C.F.; Huber, S.; Gianolini, M.E.; Pala, M.; Licona-Limon, P.; Guo, B.; Herbert, D.R.; Bulfone, A.; Trentini, F.; et al. Coexpression of CD49b and LAG-3 identifies human and mouse T regulatory type 1 cells. Nat. Med. 2013, 19, 739–746. [Google Scholar] [CrossRef] [PubMed]
- Ramsay, A.G.; Johnson, A.J.; Lee, A.M.; Gorgun, G.; Le Dieu, R.; Blum, W.; Byrd, J.C.; Gribben, J.G. Chronic lymphocytic leukemia T cells show impaired immunological synapse formation that can be reversed with an immunomodulating drug. J. Clin. Investig. 2008, 118, 2427–2437. [Google Scholar] [CrossRef]
- Vlachonikola, E.; Stamatopoulos, K.; Chatzidimitriou, A. T cells in chronic lymphocytic leukemia: A two-edged sword. Front. Immunol. 2020, 11, 612244. [Google Scholar] [CrossRef] [PubMed]
- Dustin, M.L.; Cooper, J.A. The immunological synapse and the actin cytoskeleton: Molecular hardware for T cell signaling. Nat. Immunol. 2000, 1, 23–29. [Google Scholar] [CrossRef] [Green Version]
- Mancikova, V.; Smida, M. Current State of CAR T-cell therapy in chronic lymphocytic leukemia. Int. J. Mol. Sci. 2021, 22, 5536. [Google Scholar] [CrossRef]
- Vaisitti, T.; Arruga, F.; Guerra, G.; Deaglio, S. Ectonucleotidases in blood malignancies: A tale of surface markers and therapeutic targets. Front. Immunol. 2019, 10, 2301. [Google Scholar] [CrossRef]
- Mastelic-Gavillet, B.; Navarro Rodrigo, B.; Decombaz, L.; Wang, H.; Ercolano, G.; Ahmed, R.; Lozano, L.E.; Ianaro, A.; Derre, L.; Valerio, M.; et al. Adenosine mediates functional and metabolic suppression of peripheral and tumor-infiltrating CD8(+) T cells. J. Immunother. Cancer 2019, 7, 257. [Google Scholar] [CrossRef]
- Allard, B.; Allard, D.; Buisseret, L.; Stagg, J. The adenosine pathway in immuno-oncology. Nat. Rev. Clin. Oncol. 2020, 17, 611–629. [Google Scholar] [CrossRef]
- McClanahan, F.; Hanna, B.; Miller, S.; Clear, A.J.; Lichter, P.; Gribben, J.G.; Seiffert, M. PD-L1 checkpoint blockade prevents immune dysfunction and leukemia development in a mouse model of chronic lymphocytic leukemia. Blood 2015, 126, 203–211. [Google Scholar] [CrossRef] [Green Version]
- Bottcher, M.; Bruns, H.; Volkl, S.; Lu, J.; Chartomatsidou, E.; Papakonstantinou, N.; Mentz, K.; Buttner-Herold, M.; Zenz, T.; Herling, M.; et al. Control of PD-L1 expression in CLL-cells by stromal triggering of the Notch-c-Myc-EZH2 oncogenic signaling axis. J. Immunother. Cancer 2021, 9, e001889. [Google Scholar] [CrossRef] [PubMed]
- Ramsay, A.G.; Clear, A.J.; Fatah, R.; Gribben, J.G. Multiple inhibitory ligands induce impaired T-cell immunologic synapse function in chronic lymphocytic leukemia that can be blocked with lenalidomide: Establishing a reversible immune evasion mechanism in human cancer. Blood 2012, 120, 1412–1421. [Google Scholar] [CrossRef] [PubMed]
- Scrivener, S.; Kaminski, E.R.; Demaine, A.; Prentice, A.G. Analysis of the expression of critical activation/interaction markers on peripheral blood T cells in B-cell chronic lymphocytic leukaemia: Evidence of immune dysregulation. Br. J. Haematol. 2001, 112, 959–964. [Google Scholar] [CrossRef]
