
CAR T-Cell Targeting of Macrophage Colony-Stimulating Factor Receptor



Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
2.1. Cell Lines
2.2. Human Samples
2.3. Retroviral Constructs
2.4. Transduction and Expansion of Human T-Cells
2.5. FACS Analysis
2.6. Enzyme-Linked Immunosorbent Assay
2.7. Western Blotting
2.8. Killing Assays
2.9. Statistical Analysis
3. Results
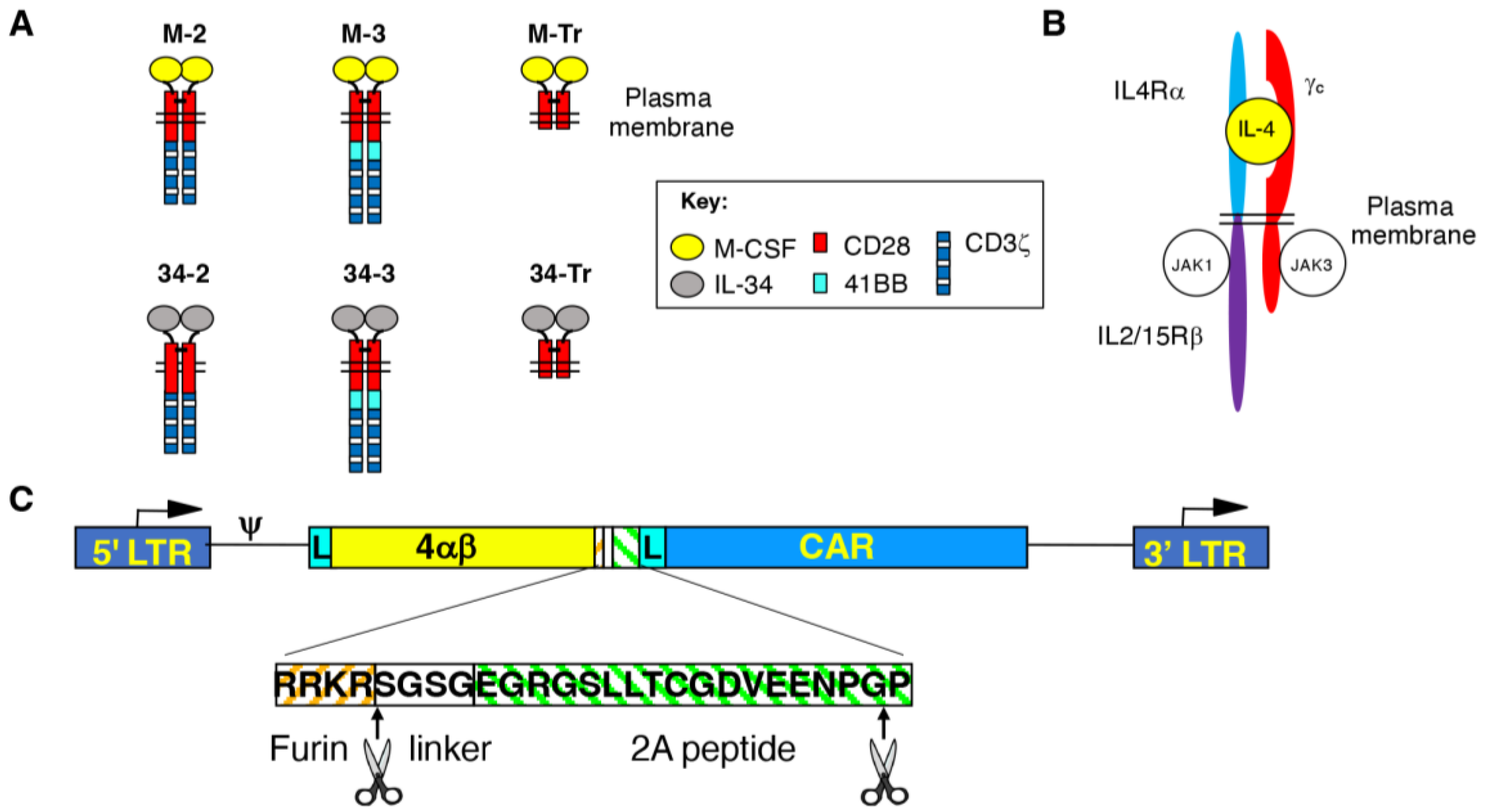
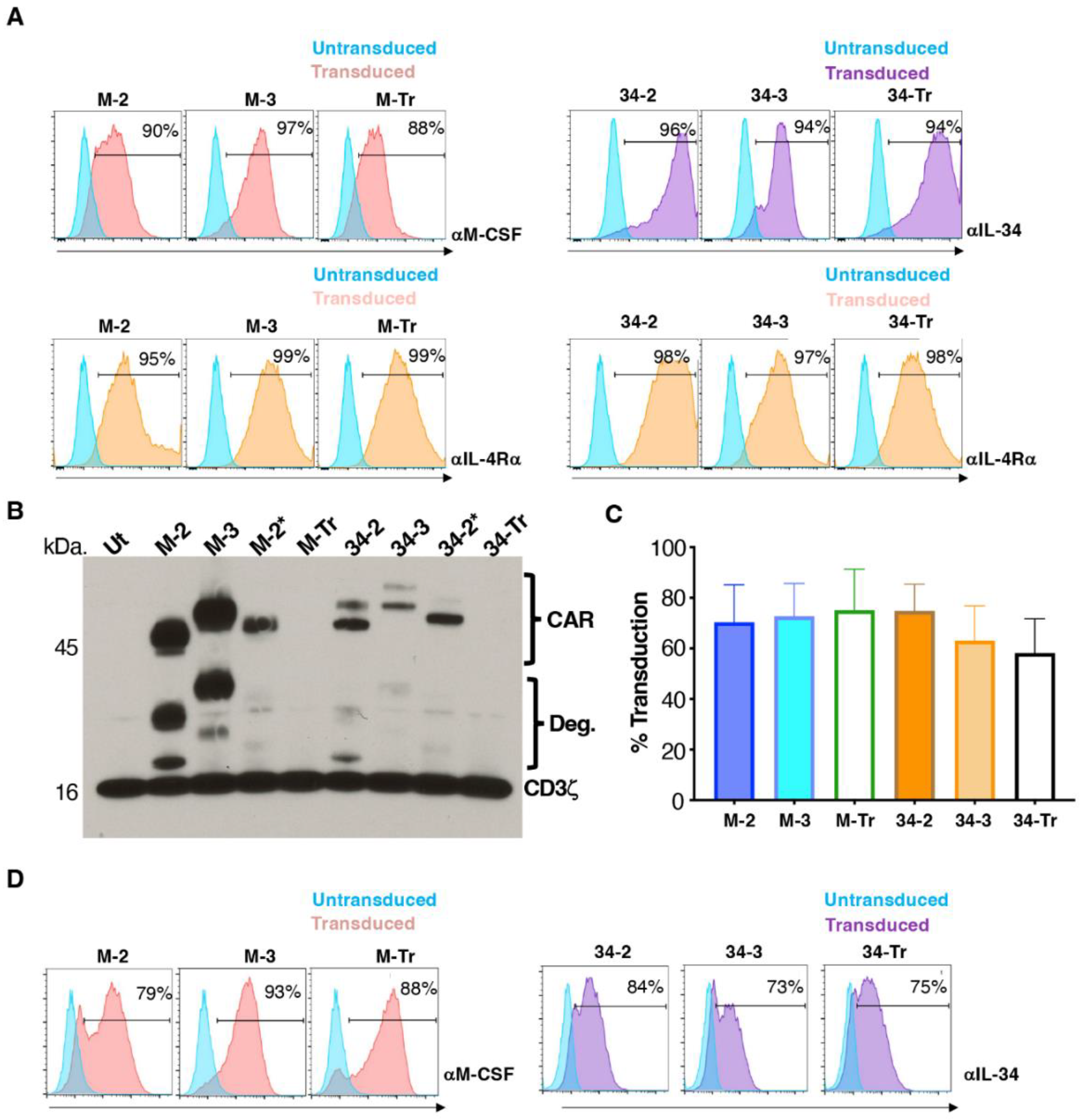
3.1. Engineering of CAR T-Cells Targeted against Macrophage Colony-Stimulating Factor Receptor
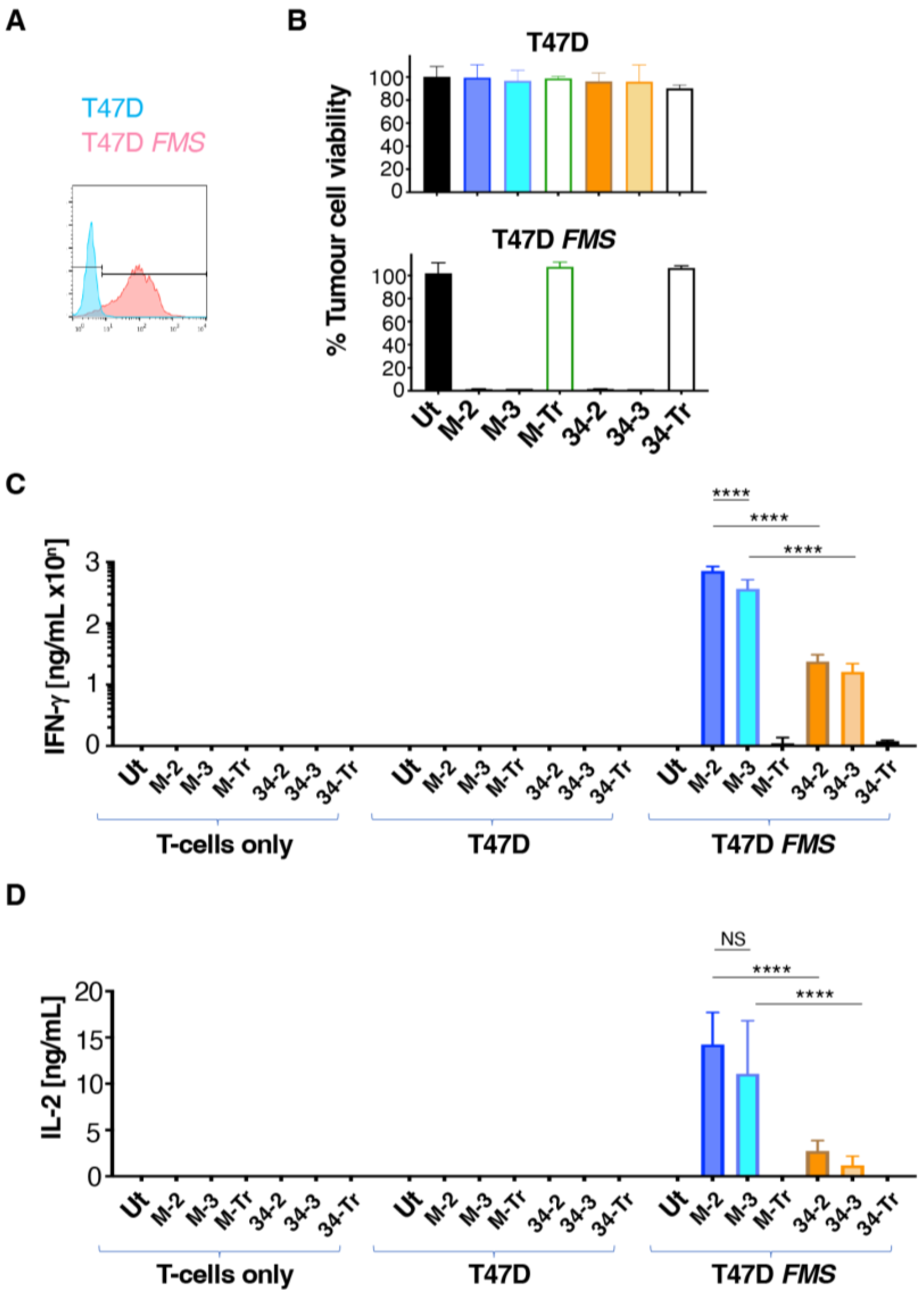
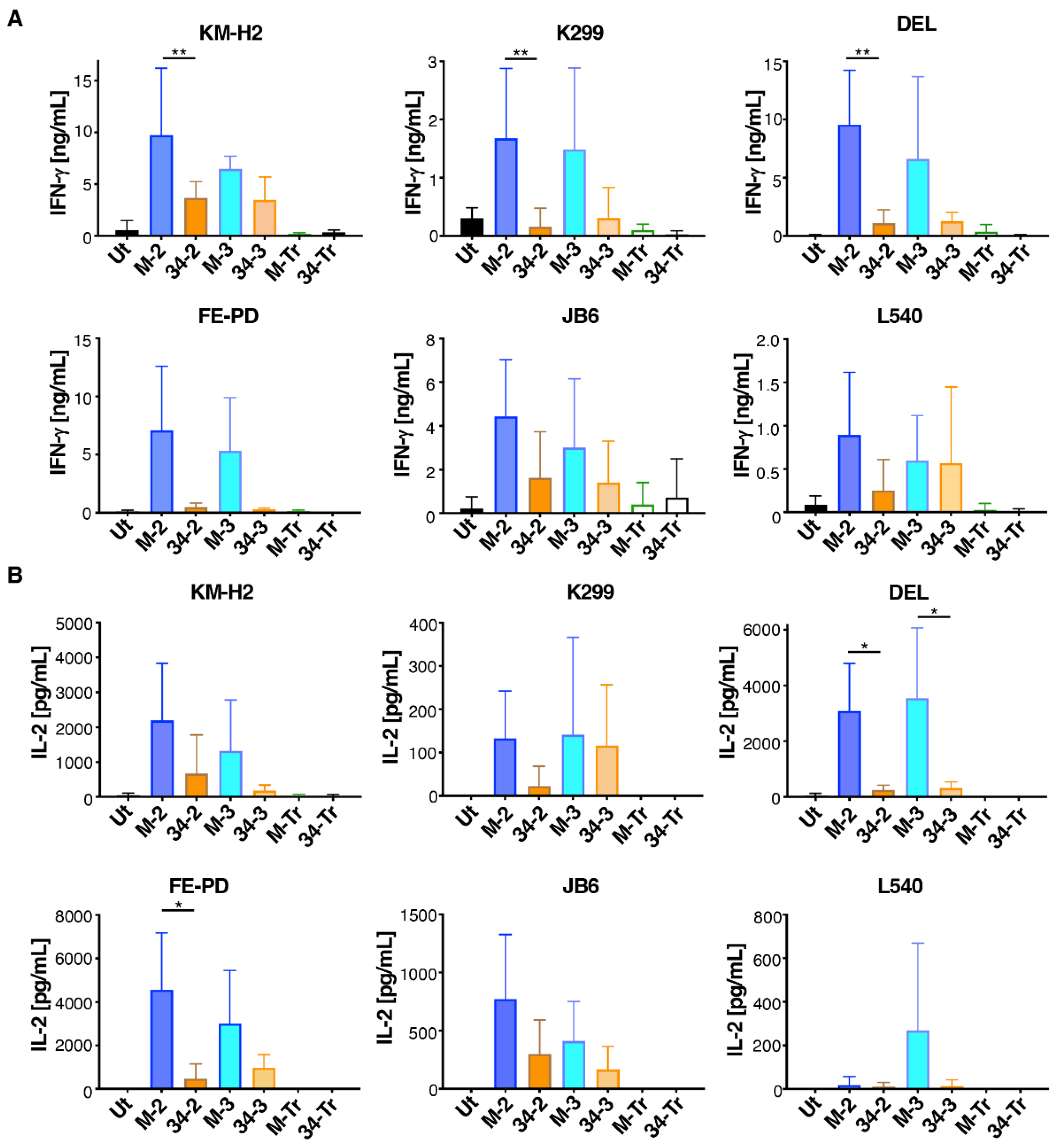
3.2. M-CSF-Targeted CAR T-Cells Mediate Enhanced Cytokine Production When Cultured with T47D FMS Tumor Cells
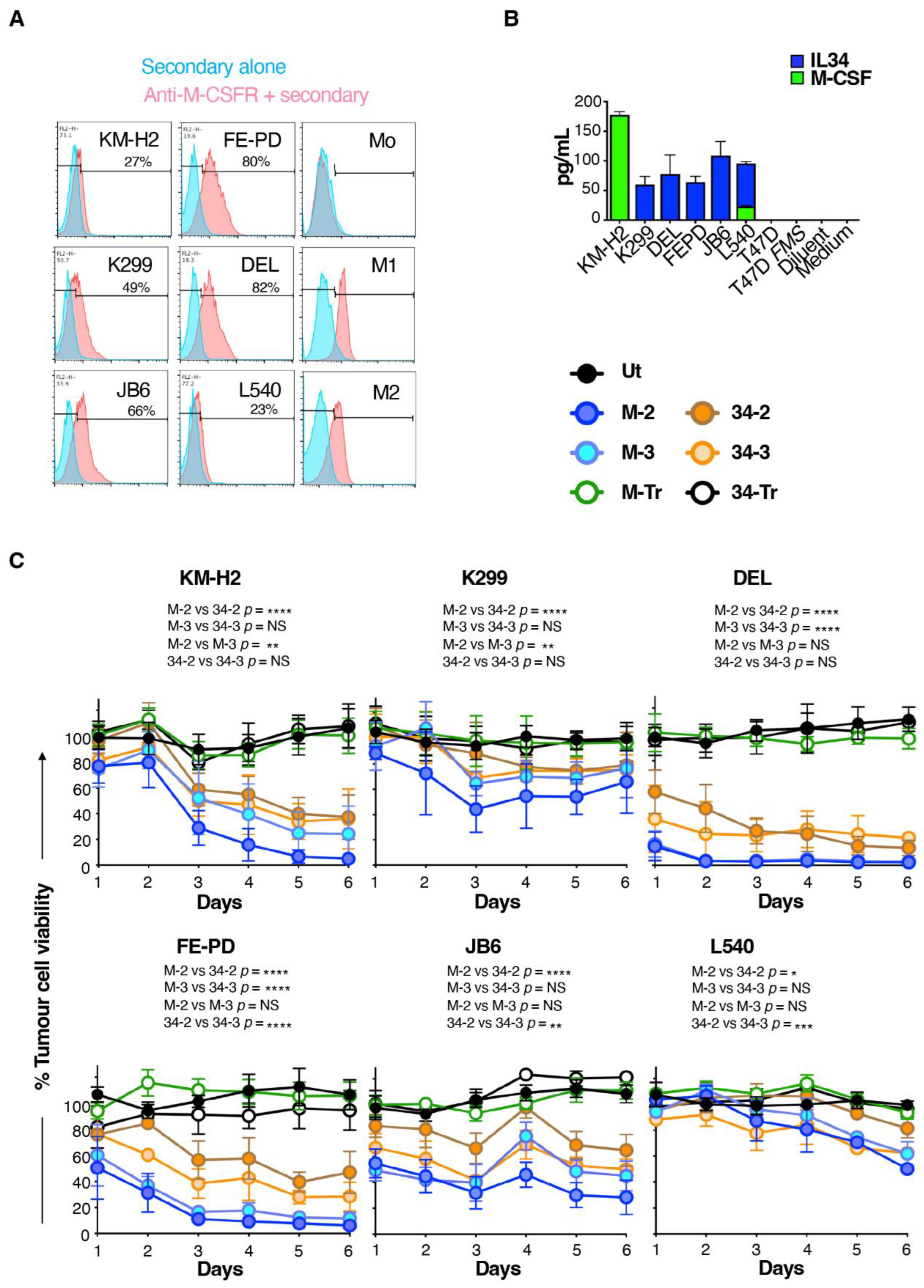
3.3. M-CSF-Targeted CAR T-Cells Mediate Enhanced Cytolytic Activity against M-CSFR+ Lymphoma Cells
3.4. M-CSF-Containing CAR T-Cells Produce Higher Cytokine Levels When Cultured with Lymphoma Cells That Express M-CSFR
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Dai, X.M.; Ryan, G.R.; Hapel, A.J.; Dominguez, M.G.; Russell, R.G.; Kapp, S.; Sylvestre, V.; Stanley, E.R. Targeted disruption of the mouse colony-stimulating factor 1 receptor gene results in osteopetrosis, mononuclear phagocyte deficiency, increased primitive progenitor cell frequencies, and reproductive defects. Blood 2002, 99, 111–120. [Google Scholar] [CrossRef]
- Stanley, E.R.; Berg, K.L.; Einstein, D.B.; Lee, P.S.; Pixley, F.J.; Wang, Y.; Yeung, Y.G. Biology and action of colony--stimulating factor-1. Mol. Reprod. Dev. 1997, 46, 4–10. [Google Scholar] [CrossRef]
- Achkova, D.; Maher, J. Role of the colony-stimulating factor (CSF)/CSF-1 receptor axis in cancer. Biochem. Soc. Trans. 2016, 44, 333–341. [Google Scholar] [CrossRef]
- Gyori, D.; Lim, E.L.; Grant, F.M.; Spensberger, D.; Roychoudhuri, R.; Shuttleworth, S.J.; Okkenhaug, K.; Stephens, L.R.; Hawkins, P.T. Compensation between CSF1R+ macrophages and Foxp3+ Treg cells drives resistance to tumor immunotherapy. JCI Insight 2018, 3, e120631. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Laoui, D.; Van Overmeire, E.; De Baetselier, P.; Van Ginderachter, J.A.; Raes, G. Functional Relationship between Tumor-Associated Macrophages and Macrophage Colony-Stimulating Factor as Contributors to Cancer Progression. Front. Immunol. 2014, 5, 489. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cannarile, M.A.; Weisser, M.; Jacob, W.; Jegg, A.M.; Ries, C.H.; Ruttinger, D. Colony-stimulating factor 1 receptor (CSF1R) inhibitors in cancer therapy. J. Immunother. Cancer 2017, 5, 53. [Google Scholar] [CrossRef] [PubMed]
