
Autophagy: A Key Regulator of Homeostasis and Disease: An Overview of Molecular Mechanisms and Modulators

 ,
,  ,
, 
 ,
,  ,
,  , and
, and 
Abstract
:
1. Introduction
2. Autophagy
2.1. Summary of Key Autophagy Players
2.2. Types of Autophagy
- Microautophagic invagination and autophagic tubes
- 2.
- Vesicle formation and expansion
- 3.
- Vesicle scission
- 4.
- Vesicle degradation and recycling.
- Substrate Selectivity
- 2.
- Recognition of Substrates
- 3.
- Unfolding of the Substrate.
- 4.
- Substrate Translocation
- 5.
- Degradation of Substrate
2.3. Mitochondrial Stress and Autophagy
2.4. Proteins Involved in the Autophagy Pathway
2.5. Description of Autophagy Proteins According to Their Participation in the Autophagy Stages
2.5.1. Induction
2.5.2. Nucleation
2.5.3. Elongation
2.6. Autophagy Is a Multistep Pathway; The Following Are the Main Steps in the Autophagy Process
2.6.1. Initiation Stage
2.6.2. Expansion (Elongation)
2.7. Mechanisms of Autophagosome or Amphysome Fusion to the Lysosome
2.8. Autophagosome Degradation and Recycling
3. Physiological Role of Autophagy during Morphogenesis
3.1. Autophagy as a Key Regulator of Embryonic Development
3.2. Physiological Role of Autophagy in the Development of the Central Nervous System
3.3. Effect of Autophagy on Neuronal Survival
4. Autophagy Modulator Factors
4.1. Trophic Factors and Autophagy
4.2. Other Modulators of Autophagy:
4.2.1. Age, Glucose, Amino Acids
4.2.2. ROS and Autophagy
4.2.3. Infections and Autophagy
5. Implications of Autophagy in Human Diseases
6. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Cardenas-Aguayo Mdel, C.; Gomez-Virgilio, L.; DeRosa, S.; Meraz-Rios, M.A. The role of tau oligomers in the onset of Alzheimer’s disease neuropathology. ACS Chem. Neurosci. 2014, 5, 1178–1191. [Google Scholar] [CrossRef] [PubMed]
- Choi, A.M.; Ryter, S.W.; Levine, B. Autophagy in human health and disease. N. Engl. J. Med. 2013, 368, 651–662. [Google Scholar] [CrossRef] [PubMed]
- Fivenson, E.M.; Lautrup, S.; Sun, N.; Scheibye-Knudsen, M.; Stevnsner, T.; Nilsen, H.; Bohr, V.A.; Fang, E.F. Mitophagy in neurodegeneration and aging. Neurochem. Int. 2017, 109, 202–209. [Google Scholar] [CrossRef] [PubMed]
- Saha, S.; Panigrahi, D.P.; Patil, S.; Bhutia, S.K. Autophagy in health and disease: A comprehensive review. Biomed. Pharmacother. Biomed. Pharmacother. 2018, 104, 485–495. [Google Scholar] [CrossRef] [PubMed]
- Kuma, A.; Mizushima, N. Physiological role of autophagy as an intracellular recycling system: With an emphasis on nutrient metabolism. Semin. Cell Dev. Biol. 2010, 21, 683–690. [Google Scholar] [CrossRef]
- Singh, R.; Cuervo, A.M. Autophagy in the cellular energetic balance. Cell Metab. 2011, 13, 495–504. [Google Scholar] [CrossRef] [Green Version]
- Kaur, J.; Debnath, J. Autophagy at the crossroads of catabolism and anabolism. Nat. Rev. Mol. Cell Biol. 2015, 16, 461–472. [Google Scholar] [CrossRef] [Green Version]
- Gross, A.S.; Graef, M. Mechanisms of Autophagy in Metabolic Stress Response. J. Mol. Biol. 2020, 432, 28–52. [Google Scholar] [CrossRef]
- Kim, J.; Lee, Y.; Jeon, T.; Kim, M.S.; Kang, C. All cells are created equal in the sight of autophagy: Selective autophagy maintains homeostasis in senescent cells. Autophagy 2021, 17, 3260–3261. [Google Scholar] [CrossRef]
- Deretic, V.; Kroemer, G. Autophagy in metabolism and quality control: Opposing, complementary or interlinked functions? Autophagy 2021, 18, 283–292. [Google Scholar] [CrossRef]
- Grumati, P.; Dikic, I. Ubiquitin signaling and autophagy. J. Biol. Chem. 2018, 293, 5404–5413. [Google Scholar] [CrossRef] [Green Version]
- Yin, Z.; Popelka, H.; Lei, Y.; Yang, Y.; Klionsky, D.J. The Roles of Ubiquitin in Mediating Autophagy. Cells 2020, 9, 2025. [Google Scholar] [CrossRef] [PubMed]
