
Epigenetic Memories in Hematopoietic Stem and Progenitor Cells


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. New NGS Technology in Hematopoiesis Research
2.1. RNA Sequencing
2.2. ATAC Sequencing
2.3. Hi-C Technique

3. Trained Immunity and Epigenetic Memory
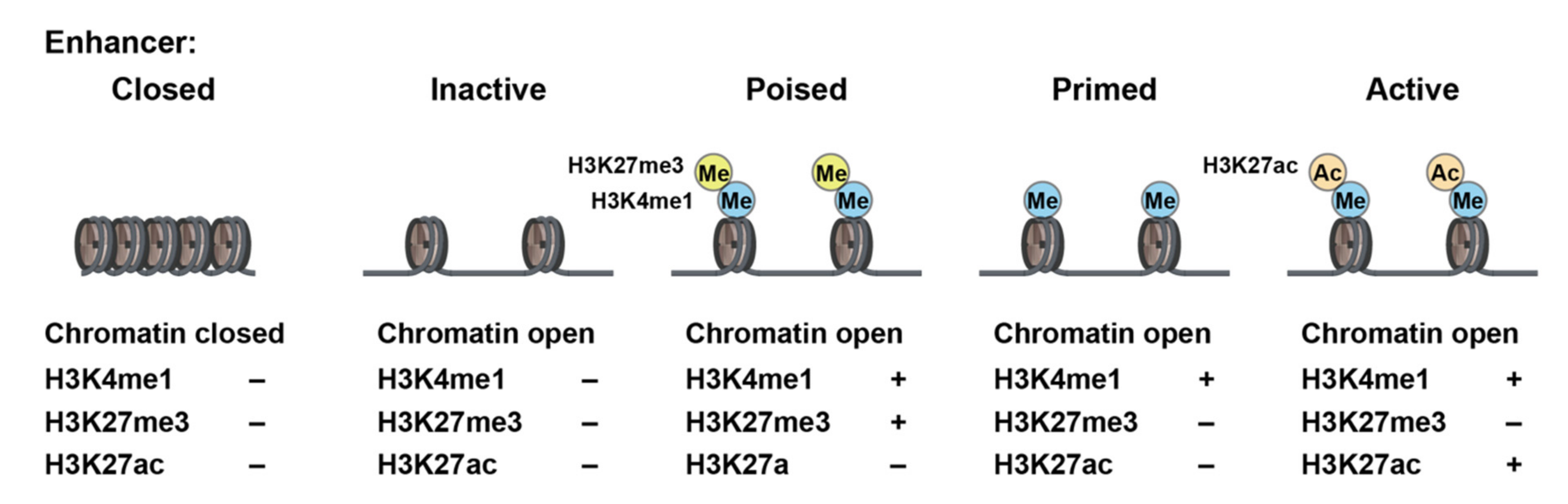
4. Epigenetic Memory in Hematopoietic Stem and Progenitor Cells
4.1. Epigenetic Memory in Hematopoietic Stem and Progenitor Cells
4.2. β-Glucan–Induced Trained Immunity in Neutrophils and BM GMPs
4.3. LPS-Induced Trained Immunity in HSCs
4.4. Epigenetic Memory in Aged HSPCs
4.5. Epigenetic Memory in Non-Immune Cells
5. Conclusions
Author Contributions
Funding
Informed Consent Statement
Conflicts of Interest
References
- Buenrostro, J.D.; Corces, M.R.; Lareau, C.A.; Wu, B.; Schep, A.N.; Aryee, M.J.; Majeti, R.; Chang, H.Y.; Greenleaf, W.J. Integrated Single-Cell Analysis Maps the Continuous Regulatory Landscape of Human Hematopoietic Differentiation. Cell 2018, 173, 1535–1548.e1516. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Collombet, S.; Ranisavljevic, N.; Nagano, T.; Varnai, C.; Shisode, T.; Leung, W.; Piolot, T.; Galupa, R.; Borensztein, M.; Servant, N.; et al. Parental-to-embryo switch of chromosome organization in early embryogenesis. Nature 2020, 580, 142–146. [Google Scholar] [CrossRef]
- Jardine, L.; Webb, S.; Goh, I.; Quiroga Londoño, M.; Reynolds, G.; Mather, M.; Olabi, B.; Stephenson, E.; Botting, R.A.; Horsfall, D.; et al. Blood and immune development in human fetal bone marrow and Down syndrome. Nature 2021, 598, 327–331. [Google Scholar] [CrossRef] [PubMed]
- Bekkering, S.; Domínguez-Andrés, J.; Joosten, L.A.B.; Riksen, N.P.; Netea, M.G. Trained Immunity: Reprogramming Innate Immunity in Health and Disease. Annu. Rev. Immunol. 2021, 39, 667–693. [Google Scholar] [CrossRef]
- Netea, M.G.; Domínguez-Andrés, J.; Barreiro, L.B.; Chavakis, T.; Divangahi, M.; Fuchs, E.; Joosten, L.A.B.; van der Meer, J.W.M.; Mhlanga, M.M.; Mulder, W.J.M.; et al. Defining trained immunity and its role in health and disease. Nat. Rev. Immunol. 2020, 20, 375–388. [Google Scholar] [CrossRef] [Green Version]
- Huang, S. Non-genetic heterogeneity of cells in development: More than just noise. Development 2009, 136, 3853–3862. [Google Scholar] [CrossRef] [Green Version]
- Li, L.; Clevers, H. Coexistence of quiescent and active adult stem cells in mammals. Science 2010, 327, 542–545. [Google Scholar] [CrossRef] [Green Version]
- Shalek, A.K.; Satija, R.; Shuga, J.; Trombetta, J.J.; Gennert, D.; Lu, D.; Chen, P.; Gertner, R.S.; Gaublomme, J.T.; Yosef, N.; et al. Single-cell RNA-seq reveals dynamic paracrine control of cellular variation. Nature 2014, 510, 363–369. [Google Scholar] [CrossRef] [Green Version]
- Eldar, A.; Elowitz, M.B. Functional roles for noise in genetic circuits. Nature 2010, 467, 167–173. [Google Scholar] [CrossRef]
- Maamar, H.; Raj, A.; Dubnau, D. Noise in gene expression determines cell fate in Bacillus subtilis. Science 2007, 317, 526–529. [Google Scholar] [CrossRef] [Green Version]
- Tang, F.; Barbacioru, C.; Wang, Y.; Nordman, E.; Lee, C.; Xu, N.; Wang, X.; Bodeau, J.; Tuch, B.B.; Siddiqui, A.; et al. mRNA-Seq whole-transcriptome analysis of a single cell. Nat. Methods 2009, 6, 377–382. [Google Scholar] [CrossRef] [PubMed]
- Chen, G.; Ning, B.; Shi, T. Single-Cell RNA-Seq Technologies and Related Computational Data Analysis. Front. Genet. 2019, 10, 317. [Google Scholar] [CrossRef]