- Maher, J. Immunotherapy of malignant disease using chimeric antigen receptor engrafted T cells. ISRN Oncol. 2012, 2012, 278093. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kershaw, M.H.; Darcy, P.K.; Hulett, M.D.; Hogarth, P.M.; Trapani, J.A.; Smyth, M.J. Redirected cytotoxic effector function. Requirements for expression of chimeric single chain high affinity immunoglobulin e receptors. J. Biol. Chem. 1996, 271, 21214–21220. [Google Scholar] [CrossRef] [Green Version]
- Finney, H.M.; Lawson, A.D.; Bebbington, C.R.; Weir, A.N. Chimeric receptors providing both primary and costimulatory signaling in T cells from a single gene product. J. Immunol. 1998, 161, 2791–2797. [Google Scholar]
- Halim, L.; Maher, J. CAR T-cell immunotherapy of B cell malignancy–the story so far. Ther. Adv. Vaccines Immunother. 2020; in press. [Google Scholar] [CrossRef]
- Simmons, G.L.; Satta, T.; Castaneda Puglianini, O. Clinical experience of CAR T cells for multiple myeloma. Best Pr. Res. Clin. Haematol. 2021, 34, 101306. [Google Scholar] [CrossRef] [PubMed]
- Pule, M.A.; Straathof, K.C.; Dotti, G.; Heslop, H.E.; Rooney, C.M.; Brenner, M.K. A chimeric T cell antigen receptor that augments cytokine release and supports clonal expansion of primary human T cells. Mol. Ther. 2005, 12, 933–941. [Google Scholar] [CrossRef] [PubMed]
- Zhang, P.; Zhao, S.; Wu, C.; Li, J.; Li, Z.; Wen, C.; Hu, S.; An, G.; Meng, H.; Zhang, X.; et al. Effects of CSF1R-targeted chimeric antigen receptor-modified NK92MI & T cells on tumor-associated macrophages. Immunotherapy 2018, 10, 935–949. [Google Scholar] [CrossRef]
- Long, A.H.; Haso, W.M.; Shern, J.F.; Wanhainen, K.M.; Murgai, M.; Ingaramo, M.; Smith, J.P.; Walker, A.J.; Kohler, M.E.; Venkateshwara, V.R.; et al. 4-1BB costimulation ameliorates T cell exhaustion induced by tonic signaling of chimeric antigen receptors. Nat. Med. 2015, 21, 581–590. [Google Scholar] [CrossRef] [Green Version]