- Scrivener, S.; Goddard, R.V.; Kaminski, E.R.; Prentice, A.G. Abnormal T-cell function in B-cell chronic lymphocytic leukaemia. Leuk. Lymphoma 2003, 44, 383–389. [Google Scholar] [CrossRef]
- Gonzalez-Rodriguez, A.P.; Contesti, J.; Huergo-Zapico, L.; Lopez-Soto, A.; Fernandez-Guizan, A.; Acebes-Huerta, A.; Gonzalez-Huerta, A.J.; Gonzalez, E.; Fernandez-Alvarez, C.; Gonzalez, S. Prognostic significance of CD8 and CD4 T cells in chronic lymphocytic leukemia. Leuk. Lymphoma 2010, 51, 1829–1836. [Google Scholar] [CrossRef]
- Nunes, C.; Wong, R.; Mason, M.; Fegan, C.; Man, S.; Pepper, C. Expansion of a CD8(+)PD-1(+) replicative senescence phenotype in early stage CLL patients is associated with inverted CD4:CD8 ratios and disease progression. Clin. Cancer Res. 2012, 18, 678–687. [Google Scholar] [CrossRef] [Green Version]
- Riches, J.C.; Davies, J.K.; McClanahan, F.; Fatah, R.; Iqbal, S.; Agrawal, S.; Ramsay, A.G.; Gribben, J.G. T cells from CLL patients exhibit features of T-cell exhaustion but retain capacity for cytokine production. Blood 2013, 121, 1612–1621. [Google Scholar] [CrossRef] [PubMed]
- Rößner, P.M.; Hanna, B.S.; Dolors, C.; Campo, E.; Stilgenbauer, S.; Lichter, P.; Seiffert, M. Exhausted PD-1hi T-Cells Are Enriched in Secondary Lymphoid Organs of CLL Patients. Blood 2017, 130, 4281. [Google Scholar] [CrossRef]
- Forconi, F.; Moss, P. Perturbation of the normal immune system in patients with CLL. Blood 2015, 126, 573–581. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gorgun, G.; Ramsay, A.G.; Holderried, T.A.; Zahrieh, D.; Le Dieu, R.; Liu, F.; Quackenbush, J.; Croce, C.M.; Gribben, J.G. E(mu)-TCL1 mice represent a model for immunotherapeutic reversal of chronic lymphocytic leukemia-induced T-cell dysfunction. Proc. Natl. Acad. Sci. USA 2009, 106, 6250–6255. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aguilar-Hernandez, M.M.; Rincon Camacho, J.C.; Galicia Garcia, G. Extracellular vesicles and their associated miRNAs as potential prognostic biomarkers in chronic lymphocytic leukemia. Curr. Oncol. Rep. 2021, 23, 66. [Google Scholar] [CrossRef] [PubMed]
- Haderk, F.; Schulz, R.; Iskar, M.; Cid, L.L.; Worst, T.; Willmund, K.V.; Schulz, A.; Warnken, U.; Seiler, J.; Benner, A.; et al. Tumor-derived exosomes modulate PD-L1 expression in monocytes. Sci. Immunol. 2017, 2, eaah5509. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Paggetti, J.; Haderk, F.; Seiffert, M.; Janji, B.; Distler, U.; Ammerlaan, W.; Kim, Y.J.; Adam, J.; Lichter, P.; Solary, E.; et al. Exosomes released by chronic lymphocytic leukemia cells induce the transition of stromal cells into cancer-associated fibroblasts. Blood 2015, 126, 1106–1117. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ma, F.; Vayalil, J.; Lee, G.; Wang, Y.; Peng, G. Emerging role of tumor-derived extracellular vesicles in T cell suppression and dysfunction in the tumor microenvironment. J. Immunother. Cancer 2021, 9, e003217. [Google Scholar] [CrossRef]
- D’Arena, G.; Vitale, C.; Coscia, M.; Festa, A.; Di Minno, N.M.D.; De Feo, V.; Caraglia, M.; Calapai, G.; Laurenti, L.; Musto, P.; et al. Regulatory T cells and their prognostic relevance in hematologic malignancies. J. Immunol. Res. 2017, 2017, 1832968. [Google Scholar] [CrossRef] [Green Version]
