
Modulation of MAPK- and PI3/AKT-Dependent Autophagy Signaling by Stavudine (D4T) in PBMC of Alzheimer’s Disease Patients

 ,
,  , ,
, ,  , ,
, , 
Abstract
:1. Introduction
2. Materials and Methods
2.1. Patients
2.2. APO ε4 Genotyping
2.3. CSF Collection and Aβ and Tau Determination
2.4. Blood Sample Collection and Processing
2.5. Cell Cultures
2.6. MTT Stavudine (D4T)
2.7. Enzyme-Linked Immunosorbent Assay (ELISA)
2.8. Image Stream Analysis by FlowSight AMNIS
2.9. Protein Extraction
2.10. Western Blot
2.11. Statistical Analysis
3. Results
3.1. Patient Characteristics
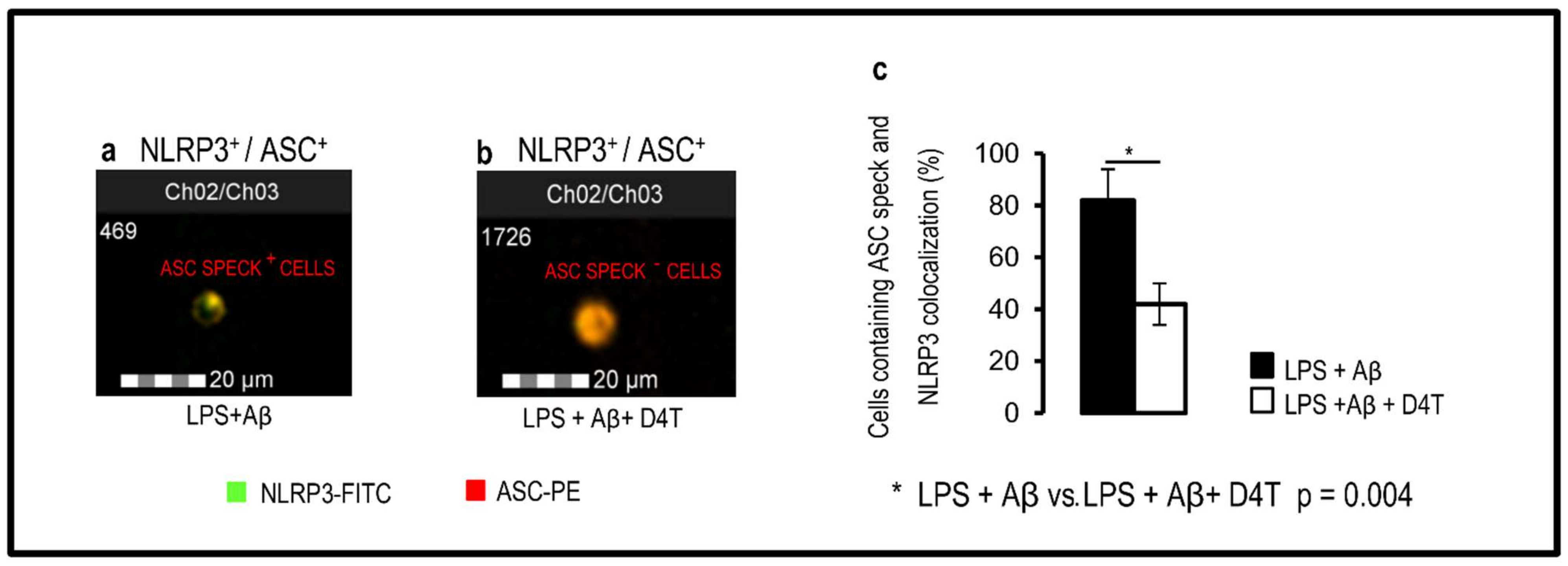
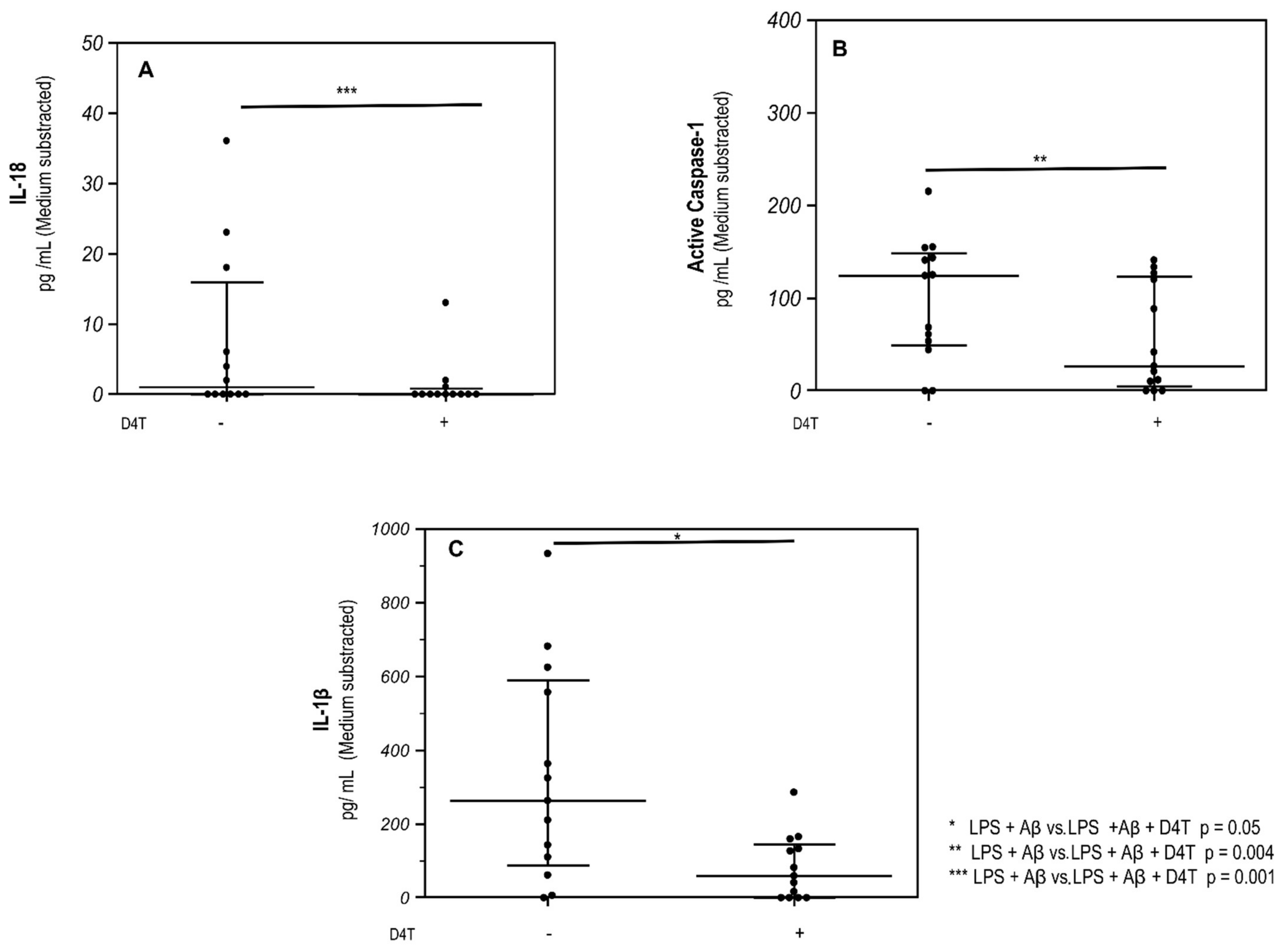
3.2. D4T Downregulates NLRP3-Complex Activation and Inflammasome-Derived Cytokines
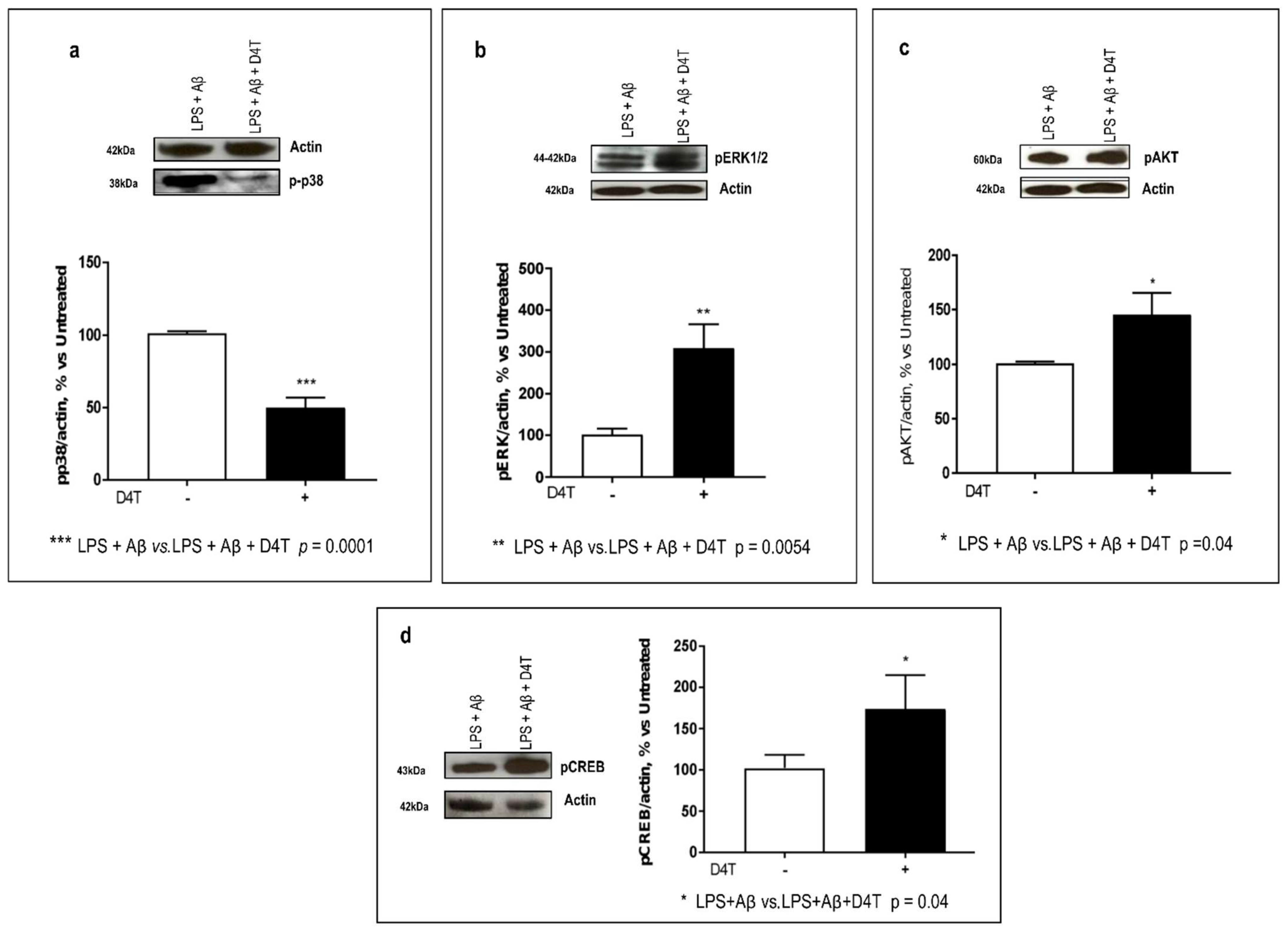
3.3. D4T Modulates MEK-(ERK and p38) and PI3/AKT-Pathways
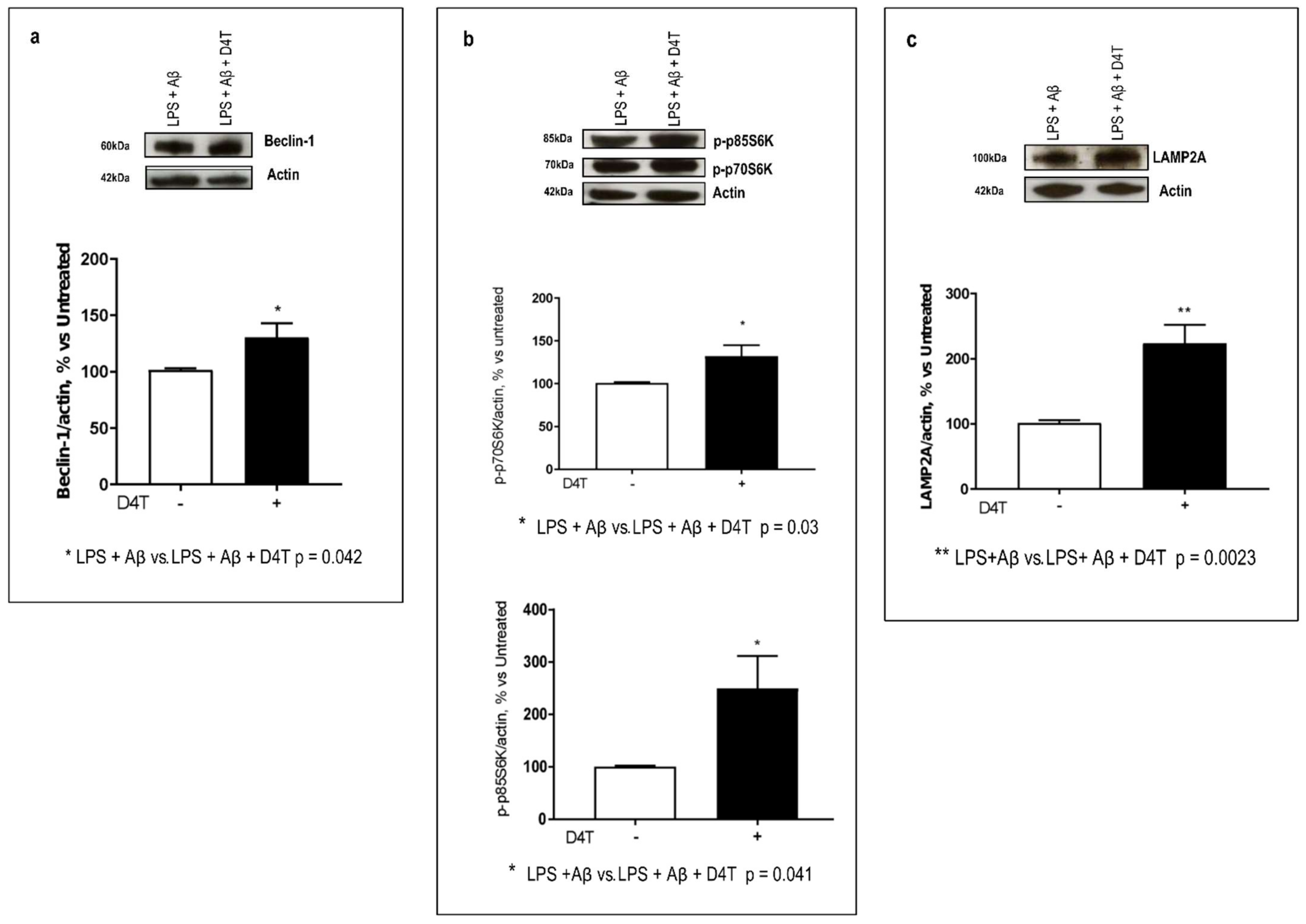
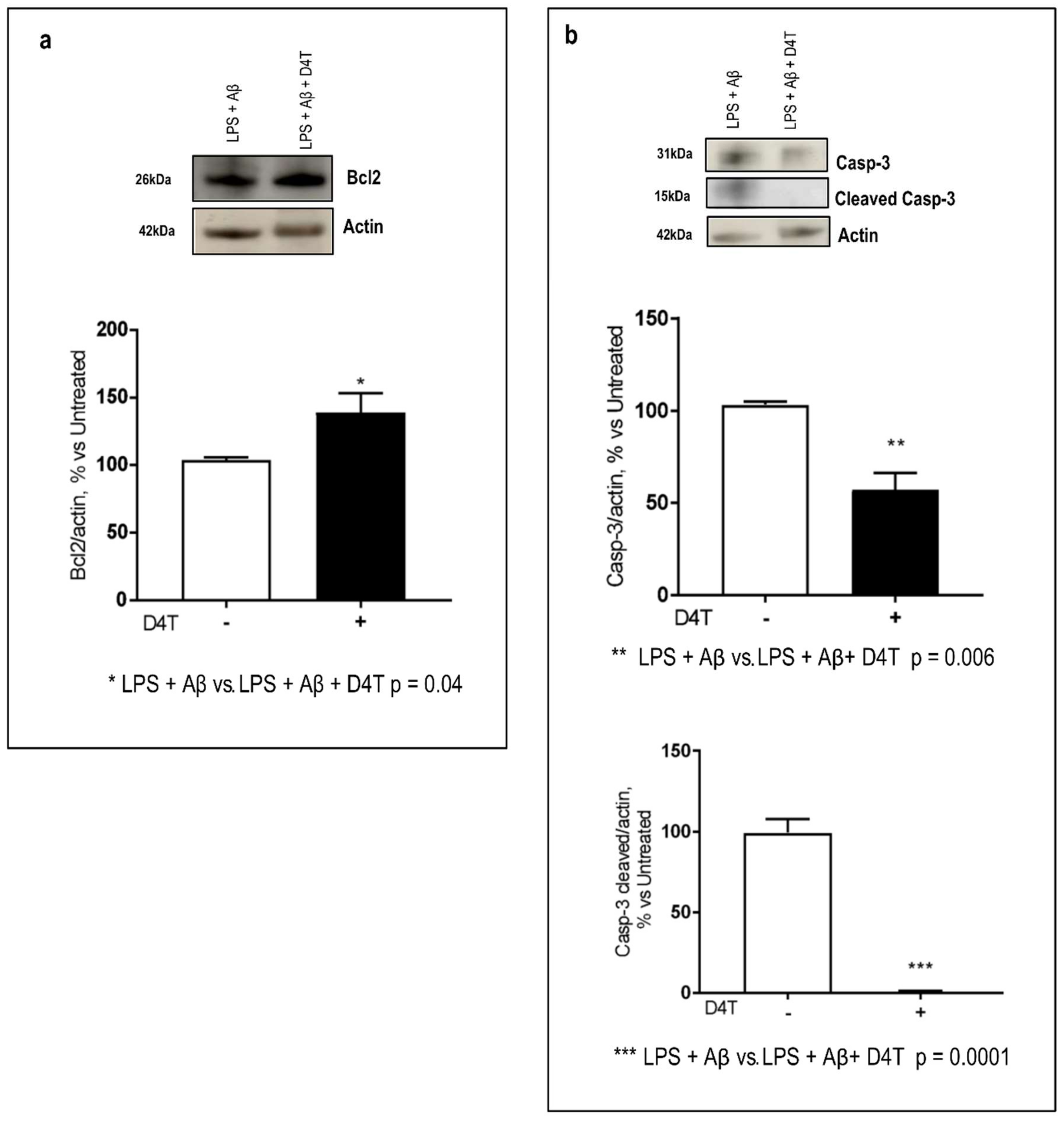
3.4. Effect of D4T on Autophagy Signaling
3.5. Beclin-1 Regulating Proteins by D4T
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Haass, C.; Selkoe, D.J. Soluble protein oligomers in neurodegeneration: Lessons from the Alzheimer’s amyloid beta-peptide. Nat. Rev. Mol. Cell Biol. 2007, 8, 101–112. [Google Scholar] [CrossRef] [PubMed]
- Cai, Z.; Hussain, M.D.; Yan, L.J. Microglia, neuroinflammation, and beta-amyloid protein in Alzheimer’s disease. Int. Neurosci. 2014, 124, 307–321. [Google Scholar] [CrossRef] [PubMed]