- Lin, H.; Lee, E.; Hestir, K.; Leo, C.; Huang, M.; Bosch, E.; Halenbeck, R.; Wu, G.; Zhou, A.; Behrens, D.; et al. Discovery of a cytokine and its receptor by functional screening of the extracellular proteome. Science 2008, 320, 807–811. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Boulakirba, S.; Pfeifer, A.; Mhaidly, R.; Obba, S.; Goulard, M.; Schmitt, T.; Chaintreuil, P.; Calleja, A.; Furstoss, N.; Orange, F.; et al. IL-34 and CSF-1 display an equivalent macrophage differentiation ability but a different polarization potential. Sci. Rep. 2018, 8, 256. [Google Scholar] [CrossRef] [PubMed]
- Freuchet, A.; Salama, A.; Remy, S.; Guillonneau, C.; Anegon, I. IL-34 and CSF-1, deciphering similarities and differences at steady state and in diseases. J. Leukoc. Biol. 2021, 110, 771–796. [Google Scholar] [CrossRef]
- Whilding, L.M.; Parente-Pereira, A.C.; Zabinski, T.; Davies, D.M.; Petrovic, R.M.G.; Kao, Y.V.; Saxena, S.A.; Romain, A.; Costa-Guerra, J.A.; Violette, S.; et al. Targeting of Aberrant alphavbeta6 Integrin Expression in Solid Tumors Using Chimeric Antigen Receptor-Engineered T Cells. Mol. Ther. 2017, 25, 259–273. [Google Scholar] [CrossRef] [Green Version]
- Lo, A.S.; Taylor, J.R.; Farzaneh, F.; Kemeny, D.M.; Dibb, N.J.; Maher, J. Harnessing the tumour-derived cytokine, CSF-1, to co-stimulate T-cell growth and activation. Mol. Immunol. 2008, 45, 1276–1287. [Google Scholar] [CrossRef]
- Davies, D.M.; Foster, J.; Van Der Stegen, S.J.; Parente-Pereira, A.C.; Chiapero-Stanke, L.; Delinassios, G.J.; Burbridge, S.E.; Kao, V.; Liu, Z.; Bosshard-Carter, L.; et al. Flexible targeting of ErbB dimers that drive tumorigenesis by using genetically engineered T cells. Mol. Med. 2012, 18, 565–576. [Google Scholar] [CrossRef]
- Wilkie, S.; Burbridge, S.E.; Chiapero-Stanke, L.; Pereira, A.C.; Cleary, S.; van der Stegen, S.J.; Spicer, J.F.; Davies, D.M.; Maher, J. Selective expansion of chimeric antigen receptor-targeted T-cells with potent effector function using interleukin-4. J. Biol. Chem. 2010, 285, 25538–25544. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Emami-Shahri, N.; Foster, J.; Kashani, R.; Gazinska, P.; Cook, C.; Sosabowski, J.; Maher, J.; Papa, S. Clinically compliant spatial and temporal imaging of chimeric antigen receptor T-cells. Nat. Commun. 2018, 9, 1081. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Whilding, L.M.; Halim, L.; Draper, B.; Parente-Pereira, A.C.; Zabinski, T.; Davies, D.M.; Maher, J. CAR T-Cells Targeting the Integrin alphavbeta6 and Co-Expressing the Chemokine Receptor CXCR2 Demonstrate Enhanced Homing and Efficacy against Several Solid Malignancies. Cancers 2019, 11, 674. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maher, J.; Brentjens, R.J.; Gunset, G.; Riviere, I.; Sadelain, M. Human T-lymphocyte cytotoxicity and proliferation directed by a single chimeric TCRzeta /CD28 receptor. Nat. Biotechnol. 2002, 20, 70–75. [Google Scholar] [CrossRef]