- Rogov, V.; Dotsch, V.; Johansen, T.; Kirkin, V. Interactions between autophagy receptors and ubiquitin-like proteins form the molecular basis for selective autophagy. Mol. Cell 2014, 53, 167–178. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hu, Y.X.; Han, X.S.; Jing, Q. Autophagy in Development and Differentiation. Adv. Exp. Med. Biol. 2019, 1206, 469–487. [Google Scholar] [CrossRef] [PubMed]
- Wu, X.; Fleming, A.; Ricketts, T.; Pavel, M.; Virgin, H.; Menzies, F.M.; Rubinsztein, D.C. Autophagy regulates Notch degradation and modulates stem cell development and neurogenesis. Nat. Commun. 2016, 7, 10533. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vessoni, A.T.; Muotri, A.R.; Okamoto, O.K. Autophagy in stem cell maintenance and differentiation. Stem Cells Dev. 2012, 21, 513–520. [Google Scholar] [CrossRef] [Green Version]
- Mizushima, N.; Komatsu, M. Autophagy: Renovation of cells and tissues. Cell 2011, 147, 728–741. [Google Scholar] [CrossRef] [Green Version]
- Mizushima, N.; Levine, B. Autophagy in mammalian development and differentiation. Nat. Cell Biol. 2010, 12, 823–830. [Google Scholar] [CrossRef]
- Mizushima, N.; Noda, T.; Yoshimori, T.; Tanaka, Y.; Ishii, T.; George, M.D.; Klionsky, D.J.; Ohsumi, M.; Ohsumi, Y. A protein conjugation system essential for autophagy. Nature 1998, 395, 395–398. [Google Scholar] [CrossRef]
- Klionsky, D.J.; Abdel-Aziz, A.K.; Abdelfatah, S.; Abdellatif, M.; Abdoli, A.; Abel, S.; Abeliovich, H.; Abildgaard, M.H.; Abudu, Y.P.; Acevedo-Arozena, A.; et al. Guidelines for the use and interpretation of assays for monitoring autophagy (4th edition). Autophagy 2021, 17, 1–382. [Google Scholar] [CrossRef]
- Kumar, A.V.; Mills, J.; Lapierre, L.R. Selective Autophagy Receptor p62/SQSTM1, a Pivotal Player in Stress and Aging. Front. Cell Dev. Biol. 2022, 10, 793328. [Google Scholar] [CrossRef] [PubMed]
- Oku, M.; Sakai, Y. Three Distinct Types of Microautophagy Based on Membrane Dynamics and Molecular Machineries. Bioessays 2018, 40, e1800008. [Google Scholar] [CrossRef]
- Scrivo, A.; Bourdenx, M.; Pampliega, O.; Cuervo, A.M. Selective autophagy as a potential therapeutic target for neurodegenerative disorders. Lancet Neurol. 2018, 17, 802–815. [Google Scholar] [CrossRef]
- Feng, Y.; He, D.; Yao, Z.; Klionsky, D.J. The machinery of macroautophagy. Cell Res. 2014, 24, 24–41. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nishida, Y.; Arakawa, S.; Fujitani, K.; Yamaguchi, H.; Mizuta, T.; Kanaseki, T.; Komatsu, M.; Otsu, K.; Tsujimoto, Y.; Shimizu, S. Discovery of Atg5/Atg7-independent alternative macroautophagy. Nature 2009, 461, 654–658. [Google Scholar] [CrossRef]
- Yu, L.; Chen, Y.; Tooze, S.A. Autophagy pathway: Cellular and molecular mechanisms. Autophagy 2018, 14, 207–215. [Google Scholar] [CrossRef] [Green Version]
- Almaguer-Maderos, L.; Almaguer-Gotay, D.; Cuello-Almarales, D.; Aguilera-Rodríguez, R. Autofagia en enfermedades poliglutamínicas: Roles e implicaciones terapéuticas. Rev. Mex. Neurocienc. 2016, 17, 76–90. [Google Scholar]
- Parzych, K.R.; Klionsky, D.J. An overview of autophagy: Morphology, mechanism, and regulation. Antioxid. Redox Signal. 2014, 20, 460–473. [Google Scholar] [CrossRef] [Green Version]
- Cerri, S.; Blandini, F. Role of Autophagy in Parkinson’s Disease. Curr. Med. Chem. 2019, 26, 3702–3718. [Google Scholar] [CrossRef]
- Glick, D.; Barth, S.; Macleod, K.F. Autophagy: Cellular and molecular mechanisms. J. Pathol. 2010, 221, 3–12. [Google Scholar] [CrossRef] [Green Version]
- Tekirdag, K.; Cuervo, A.M. Chaperone-mediated autophagy and endosomal microautophagy: Joint by a chaperone. J. Biol. Chem. 2018, 293, 5414–5424. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Uttenweiler, A.; Schwarz, H.; Mayer, A. Microautophagic vacuole invagination requires calmodulin in a Ca2+-independent function. J. Biol. Chem. 2005, 280, 33289–33297. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Muller, O.; Sattler, T.; Flotenmeyer, M.; Schwarz, H.; Plattner, H.; Mayer, A. Autophagic tubes: Vacuolar invaginations involved in lateral membrane sorting and inverse vesicle budding. J. Cell Biol. 2000, 151, 519–528. [Google Scholar] [CrossRef] [PubMed]