- Jitschin, R.; Braun, M.; Büttner, M.; Dettmer-Wilde, K.; Bricks, J.; Berger, J.; Eckart, M.J.; Krause, S.W.; Oefner, P.J.; Le Blanc, K.; et al. CLL-cells induce IDOhi CD14+HLA-DRlo myeloid-derived suppressor cells that inhibit T-cell responses and promote TRegs. Blood 2014, 124, 750–760. [Google Scholar] [CrossRef] [Green Version]
- Fu, S.; Zhang, N.; Yopp, A.C.; Chen, D.; Mao, M.; Chen, D.; Zhang, H.; Ding, Y.; Bromberg, J.S. TGF-beta induces Foxp3+ T-regulatory cells from CD4+ CD25− precursors. Am. J. Transplant. 2004, 4, 1614–1627. [Google Scholar] [CrossRef]
- Serra, S.; Horenstein, A.L.; Vaisitti, T.; Brusa, D.; Rossi, D.; Laurenti, L.; D’Arena, G.; Coscia, M.; Tripodo, C.; Inghirami, G.; et al. CD73-generated extracellular adenosine in chronic lymphocytic leukemia creates local conditions counteracting drug-induced cell death. Blood 2011, 118, 6141–6152. [Google Scholar] [CrossRef]
- Vigano, S.; Alatzoglou, D.; Irving, M.; Menetrier-Caux, C.; Caux, C.; Romero, P.; Coukos, G. Targeting adenosine in cancer immunotherapy to enhance T-cell function. Front. Immunol. 2019, 10, 925. [Google Scholar] [CrossRef] [Green Version]
- Sorrentino, C.; Hossain, F.; Rodriguez, P.C.; Sierra, R.A.; Pannuti, A.; Osborne, B.A.; Minter, L.M.; Miele, L.; Morello, S. Adenosine A2A receptor stimulation inhibits TCR-induced notch1 activation in CD8+T-cells. Front. Immunol. 2019, 10, 162. [Google Scholar] [CrossRef]
- Ohta, A.; Kini, R.; Ohta, A.; Subramanian, M.; Madasu, M.; Sitkovsky, M. The development and immunosuppressive functions of CD4(+) CD25(+) FoxP3(+) regulatory T cells are under influence of the adenosine-A2A adenosine receptor pathway. Front. Immunol. 2012, 3, 190. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shi, L.; Feng, M.; Du, S.; Wei, X.; Song, H.; Yixin, X.; Song, J.; Wenxian, G. Adenosine generated by regulatory T cells induces CD8(+) T cell exhaustion in gastric cancer through A2aR pathway. BioMed Res. Int. 2019, 2019, 4093214. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sun, C.; Mezzadra, R.; Schumacher, T.N. Regulation and function of the PD-L1 checkpoint. Immunity 2018, 48, 434–452. [Google Scholar] [CrossRef] [Green Version]
- Xu-Monette, Z.Y.; Zhou, J.; Young, K.H. PD-1 expression and clinical PD-1 blockade in B-cell lymphomas. Blood 2018, 131, 68–83. [Google Scholar] [CrossRef] [Green Version]
- Brusa, D.; Serra, S.; Coscia, M.; Rossi, D.; D’Arena, G.; Laurenti, L.; Jaksic, O.; Fedele, G.; Inghirami, G.; Gaidano, G.; et al. The PD-1/PD-L1 axis contributes to T-cell dysfunction in chronic lymphocytic leukemia. Haematologica 2013, 98, 953–963. [Google Scholar] [CrossRef] [PubMed]
- Ding, W.; LaPlant, B.R.; Call, T.G.; Parikh, S.A.; Leis, J.F.; He, R.; Shanafelt, T.D.; Sinha, S.; Le-Rademacher, J.; Feldman, A.L.; et al. Pembrolizumab in patients with CLL and Richter transformation or with relapsed CLL. Blood 2017, 129, 3419–3427. [Google Scholar] [CrossRef]