- Larcombe-Young, D.; Whilding, L.; Davies, D.M.; Draper, B.; Bechman, N.; Maher, J. Generation of human parallel chimeric antigen receptor (pCAR) T cells to achieve synergistic T cell co-stimulation. STAR Protoc. 2022, 3, 101414. [Google Scholar] [CrossRef]
- Parente-Pereira, A.C.; Beatson, R.E.; Davies, D.M.; Hull, C.; Whilding, L.M.; Porter, J.C.; Maher, J. Generation and application of TGFbeta-educated human Vgamma9Vdelta2 T cells. STAR Protoc. 2022, 3, 101319. [Google Scholar] [CrossRef]
- Felix, J.; Elegheert, J.; Gutsche, I.; Shkumatov, A.V.; Wen, Y.; Bracke, N.; Pannecoucke, E.; Vandenberghe, I.; Devreese, B.; Svergun, D.I.; et al. Human IL-34 and CSF-1 establish structurally similar extracellular assemblies with their common hematopoietic receptor. Structure 2013, 21, 528–539. [Google Scholar] [CrossRef] [Green Version]
- Patsialou, A.; Wang, Y.; Pignatelli, J.; Chen, X.; Entenberg, D.; Oktay, M.; Condeelis, J.S. Autocrine CSF1R signaling mediates switching between invasion and proliferation downstream of TGFbeta in claudin-low breast tumor cells. Oncogene 2015, 34, 2721–2731. [Google Scholar] [CrossRef] [Green Version]
- Lamprecht, B.; Walter, K.; Kreher, S.; Kumar, R.; Hummel, M.; Lenze, D.; Kochert, K.; Bouhlel, M.A.; Richter, J.; Soler, E.; et al. Derepression of an endogenous long terminal repeat activates the CSF1R proto-oncogene in human lymphoma. Nat. Med. 2010, 16, 571–579. [Google Scholar] [CrossRef]
- Ullrich, K.; Wurster, K.D.; Lamprecht, B.; Kochert, K.; Engert, A.; Dorken, B.; Janz, M.; Mathas, S. BAY 43-9006/Sorafenib blocks CSF1R activity and induces apoptosis in various classical Hodgkin lymphoma cell lines. Br. J. Haematol. 2011, 155, 398–402. [Google Scholar] [CrossRef]
- Sun, L.; Gao, F.; Gao, Z.; Ao, L.; Li, N.; Ma, S.; Jia, M.; Li, N.; Lu, P.; Sun, B.; et al. Shed antigen-induced blocking effect on CAR-T cells targeting Glypican-3 in Hepatocellular Carcinoma. J. Immunother. Cancer 2021, 9, e001875. [Google Scholar] [CrossRef]