- Sattler, T.; Mayer, A. Cell-free reconstitution of microautophagic vacuole invagination and vesicle formation. J. Cell Biol. 2000, 151, 529–538. [Google Scholar] [CrossRef] [PubMed]
- Uttenweiler, A.; Schwarz, H.; Neumann, H.; Mayer, A. The vacuolar transporter chaperone (VTC) complex is required for microautophagy. Mol. Biol. Cell 2007, 18, 166–175. [Google Scholar] [CrossRef] [Green Version]
- Epple, U.D.; Suriapranata, I.; Eskelinen, E.L.; Thumm, M. Aut5/Cvt17p, a putative lipase essential for disintegration of autophagic bodies inside the vacuole. J. Bacteriol. 2001, 183, 5942–5955. [Google Scholar] [CrossRef] [Green Version]
- Yang, Z.; Klionsky, D.J. Permeases recycle amino acids resulting from autophagy. Autophagy 2007, 3, 149–150. [Google Scholar] [CrossRef] [Green Version]
- Schuck, S. Microautophagy—Distinct molecular mechanisms handle cargoes of many sizes. J. Cell Sci. 2020, 133. [Google Scholar] [CrossRef]
- Wang, L.; Seeley, E.S.; Wickner, W.; Merz, A.J. Vacuole fusion at a ring of vertex docking sites leaves membrane fragments within the organelle. Cell 2002, 108, 357–369. [Google Scholar] [CrossRef] [Green Version]
- Tabone, M.D.; Kalifa, C.; Rodary, C.; Raquin, M.; Valteau-Couanet, D.; Lemerle, J. Osteosarcoma recurrences in pediatric patients previously treated with intensive chemotherapy. J. Clin Oncol. 1994, 12, 2614–2620. [Google Scholar] [CrossRef]
- Yang, Q.; Wang, R.; Zhu, L. Chaperone-Mediated Autophagy. Adv. Exp. Med. Biol. 2019, 1206, 435–452. [Google Scholar] [CrossRef] [PubMed]
- Kaushik, S.; Cuervo, A.M. The coming of age of chaperone-mediated autophagy. Nat. Rev. Mol. Cell Biol. 2018, 19, 365–381. [Google Scholar] [CrossRef] [PubMed]
- Salvador, N.; Aguado, C.; Horst, M.; Knecht, E. Import of a cytosolic protein into lysosomes by chaperone-mediated autophagy depends on its folding state. J. Biol. Chem. 2000, 275, 27447–27456. [Google Scholar] [CrossRef]
- Cuervo, A.M.; Dice, J.F. A receptor for the selective uptake and degradation of proteins by lysosomes. Science 1996, 273, 501–503. [Google Scholar] [CrossRef]
- Bandyopadhyay, U.; Kaushik, S.; Varticovski, L.; Cuervo, A.M. The chaperone-mediated autophagy receptor organizes in dynamic protein complexes at the lysosomal membrane. Mol. Cell Biol. 2008, 28, 5747–5763. [Google Scholar] [CrossRef] [Green Version]
- Cuervo, A.M.; Mann, L.; Bonten, E.J.; d’Azzo, A.; Dice, J.F. Cathepsin A regulates chaperone-mediated autophagy through cleavage of the lysosomal receptor. EMBO J. 2003, 22, 47–59. [Google Scholar] [CrossRef] [Green Version]
- Kaushik, S.; Massey, A.C.; Cuervo, A.M. Lysosome membrane lipid microdomains: Novel regulators of chaperone-mediated autophagy. EMBO J. 2006, 25, 3921–3933. [Google Scholar] [CrossRef] [Green Version]
- Schneider, J.L.; Villarroya, J.; Diaz-Carretero, A.; Patel, B.; Urbanska, A.M.; Thi, M.M.; Villarroya, F.; Santambrogio, L.; Cuervo, A.M. Loss of hepatic chaperone-mediated autophagy accelerates proteostasis failure in aging. Aging Cell 2015, 14, 249–264. [Google Scholar] [CrossRef]
- Cuervo, A.M.; Knecht, E.; Terlecky, S.R.; Dice, J.F. Activation of a selective pathway of lysosomal proteolysis in rat liver by prolonged starvation. Am. J. Physiol. 1995, 269, C1200–C1208. [Google Scholar] [CrossRef]
- Schneider, J.L.; Suh, Y.; Cuervo, A.M. Deficient chaperone-mediated autophagy in liver leads to metabolic dysregulation. Cell Metab. 2014, 20, 417–432. [Google Scholar] [CrossRef] [Green Version]