- Hwang, B.; Lee, J.H.; Bang, D. Single-cell RNA sequencing technologies and bioinformatics pipelines. Exp. Mol. Med. 2018, 50, 1–14. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Suvà, M.L.; Tirosh, I. Single-Cell RNA Sequencing in Cancer: Lessons Learned and Emerging Challenges. Mol. Cell 2019, 75, 7–12. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Wang, D.; Peng, M.; Tang, L.; Ouyang, J.; Xiong, F.; Guo, C.; Tang, Y.; Zhou, Y.; Liao, Q.; et al. Single-cell RNA sequencing in cancer research. J. Exp. Clin. Cancer Res. 2021, 40, 81. [Google Scholar] [CrossRef]
- Shaffer, S.M.; Dunagin, M.C.; Torborg, S.R.; Torre, E.A.; Emert, B.; Krepler, C.; Beqiri, M.; Sproesser, K.; Brafford, P.A.; Xiao, M.; et al. Rare cell variability and drug-induced reprogramming as a mode of cancer drug resistance. Nature 2017, 546, 431–435. [Google Scholar] [CrossRef] [Green Version]
- Haque, A.; Engel, J.; Teichmann, S.A.; Lönnberg, T. A practical guide to single-cell RNA-sequencing for biomedical research and clinical applications. Genome Med. 2017, 9, 75. [Google Scholar] [CrossRef]
- Shalek, A.K.; Satija, R.; Adiconis, X.; Gertner, R.S.; Gaublomme, J.T.; Raychowdhury, R.; Schwartz, S.; Yosef, N.; Malboeuf, C.; Lu, D.; et al. Single-cell transcriptomics reveals bimodality in expression and splicing in immune cells. Nature 2013, 498, 236–240. [Google Scholar] [CrossRef] [Green Version]
- Trapnell, C.; Cacchiarelli, D.; Grimsby, J.; Pokharel, P.; Li, S.; Morse, M.; Lennon, N.J.; Livak, K.J.; Mikkelsen, T.S.; Rinn, J.L. The dynamics and regulators of cell fate decisions are revealed by pseudotemporal ordering of single cells. Nat. Biotechnol. 2014, 32, 381–386. [Google Scholar] [CrossRef] [Green Version]
- Petropoulos, S.; Edsgärd, D.; Reinius, B.; Deng, Q.; Panula, S.P.; Codeluppi, S.; Reyes, A.P.; Linnarsson, S.; Sandberg, R.; Lanner, F. Single-Cell RNA-Seq Reveals Lineage and X Chromosome Dynamics in Human Preimplantation Embryos. Cell 2016, 167, 285. [Google Scholar] [CrossRef] [Green Version]
- Stubbington, M.J.T.; Lönnberg, T.; Proserpio, V.; Clare, S.; Speak, A.O.; Dougan, G.; Teichmann, S.A. T cell fate and clonality inference from single-cell transcriptomes. Nat. Methods 2016, 13, 329–332. [Google Scholar] [CrossRef] [Green Version]
- Yu, V.W.C.; Yusuf, R.Z.; Oki, T.; Wu, J.; Saez, B.; Wang, X.; Cook, C.; Baryawno, N.; Ziller, M.J.; Lee, E.; et al. Epigenetic Memory Underlies Cell-Autonomous Heterogeneous Behavior of Hematopoietic Stem Cells. Cell 2017, 168, 944–945. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schulte-Schrepping, J.; Reusch, N.; Paclik, D.; Baßler, K.; Schlickeiser, S.; Zhang, B.; Krämer, B.; Krammer, T.; Brumhard, S.; Bonaguro, L.; et al. Severe COVID-19 Is Marked by a Dysregulated Myeloid Cell Compartment. Cell 2020, 182, 1419–1440.e23. [Google Scholar] [CrossRef] [PubMed]
- Buenrostro, J.D.; Giresi, P.G.; Zaba, L.C.; Chang, H.Y.; Greenleaf, W.J. Transposition of native chromatin for fast and sensitive epigenomic profiling of open chromatin, DNA-binding proteins and nucleosome position. Nat. Methods 2013, 10, 1213–1218. [Google Scholar] [CrossRef] [PubMed]
- Buenrostro, J.D.; Wu, B.; Chang, H.Y.; Greenleaf, W.J. ATAC-seq: A Method for Assaying Chromatin Accessibility Genome-Wide. Curr. Protoc. Mol. Biol. 2015, 109, 21.29.21–21.29.29. [Google Scholar] [CrossRef]
- Shashikant, T.; Ettensohn, C.A. Genome-wide analysis of chromatin accessibility using ATAC-seq. Methods Cell Biol. 2019, 151, 219–235. [Google Scholar] [CrossRef]