- Kahlon, K.S.; Brown, C.; Cooper, L.J.; Raubitschek, A.; Forman, S.J.; Jensen, M.C. Specific recognition and killing of glioblastoma multiforme by interleukin 13-zetakine redirected cytolytic T cells. Cancer Res. 2004, 64, 9160–9166. [Google Scholar] [CrossRef] [Green Version]
- Zhang, T.; Lemoi, B.A.; Sentman, C.L. Chimeric NK-receptor-bearing T cells mediate antitumor immunotherapy. Blood 2005, 106, 1544–1551. [Google Scholar] [CrossRef]
- Guest, R.D.; Hawkins, R.E.; Kirillova, N.; Cheadle, E.J.; Arnold, J.; O’Neill, A.; Irlam, J.; Chester, K.A.; Kemshead, J.T.; Shaw, D.M.; et al. The role of extracellular spacer regions in the optimal design of chimeric immune receptors: Evaluation of four different scFvs and antigens. J. Immunother. 2005, 28, 203–211. [Google Scholar] [CrossRef]
- Schmid, D.A.; Irving, M.B.; Posevitz, V.; Hebeisen, M.; Posevitz-Fejfar, A.; Sarria, J.C.; Gomez-Eerland, R.; Thome, M.; Schumacher, T.N.; Romero, P.; et al. Evidence for a TCR affinity threshold delimiting maximal CD8 T cell function. J. Immunol. 2010, 184, 4936–4946. [Google Scholar] [CrossRef]
- Thomas, S.; Xue, S.A.; Bangham, C.R.; Jakobsen, B.K.; Morris, E.C.; Stauss, H.J. Human T cells expressing affinity-matured TCR display accelerated responses but fail to recognize low density of MHC-peptide antigen. Blood 2011, 118, 319–329. [Google Scholar] [CrossRef]
- Chervin, A.S.; Stone, J.D.; Soto, C.M.; Engels, B.; Schreiber, H.; Roy, E.J.; Kranz, D.M. Design of T-cell receptor libraries with diverse binding properties to examine adoptive T-cell responses. Gene Ther. 2013, 20, 634–644. [Google Scholar] [CrossRef]
- Chmielewski, M.; Hombach, A.; Heuser, C.; Adams, G.P.; Abken, H. T cell activation by antibody-like immunoreceptors: Increase in affinity of the single-chain fragment domain above threshold does not increase T cell activation against antigen-positive target cells but decreases selectivity. J. Immunol. 2004, 173, 7647–7653. [Google Scholar] [CrossRef]
- Liu, X.; Jiang, S.; Fang, C.; Yang, S.; Olalere, D.; Pequignot, E.C.; Cogdill, A.P.; Li, N.; Ramones, M.; Granda, B.; et al. Affinity-Tuned ErbB2 or EGFR Chimeric Antigen Receptor T Cells Exhibit an Increased Therapeutic Index against Tumors in Mice. Cancer Res. 2015, 75, 3596–3607. [Google Scholar] [CrossRef] [Green Version]
- Caruso, H.G.; Hurton, L.V.; Najjar, A.; Rushworth, D.; Ang, S.; Olivares, S.; Mi, T.; Switzer, K.; Singh, H.; Huls, H.; et al. Tuning Sensitivity of CAR to EGFR Density Limits Recognition of Normal Tissue While Maintaining Potent Antitumor Activity. Cancer Res. 2015, 75, 3505–3518. [Google Scholar] [CrossRef] [Green Version]
