
A Mechanistic Model for Cell Cycle Control in Which CDKs Act as Switches of Disordered Protein Phase Separation


Abstract
:
{kind=link}
{kind=link}
{kind=link}
1. What Dictates CDK Substrate Specificity?
2. Structural Effects of Protein Phosphorylation
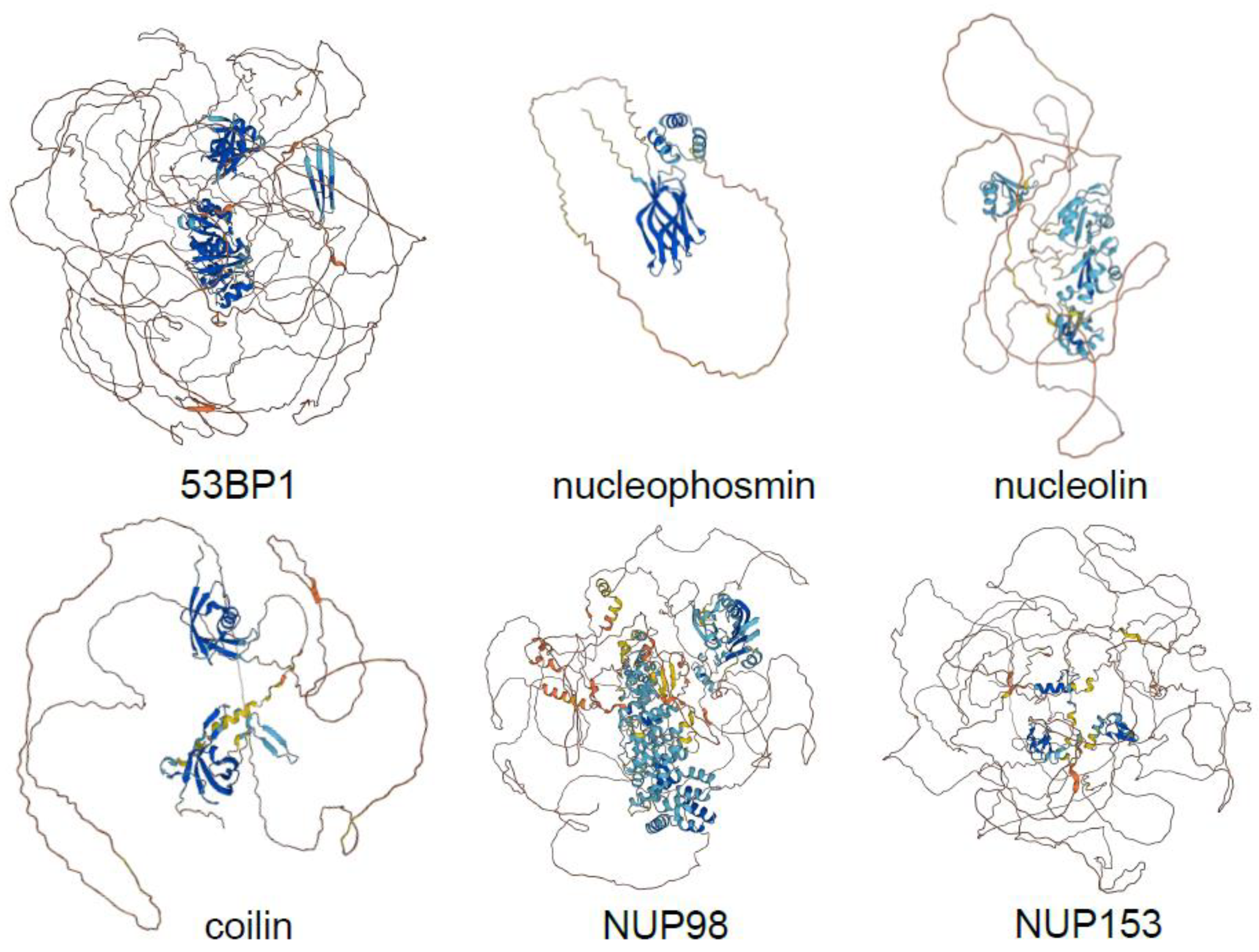
3. CDKs Mainly Phosphorylate Disordered Proteins of Membraneless Organelles
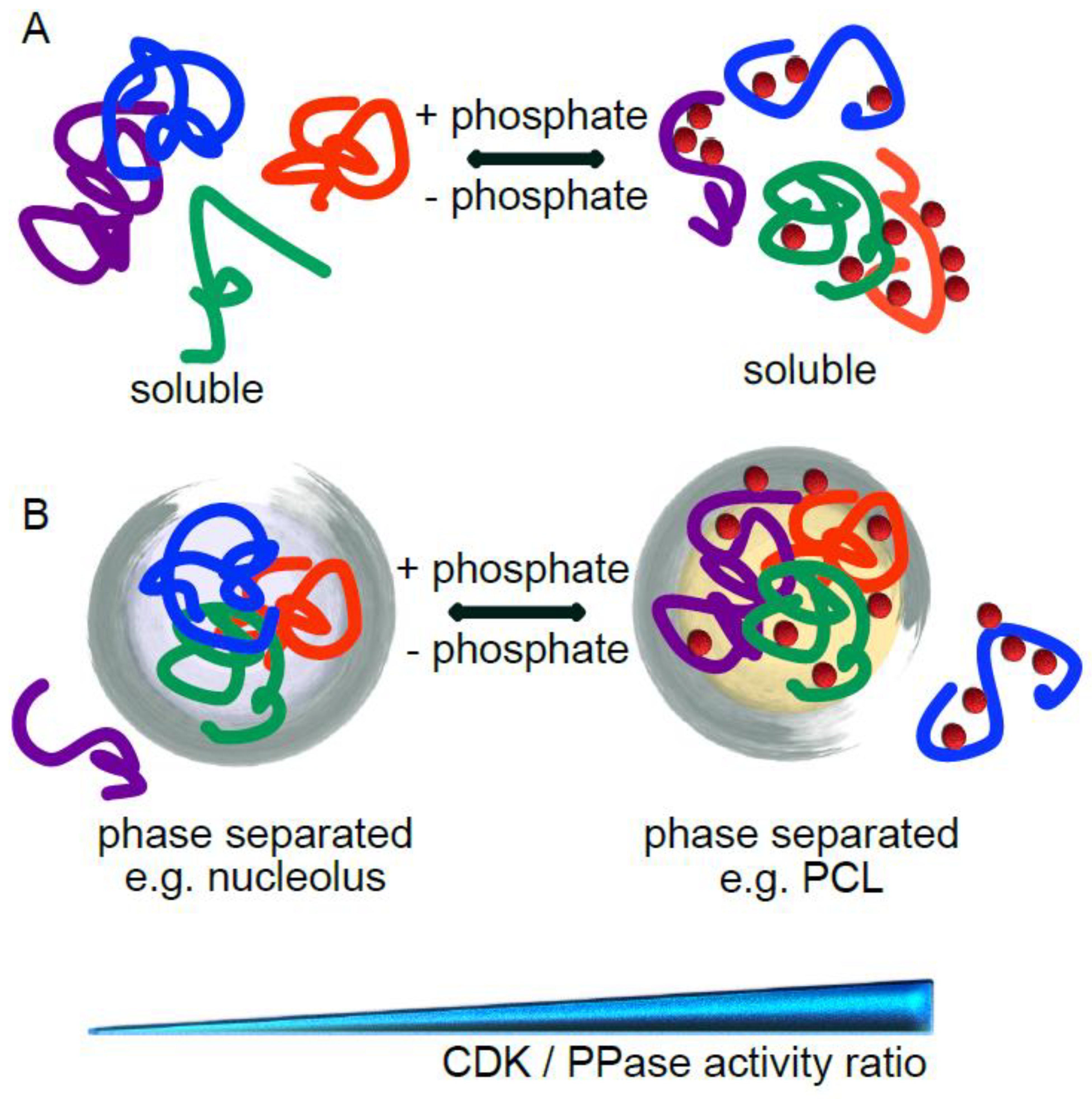
4. CDK-Mediated Phosphorylation Can Promote or Inhibit Phase Separation
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Michowski, W.; Chick, J.M.; Chu, C.; Kolodziejczyk, A.; Wang, Y.; Suski, J.M.; Abraham, B.; Anders, L.; Day, D.; Dunkl, L.M.; et al. Cdk1 Controls Global Epigenetic Landscape in Embryonic Stem Cells. Mol. Cell 2020, 78, 459–476.e13. [Google Scholar] [CrossRef] [PubMed]
- Holt, L.J.; Tuch, B.B.; Villen, J.; Johnson, A.D.; Gygi, S.P.; Morgan, D.O. Global Analysis of Cdk1 Substrate Phosphorylation Sites Provides Insights into Evolution. Science 2009, 325, 1682–1686. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chi, Y.; Carter, J.H.; Swanger, J.; Mazin, A.V.; Moritz, R.L.; Clurman, B.E. A Novel Landscape of Nuclear Human CDK2 Substrates Revealed by in Situ Phosphorylation. Sci. Adv. 2020, 6, eaaz9899. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Blethrow, J.D.; Glavy, J.S.; Morgan, D.O.; Shokat, K.M. Covalent Capture of Kinase-Specific Phosphopeptides Reveals Cdk1-Cyclin B Substrates. Proc. Natl. Acad. Sci. USA 2008, 105, 1442–1447. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Holmes, J.K.; Solomon, M.J. A Predictive Scale for Evaluating Cyclin-Dependent Kinase Substrates. A Comparison of P34cdc2 and P33cdk2. J. Biol. Chem. 1996, 271, 25240–25246. [Google Scholar] [CrossRef] [Green Version]
- Minshull, J.; Golsteyn, R.; Hill, C.S.; Hunt, T. The A- and B-Type Cyclin Associated Cdc2 Kinases in Xenopus Turn on and off at Different Times in the Cell Cycle. Embo. J. 1990, 9, 2865–2875. [Google Scholar] [CrossRef]
- Songyang, Z.; Blechner, S.; Hoagland, N.; Hoekstra, M.F.; Piwnica-Worms, H.; Cantley, L.C. Use of an Oriented Peptide Library to Determine the Optimal Substrates of Protein Kinases. Curr. Biol. 1994, 4, 973–982. [Google Scholar] [CrossRef]
- Brown, N.R.; Korolchuk, S.; Martin, M.P.; Stanley, W.A.; Moukhametzianov, R.; Noble, M.E.M.; Endicott, J.A. CDK1 Structures Reveal Conserved and Unique Features of the Essential Cell Cycle CDK. Nat. Commun. 2015, 6, 6769. [Google Scholar] [CrossRef]