- Sun, Y.; Miao, N.; Sun, T. Detect accessible chromatin using ATAC-sequencing, from principle to applications. Hereditas 2019, 156, 29. [Google Scholar] [CrossRef] [Green Version]
- Yan, F.; Powell, D.R.; Curtis, D.J.; Wong, N.C. From reads to insight: A hitchhiker’s guide to ATAC-seq data analysis. Genome Biol. 2020, 21, 22. [Google Scholar] [CrossRef]
- Boyle, A.P.; Davis, S.; Shulha, H.P.; Meltzer, P.; Margulies, E.H.; Weng, Z.; Furey, T.S.; Crawford, G.E. High-resolution mapping and characterization of open chromatin across the genome. Cell 2008, 132, 311–322. [Google Scholar] [CrossRef] [Green Version]
- Song, L.; Crawford, G.E. DNase-seq: A high-resolution technique for mapping active gene regulatory elements across the genome from mammalian cells. Cold Spring Harb. Protoc. 2010, 2010, pdb.prot5384. [Google Scholar] [CrossRef] [Green Version]
- Thurman, R.E.; Rynes, E.; Humbert, R.; Vierstra, J.; Maurano, M.T.; Haugen, E.; Sheffield, N.C.; Stergachis, A.B.; Wang, H.; Vernot, B.; et al. The accessible chromatin landscape of the human genome. Nature 2012, 489, 75–82. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Giresi, P.G.; Kim, J.; McDaniell, R.M.; Iyer, V.R.; Lieb, J.D. FAIRE (Formaldehyde-Assisted Isolation of Regulatory Elements) isolates active regulatory elements from human chromatin. Genome Res. 2007, 17, 877–885. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Simon, J.M.; Giresi, P.G.; Davis, I.J.; Lieb, J.D. Using formaldehyde-assisted isolation of regulatory elements (FAIRE) to isolate active regulatory DNA. Nat. Protoc. 2012, 7, 256–267. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barski, A.; Cuddapah, S.; Cui, K.; Roh, T.Y.; Schones, D.E.; Wang, Z.; Wei, G.; Chepelev, I.; Zhao, K. High-resolution profiling of histone methylations in the human genome. Cell 2007, 129, 823–837. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Johnson, D.S.; Mortazavi, A.; Myers, R.M.; Wold, B. Genome-wide mapping of in vivo protein-DNA interactions. Science 2007, 316, 1497–1502. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- O’Geen, H.; Echipare, L.; Farnham, P.J. Using ChIP-seq technology to generate high-resolution profiles of histone modifications. Methods Mol. Biol. 2011, 791, 265–286. [Google Scholar] [CrossRef] [Green Version]
- Raha, D.; Hong, M.; Snyder, M. ChIP-Seq: A method for global identification of regulatory elements in the genome. Curr. Protoc. Mol. Biol. 2010, 21, 21.19.1–21.19.14. [Google Scholar] [CrossRef]
- Cui, K.; Zhao, K. Genome-wide approaches to determining nucleosome occupancy in metazoans using MNase-Seq. Methods Mol. Biol. 2012, 833, 413–419. [Google Scholar] [CrossRef] [Green Version]
- Schones, D.E.; Cui, K.; Cuddapah, S.; Roh, T.Y.; Barski, A.; Wang, Z.; Wei, G.; Zhao, K. Dynamic regulation of nucleosome positioning in the human genome. Cell 2008, 132, 887–898. [Google Scholar] [CrossRef] [Green Version]
- Buenrostro, J.D.; Wu, B.; Litzenburger, U.M.; Ruff, D.; Gonzales, M.L.; Snyder, M.P.; Chang, H.Y.; Greenleaf, W.J. Single-cell chromatin accessibility reveals principles of regulatory variation. Nature 2015, 523, 486–490. [Google Scholar] [CrossRef]
- Cusanovich, D.A.; Daza, R.; Adey, A.; Pliner, H.A.; Christiansen, L.; Gunderson, K.L.; Steemers, F.J.; Trapnell, C.; Shendure, J. Multiplex single cell profiling of chromatin accessibility by combinatorial cellular indexing. Science 2015, 348, 910–914. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mezger, A.; Klemm, S.; Mann, I.; Brower, K.; Mir, A.; Bostick, M.; Farmer, A.; Fordyce, P.; Linnarsson, S.; Greenleaf, W. High-throughput chromatin accessibility profiling at single-cell resolution. Nat. Commun. 2018, 9, 3647. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, K.; Hocker, J.D.; Miller, M.; Hou, X.; Chiou, J.; Poirion, O.B.; Qiu, Y.; Li, Y.E.; Gaulton, K.J.; Wang, A.; et al. A single-cell atlas of chromatin accessibility in the human genome. Cell 2021, 184, 5985–6001.e19. [Google Scholar] [CrossRef] [PubMed]