- Castellarin, M.; Sands, C.; Da, T.; Scholler, J.; Graham, K.; Buza, E.; Fraietta, J.A.; Zhao, Y.; June, C.H. A rational mouse model to detect on-target, off-tumor CAR T cell toxicity. JCI Insight 2020, 5, e136012. [Google Scholar] [CrossRef]
- Hudecek, M.; Lupo-Stanghellini, M.T.; Kosasih, P.L.; Sommermeyer, D.; Jensen, M.C.; Rader, C.; Riddell, S.R. Receptor affinity and extracellular domain modifications affect tumor recognition by ROR1-specific chimeric antigen receptor T cells. Clin. Cancer Res. 2013, 19, 3153–3164. [Google Scholar] [CrossRef] [Green Version]
- Sharma, P.; Marada, V.; Cai, Q.; Kizerwetter, M.; He, Y.; Wolf, S.P.; Schreiber, K.; Clausen, H.; Schreiber, H.; Kranz, D.M. Structure-guided engineering of the affinity and specificity of CARs against Tn-glycopeptides. Proc. Natl. Acad. Sci. USA 2020, 117, 15148–15159. [Google Scholar] [CrossRef]
- Turatti, F.; Figini, M.; Balladore, E.; Alberti, P.; Casalini, P.; Marks, J.D.; Canevari, S.; Mezzanzanica, D. Redirected activity of human antitumor chimeric immune receptors is governed by antigen and receptor expression levels and affinity of interaction. J. Immunother. 2007, 30, 684–693. [Google Scholar] [CrossRef]
- Park, S.; Shevlin, E.; Vedvyas, Y.; Zaman, M.; Park, S.; Hsu, Y.S.; Min, I.M.; Jin, M.M. Micromolar affinity CAR T cells to ICAM-1 achieves rapid tumor elimination while avoiding systemic toxicity. Sci. Rep. 2017, 7, 14366. [Google Scholar] [CrossRef] [Green Version]
- Drent, E.; Themeli, M.; Poels, R.; de Jong-Korlaar, R.; Yuan, H.; de Bruijn, J.; Martens, A.C.M.; Zweegman, S.; van de Donk, N.; Groen, R.W.J.; et al. A Rational Strategy for Reducing On-Target Off-Tumor Effects of CD38-Chimeric Antigen Receptors by Affinity Optimization. Mol. Ther. 2017, 25, 1946–1958. [Google Scholar] [CrossRef]
- Ghorashian, S.; Kramer, A.M.; Onuoha, S.; Wright, G.; Bartram, J.; Richardson, R.; Albon, S.J.; Casanovas-Company, J.; Castro, F.; Popova, B.; et al. Enhanced CAR T cell expansion and prolonged persistence in pediatric patients with ALL treated with a low-affinity CD19 CAR. Nat. Med. 2019, 25, 1408–1414. [Google Scholar] [CrossRef]