- Heneka, M.T. Macrophages derived from infiltrating monocytes mediate autoimmune myelin destruction. J. Exp. Med. 2014, 211, 1500. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, F.; Jiang, L. Neuroinflammation in Alzheimer’s disease. Neuropsychiatr. Dis. Treat. 2015, 11, 243–256. [Google Scholar] [CrossRef] [Green Version]
- Van Dyck, C.H. Anti-Amyloid-β Monoclonal Antibodies for Alzheimer’s Disease: Pitfalls and Promise. Biol. Psychiatry 2018, 83, 311–319. [Google Scholar] [CrossRef] [Green Version]
- Agostini, L.; Martinon, F.; Burns, K.; McDermott, M.F.; Hawkins, P.N.; Tschopp, J. NALP3 forms an IL-1beta-processing inflammasome with increased activity in Muckle-Wells autoinflammatory disorder. Immunity 2004, 20, 319–325. [Google Scholar] [CrossRef] [Green Version]
- Sagulenko, V.; Thygesen, S.J.; Sester, D.P.; Idris, A.; Cridland, J.A.; Vajjhala, P.R.; Roberts, T.L.; Schroder, K.; Vince, J.E.; Hill, J.M.; et al. AIM2 and NLRP3 inflammasomes activate both apoptotic and pyroptotic death pathways via ASC. Cell Death Differ. 2013, 20, 1149–1160. [Google Scholar] [CrossRef] [Green Version]
- Masumoto, J.; Taniguchi, S.; Ayukawa, K.; Sarvotham, H.; Kishino, T.; Niikawa, N.; Hidaka, E.; Katsuyama, T.; Higuchi, T.; Sagara, J. ASC, a novel 22-kDa protein, aggregates during apoptosis of human promyelocytic leukemia HL-60 cells. J. Biol. Chem. 1999, 26, 33835–33838. [Google Scholar] [CrossRef] [Green Version]