- Corces, M.R.; Granja, J.M.; Shams, S.; Louie, B.H.; Seoane, J.A.; Zhou, W.; Silva, T.C.; Groeneveld, C.; Wong, C.K.; Cho, S.W.; et al. The chromatin accessibility landscape of primary human cancers. Science 2018, 362, eaav1898. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cusanovich, D.A.; Hill, A.J.; Aghamirzaie, D.; Daza, R.M.; Pliner, H.A.; Berletch, J.B.; Filippova, G.N.; Huang, X.; Christiansen, L.; De Witt, W.S.; et al. A Single-Cell Atlas of In Vivo Mammalian Chromatin Accessibility. Cell 2018, 174, 1309–1324.e18. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ranzoni, A.M.; Tangherloni, A.; Berest, I.; Riva, S.G.; Myers, B.; Strzelecka, P.M.; Xu, J.; Panada, E.; Mohorianu, I.; Zaugg, J.B.; et al. Integrative Single-Cell RNA-Seq and ATAC-Seq Analysis of Human Developmental Hematopoiesis. Cell Stem Cell 2021, 28, 472–487.e7. [Google Scholar] [CrossRef]
- Dekker, J.; Marti-Renom, M.A.; Mirny, L.A. Exploring the three-dimensional organization of genomes: Interpreting chromatin interaction data. Nat. Rev. Genet. 2013, 14, 390–403. [Google Scholar] [CrossRef] [Green Version]
- Dixon, J.R.; Selvaraj, S.; Yue, F.; Kim, A.; Li, Y.; Shen, Y.; Hu, M.; Liu, J.S.; Ren, B. Topological domains in mammalian genomes identified by analysis of chromatin interactions. Nature 2012, 485, 376–380. [Google Scholar] [CrossRef] [Green Version]
- Furlan-Magaril, M.; Várnai, C.; Nagano, T.; Fraser, P. 3D genome architecture from populations to single cells. Curr. Opin. Genet. Dev. 2015, 31, 36–41. [Google Scholar] [CrossRef]
- Sexton, T.; Yaffe, E.; Kenigsberg, E.; Bantignies, F.; Leblanc, B.; Hoichman, M.; Parrinello, H.; Tanay, A.; Cavalli, G. Three-dimensional folding and functional organization principles of the Drosophila genome. Cell 2012, 148, 458–472. [Google Scholar] [CrossRef] [Green Version]
- Nagano, T.; Lubling, Y.; Stevens, T.J.; Schoenfelder, S.; Yaffe, E.; Dean, W.; Laue, E.D.; Tanay, A.; Fraser, P. Single-cell Hi-C reveals cell-to-cell variability in chromosome structure. Nature 2013, 502, 59–64. [Google Scholar] [CrossRef] [Green Version]
- Nagano, T.; Lubling, Y.; Yaffe, E.; Wingett, S.W.; Dean, W.; Tanay, A.; Fraser, P. Single-cell Hi-C for genome-wide detection of chromatin interactions that occur simultaneously in a single cell. Nat. Protoc. 2015, 10, 1986–2003. [Google Scholar] [CrossRef] [PubMed]
- Nagano, T.; Lubling, Y.; Várnai, C.; Dudley, C.; Leung, W.; Baran, Y.; Mendelson Cohen, N.; Wingett, S.; Fraser, P.; Tanay, A. Cell-cycle dynamics of chromosomal organization at single-cell resolution. Nature 2017, 547, 61–67. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- van Berkum, N.L.; Lieberman-Aiden, E.; Williams, L.; Imakaev, M.; Gnirke, A.; Mirny, L.A.; Dekker, J.; Lander, E.S. Hi-C: A method to study the three-dimensional architecture of genomes. J. Vis. Exp. 2010, 6, 1869. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Netea, M.G.; Joosten, L.A.; Latz, E.; Mills, K.H.; Natoli, G.; Stunnenberg, H.G.; O’Neill, L.A.; Xavier, R.J. Trained immunity: A program of innate immune memory in health and disease. Science 2016, 352, aaf1098. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Domínguez-Andrés, J.; Joosten, L.A.; Netea, M.G. Induction of innate immune memory: The role of cellular metabolism. Curr. Opin. Immunol. 2019, 56, 10–16. [Google Scholar] [CrossRef] [PubMed]