- Nicholson, I.C.; Lenton, K.A.; Little, D.J.; Decorso, T.; Lee, F.T.; Scott, A.M.; Zola, H.; Hohmann, A.W. Construction and characterisation of a functional CD19 specific single chain Fv fragment for immunotherapy of B lineage leukaemia and lymphoma. Mol. Immunol. 1997, 34, 1157–1165. [Google Scholar] [CrossRef]
- Davenport, A.J.; Cross, R.S.; Watson, K.A.; Liao, Y.; Shi, W.; Prince, H.M.; Beavis, P.A.; Trapani, J.A.; Kershaw, M.H.; Ritchie, D.S.; et al. Chimeric antigen receptor T cells form nonclassical and potent immune synapses driving rapid cytotoxicity. Proc. Natl. Acad. Sci. USA 2018, 115, E2068–E2076. [Google Scholar] [CrossRef] [Green Version]
- Wang, J.; Jensen, M.; Lin, Y.; Sui, X.; Chen, E.; Lindgren, C.G.; Till, B.; Raubitschek, A.; Forman, S.J.; Qian, X.; et al. Optimizing adoptive polyclonal T cell immunotherapy of lymphomas, using a chimeric T cell receptor possessing CD28 and CD137 costimulatory domains. Hum. Gene Ther. 2007, 18, 712–725. [Google Scholar] [CrossRef]
- Zhong, X.S.; Matsushita, M.; Plotkin, J.; Riviere, I.; Sadelain, M. Chimeric antigen receptors combining 4-1BB and CD28 signaling domains augment PI3kinase/AKT/Bcl-XL activation and CD8+ T cell-mediated tumor eradication. Mol. Ther. 2010, 18, 413–420. [Google Scholar] [CrossRef]
- Tammana, S.; Huang, X.; Wong, M.; Milone, M.C.; Ma, L.; Levine, B.L.; June, C.H.; Wagner, J.E.; Blazar, B.R.; Zhou, X. 4-1BB and CD28 signaling plays a synergistic role in redirecting umbilical cord blood T cells against B-cell malignancies. Hum. Gene Ther. 2010, 21, 75–86. [Google Scholar] [CrossRef]
- Karlsson, H.; Svensson, E.; Gigg, C.; Jarvius, M.; Olsson-Stromberg, U.; Savoldo, B.; Dotti, G.; Loskog, A. Evaluation of Intracellular Signaling Downstream Chimeric Antigen Receptors. PLoS ONE 2015, 10, e0144787. [Google Scholar] [CrossRef]
- Guedan, S.; Posey, A.D., Jr.; Shaw, C.; Wing, A.; Da, T.; Patel, P.R.; McGettigan, S.E.; Casado-Medrano, V.; Kawalekar, O.U.; Uribe-Herranz, M.; et al. Enhancing CAR T cell persistence through ICOS and 4-1BB costimulation. JCI Insight 2018, 3, e96976. [Google Scholar] [CrossRef] [Green Version]
- Wilkie, S.; Picco, G.; Foster, J.; Davies, D.M.; Julien, S.; Cooper, L.; Arif, S.; Mather, S.J.; Taylor-Papadimitriou, J.; Burchell, J.M.; et al. Retargeting of human T cells to tumor-associated MUC1: The evolution of a chimeric antigen receptor. J. Immunol. 2008, 180, 4901–4909. [Google Scholar] [CrossRef] [Green Version]