- Valdor, R.; Mocholi, E.; Botbol, Y.; Guerrero-Ros, I.; Chandra, D.; Koga, H.; Gravekamp, C.; Cuervo, A.M.; Macian, F. Chaperone-mediated autophagy regulates T cell responses through targeted degradation of negative regulators of T cell activation. Nat. Immunol. 2014, 15, 1046–1054. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Park, C.; Suh, Y.; Cuervo, A.M. Regulated degradation of Chk1 by chaperone-mediated autophagy in response to DNA damage. Nat. Commun. 2015, 6, 6823. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Koga, H.; Martinez-Vicente, M.; Macian, F.; Verkhusha, V.V.; Cuervo, A.M. A photoconvertible fluorescent reporter to track chaperone-mediated autophagy. Nat. Commun. 2011, 2, 386. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kiffin, R.; Christian, C.; Knecht, E.; Cuervo, A.M. Activation of chaperone-mediated autophagy during oxidative stress. Mol. Biol. Cell 2004, 15, 4829–4840. [Google Scholar] [CrossRef] [Green Version]
- Dohi, E.; Tanaka, S.; Seki, T.; Miyagi, T.; Hide, I.; Takahashi, T.; Matsumoto, M.; Sakai, N. Hypoxic stress activates chaperone-mediated autophagy and modulates neuronal cell survival. Neurochem. Int. 2012, 60, 431–442. [Google Scholar] [CrossRef]
- Arias, E.; Koga, H.; Diaz, A.; Mocholi, E.; Patel, B.; Cuervo, A.M. Lysosomal mTORC2/PHLPP1/Akt Regulate Chaperone-Mediated Autophagy. Mol. Cell 2015, 59, 270–284. [Google Scholar] [CrossRef] [Green Version]
- Massey, A.C.; Kaushik, S.; Cuervo, A.M. Lysosomal chat maintains the balance. Autophagy 2006, 2, 325–327. [Google Scholar] [CrossRef]
- Massey, A.C.; Kaushik, S.; Sovak, G.; Kiffin, R.; Cuervo, A.M. Consequences of the selective blockage of chaperone-mediated autophagy. Proc. Natl. Acad. Sci. USA 2006, 103, 5805–5810. [Google Scholar] [CrossRef] [Green Version]
- Xilouri, M.; Vogiatzi, T.; Vekrellis, K.; Park, D.; Stefanis, L. Abberant alpha-synuclein confers toxicity to neurons in part through inhibition of chaperone-mediated autophagy. PLoS ONE 2009, 4, e5515. [Google Scholar] [CrossRef] [Green Version]
- Kaushik, S.; Massey, A.C.; Mizushima, N.; Cuervo, A.M. Constitutive activation of chaperone-mediated autophagy in cells with impaired macroautophagy. Mol. Biol. Cell 2008, 19, 2179–2192. [Google Scholar] [CrossRef] [Green Version]
- Wang, Y.; Singh, R.; Xiang, Y.; Czaja, M.J. Macroautophagy and chaperone-mediated autophagy are required for hepatocyte resistance to oxidant stress. Hepatology 2010, 52, 266–277. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Douglas, P.M.; Dillin, A. Protein homeostasis and aging in neurodegeneration. J. Cell Biol. 2010, 190, 719–729. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, C.Y.; Kang, W.Y.; Chen, Y.M.; Jiang, T.F.; Zhang, J.; Zhang, L.N.; Ding, J.Q.; Liu, J.; Chen, S.D. DJ-1 Inhibits alpha-Synuclein Aggregation by Regulating Chaperone-Mediated Autophagy. Front. Aging Neurosci. 2017, 9, 308. [Google Scholar] [CrossRef] [Green Version]
- Caballero, B.; Wang, Y.; Diaz, A.; Tasset, I.; Juste, Y.R.; Stiller, B.; Mandelkow, E.M.; Mandelkow, E.; Cuervo, A.M. Interplay of pathogenic forms of human tau with different autophagic pathways. Aging Cell 2018, 17, 12692. [Google Scholar] [CrossRef] [Green Version]
- Bourdenx, M.; Martin-Segura, A.; Scrivo, A.; Rodriguez-Navarro, J.A.; Kaushik, S.; Tasset, I.; Diaz, A.; Storm, N.J.; Xin, Q.; Juste, Y.R.; et al. Chaperone-mediated autophagy prevents collapse of the neuronal metastable proteome. Cell 2021, 184, 2696–2714.e2625. [Google Scholar] [CrossRef]
- Mathys, H.; Davila-Velderrain, J.; Peng, Z.; Gao, F.; Mohammadi, S.; Young, J.Z.; Menon, M.; He, L.; Abdurrob, F.; Jiang, X.; et al. Single-cell transcriptomic analysis of Alzheimer’s disease. Nature 2019, 570, 332–337. [Google Scholar] [CrossRef] [PubMed]
- Bellver-Landete, A. Mitofagia en Enfermedades Neurodegenerativas; Universidad Complutense de Madrid: Madrid, Spain, 2019. [Google Scholar]
- Li, X.; Huang, L.; Lan, J.; Feng, X.; Li, P.; Wu, L.; Peng, Y. Molecular mechanisms of mitophagy and its roles in neurodegenerative diseases. Pharmacol. Res. 2021, 163, 105240. [Google Scholar] [CrossRef]
- Angelova, P.R.; Abramov, A.Y. Role of mitochondrial ROS in the brain: From physiology to neurodegeneration. FEBS Lett. 2018, 592, 692–702. [Google Scholar] [CrossRef]