- Chou, Y.H.; Ngai, K.L.; Goldman, R. The Regulation of Intermediate Filament Reorganization in Mitosis. P34cdc2 Phosphorylates Vimentin at a Unique N-Terminal Site. J. Biol. Chem. 1991, 266, 7325–7328. [Google Scholar] [CrossRef]
- Leng, X.; Noble, M.; Adams, P.D.; Qin, J.; Harper, J.W. Reversal of Growth Suppression by P107 via Direct Phosphorylation by Cyclin D1/Cyclin-Dependent Kinase 4. Mol. Cell. Biol. 2002, 22, 2242–2254. [Google Scholar] [CrossRef] [Green Version]
- Harvey, S.L.; Charlet, A.; Haas, W.; Gygi, S.P.; Kellogg, D.R. Cdk1-Dependent Regulation of the Mitotic Inhibitor Wee1. Cell 2005, 122, 407–420. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McCusker, D.; Denison, C.; Anderson, S.; Egelhofer, T.A.; Yates, J.R.; Gygi, S.P.; Kellogg, D.R. Cdk1 Coordinates Cell-Surface Growth with the Cell Cycle. Nat. Cell Biol. 2007, 9, 506–515. [Google Scholar] [CrossRef] [PubMed]
- Altelaar, M.; Valverde, J.; Dubra, G.; den Toorn, H.W.P.V.; van Mierlo, G.; Vermeulen, M.; Heck, A.; Elena-Real, C.; Fournet, A.; Ghoul, E.A.; et al. A CDK-Mediated Phosphorylation Switch of Disordered Protein Condensation. Available online: https://assets.researchsquare.com/files/rs-1370895/v1_covered.pdf?c=1645716495 (accessed on 8 June 2022).