- Nakahira, K.; Haspel, J.A.; Rathinam, V.A.; Lee, S.J.; Dolinay, T.; Lam, H.C.; Englert, J.A.; Rabinovitch, M.; Cernadas, M.; Kim, H.P.; et al. Autophagy proteins regulate innate immune responses by inhibiting the release of mitochondrial DNA mediated by the NALP3 inflammasome. Nat. Immunol. 2011, 12, 222–230. [Google Scholar] [CrossRef] [Green Version]
- Kiffin, R.; Christian, C.; Knecht, E.; Cuervo, A.M. Activation of chaperone-mediated autophagy during oxidative stress. Mol. Biol. Cell 2011, 15, 4829–4840. [Google Scholar] [CrossRef] [Green Version]
- Martinez-Lopez, N.; Athonvarangkul, D.; Mishall, P.; Sahu, S.; Singh, R. Autophagy protein regulate ERK phosphorylation. Nat. Commun. 2013, 4, 2799. [Google Scholar] [CrossRef]
- Harris, J.; Lang, T.; Thomas, J.P.W.; Sukkar, M.B.; Nabar, N.R.; Kehrl, J.H. Autophagy and inflammasomes. Mol. Immunol. 2017, 86, 10–15. [Google Scholar] [CrossRef]
- Yu, L.; Chen, Y. Autophagy pathway: Cellular and molecular mechanisms. Autophagy 2018, 2, 207–215. [Google Scholar] [CrossRef] [Green Version]
- Shibutani, S.T.; Saitoh, T.; Nowag, H.; Münz, C.; Yoshimori, T. Autophagy and autophagy-related proteins in the immune system. Nat. Immunol. 2015, 16, 1014–1024. [Google Scholar] [CrossRef]