- Yan, M.H.; Wang, X.; Zhu, X. Mitochondrial defects and oxidative stress in Alzheimer disease and Parkinson disease. Free Radic. Biol. Med. 2013, 62, 90–101. [Google Scholar] [CrossRef] [Green Version]
- Nthiga, T.M.; Kumar Shrestha, B.; Lamark, T.; Johansen, T. The soluble reticulophagy receptor CALCOCO1 is also a Golgiphagy receptor. Autophagy 2021, 17, 2051–2052. [Google Scholar] [CrossRef]
- Nthiga, T.M.; Shrestha, B.K.; Bruun, J.A.; Larsen, K.B.; Lamark, T.; Johansen, T. Regulation of Golgi turnover by CALCOCO1-mediated selective autophagy. J. Cell Biol. 2021, 220, e202006128. [Google Scholar] [CrossRef] [PubMed]
- Lu, L.Q.; Tang, M.Z.; Qi, Z.H.; Huang, S.F.; He, Y.Q.; Li, D.K.; Li, L.F.; Chen, L.X. Regulation of the Golgi apparatus via GOLPH3-mediated new selective autophagy. Life Sci. 2020, 253, 117700. [Google Scholar] [CrossRef] [PubMed]
- Jetto, C.T.; Nambiar, A.; Manjithaya, R. Mitophagy and Neurodegeneration: Between the Knowns and the Unknowns. Front. Cell Dev. Biol. 2022, 10, 837337. [Google Scholar] [CrossRef] [PubMed]
- Rahman, A.; Lorincz, P.; Gohel, R.; Nagy, A.; Csordas, G.; Zhang, Y.; Juhasz, G.; Nezis, I.P. GMAP is an Atg8a-interacting protein that regulates Golgi turnover in Drosophila. Cell Rep. 2022, 39, 110903. [Google Scholar] [CrossRef]
- Ravikumar, B.; Futter, M.; Jahreiss, L.; Korolchuk, V.I.; Lichtenberg, M.; Luo, S.; Massey, D.C.; Menzies, F.M.; Narayanan, U.; Renna, M.; et al. Mammalian macroautophagy at a glance. J. Cell Sci. 2009, 122, 1707–1711. [Google Scholar] [CrossRef] [Green Version]
- Klionsky, D.J.; Codogno, P. The mechanism and physiological function of macroautophagy. J. Innate Immun. 2013, 5, 427–433. [Google Scholar] [CrossRef]
- Li, W.W.; Li, J.; Bao, J.K. Microautophagy: Lesser-known self-eating. Cell Mol. Life Sci. 2012, 69, 1125–1136. [Google Scholar] [CrossRef]
- Cuervo, A.M.; Wong, E. Chaperone-mediated autophagy: Roles in disease and aging. Cell Res. 2014, 24, 92–104. [Google Scholar] [CrossRef] [Green Version]
- Juste, Y.R.; Cuervo, A.M. Analysis of Chaperone-Mediated Autophagy. Methods Mol. Biol. 2019, 1880, 703–727. [Google Scholar] [CrossRef]
- Ding, W.X.; Yin, X.M. Mitophagy: Mechanisms, pathophysiological roles, and analysis. Biol. Chem. 2012, 393, 547–564. [Google Scholar] [CrossRef] [Green Version]
- Ashrafi, G.; Schwarz, T.L. The pathways of mitophagy for quality control and clearance of mitochondria. Cell Death Differ. 2013, 20, 31–42. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pickles, S.; Vigie, P.; Youle, R.J. Mitophagy and Quality Control Mechanisms in Mitochondrial Maintenance. Curr. Biol. 2018, 28, R170–R185. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Quinlan, C.L.; Perevoshchikova, I.V.; Hey-Mogensen, M.; Orr, A.L.; Brand, M.D. Sites of reactive oxygen species generation by mitochondria oxidizing different substrates. Redox. Biol. 2013, 1, 304–312. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Parihar, M.S.; Nazarewicz, R.R.; Kincaid, E.; Bringold, U.; Ghafourifar, P. Association of mitochondrial nitric oxide synthase activity with respiratory chain complex I. Biochem. Biophys. Res. Commun. 2008, 366, 23–28. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Salcher, S.; Hermann, M.; Kiechl-Kohlendorfer, U.; Ausserlechner, M.J.; Obexer, P. C10ORF10/DEPP-mediated ROS accumulation is a critical modulator of FOXO3-induced autophagy. Mol. Cancer 2017, 16, 95. [Google Scholar] [CrossRef] [Green Version]
- Nah, J.; Yoo, S.M.; Jung, S.; Jeong, E.I.; Park, M.; Kaang, B.K.; Jung, Y.K. Phosphorylated CAV1 activates autophagy through an interaction with BECN1 under oxidative stress. Cell Death Dis. 2017, 8, e2822. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, X.; Cheng, X.; Yu, L.; Yang, J.; Calvo, R.; Patnaik, S.; Hu, X.; Gao, Q.; Yang, M.; Lawas, M.; et al. MCOLN1 is a ROS sensor in lysosomes that regulates autophagy. Nat. Commun. 2016, 7, 12109. [Google Scholar] [CrossRef] [Green Version]