- Fanucchi, S.; Domínguez-Andrés, J.; Joosten, L.A.B.; Netea, M.G.; Mhlanga, M.M. The Intersection of Epigenetics and Metabolism in Trained Immunity. Immunity 2021, 54, 32–43. [Google Scholar] [CrossRef]
- Divangahi, M.; Desjardins, D.; Nunes-Alves, C.; Remold, H.G.; Behar, S.M. Eicosanoid pathways regulate adaptive immunity to Mycobacterium tuberculosis. Nat. Immunol. 2010, 11, 751–758. [Google Scholar] [CrossRef] [Green Version]
- Calo, E.; Wysocka, J. Modification of enhancer chromatin: What, how, and why? Mol. Cell 2013, 49, 825–837. [Google Scholar] [CrossRef] [Green Version]
- An integrated encyclopedia of DNA elements in the human genome. Nature 2012, 489, 57–74. [CrossRef]
- Yao, J.; Chen, J.; Li, L.Y.; Wu, M. Epigenetic plasticity of enhancers in cancer. Transcription 2020, 11, 26–36. [Google Scholar] [CrossRef] [PubMed]
- Ren, B.; Yue, F. Transcriptional Enhancers: Bridging the Genome and Phenome. Cold Spring Harb. Symp. Quant. Biol. 2015, 80, 17–26. [Google Scholar] [CrossRef] [Green Version]
- Smith, E.; Shilatifard, A. Enhancer biology and enhanceropathies. Nat. Struct. Mol. Biol. 2014, 21, 210–219. [Google Scholar] [CrossRef]
- Fujisawa, T.; Filippakopoulos, P. Functions of bromodomain-containing proteins and their roles in homeostasis and cancer. Nat. Rev. Mol. Cell Biol. 2017, 18, 246–262. [Google Scholar] [CrossRef] [PubMed]
- Jang, M.K.; Mochizuki, K.; Zhou, M.; Jeong, H.S.; Brady, J.N.; Ozato, K. The bromodomain protein Brd4 is a positive regulatory component of P-TEFb and stimulates RNA polymerase II-dependent transcription. Mol. Cell 2005, 19, 523–534. [Google Scholar] [CrossRef] [PubMed]
- Yang, Z.; Yik, J.H.; Chen, R.; He, N.; Jang, M.K.; Ozato, K.; Zhou, Q. Recruitment of P-TEFb for stimulation of transcriptional elongation by the bromodomain protein Brd4. Mol. Cell 2005, 19, 535–545. [Google Scholar] [CrossRef]
- Tafessu, A.; Banaszynski, L.A. Establishment and function of chromatin modification at enhancers. Open Biol. 2020, 10, 200255. [Google Scholar] [CrossRef]
- Chen, L.; Ozato, K. Innate Immune Memory in Hematopoietic Stem/Progenitor Cells: Myeloid-Biased Differentiation and the Role of Interferon. Front. Immunol. 2021, 12, 621333. [Google Scholar] [CrossRef]
- Kaufmann, E.; Sanz, J.; Dunn, J.L.; Khan, N.; Mendonça, L.E.; Pacis, A.; Tzelepis, F.; Pernet, E.; Dumaine, A.; Grenier, J.C.; et al. BCG Educates Hematopoietic Stem Cells to Generate Protective Innate Immunity against Tuberculosis. Cell 2018, 172, 176–190.e119. [Google Scholar] [CrossRef] [Green Version]
- Khan, N.; Downey, J.; Sanz, J.; Kaufmann, E.; Blankenhaus, B.; Pacis, A.; Pernet, E.; Ahmed, E.; Cardoso, S.; Nijnik, A.; et al. M. tuberculosis Reprograms Hematopoietic Stem Cells to Limit Myelopoiesis and Impair Trained Immunity. Cell 2020, 183, 752–770.e722. [Google Scholar] [CrossRef]
- de Laval, B.; Maurizio, J.; Kandalla, P.K.; Brisou, G.; Simonnet, L.; Huber, C.; Gimenez, G.; Matcovitch-Natan, O.; Reinhardt, S.; David, E.; et al. C/EBPβ-Dependent Epigenetic Memory Induces Trained Immunity in Hematopoietic Stem Cells. Cell Stem Cell 2020, 26, 657–674.e658. [Google Scholar] [CrossRef] [PubMed]