- Suzuki, K.; Sako, K.; Akiyama, K.; Isoda, M.; Senoo, C.; Nakajo, N.; Sagata, N. Identification of Non-Ser/Thr-Pro Consensus Motifs for Cdk1 and Their Roles in Mitotic Regulation of C2H2 Zinc Finger Proteins and Ect2. Sci. Rep. 2015, 5, 7929. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Koivomagi, M.; Valk, E.; Venta, R.; Iofik, A.; Lepiku, M.; Morgan, D.O.; Loog, M. Dynamics of Cdk1 Substrate Specificity during the Cell Cycle. Mol. Cell 2011, 42, 610–623. [Google Scholar] [CrossRef]
- Kõivomägi, M.; Ord, M.; Iofik, A.; Valk, E.; Venta, R.; Faustova, I.; Kivi, R.; Balog, E.R.M.; Rubin, S.M.; Loog, M. Multisite Phosphorylation Networks as Signal Processors for Cdk1. Nat. Struct. Mol. Biol. 2013, 20, 1415–1424. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Örd, M.; Möll, K.; Agerova, A.; Kivi, R.; Faustova, I.; Venta, R.; Valk, E.; Loog, M. Multisite Phosphorylation Code of CDK. Nat. Struct. Mol. Biol. 2019, 26, 649–658. [Google Scholar] [CrossRef]
- Fisher, D.; Krasinska, L.; Coudreuse, D.; Novak, B. Phosphorylation Network Dynamics in Control of Cell Cycle Transitions. J. Cell. Sci. 2012, 125, 4703–4711. [Google Scholar] [CrossRef] [Green Version]
- Fisher, D.; Krasinska, L. Explaining Redundancy in CDK-Mediated Control of the Cell Cycle: Unifying the Continuum and Quantitative Models. Cells 2022, 11, 2019. [Google Scholar] [CrossRef]
- Errico, A.; Deshmukh, K.; Tanaka, Y.; Pozniakovsky, A.; Hunt, T. Identification of Substrates for Cyclin Dependent Kinases. Adv. Enzyme Regul. 2010, 50, 375–399. [Google Scholar] [CrossRef]
- Pagliuca, F.W.; Collins, M.O.; Lichawska, A.; Zegerman, P.; Choudhary, J.S.; Pines, J. Quantitative Proteomics Reveals the Basis for the Biochemical Specificity of the Cell-Cycle Machinery. Mol. Cell 2011, 43, 406–417. [Google Scholar] [CrossRef] [Green Version]
- Coudreuse, D.; Nurse, P. Driving the Cell Cycle with a Minimal CDK Control Network. Nature 2010, 468, 1074–1079. [Google Scholar] [CrossRef]
- Krasinska, L.; Domingo-Sananes, M.R.; Kapuy, O.; Parisis, N.; Harker, B.; Moorhead, G.; Rossignol, M.; Novak, B.; Fisher, D. Protein Phosphatase 2A Controls the Order and Dynamics of Cell-Cycle Transitions. Mol. Cell 2011, 44, 437–450. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Santamaria, D.; Barriere, C.; Cerqueira, A.; Hunt, S.; Tardy, C.; Newton, K.; Caceres, J.F.; Dubus, P.; Malumbres, M.; Barbacid, M. Cdk1 Is Sufficient to Drive the Mammalian Cell Cycle. Nature 2007, 448, 811–815. [Google Scholar] [CrossRef] [PubMed]
- Krasinska, L.; Besnard, E.; Cot, E.; Dohet, C.; Mechali, M.; Lemaitre, J.M.; Fisher, D. Cdk1 and Cdk2 Activity Levels Determine the Efficiency of Replication Origin Firing in Xenopus. EMBO J. 2008, 27, 758–769. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Suski, J.M.; Ratnayeke, N.; Braun, M.; Zhang, T.; Strmiska, V.; Michowski, W.; Can, G.; Simoneau, A.; Snioch, K.; Cup, M.; et al. CDC7-Independent G1/S Transition Revealed by Targeted Protein Degradation. Nature 2022, 605, 357–365. [Google Scholar] [CrossRef]
- Hégarat, N.; Crncec, A.; Suarez Peredo Rodriguez, M.F.; Echegaray Iturra, F.; Gu, Y.; Busby, O.; Lang, P.F.; Barr, A.R.; Bakal, C.; Kanemaki, M.T.; et al. Cyclin A Triggers Mitosis Either via the Greatwall Kinase Pathway or Cyclin B. EMBO J. 2020, 39, e104419. [Google Scholar] [CrossRef]