- Sui, X.; Kong, N.; Ye, L.; Han, W.; Zhou, J.; Zhang, Q.; He, C.; Pan, H. p38 and JNK MAPK pathways control the balance of apoptosis and autophagy in response to chemotherapeutic agents. Cancer Lett. 2014, 344, 174–179. [Google Scholar] [CrossRef]
- Shen, T.; Miao, Y.; Ding, C.; Fan, W.; Liu, S.; Lv, Y.; Gao, X.; De Boevre, M.; Yan, L.; Okoth, S.; et al. Activation of the p38/MAPK pathway regulates autophagy in response to the CYPOR-dependent oxidative stress induced by zearalenone in porcine intestinal epithelial cells. Food Chem. Toxicol. 2019, 131, 110527. [Google Scholar] [CrossRef]
- Wang, X.; Fu, Y.F.; Liu, X.; Feng, G.; Xiong, D.; Mu, G.F.; Chen, F.P. ROS Promote Ox-LDL-Induced Platelet Activation by Up-Regulating Autophagy Through the Inhibition of the PI3K/AKT/mTOR Pathway. Cell Physiol. Biochem. 2018, 50, 1779–1793. [Google Scholar] [CrossRef]
- Cho, Y.L.; Tan, H.W.S.; Saquib, Q.; Ren, Y.; Ahmad, J.; Wahab, R.; He, W.; Bay, B.H.; Shen, H.M. Dual role of oxidative stress-JNK activation in autophagy and apoptosis induced by nickel oxide nanoparticles in human cancer cells. Free Radic. Biol. Med. 2020, 153, 173–186. [Google Scholar] [CrossRef] [PubMed]
- Perez-Perez, M.E.; Zaffagnini, M.; Marchand, C.H.; Crespo, J.L.; Lemaire, S.D. The yeast autophagy protease Atg4 is regulated by thioredoxin. Autophagy 2014, 10, 1953–1964. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Scherz-Shouval, R.; Shvets, E.; Fass, E.; Shorer, H.; Gil, L.; Elazar, Z. Reactive oxygen species are essential for autophagy and specifically regulate the activity of Atg4. EMBO J. 2007, 26, 1749–1760. [Google Scholar] [CrossRef] [PubMed]
- Tripathi, D.N.; Chowdhury, R.; Trudel, L.J.; Tee, A.R.; Slack, R.S.; Walker, C.L.; Wogan, G.N. Reactive nitrogen species regulate autophagy through ATM-AMPK-TSC2-mediated suppression of mTORC1. Proc. Natl. Acad. Sci. USA 2013, 110, E2950–E2957. [Google Scholar] [CrossRef] [Green Version]
- Chakraborty, S.; Datta, S.; Ghosh, S. Induction of autophagy under nitrosative stress: A complex regulatory interplay between SIRT1 and AMPK in MCF7 cells. Cell Signal 2019, 64, 109411. [Google Scholar] [CrossRef] [PubMed]
- Datta, S.; Chakraborty, S.; Panja, C.; Ghosh, S. Reactive nitrogen species control apoptosis and autophagy in K562 cells: Implication of TAp73alpha induction in controlling autophagy. Free Radic. Res. 2018, 52, 491–506. [Google Scholar] [CrossRef]
- Xiao, B.; Goh, J.Y.; Xiao, L.; Xian, H.; Lim, K.L.; Liou, Y.C. Reactive oxygen species trigger Parkin/PINK1 pathway-dependent mitophagy by inducing mitochondrial recruitment of Parkin. J. Biol. Chem. 2017, 292, 16697–16708. [Google Scholar] [CrossRef] [Green Version]
- Han, J.Y.; Kang, M.J.; Kim, K.H.; Han, P.L.; Kim, H.S.; Ha, J.Y.; Son, J.H. Nitric oxide induction of Parkin translocation in PTEN-induced putative kinase 1 (PINK1) deficiency: Functional role of neuronal nitric oxide synthase during mitophagy. J. Biol. Chem. 2015, 290, 10325–10335. [Google Scholar] [CrossRef] [Green Version]
- Morton, H.; Kshirsagar, S.; Orlov, E.; Bunquin, L.E.; Sawant, N.; Boleng, L.; George, M.; Basu, T.; Ramasubramanian, B.; Pradeepkiran, J.A.; et al. Defective mitophagy and synaptic degeneration in Alzheimer’s disease: Focus on aging, mitochondria and synapse. Free Radic. Biol. Med. 2021, 172, 652–667. [Google Scholar] [CrossRef]
- Amadoro, G.; Corsetti, V.; Florenzano, F.; Atlante, A.; Bobba, A.; Nicolin, V.; Nori, S.L.; Calissano, P. Morphological and bioenergetic demands underlying the mitophagy in post-mitotic neurons: The pink-parkin pathway. Front. Aging Neurosci. 2014, 6, 18. [Google Scholar] [CrossRef]
- Goswami, A.; Dikshit, P.; Mishra, A.; Mulherkar, S.; Nukina, N.; Jana, N.R. Oxidative stress promotes mutant huntingtin aggregation and mutant huntingtin-dependent cell death by mimicking proteasomal malfunction. Biochem. Biophys. Res. Commun. 2006, 342, 184–190. [Google Scholar] [CrossRef] [PubMed]