- Kalafati, L.; Kourtzelis, I.; Schulte-Schrepping, J.; Li, X.; Hatzioannou, A.; Grinenko, T.; Hagag, E.; Sinha, A.; Has, C.; Dietz, S.; et al. Innate Immune Training of Granulopoiesis Promotes Anti-tumor Activity. Cell 2020, 183, 771–785.e712. [Google Scholar] [CrossRef] [PubMed]
- Larsen, S.B.; Cowley, C.J.; Sajjath, S.M.; Barrows, D.; Yang, Y.; Carroll, T.S.; Fuchs, E. Establishment, maintenance, and recall of inflammatory memory. Cell Stem Cell 2021, 28, 1758–1774.e8. [Google Scholar] [CrossRef] [PubMed]
- Behar, S.M.; Divangahi, M.; Remold, H.G. Evasion of innate immunity by Mycobacterium tuberculosis: Is death an exit strategy? Nat. Rev. Microbiol. 2010, 8, 668–674. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Divangahi, M.; Chen, M.; Gan, H.; Desjardins, D.; Hickman, T.T.; Lee, D.M.; Fortune, S.; Behar, S.M.; Remold, H.G. Mycobacterium tuberculosis evades macrophage defenses by inhibiting plasma membrane repair. Nat. Immunol. 2009, 10, 899–906. [Google Scholar] [CrossRef] [Green Version]
- Cirovic, B.; de Bree, L.C.J.; Groh, L.; Blok, B.A.; Chan, J.; van der Velden, W.; Bremmers, M.E.J.; van Crevel, R.; Händler, K.; Picelli, S.; et al. BCG Vaccination in Humans Elicits Trained Immunity via the Hematopoietic Progenitor Compartment. Cell Host Microbe 2020, 28, 322–334.e5. [Google Scholar] [CrossRef]
- Das, B.; Kashino, S.S.; Pulu, I.; Kalita, D.; Swami, V.; Yeger, H.; Felsher, D.W.; Campos-Neto, A. CD271(+) bone marrow mesenchymal stem cells may provide a niche for dormant Mycobacterium tuberculosis. Sci. Transl. Med. 2013, 5, 170ra113. [Google Scholar] [CrossRef] [Green Version]
- Baldridge, M.T.; King, K.Y.; Boles, N.C.; Weksberg, D.C.; Goodell, M.A. Quiescent haematopoietic stem cells are activated by IFN-gamma in response to chronic infection. Nature 2010, 465, 793–797. [Google Scholar] [CrossRef]
- Belyaev, N.N.; Brown, D.E.; Diaz, A.I.; Rae, A.; Jarra, W.; Thompson, J.; Langhorne, J.; Potocnik, A.J. Induction of an IL7-R(+)c-Kit(hi) myelolymphoid progenitor critically dependent on IFN-gamma signaling during acute malaria. Nat. Immunol. 2010, 11, 477–485. [Google Scholar] [CrossRef]
- Pietras, E.M.; Lakshminarasimhan, R.; Techner, J.M.; Fong, S.; Flach, J.; Binnewies, M.; Passegué, E. Re-entry into quiescence protects hematopoietic stem cells from the killing effect of chronic exposure to type I interferons. J. Exp. Med. 2014, 211, 245–262. [Google Scholar] [CrossRef]
- Smith, J.N.P.; Zhang, Y.; Li, J.J.; McCabe, A.; Jo, H.J.; Maloney, J.; MacNamara, K.C. Type I IFNs drive hematopoietic stem and progenitor cell collapse via impaired proliferation and increased RIPK1-dependent cell death during shock-like ehrlichial infection. PLoS Pathog. 2018, 14, e1007234. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alexander, M.P.; Fiering, S.N.; Ostroff, G.R.; Cramer, R.A.; Mullins, D.W. Beta-glucan-induced inflammatory monocytes mediate antitumor efficacy in the murine lung. Cancer Immunol. Immunother. 2018, 67, 1731–1742. [Google Scholar] [CrossRef] [PubMed]
- Cheung, N.K.; Modak, S.; Vickers, A.; Knuckles, B. Orally administered beta-glucans enhance anti-tumor effects of monoclonal antibodies. Cancer Immunol. Immunother. 2002, 51, 557–564. [Google Scholar] [CrossRef] [PubMed]
- Li, B.; Cai, Y.; Qi, C.; Hansen, R.; Ding, C.; Mitchell, T.C.; Yan, J. Orally administered particulate beta-glucan modulates tumor-capturing dendritic cells and improves antitumor T-cell responses in cancer. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2010, 16, 5153–5164. [Google Scholar] [CrossRef] [Green Version]