- Hara, T.; Nakamura, K.; Matsui, M.; Yamamoto, A.; Nakahara, Y.; Suzuki-Migishima, R.; Yokoyama, M.; Mishima, K.; Saito, I.; Okano, H.; et al. Suppression of basal autophagy in neural cells causes neurodegenerative disease in mice. Nature 2006, 15, 885–889. [Google Scholar] [CrossRef]
- Jessop, F.; Hamilton, R.F.; Rhoderick, J.F.; Shaw, P.K.; Holian, A. Autophagy deficiency in macrophages enhances NLRP3 inflammasome activity and chronic lung disease following silica exposure. Toxicol. Appl. Pharmacol. 2016, 309, 101–110. [Google Scholar] [CrossRef] [Green Version]
- Fowler, B.J.; Gelfand, B.D.; Kim, Y.; Kerur, N.; Tarallo, V.; Hiran Amarnath, S.; Fowler, D.H.; Radwan, M.; Young, M.T. Nucleoside reverse transcriptase inhibitors possess intrinsic anti-inflammatory activity. Science 2014, 346, 1000–1003. [Google Scholar] [CrossRef] [Green Version]
- Cai, R.; Liu, L.; Luo, B.; Wang, J.; Shen, J.; Shen, Y.; Zhang, R.; Chen, J.; Lu, H. Caspase-1 Activity in CD4 T Cells Is Downregulated Following Antiretroviral Therapy for HIV-1 Infection. AIDS Res. Hum. Retrovir. 2017, 33, 164–171. [Google Scholar] [CrossRef] [Green Version]
- La Rosa, F.; Saresella, M.; Marventano, I.; Piancone, F.; Ripamonti, E.; Al-Daghri, N.; Bazzini, C.; Zoia, C.P.; Conti, E.; Ferrarese, C.; et al. Stavudine Reduces NLRP3 Inflammasome Activation and Modulates Amyloid-β Autophagy. J. Alzheimers Dis. 2019, 72, 401–412. [Google Scholar] [CrossRef]