- Lau, H.W.; Ma, H.T.; Yeung, T.K.; Tam, M.Y.; Zheng, D.; Chu, S.K.; Poon, R.Y.C. Quantitative Differences between Cyclin-Dependent Kinases Underlie the Unique Functions of CDK1 in Human Cells. Cell. Rep. 2021, 37, 109808. [Google Scholar] [CrossRef] [PubMed]
- Moses, A.M.; Hériché, J.-K.; Durbin, R. Clustering of Phosphorylation Site Recognition Motifs Can Be Exploited to Predict the Targets of Cyclin-Dependent Kinase. Genome Biol. 2007, 8, R23. [Google Scholar] [CrossRef] [Green Version]
- Brown, C.J.; Johnson, A.K.; Dunker, A.K.; Daughdrill, G.W. Evolution and Disorder. Curr. Opin. Struct. Biol. 2011, 21, 441–446. [Google Scholar] [CrossRef]
- Schaefer, C.; Schlessinger, A.; Rost, B. Protein Secondary Structure Appears to Be Robust under in Silico Evolution While Protein Disorder Appears Not to Be. Bioinformatics 2010, 26, 625–631. [Google Scholar] [CrossRef]
- Tan, C.S.H.; Jørgensen, C.; Linding, R. Roles of “Junk Phosphorylation” in Modulating Biomolecular Association of Phosphorylated Proteins? Cell Cycle 2010, 9, 1276–1280. [Google Scholar] [CrossRef] [PubMed]
- Patil, A.; Nakamura, H. Disordered Domains and High Surface Charge Confer Hubs with the Ability to Interact with Multiple Proteins in Interaction Networks. FEBS Lett. 2006, 580, 2041–2045. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mittag, T.; Kay, L.E.; Forman-Kay, J.D. Protein Dynamics and Conformational Disorder in Molecular Recognition. J. Mol. Recognit. 2010, 23, 105–116. [Google Scholar] [CrossRef] [PubMed]
- Huihui, J.; Ghosh, K. An Analytical Theory to Describe Sequence-Specific Inter-Residue Distance Profiles for Polyampholytes and Intrinsically Disordered Proteins. J. Chem. Phys.. 2020, 152, 161102. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huihui, J.; Ghosh, K. Intrachain Interaction Topology Can Identify Functionally Similar Intrinsically Disordered Proteins. Biophys. J. 2021, 120, 1860–1868. [Google Scholar] [CrossRef] [PubMed]
- Das, R.K.; Pappu, R.V. Conformations of Intrinsically Disordered Proteins Are Influenced by Linear Sequence Distributions of Oppositely Charged Residues. Proc. Natl. Acad. Sci. USA 2013, 110, 13392–13397. [Google Scholar] [CrossRef] [Green Version]
- Martin, E.W.; Holehouse, A.S.; Grace, C.R.; Hughes, A.; Pappu, R.V.; Mittag, T. Sequence Determinants of the Conformational Properties of an Intrinsically Disordered Protein Prior to and upon Multisite Phosphorylation. J. Am. Chem. Soc. 2016, 138, 15323–15335. [Google Scholar] [CrossRef] [Green Version]
- Bah, A.; Forman-Kay, J.D. Modulation of Intrinsically Disordered Protein Function by Post-Translational Modifications. J. Biol. Chem. 2016, 291, 6696–6705. [Google Scholar] [CrossRef] [Green Version]
- Rieloff, E.; Skepö, M. The Effect of Multisite Phosphorylation on the Conformational Properties of Intrinsically Disordered Proteins. Int. J. Mol. Sci. 2021, 22, 11058. [Google Scholar] [CrossRef]
- Schwalbe, M.; Kadavath, H.; Biernat, J.; Ozenne, V.; Blackledge, M.; Mandelkow, E.; Zweckstetter, M. Structural Impact of Tau Phosphorylation at Threonine 231. Structure 2015, 23, 1448–1458. [Google Scholar] [CrossRef] [Green Version]
- Bah, A.; Vernon, R.M.; Siddiqui, Z.; Krzeminski, M.; Muhandiram, R.; Zhao, C.; Sonenberg, N.; Kay, L.E.; Forman-Kay, J.D. Folding of an Intrinsically Disordered Protein by Phosphorylation as a Regulatory Switch. Nature 2015, 519, 106–109. [Google Scholar] [CrossRef] [PubMed]
- Uversky, V.N. Unusual Biophysics of Intrinsically Disordered Proteins. Biochim. Biophys. Acta 2013, 1834, 932–951. [Google Scholar] [CrossRef]