- Masuda, Y.; Nawa, D.; Nakayama, Y.; Konishi, M.; Nanba, H. Soluble β-glucan from Grifola frondosa induces tumor regression in synergy with TLR9 agonist via dendritic cell-mediated immunity. J. Leukoc Biol. 2015, 98, 1015–1025. [Google Scholar] [CrossRef] [PubMed]
- Xu, H.; Zou, S.; Xu, X.; Zhang, L. Anti-tumor effect of β-glucan from Lentinus edodes and the underlying mechanism. Sci. Rep. 2016, 6, 28802. [Google Scholar] [CrossRef] [Green Version]
- Nagai, Y.; Garrett, K.P.; Ohta, S.; Bahrun, U.; Kouro, T.; Akira, S.; Takatsu, K.; Kincade, P.W. Toll-like receptors on hematopoietic progenitor cells stimulate innate immune system replenishment. Immunity 2006, 24, 801–812. [Google Scholar] [CrossRef] [Green Version]
- Takizawa, H.; Fritsch, K.; Kovtonyuk, L.V.; Saito, Y.; Yakkala, C.; Jacobs, K.; Ahuja, A.K.; Lopes, M.; Hausmann, A.; Hardt, W.D.; et al. Pathogen-Induced TLR4-TRIF Innate Immune Signaling in Hematopoietic Stem Cells Promotes Proliferation but Reduces Competitive Fitness. Cell Stem Cell 2017, 21, 225–240.e5. [Google Scholar] [CrossRef]
- Trumpp, A.; Essers, M.; Wilson, A. Awakening dormant haematopoietic stem cells. Nat. Rev. Immunol. 2010, 10, 201–209. [Google Scholar] [CrossRef]
- Zhao, J.L.; Baltimore, D. Regulation of stress-induced hematopoiesis. Curr. Opin. Hematol. 2015, 22, 286–292. [Google Scholar] [CrossRef] [Green Version]
- Pietras, E.M.; Mirantes-Barbeito, C.; Fong, S.; Loeffler, D.; Kovtonyuk, L.V.; Zhang, S.; Lakshminarasimhan, R.; Chin, C.P.; Techner, J.M.; Will, B.; et al. Chronic interleukin-1 exposure drives haematopoietic stem cells towards precocious myeloid differentiation at the expense of self-renewal. Nat. Cell Biol. 2016, 18, 607–618. [Google Scholar] [CrossRef] [PubMed]
- Hérault, A.; Binnewies, M.; Leong, S.; Calero-Nieto, F.J.; Zhang, S.Y.; Kang, Y.A.; Wang, X.; Pietras, E.M.; Chu, S.H.; Barry-Holson, K.; et al. Myeloid progenitor cluster formation drives emergency and leukaemic myelopoiesis. Nature 2017, 544, 53–58. [Google Scholar] [CrossRef] [PubMed]
- Manz, M.G.; Boettcher, S. Emergency granulopoiesis. Nat. Rev. Immunol. 2014, 14, 302–314. [Google Scholar] [CrossRef] [PubMed]
- Rosenbauer, F.; Tenen, D.G. Transcription factors in myeloid development: Balancing differentiation with transformation. Nat. Rev. Immunol. 2007, 7, 105–117. [Google Scholar] [CrossRef]
- Issa, J.P. Aging and epigenetic drift: A vicious cycle. J. Clin. Investig. 2014, 124, 24–29. [Google Scholar] [CrossRef] [Green Version]
- López-Otín, C.; Blasco, M.A.; Partridge, L.; Serrano, M.; Kroemer, G. The hallmarks of aging. Cell 2013, 153, 1194–1217. [Google Scholar] [CrossRef] [Green Version]
- Adelman, E.R.; Huang, H.T.; Roisman, A.; Olsson, A.; Colaprico, A.; Qin, T.; Lindsley, R.C.; Bejar, R.; Salomonis, N.; Grimes, H.L.; et al. Aging Human Hematopoietic Stem Cells Manifest Profound Epigenetic Reprogramming of Enhancers That May Predispose to Leukemia. Cancer Discov. 2019, 9, 1080–1101. [Google Scholar] [CrossRef]
- Beerman, I.; Bock, C.; Garrison, B.S.; Smith, Z.D.; Gu, H.; Meissner, A.; Rossi, D.J. Proliferation-dependent alterations of the DNA methylation landscape underlie hematopoietic stem cell aging. Cell Stem Cell 2013, 12, 413–425. [Google Scholar] [CrossRef] [Green Version]
- Sun, D.; Luo, M.; Jeong, M.; Rodriguez, B.; Xia, Z.; Hannah, R.; Wang, H.; Le, T.; Faull, K.F.; Chen, R.; et al. Epigenomic profiling of young and aged HSCs reveals concerted changes during aging that reinforce self-renewal. Cell Stem Cell 2014, 14, 673–688. [Google Scholar] [CrossRef] [Green Version]