- Ganguly, G.; Chakrabarti, S.; Chatterjee, U.; Saso, L. Proteinopathy, oxidative stress and mitochondrial dysfunction: Cross talk in Alzheimer’s disease and Parkinson’s disease. Drug Des. Devel. 2017, 11, 797–810. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nixon, R.A.; Wegiel, J.; Kumar, A.; Yu, W.H.; Peterhoff, C.; Cataldo, A.; Cuervo, A.M. Extensive involvement of autophagy in Alzheimer disease: An immuno-electron microscopy study. J. Neuropathol. Exp. Neurol. 2005, 64, 113–122. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Anglade, P.; Vyas, S.; Javoy-Agid, F.; Herrero, M.T.; Michel, P.P.; Marquez, J.; Mouatt-Prigent, A.; Ruberg, M.; Hirsch, E.C.; Agid, Y. Apoptosis and autophagy in nigral neurons of patients with Parkinson’s disease. Histol. Histopathol. 1997, 12, 25–31. [Google Scholar]
- Hara, T.; Nakamura, K.; Matsui, M.; Yamamoto, A.; Nakahara, Y.; Suzuki-Migishima, R.; Yokoyama, M.; Mishima, K.; Saito, I.; Okano, H.; et al. Suppression of basal autophagy in neural cells causes neurodegenerative disease in mice. Nature 2006, 441, 885–889. [Google Scholar] [CrossRef]
- Cha, M.Y.; Kim, D.K.; Mook-Jung, I. The role of mitochondrial DNA mutation on neurodegenerative diseases. Exp. Mol. Med. 2015, 47, e150. [Google Scholar] [CrossRef] [Green Version]
- Briston, T.; Hicks, A.R. Mitochondrial dysfunction and neurodegenerative proteinopathies: Mechanisms and prospects for therapeutic intervention. Biochem. Soc. Trans. 2018, 46, 829–842. [Google Scholar] [CrossRef] [Green Version]
- Wang, W.; Zhao, F.; Ma, X.; Perry, G.; Zhu, X. Mitochondria dysfunction in the pathogenesis of Alzheimer’s disease: Recent advances. Mol. Neurodegener. 2020, 15, 30. [Google Scholar] [CrossRef]
- Cristina, P.M. La Autofagia Celular y su Implicación en la Efermedad de Alzheimer; Universidad Autónoma de Barcelona: Barcelona, Spain, 2016. [Google Scholar]
- Garcia-Huerta, P.; Troncoso-Escudero, P.; Jerez, C.; Hetz, C.; Vidal, R.L. The intersection between growth factors, autophagy and ER stress: A new target to treat neurodegenerative diseases? Brain Res. 2016, 1649, 173–180. [Google Scholar] [CrossRef]
- Villarejo-Zori, B. Autofagia Selectiva en la Retina: Fisiología y Patología. Ph.D. Thesis, Centro de Investigaciones Biológicas Margarita Salas (CIB), Universidad Complutense de Madrid, Madrid, Spain, 2019; pp. 1–244. Available online: https://eprints.ucm.es/id/eprint/59664/ (accessed on 1 May 2022).
- Herrero, G. Mecanismos Moleculares de Inducción de Autofagia por trail e Celular Epiteliales de Mama y su Relación con Apoptosis y Transformación Oncogénica. Ph.D. Thesis, Centro Andaluz de Biología Molecular y Medicina Regenerativa del CSIC, Universidad de Granada, Granada, Spain, 2008; pp. 1–300. Available online: https://digital.csic.es/handle/10261/131190 (accessed on 1 May 2022).
- Poma, J. Análisis de Polimorfismos de Genes Relacionados a la Autofagia en Pacientes con Colitis Ulcerosa. Ph.D. Thesis, Instituto Universitario de Iologia Molecular y Celular del Cancer, Universidad de Salamanca, Salamanca, Spain, 2017. [Google Scholar]
- Noda, N.N.; Fujioka, Y. Atg1 family kinases in autophagy initiation. Cell Mol. Life Sci. 2015, 72, 3083–3096. [Google Scholar] [CrossRef] [Green Version]
- Matsuura, A.; Tsukada, M.; Wada, Y.; Ohsumi, Y. Apg1p, a novel protein kinase required for the autophagic process in Saccharomyces cerevisiae. Gene 1997, 192, 245–250. [Google Scholar] [CrossRef]
- Kawamata, T.; Kamada, Y.; Kabeya, Y.; Sekito, T.; Ohsumi, Y. Organization of the pre-autophagosomal structure responsible for autophagosome formation. Mol. Biol. Cell 2008, 19, 2039–2050. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kabeya, Y.; Kawamata, T.; Suzuki, K.; Ohsumi, Y. Cis1/Atg31 is required for autophagosome formation in Saccharomyces cerevisiae. Biochem. Biophys. Res. Commun. 2007, 356, 405–410. [Google Scholar] [CrossRef] [PubMed]
- Ganley, I.G.; Lamdu, H.; Wang, J.; Ding, X.; Chen, S.; Jiang, X. ULK1.ATG13.FIP200 complex mediates mTOR signaling and is essential for autophagy. J. Biol. Chem. 2009, 284, 12297–12305. [Google Scholar] [CrossRef] [Green Version]