- Kirouac, L.; Rajic, A.J.; Cribbs, D.H.; Padmanabhan, J. Activation of Ras-ERK Signaling and GSK-3 by Amyloid Precursor Protein and Amyloid Beta Facilitates Neurodegeneration in Alzheimer’s Disease. eNeuro 2017, 4. [Google Scholar] [CrossRef] [Green Version]
- Heras-Sandoval, D.; Ferrera, P.; Arias, C. Amyloid-β protein modulates insulin signaling in presynaptic terminals. Neurochem. Res. 2012, 37, 1879–1885. [Google Scholar] [CrossRef]
- Lin, M.W.; Chen, Y.H.; Yang, H.B.; Lin, C.C.; Hung, S.Y. Galantamine Inhibits Aβ1-42-Induced Neurotoxicity by Enhancing α7nAChR Expression as a Cargo Carrier for LC3 Binding and Aβ1-42 Engulfment During Autophagic Degradation. Neurotherapeutics 2019, 10, 676–689. [Google Scholar] [CrossRef]
- Wang, X.; Wang, Y.; Zhu, Y.; Yan, L.; Zhao, L. Neuroprotective Effect of S-trans, Trans-farnesylthiosalicylic Acid via Inhibition of RAS/ERK Pathway for the Treatment of Alzheimer’s Disease. Drug Des. Dev. Ther. 2019, 29, 4053–4063. [Google Scholar] [CrossRef] [Green Version]
- McKhann, G.; Drachman, D.; Folstein, M.; Katzman, R.; Price, D.; Stadlan, E.M. Clinical diagnosis of Alzheimer’s Disease: Report of the NINCDS-ADRDA Work Group under the auspices of Department of Health and Human Service Task Force on Alzheimer’s Disease. Neurology 1984, 34, 939–944. [Google Scholar] [CrossRef] [Green Version]
- American Psychiatric Association. Diagnostic and Statistical Manual of Mental Disorders DSM-IV-R. 1994. Available online: http://www.psychiatryonline.com/DSMPDF/dsm-iv.pdf (accessed on 20 May 2021).