- Younes, A.; Brody, J.; Carpio, C.; Lopez-Guillermo, A.; Ben-Yehuda, D.; Ferhanoglu, B.; Nagler, A.; Ozcan, M.; Avivi, I.; Bosch, F.; et al. Safety and activity of ibrutinib in combination with nivolumab in patients with relapsed non-Hodgkin lymphoma or chronic lymphocytic leukaemia: A phase 1/2a study. Lancet Haematol. 2019, 6, e67–e78. [Google Scholar] [CrossRef]
- Yang, R.; Sun, L.; Li, C.F.; Wang, Y.H.; Yao, J.; Li, H.; Yan, M.; Chang, W.C.; Hsu, J.M.; Cha, J.H.; et al. Galectin-9 interacts with PD-1 and TIM-3 to regulate T cell death and is a target for cancer immunotherapy. Nat. Commun. 2021, 12, 832. [Google Scholar] [CrossRef]
- Chauvin, J.M.; Zarour, H.M. TIGIT in cancer immunotherapy. J. Immunother. Cancer 2020, 8, e000957. [Google Scholar] [CrossRef]
- Wierz, M.; Pierson, S.; Guyonnet, L.; Viry, E.; Lequeux, A.; Oudin, A.; Niclou, S.P.; Ollert, M.; Berchem, G.; Janji, B.; et al. Dual PD1/LAG3 immune checkpoint blockade limits tumor development in a murine model of chronic lymphocytic leukemia. Blood 2018, 131, 1617–1621. [Google Scholar] [CrossRef] [Green Version]
- Yin, Z.; Yu, M.; Ma, T.; Zhang, C.; Huang, S.; Karimzadeh, M.R.; Momtazi-Borojeni, A.A.; Chen, S. Mechanisms underlying low-clinical responses to PD-1/PD-L1 blocking antibodies in immunotherapy of cancer: A key role of exosomal PD-L1. J. Immunother. Cancer 2021, 9, e001698. [Google Scholar] [CrossRef] [PubMed]
- Vautrot, V.; Bentayeb, H.; Causse, S.; Garrido, C.; Gobbo, J. Tumor-derived exosomes: Hidden players in PD-1/PD-L1 resistance. Cancers 2021, 13, 4537. [Google Scholar] [CrossRef] [PubMed]
- Sagiv-Barfi, I.; Kohrt, H.E.; Czerwinski, D.K.; Ng, P.P.; Chang, B.Y.; Levy, R. Therapeutic antitumor immunity by checkpoint blockade is enhanced by ibrutinib, an inhibitor of both BTK and ITK. Proc. Natl. Acad. Sci. USA 2015, 112, E966–E972. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hanna, B.S.; Yazdanparast, H.; Demerdash, Y.; Roessner, P.M.; Schulz, R.; Lichter, P.; Stilgenbauer, S.; Seiffert, M. Combining ibrutinib and checkpoint blockade improves CD8+ T-cell function and control of chronic lymphocytic leukemia in Em-TCL1 mice. Haematologica 2021, 106, 968–977. [Google Scholar] [CrossRef] [Green Version]
- Chavez, J.C.; Grajales-Cruz, A.F.; Volpe, V.O.; Turba, E.P.; Nodzon, L.; Sahakian, E.; Rozario, N.; Ibarz, J.P. A phase II trial of ibrutinib and PD-1 blockade in asymptomatic high risk chronic lymphocytic leukemia to improve immune function. Blood 2019, 134, 5483. [Google Scholar] [CrossRef]
- Gauthier, J.; Hirayama, A.V.; Hay, K.A.; Li, D.; Lymp, J.; Sheih, A.; Purushe, J.; Pender, B.S.; Hawkins, R.M.; Vakil, A.; et al. Efficacy and toxicity of CD19-specific chimeric antigen receptor T cells alone or in combination with ibrutinib for relapsed and/or refractory CLL. Biol. Blood Marrow Transplant. 2019, 25, S9–S10. [Google Scholar] [CrossRef] [Green Version]
- Porter, D.L.; Hwang, W.T.; Frey, N.V.; Lacey, S.F.; Shaw, P.A.; Loren, A.W.; Bagg, A.; Marcucci, K.T.; Shen, A.; Gonzalez, V.; et al. Chimeric antigen receptor T cells persist and induce sustained remissions in relapsed refractory chronic lymphocytic leukemia. Sci. Transl. Med. 2015, 7, 303ra139. [Google Scholar] [CrossRef] [Green Version]