- Bozoky, Z.; Krzeminski, M.; Chong, P.A.; Forman-Kay, J.D. Structural Changes of CFTR R Region upon Phosphorylation: A Plastic Platform for Intramolecular and Intermolecular Interactions. FEBS J. 2013, 280, 4407–4416. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Della Sala, A.; Prono, G.; Hirsch, E.; Ghigo, A. Role of Protein Kinase A-Mediated Phosphorylation in CFTR Channel Activity Regulation. Front. Physiol. 2021, 12, 690247. [Google Scholar] [CrossRef] [PubMed]
- Liu, F.; Zhang, Z.; Csanády, L.; Gadsby, D.C.; Chen, J. Molecular Structure of the Human CFTR Ion Channel. Cell 2017, 169, 85–95.e8. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Z.; Liu, F.; Chen, J. Conformational Changes of CFTR upon Phosphorylation and ATP Binding. Cell 2017, 170, 483–491.e8. [Google Scholar] [CrossRef] [Green Version]
- Gao, J.; Xu, D. Correlation between Posttranslational Modification and Intrinsic Disorder in Protein. Pac. Symp. Biocomput. 2012, 94–103. [Google Scholar]
- Iakoucheva, L.M.; Radivojac, P.; Brown, C.J.; O’Connor, T.R.; Sikes, J.G.; Obradovic, Z.; Dunker, A.K. The Importance of Intrinsic Disorder for Protein Phosphorylation. Nucleic Acids Res. 2004, 32, 1037–1049. [Google Scholar] [CrossRef] [Green Version]
- Uversky, V.N. Intrinsically Disordered Proteins in Overcrowded Milieu: Membrane-Less Organelles, Phase Separation, and Intrinsic Disorder. Curr. Opin. Struct. Biol. 2017, 44, 18–30. [Google Scholar] [CrossRef]
- Banani, S.F.; Lee, H.O.; Hyman, A.A.; Rosen, M.K. Biomolecular Condensates: Organizers of Cellular Biochemistry. Nat. Rev. Mol. Cell Biol. 2017, 18, 285–298. [Google Scholar] [CrossRef]
- Shin, Y.; Brangwynne, C.P. Liquid Phase Condensation in Cell Physiology and Disease. Science 2017, 357, eaaf4382. [Google Scholar] [CrossRef] [Green Version]
- Alberti, S.; Hyman, A.A. Biomolecular Condensates at the Nexus of Cellular Stress, Protein Aggregation Disease and Ageing. Nat. Rev. Mol. Cell Biol. 2021, 22, 196–213. [Google Scholar] [CrossRef] [PubMed]
- Choi, J.-M.; Holehouse, A.S.; Pappu, R.V. Physical Principles Underlying the Complex Biology of Intracellular Phase Transitions. Annu. Rev. Biophys. 2020, 49, 107–133. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Misteli, T.; Cáceres, J.F.; Clement, J.Q.; Krainer, A.R.; Wilkinson, M.F.; Spector, D.L. Serine Phosphorylation of SR Proteins Is Required for Their Recruitment to Sites of Transcription in Vivo. J. Cell Biol. 1998, 143, 297–307. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kedersha, N.L.; Gupta, M.; Li, W.; Miller, I.; Anderson, P. RNA-Binding Proteins TIA-1 and TIAR Link the Phosphorylation of EIF-2 Alpha to the Assembly of Mammalian Stress Granules. J. Cell Biol. 1999, 147, 1431–1442. [Google Scholar] [CrossRef] [PubMed]
- Petri, S.; Grimmler, M.; Over, S.; Fischer, U.; Gruss, O.J. Dephosphorylation of Survival Motor Neurons (SMN) by PPM1G/PP2Cgamma Governs Cajal Body Localization and Stability of the SMN Complex. J. Cell Biol. 2007, 179, 451–465. [Google Scholar] [CrossRef] [Green Version]
- Han, T.W.; Kato, M.; Xie, S.; Wu, L.C.; Mirzaei, H.; Pei, J.; Chen, M.; Xie, Y.; Allen, J.; Xiao, G.; et al. Cell-Free Formation of RNA Granules: Bound RNAs Identify Features and Components of Cellular Assemblies. Cell 2012, 149, 768–779. [Google Scholar] [CrossRef] [Green Version]