- Folstein, M.F.; Folstein, S.E.; McHugh, P.R. Mini-mental state. A practical method for grading the cognitive state of patients for the clinicians. J. Psychiatric Res. 1975, 12, 189–198. [Google Scholar] [CrossRef]
- Hughes, C.P.; Berg, L.; Danziger, W.L.; Coben, L.A.; Martin, R.L. A new clinical scale for staging of dementia. Br. J. Psychiatry 1982, 140, 566–572. [Google Scholar] [CrossRef]
- Koch, W.; Ehrenhaft, A.; Griesser, K.; Pfeufer, A.; Müller, J.; Schomig, A. Taqman systems for geno-typing of disease-related polymorphisms present in the gene encoding Apolipoprotein, E. Clin. Chem. Lab. Med. 2002, 40, 1123–1131. [Google Scholar] [CrossRef]
- Gray, L.R.; Turville, S.G.; HItchen, T.L.; Cheng, W.J.; Ellett, A.M.; Salimi, H.; Roche, M.J.; Wesselingh, S.L.; Gorry, P.R.; Churchill, M.J. HIV-1 Entry and Trans-Infection of Astrocytes Involves CD81 Vesicles. PLoS ONE 2014, 9, e90620. [Google Scholar] [CrossRef] [PubMed]
- Mosmann, T. Rapid colorimetric assay for cellular growth and survival: Application to proliferation and cytotoxicity assays. J. Immunol. Methods 1983, 65, 55–63. [Google Scholar] [CrossRef]
- Sjögren, M.; Vanderstichele, H.; Agren, H.; Zachrisson, O.; Edsbagge, M.; Wikkelsø, C.; Skoog, I.; Wallin, A.; Wahlund, L.O.; Marcusson, J. Tau and Abeta42 in cerebrospinal fluid from healthy adults 21–93 years of age: Establishment of reference values. Clin. Chem. 2001, 47, 1776–1781. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Halle, A.; Hornung, V.; Petzold, G.C.; Stewart, C.R.; Monks, B.G.; Reinheckel, T.; Fitzgerald, K.A.; Latz, E.; Moore, K.J.; Golenbock, D.T. The NALP3 inflammasome is involved in the innate immune response to amyloid-beta. Nat. Immunol. 2008, 9, 857–865. [Google Scholar] [CrossRef] [Green Version]
- Heneka, M.T.; Kummer, M.P.; Stutz, A.; Delekate, A.; Schwartz, S.; Vieira-Saecker, A.; Griep, A.; Axt, D.; Remus, A.; Tzeng, T.C.; et al. NLRP3 is activated in Alzheimer’s disease and contributes to pathology in APP/PS1 mice. Nature 2013, 31, 674–678. [Google Scholar] [CrossRef]
- Saresella, M.; La Rosa, F.; Piancone, F.; Zoppis, M.; Marventano, I.; Calabrese, E.; Rainone, V.; Nemni, R.; Mancuso, R.; Clerici, M. The NLRP3 and NLRP1 inflammasomes are activated in Alzheimer’s disease. Mol. Neurodegener. 2016, 3, 23. [Google Scholar] [CrossRef] [Green Version]
- Semmler, A.; Okulla, T.; Sastre, M.; Dumitrescu-Ozimek, L.; Heneka, M.T. Systemic inflammation induces apoptosis with variable vulnerability of different brain regions. J. Chem. Neuroanat. 2005, 30, 144–157. [Google Scholar] [CrossRef]
- Semmler, A.; Frisch, C.; Debeir, T.; Ramanathan, M.; Okulla, T.; Klockgether, T.; Heneka, M.T. Long-term cognitive impairment, neuronal loss and reduced cortical cholinergic innervation after recovery from sepsis in a rodent model. Exp. Neurol. 2017, 204, 733–740. [Google Scholar] [CrossRef]
- Weberpals, M.; Hermes, M.; Hermann, S.; Kummer, M.P.; Terwel, D.; Semmler, A.; Berger, M.; Schäfers, M.; Heneka, M.T. NOS2 gene deficiency protects from sepsis-induced long-term cognitive deficits. J. Neurosci. 2009, 29, 14177–14184. [Google Scholar] [CrossRef] [Green Version]
- Qin, L.; Wu, X.; Block, M.L.; Liu, Y.; Breese, G.R.; Hong, J.; Knapp, D.J.; Crews, F.T. Systemic LPS causes chronic neuroinflammation and progressive neurodegeneration. Glia 2007, 462, 453–462. [Google Scholar] [CrossRef] [Green Version]
- Semmler, A.; Hermann, S.; Mormann, F.; Weberpals, M.; Paxian, S.A.; Okulla, T.; Schäfers, M.; Kummer, M.P.; Klockgether, T.; Heneka, M.T. Sepsis causes neuroinflammation and concomitant decrease of cerebral metabolism. J. Neuroinflamm. 2008, 5, 38. [Google Scholar] [CrossRef] [Green Version]
- Semmler, A.; Widmann, C.N.; Okulla, T.; Urbach, H.; Kaiser, M.; Widman, G.; Mormann, F.; Weide, J.; Fliessbach, K.; Hoeft, A.; et al. Persistent cognitive impairment, hippocampal atrophy and EEG changes in sepsis survivors. J. Neurol. Neurosurg. Psychiatry 2013, 84, 62–69. [Google Scholar] [CrossRef] [Green Version]