- Corces, M.R.; Buenrostro, J.D.; Wu, B.; Greenside, P.G.; Chan, S.M.; Koenig, J.L.; Snyder, M.P.; Pritchard, J.K.; Kundaje, A.; Greenleaf, W.J.; et al. Lineage-specific and single-cell chromatin accessibility charts human hematopoiesis and leukemia evolution. Nat. Genet. 2016, 48, 1193–1203. [Google Scholar] [CrossRef] [Green Version]
- Ucar, D.; Márquez, E.J.; Chung, C.H.; Marches, R.; Rossi, R.J.; Uyar, A.; Wu, T.C.; George, J.; Stitzel, M.L.; Palucka, A.K.; et al. The chromatin accessibility signature of human immune aging stems from CD8(+) T cells. J. Exp. Med. 2017, 214, 3123–3144. [Google Scholar] [CrossRef] [PubMed]
- Itokawa, N.; Oshima, M.; Koide, S.; Takayama, N.; Kuribayashi, W.; Nakajima-Takagi, Y.; Aoyama, K.; Yamazaki, S.; Yamaguchi, K.; Furukawa, Y.; et al. Epigenetic traits inscribed in chromatin accessibility in aged hematopoietic stem cells. Nat. Commun. 2022, 13, 2691. [Google Scholar] [CrossRef] [PubMed]
- Hayes, J.D.; Dinkova-Kostova, A.T. The Nrf2 regulatory network provides an interface between redox and intermediary metabolism. Trends Biochem. Sci. 2014, 39, 199–218. [Google Scholar] [CrossRef] [PubMed]
- Salas, A.; Hernandez-Rocha, C.; Duijvestein, M.; Faubion, W.; McGovern, D.; Vermeire, S.; Vetrano, S.; Vande Casteele, N. JAK-STAT pathway targeting for the treatment of inflammatory bowel disease. Nat. Rev. Gastroenterol. Hepatol. 2020, 17, 323–337. [Google Scholar] [CrossRef] [PubMed]
- Seong, K.H.; Maekawa, T.; Ishii, S. Inheritance and memory of stress-induced epigenome change: Roles played by the ATF-2 family of transcription factors. Genes Cells 2012, 17, 249–263. [Google Scholar] [CrossRef] [Green Version]
- Fanucchi, S.; Mhlanga, M.M. Lnc-ing Trained Immunity to Chromatin Architecture. Front. Cell Dev. Biol. 2019, 7, 2. [Google Scholar] [CrossRef]
- Kim, M.Y.; Lee, J.E.; Kim, L.K.; Kim, T. Epigenetic memory in gene regulation and immune response. BMB Rep. 2019, 52, 127–132. [Google Scholar] [CrossRef] [Green Version]
- van der Heijden, C.; Noz, M.P.; Joosten, L.A.B.; Netea, M.G.; Riksen, N.P.; Keating, S.T. Epigenetics and Trained Immunity. Antioxid. Redox Signal. 2018, 29, 1023–1040. [Google Scholar] [CrossRef]
- Zhang, Q.; Cao, X. Epigenetic Remodeling in Innate Immunity and Inflammation. Annu. Rev. Immunol. 2021, 39, 279–311. [Google Scholar] [CrossRef]
- van der Fits, L.; Mourits, S.; Voerman, J.S.; Kant, M.; Boon, L.; Laman, J.D.; Cornelissen, F.; Mus, A.M.; Florencia, E.; Prens, E.P.; et al. Imiquimod-induced psoriasis-like skin inflammation in mice is mediated via the IL-23/IL-17 axis. J. Immunol. 2009, 182, 5836–5845. [Google Scholar] [CrossRef]
- Gonzales, K.A.U.; Polak, L.; Matos, I.; Tierney, M.T.; Gola, A.; Wong, E.; Infarinato, N.R.; Nikolova, M.; Luo, S.; Liu, S.; et al. Stem cells expand potency and alter tissue fitness by accumulating diverse epigenetic memories. Science 2021, 374, eabh2444. [Google Scholar] [CrossRef] [PubMed]



Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Aoyama, K.; Itokawa, N.; Oshima, M.; Iwama, A. Epigenetic Memories in Hematopoietic Stem and Progenitor Cells. Cells 2022, 11, 2187. https://doi.org/10.3390/cells11142187
Aoyama K, Itokawa N, Oshima M, Iwama A. Epigenetic Memories in Hematopoietic Stem and Progenitor Cells. Cells. 2022; 11(14):2187. https://doi.org/10.3390/cells11142187
Chicago/Turabian StyleAoyama, Kazumasa, Naoki Itokawa, Motohiko Oshima, and Atsushi Iwama. 2022. "Epigenetic Memories in Hematopoietic Stem and Progenitor Cells" Cells 11, no. 14: 2187. https://doi.org/10.3390/cells11142187