- Milone, M.C.; Fish, J.D.; Carpenito, C.; Carroll, R.G.; Binder, G.K.; Teachey, D.; Samanta, M.; Lakhal, M.; Gloss, B.; Danet-Desnoyers, G.; et al. Chimeric receptors containing CD137 signal transduction domains mediate enhanced survival of T cells and increased antileukemic efficacy in vivo. Mol. Ther. 2009, 17, 1453–1464. [Google Scholar] [CrossRef]
- Carpenito, C.; Milone, M.C.; Hassan, R.; Simonet, J.C.; Lakhal, M.; Suhoski, M.M.; Varela-Rohena, A.; Haines, K.M.; Heitjan, D.F.; Albelda, S.M.; et al. Control of large, established tumor xenografts with genetically retargeted human T cells containing CD28 and CD137 domains. Proc. Natl. Acad. Sci. USA 2009, 106, 3360–3365. [Google Scholar] [CrossRef] [Green Version]
- Hombach, A.A.; Rappl, G.; Abken, H. Arming cytokine-induced killer cells with chimeric antigen receptors: CD28 outperforms combined CD28-OX40 “super-stimulation”. Mol. Ther. 2013, 21, 2268–2277. [Google Scholar] [CrossRef] [Green Version]
- Abate-Daga, D.; Lagisetty, K.H.; Tran, E.; Zheng, Z.; Gattinoni, L.; Yu, Z.; Burns, W.R.; Miermont, A.M.; Teper, Y.; Rudloff, U.; et al. A novel chimeric antigen receptor against prostate stem cell antigen mediates tumor destruction in a humanized mouse model of pancreatic cancer. Hum. Gene Ther. 2014, 25, 1003–1012. [Google Scholar] [CrossRef]
- Muliaditan, T.; Halim, L.; Whilding, L.M.; Draper, B.; Achkova, D.Y.; Kausar, F.; Glover, M.; Bechman, N.; Arulappu, A.; Sanchez, J.; et al. Synergistic T-cell signaling by 41BB and CD28 is optimally achieved by membrane proximal positioning within parallel chimeric antigen receptors. Cell Rep. Med. 2021, 2, 100457. [Google Scholar] [CrossRef]
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Achkova, D.Y.; Beatson, R.E.; Maher, J. CAR T-Cell Targeting of Macrophage Colony-Stimulating Factor Receptor. Cells 2022, 11, 2190. https://doi.org/10.3390/cells11142190
Achkova DY, Beatson RE, Maher J. CAR T-Cell Targeting of Macrophage Colony-Stimulating Factor Receptor. Cells. 2022; 11(14):2190. https://doi.org/10.3390/cells11142190
Chicago/Turabian StyleAchkova, Daniela Yordanova, Richard Esmond Beatson, and John Maher. 2022. "CAR T-Cell Targeting of Macrophage Colony-Stimulating Factor Receptor" Cells 11, no. 14: 2190. https://doi.org/10.3390/cells11142190
APA StyleAchkova, D. Y., Beatson, R. E., & Maher, J. (2022). CAR T-Cell Targeting of Macrophage Colony-Stimulating Factor Receptor. Cells, 11(14), 2190. https://doi.org/10.3390/cells11142190