- Iwashyna, T.J.; Ely, E.W.; Smith, D.M.; Langa, K.M. Long-term cognitive impairment and functional disability among survivors of severe sepsis. JAMA 2010, 304, 1787. [Google Scholar] [CrossRef] [Green Version]
- Gyoneva, S.; Swanger, S.A.; Zhang, J.; Weinshenker, D.; Traynelis, S.F. Altered motility of plaque-associated microglia in a model of Alzheimer’s disease. Neuroscience 2016, 330, 410–420. [Google Scholar] [CrossRef] [Green Version]
- Widmann, C.N.; Heneka, M.T. Long-term cerebral consequences of sepsis. Lancet Neurol. 2014, 13, 630–636. [Google Scholar] [CrossRef]
- Tejera, D.; Mercan, D.; Sanchez-Caro, J.M.; Hanan, M.; Greenberg, D.; Soreq, H.; Latz, E.; Golenbock, D.; Heneka, M.T. Systemic inflammation impairs microglial Aβ clearance through NLRP3 inflammasome. Embo J. 2019, 2, e101064. [Google Scholar]
- Fésüs, L.; Demény Ma Petrovski, G. Autophagy shapes inflammation. Antioxid. Redox Signal. 2011, 14, 11. [Google Scholar] [CrossRef]
- Erivan, S.R.J.; Morandini, A.C. Gasdermin: A new player to the inflammasome game. Biomed. J. 2017, 40, 301–368. [Google Scholar] [CrossRef]
- Shi, J.; Gao, W.; Shao, F. Pyroptosis: Gasdermin-mediated programmed necrotic cell death. Trends Biochem. Sci. 2017, 42, 245–254. [Google Scholar] [CrossRef]
- Deretic, V.; Levine, B. Autophagy balances inflammation in innate immunity. Autophagy 2018, 14, 243–251. [Google Scholar] [CrossRef] [Green Version]
- Netea-Maier, R.T.; Plantinga, T.S.; Van De Veerdonk, F.L.; Smit, J.W.; Netea, M.G. Modulation of inflammation by autophagy: Consequences for human disease. Autophagy 2016, 12, 245–260. [Google Scholar] [CrossRef] [Green Version]
- Mizushima, N.; Levine, B.; Cuervo, A.M.; Klionsky, D.J. Autophagy fights disease through cellular self-digestion. Nature 2008, 28, 1069–1075. [Google Scholar] [CrossRef] [Green Version]
- Bejarano, E.; Cuervo, A.M. Chaperone-Mediated Autophagy. Proc. Am. Thorac. Soc. 2010, 15, 29–39. [Google Scholar] [CrossRef]
- Kaushik, S.; Cuervo, A. Chaperone-mediated autophagy: A unique way to enter the lysosome world. Trends Cell Biol. 2012, 22, 407–417. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Klionsky, D.J.; Meijer, A.J.; Codogno, P.; Neufeld, T.P.; Scott, R.C. Autophagy and p70S6 Kinase. Autophagy 2015, 1, 59–61. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Decuypere, J.P.; Parys, J.P.; Bultynck, G. Regulation of the Autophagic Bcl-2/Beclin 1 Interaction. Cells 2012, 1, 284–312. [Google Scholar] [CrossRef] [PubMed]
- Pattingre, S.; Tassa, A.; Qu, X.; Garuti, R.; Liang, X.H.; Mizushima, N.; Packer, M.; Schneider, M.D.; Levine, B. Bcl-2 Antiapoptotic Proteins Inhibit Beclin 1-Dependent Autophagy. Cell 2005, 122, 927–939. [Google Scholar] [CrossRef] [Green Version]
- Zhu, Y.; Zhao, L.; Liu, L.; Gao, P.; Tian, W.; Wang, X.; Jin, H.; Xu, H.; Chen, Q. Beclin 1 cleavage by caspase-3 inactivates autophagy and promotes apoptosis. Protein Cells 2010, 1, 468–477. [Google Scholar] [CrossRef] [Green Version]
- Mayr, B.; Montminy, M. Transcriptional regulation by the phosphorylation-dependent factor CREB. Nat. Rev. Mol. Cell Biol. 2008, 2, 599–609. [Google Scholar] [CrossRef]
- Carlezon, W.A.; Duman, R.S.; Nestler, E.J. The many faces of CREB. Trends Neurosci. 2005, 28, 436–445. [Google Scholar] [CrossRef]
- Bartolotti, N.; Bennett, D.A.; Lazarov, O. Reduced pCREB in Alzheimer’s disease prefrontal cortex is reflected in peripheral blood mononuclear cells. Mol. Psychiatry 2016, 21, 1158–1166. [Google Scholar] [CrossRef]
- Bartolotti, N.; Lazarov, O. CREB signals as PBMC-based biomarkers of cognitive dysfunction: A novel perspective of the brain-immune axis. Brain Behav. Immun. 2019, 78, 9–20. [Google Scholar] [CrossRef]