- Fraietta, J.A.; Lacey, S.F.; Orlando, E.J.; Pruteanu-Malinici, I.; Gohil, M.; Lundh, S.; Boesteanu, A.C.; Wang, Y.; O’Connor, R.S.; Hwang, W.T.; et al. Determinants of response and resistance to CD19 chimeric antigen receptor (CAR) T cell therapy of chronic lymphocytic leukemia. Nat. Med. 2018, 24, 563–571. [Google Scholar] [CrossRef]
- Sotillo, E.; Barrett, D.M.; Black, K.L.; Bagashev, A.; Oldridge, D.; Wu, G.; Sussman, R.; Lanauze, C.; Ruella, M.; Gazzara, M.R.; et al. Convergence of acquired mutations and alternative splicing of CD19 enables resistance to CART-19 immunotherapy. Cancer Discov. 2015, 5, 1282–1295. [Google Scholar] [CrossRef] [Green Version]
- Long, A.H.; Haso, W.M.; Shern, J.F.; Wanhainen, K.M.; Murgai, M.; Ingaramo, M.; Smith, J.P.; Walker, A.J.; Kohler, M.E.; Venkateshwara, V.R.; et al. 4-1BB costimulation ameliorates T cell exhaustion induced by tonic signaling of chimeric antigen receptors. Nat. Med. 2015, 21, 581–590. [Google Scholar] [CrossRef] [Green Version]
- Can, I.; Cox, M.J.; Siegler, E.L.; Sakemura, R.; Kenderian, S.S. Challenges of chimeric antigen receptor T-cell therapy in chronic lymphocytic leukemia: Lessons learned. Exp. Hematol. 2022, 108, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Hoffmann, J.M.; Schubert, M.L.; Wang, L.; Huckelhoven, A.; Sellner, L.; Stock, S.; Schmitt, A.; Kleist, C.; Gern, U.; Loskog, A.; et al. Differences in expansion potential of naive chimeric antigen receptor T cells from healthy donors and untreated chronic lymphocytic leukemia patients. Front. Immunol. 2017, 8, 1956. [Google Scholar] [CrossRef] [PubMed] [Green Version]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Böttcher, M.; Böttcher-Loschinski, R.; Kahlfuss, S.; Aigner, M.; Gießl, A.; Mackensen, A.; Schlötzer-Schrehardt, U.; Tüting, T.; Bruns, H.; Mougiakakos, D. CLL-Derived Extracellular Vesicles Impair T-Cell Activation and Foster T-Cell Exhaustion via Multiple Immunological Checkpoints. Cells 2022, 11, 2176. https://doi.org/10.3390/cells11142176
Böttcher M, Böttcher-Loschinski R, Kahlfuss S, Aigner M, Gießl A, Mackensen A, Schlötzer-Schrehardt U, Tüting T, Bruns H, Mougiakakos D. CLL-Derived Extracellular Vesicles Impair T-Cell Activation and Foster T-Cell Exhaustion via Multiple Immunological Checkpoints. Cells. 2022; 11(14):2176. https://doi.org/10.3390/cells11142176
Chicago/Turabian StyleBöttcher, Martin, Romy Böttcher-Loschinski, Sascha Kahlfuss, Michael Aigner, Andreas Gießl, Andreas Mackensen, Ursula Schlötzer-Schrehardt, Thomas Tüting, Heiko Bruns, and Dimitrios Mougiakakos. 2022. "CLL-Derived Extracellular Vesicles Impair T-Cell Activation and Foster T-Cell Exhaustion via Multiple Immunological Checkpoints" Cells 11, no. 14: 2176. https://doi.org/10.3390/cells11142176
APA StyleBöttcher, M., Böttcher-Loschinski, R., Kahlfuss, S., Aigner, M., Gießl, A., Mackensen, A., Schlötzer-Schrehardt, U., Tüting, T., Bruns, H., & Mougiakakos, D. (2022). CLL-Derived Extracellular Vesicles Impair T-Cell Activation and Foster T-Cell Exhaustion via Multiple Immunological Checkpoints. Cells, 11(14), 2176. https://doi.org/10.3390/cells11142176