- Wippich, F.; Bodenmiller, B.; Trajkovska, M.G.; Wanka, S.; Aebersold, R.; Pelkmans, L. Dual Specificity Kinase DYRK3 Couples Stress Granule Condensation/Dissolution to MTORC1 Signaling. Cell 2013, 152, 791–805. [Google Scholar] [CrossRef] [Green Version]
- Wang, J.T.; Smith, J.; Chen, B.-C.; Schmidt, H.; Rasoloson, D.; Paix, A.; Lambrus, B.G.; Calidas, D.; Betzig, E.; Seydoux, G. Regulation of RNA Granule Dynamics by Phosphorylation of Serine-Rich, Intrinsically Disordered Proteins in C. Elegans. Elife 2014, 3, e04591. [Google Scholar] [CrossRef]
- Rai, A.K.; Chen, J.-X.; Selbach, M.; Pelkmans, L. Kinase-Controlled Phase Transition of Membraneless Organelles in Mitosis. Nature 2018, 559, 211–216. [Google Scholar] [CrossRef] [Green Version]
- Frey, S.; Richter, R.P.; Görlich, D. FG-Rich Repeats of Nuclear Pore Proteins Form a Three-Dimensional Meshwork with Hydrogel-like Properties. Science 2006, 314, 815–817. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Celetti, G.; Paci, G.; Caria, J.; VanDelinder, V.; Bachand, G.; Lemke, E.A. The Liquid State of FG-Nucleoporins Mimics Permeability Barrier Properties of Nuclear Pore Complexes. J. Cell Biol. 2019, 219, e201907157. [Google Scholar] [CrossRef] [PubMed]
- Labokha, A.A.; Gradmann, S.; Frey, S.; Hülsmann, B.B.; Urlaub, H.; Baldus, M.; Görlich, D. Systematic Analysis of Barrier-Forming FG Hydrogels from Xenopus Nuclear Pore Complexes. EMBO J. 2013, 32, 204–218. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hulsmann, B.B.; Labokha, A.A.; Gorlich, D. The Permeability of Reconstituted Nuclear Pores Provides Direct Evidence for the Selective Phase Model. Cell 2012, 150, 738–751. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Laurell, E.; Beck, K.; Krupina, K.; Theerthagiri, G.; Bodenmiller, B.; Horvath, P.; Aebersold, R.; Antonin, W.; Kutay, U. Phosphorylation of Nup98 by Multiple Kinases Is Crucial for NPC Disassembly during Mitotic Entry. Cell 2011, 144, 539–550. [Google Scholar] [CrossRef] [Green Version]
- Yahya, G.; Pérez, A.P.; Mendoza, M.B.; Parisi, E.; Moreno, D.F.; Artés, M.H.; Gallego, C.; Aldea, M. Stress Granules Display Bistable Dynamics Modulated by Cdk. J. Cell Biol. 2021, 220, e202005102. [Google Scholar] [CrossRef]
- Protter, D.S.W.; Parker, R. Principles and Properties of Stress Granules. Trends Cell Biol. 2016, 26, 668–679. [Google Scholar] [CrossRef] [Green Version]
- Parker, M.W.; Bell, M.; Mir, M.; Kao, J.A.; Darzacq, X.; Botchan, M.R.; Berger, J.M. A New Class of Disordered Elements Controls DNA Replication through Initiator Self-Assembly. Elife 2019, 8, e48562. [Google Scholar] [CrossRef]
- Parker, M.W.; Kao, J.A.; Huang, A.; Berger, J.M.; Botchan, M.R. Molecular Determinants of Phase Separation for Drosophila DNA Replication Licensing Factors. Elife 2021, 10, e70535. [Google Scholar] [CrossRef]
- Hossain, M.; Bhalla, K.; Stillman, B. Multiple, Short Protein Binding Motifs in ORC1 and CDC6 Control the Initiation of DNA Replication. Mol. Cell 2021, 81, 1951–1969.e6. [Google Scholar] [CrossRef]
- Hendrickson, M.; Madine, M.; Dalton, S.; Gautier, J. Phosphorylation of MCM4 by Cdc2 Protein Kinase Inhibits the Activity of the Minichromosome Maintenance Complex. Proc. Natl. Acad. Sci. USA 1996, 93, 12223–12228. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Findeisen, M.; El-Denary, M.; Kapitza, T.; Graf, R.; Strausfeld, U. Cyclin A-Dependent Kinase Activity Affects Chromatin Binding of ORC, Cdc6, and MCM in Egg Extracts of Xenopus Laevis. Eur. J. Biochem. 1999, 264, 415–426. [Google Scholar] [CrossRef] [PubMed]