- Long, H.Z.; Cheng, Y.; Zhou, Z.W.; Luo, H.Y.; Wen, D.D.; Gao, L.C. PI3K/AKT Signal Pathway: A Target of Natural Products in the Prevention and Treatment of Alzheimer’s Disease and Parkinson’s Disease. Front. Pharmacol. 2021, 12, 648636. [Google Scholar] [CrossRef]
- Singh, R.K.; Diwan, M.; Dastidar, S.G.; Najmi, A.K. Differential effect of p38 and MK2 kinase inhibitors on the inflammatory and toxicity biomarkers in vitro. Hum. Exp. Toxicol. 2018, 37, 521–531. [Google Scholar] [CrossRef]
- Ronkina, N.; Menon, M.B.; Schwermann, J. Map kap kinases Mk2 and Mk3 in inflammation: Complex regulation of tnf biosynthesis via expression and phosphorylation of tristetraprolin. Biochem. Pharmacol. 2010, 80, 1915–1920. [Google Scholar] [CrossRef]
- Gaestel, M.; Kotlyarov, A.; Kracht, M. Targeting innate immunity protein kinase signalling in inflammation. Nat. Rev. Drug Discov. 2009, 8, 480–499. [Google Scholar] [CrossRef]
- Ruiz-Medina, B.E.; Lerma, D.; Hwang, M.; Ross, J.A.; Skouta, R.; Aguilera, R. Green barley mitigates cytotoxicity in human lymphocytes undergoing aggressive oxidative stress, via activation of both the Lyn/PI3K/Akt and MAPK/ERK pathways. Sci. Rep. 2018, 9, 6005. [Google Scholar] [CrossRef]
- Abdelaziz, D.H.; Khalil, H.; Cormet-Boyaka, E.; Amer, A.O. The cooperation between the autophagy machinery and the inflammasome to implement an appropriate innate immune response: Do they regulate each other? Immunol. Rev. 2015, 265, 194–204. [Google Scholar] [CrossRef] [Green Version]
- Majumder, S.; Richardson, A.; Strong, R.; Oddo, S. Inducing autophagy by rapamycin before, but not after, the formation of plaques and tangles ameliorates cognitive deficits. PLoS ONE 2016, 6, e25416. [Google Scholar] [CrossRef] [Green Version]
- Salminen, A.; Kaarniranta, K.; Kauppinen, A.; Ojala, J.; Haapasalo, A.; Soininen, H.; Hiltunen, M. Impaired autophagy and APP processing in Alzheimer’s disease: The potential role of Beclin 1 interactome. Prog. Neurobiol. 2013, 106–107, 33–54. [Google Scholar] [CrossRef]
- Zhang, Y.; Dong, Z.; Song, W. NLRP3 inflammasome as a novel therapeutic target for Alzheimer’s disease. Signal Transduct. Target. Therapy 2020, 5, 37. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Alzheimer’s Disease Patients (AD) | |
|---|---|
| N | 13 |
| Gender (M:F) | 5:8 |
| Age (years) | 77.20 ± 6.34 |
| MMSE | 17.07 ± 4.51 |
| Aβ (pg/mL) | 530 ± 70.50 |
| Total-τ (pg/mL) | 647.31 ± 300 |
| Phospo-τ (pg/mL) | 88.62 ± 21.89 |
| Apo ε4 | 30% |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
La Rosa, F.; Zoia, C.P.; Bazzini, C.; Bolognini, A.; Saresella, M.; Conti, E.; Ferrarese, C.; Piancone, F.; Marventano, I.; Galimberti, D.; et al. Modulation of MAPK- and PI3/AKT-Dependent Autophagy Signaling by Stavudine (D4T) in PBMC of Alzheimer’s Disease Patients. Cells 2022, 11, 2180. https://doi.org/10.3390/cells11142180
La Rosa F, Zoia CP, Bazzini C, Bolognini A, Saresella M, Conti E, Ferrarese C, Piancone F, Marventano I, Galimberti D, et al. Modulation of MAPK- and PI3/AKT-Dependent Autophagy Signaling by Stavudine (D4T) in PBMC of Alzheimer’s Disease Patients. Cells. 2022; 11(14):2180. https://doi.org/10.3390/cells11142180
Chicago/Turabian StyleLa Rosa, Francesca, Chiara Paola Zoia, Chiara Bazzini, Alessandra Bolognini, Marina Saresella, Elisa Conti, Carlo Ferrarese, Federica Piancone, Ivana Marventano, Daniela Galimberti, and et al. 2022. "Modulation of MAPK- and PI3/AKT-Dependent Autophagy Signaling by Stavudine (D4T) in PBMC of Alzheimer’s Disease Patients" Cells 11, no. 14: 2180. https://doi.org/10.3390/cells11142180
APA StyleLa Rosa, F., Zoia, C. P., Bazzini, C., Bolognini, A., Saresella, M., Conti, E., Ferrarese, C., Piancone, F., Marventano, I., Galimberti, D., Fenoglio, C., Scarpini, E., & Clerici, M. (2022). Modulation of MAPK- and PI3/AKT-Dependent Autophagy Signaling by Stavudine (D4T) in PBMC of Alzheimer’s Disease Patients. Cells, 11(14), 2180. https://doi.org/10.3390/cells11142180