- Remus, D.; Blanchette, M.; Rio, D.C.; Botchan, M.R. CDK Phosphorylation Inhibits the DNA-Binding and ATP-Hydrolysis Activities of the Drosophila Origin Recognition Complex. J. Biol. Chem. 2005, 280, 39740–39751. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, S.; Bell, S.P. CDK Prevents Mcm2-7 Helicase Loading by Inhibiting Cdt1 Interaction with Orc6. Genes Dev. 2011, 25, 363–372. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sabari, B.R.; Dall’Agnese, A.; Boija, A.; Klein, I.A.; Coffey, E.L.; Shrinivas, K.; Abraham, B.J.; Hannett, N.M.; Zamudio, A.V.; Manteiga, J.C.; et al. Coactivator Condensation at Super-Enhancers Links Phase Separation and Gene Control. Science 2018, 361, eaar3958. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Remnant, L.; Kochanova, N.Y.; Reid, C.; Cisneros-Soberanis, F.; Earnshaw, W.C. The Intrinsically Disorderly Story of Ki-67. Open Biol. 2021, 11, 210120. [Google Scholar] [CrossRef]
- Andrés-Sánchez, N.; Fisher, D.; Krasinska, L. Physiological Functions and Roles in Cancer of the Proliferation Marker Ki-67. J. Cell Sci. 2022, 135, jcs258932. [Google Scholar] [CrossRef]
- Sobecki, M.; Mrouj, K.; Camasses, A.; Parisis, N.; Nicolas, E.; Llères, D.; Gerbe, F.; Prieto, S.; Krasinska, L.; David, A.; et al. The Cell Proliferation Antigen Ki-67 Organises Heterochromatin. Elife 2016, 5, e13722. [Google Scholar] [CrossRef]
- Booth, D.G.; Takagi, M.; Sanchez-Pulido, L.; Petfalski, E.; Vargiu, G.; Samejima, K.; Imamoto, N.; Ponting, C.P.; Tollervey, D.; Earnshaw, W.C.; et al. Ki-67 Is a PP1-Interacting Protein That Organises the Mitotic Chromosome Periphery. Elife 2014, 3, e01641. [Google Scholar] [CrossRef]
- Takagi, M.; Natsume, T.; Kanemaki, M.T.; Imamoto, N. Perichromosomal Protein Ki67 Supports Mitotic Chromosome Architecture. Genes Cells 2016, 21, 1113–1124. [Google Scholar] [CrossRef]
- Shin, Y.; Berry, J.; Pannucci, N.; Haataja, M.P.; Toettcher, J.E.; Brangwynne, C.P. Spatiotemporal Control of Intracellular Phase Transitions Using Light-Activated OptoDroplets. Cell 2017, 168, 159–171.e14. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Negi, S.S.; Olson, M.O.J. Effects of Interphase and Mitotic Phosphorylation on the Mobility and Location of Nucleolar Protein B23. J. Cell Sci. 2006, 119, 3676–3685. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yamazaki, H.; Takagi, M.; Kosako, H.; Hirano, T.; Yoshimura, S.H. Cell Cycle-Specific Phase Separation Regulated by Protein Charge Blockiness. Nat. Cell Biol. 2022, 24, 625–632. [Google Scholar] [CrossRef] [PubMed]
- Das, S.; Eisen, A.; Lin, Y.-H.; Chan, H.S. A Lattice Model of Charge-Pattern-Dependent Polyampholyte Phase Separation. J. Phys. Chem. B 2018, 122, 5418–5431. [Google Scholar] [CrossRef] [Green Version]
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Krasinska, L.; Fisher, D. A Mechanistic Model for Cell Cycle Control in Which CDKs Act as Switches of Disordered Protein Phase Separation. Cells 2022, 11, 2189. https://doi.org/10.3390/cells11142189
Krasinska L, Fisher D. A Mechanistic Model for Cell Cycle Control in Which CDKs Act as Switches of Disordered Protein Phase Separation. Cells. 2022; 11(14):2189. https://doi.org/10.3390/cells11142189
Chicago/Turabian StyleKrasinska, Liliana, and Daniel Fisher. 2022. "A Mechanistic Model for Cell Cycle Control in Which CDKs Act as Switches of Disordered Protein Phase Separation" Cells 11, no. 14: 2189. https://doi.org/10.3390/cells11142189