- Hosokawa, N.; Sasaki, T.; Iemura, S.; Natsume, T.; Hara, T.; Mizushima, N. Atg101, a novel mammalian autophagy protein interacting with Atg13. Autophagy 2009, 5, 973–979. [Google Scholar] [CrossRef] [Green Version]
- Stephan, J.S.; Yeh, Y.Y.; Ramachandran, V.; Deminoff, S.J.; Herman, P.K. The Tor and PKA signaling pathways independently target the Atg1/Atg13 protein kinase complex to control autophagy. Proc. Natl. Acad. Sci. USA 2009, 106, 17049–17054. [Google Scholar] [CrossRef] [Green Version]
- Fujioka, Y.; Suzuki, S.W.; Yamamoto, H.; Kondo-Kakuta, C.; Kimura, Y.; Hirano, H.; Akada, R.; Inagaki, F.; Ohsumi, Y.; Noda, N.N. Structural basis of starvation-induced assembly of the autophagy initiation complex. Nat. Struct. Mol. Biol. 2014, 21, 513–521. [Google Scholar] [CrossRef]
- Puente, C.; Hendrickson, R.C.; Jiang, X. Nutrient-regulated Phosphorylation of ATG13 Inhibits Starvation-induced Autophagy. J. Biol. Chem. 2016, 291, 6026–6035. [Google Scholar] [CrossRef] [Green Version]
- Kabeya, Y.; Kamada, Y.; Baba, M.; Takikawa, H.; Sasaki, M.; Ohsumi, Y. Atg17 functions in cooperation with Atg1 and Atg13 in yeast autophagy. Mol. Biol. Cell 2005, 16, 2544–2553. [Google Scholar] [CrossRef] [Green Version]
- Cheong, H.; Yorimitsu, T.; Reggiori, F.; Legakis, J.E.; Wang, C.W.; Klionsky, D.J. Atg17 regulates the magnitude of the autophagic response. Mol. Biol. Cell 2005, 16, 3438–3453. [Google Scholar] [CrossRef]
- Kametaka, S.; Okano, T.; Ohsumi, M.; Ohsumi, Y. Apg14p and Apg6/Vps30p form a protein complex essential for autophagy in the yeast, Saccharomyces cerevisiae. J. Biol. Chem. 1998, 273, 22284–22291. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liang, X.H.; Yu, J.; Brown, K.; Levine, B. Beclin 1 contains a leucine-rich nuclear export signal that is required for its autophagy and tumor suppressor function. Cancer Res. 2001, 61, 3443–3449. [Google Scholar] [PubMed]
- Tassa, A.; Roux, M.P.; Attaix, D.; Bechet, D.M. Class III phosphoinositide 3-kinase--Beclin1 complex mediates the amino acid-dependent regulation of autophagy in C2C12 myotubes. Biochem. J. 2003, 376, 577–586. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kang, R.; Zeh, H.J.; Lotze, M.T.; Tang, D. The Beclin 1 network regulates autophagy and apoptosis. Cell Death Differ. 2011, 18, 571–580. [Google Scholar] [CrossRef] [PubMed]
- Furuya, N.; Yu, J.; Byfield, M.; Pattingre, S.; Levine, B. The evolutionarily conserved domain of Beclin 1 is required for Vps34 binding, autophagy and tumor suppressor function. Autophagy 2005, 1, 46–52. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Noda, T.; Kim, J.; Huang, W.P.; Baba, M.; Tokunaga, C.; Ohsumi, Y.; Klionsky, D.J. Apg9p/Cvt7p is an integral membrane protein required for transport vesicle formation in the Cvt and autophagy pathways. J. Cell Biol. 2000, 148, 465–480. [Google Scholar] [CrossRef]
- Mari, M.; Griffith, J.; Rieter, E.; Krishnappa, L.; Klionsky, D.J.; Reggiori, F. An Atg9-containing compartment that functions in the early steps of autophagosome biogenesis. J. Cell Biol. 2010, 190, 1005–1022. [Google Scholar] [CrossRef] [Green Version]
- Matoba, K.; Kotani, T.; Tsutsumi, A.; Tsuji, T.; Mori, T.; Noshiro, D.; Sugita, Y.; Nomura, N.; Iwata, S.; Ohsumi, Y.; et al. Atg9 is a lipid scramblase that mediates autophagosomal membrane expansion. Nat. Struct. Mol. Biol. 2020, 27, 1185–1193. [Google Scholar] [CrossRef]
- Diao, J.; Liu, R.; Rong, Y.; Zhao, M.; Zhang, J.; Lai, Y.; Zhou, Q.; Wilz, L.M.; Li, J.; Vivona, S.; et al. ATG14 promotes membrane tethering and fusion of autophagosomes to endolysosomes. Nature 2015, 520, 563–566. [Google Scholar] [CrossRef] [Green Version]
- Nakatogawa, H.; Ichimura, Y.; Ohsumi, Y. Atg8, a ubiquitin-like protein required for autophagosome formation, mediates membrane tethering and hemifusion. Cell 2007, 130, 165–178. [Google Scholar] [CrossRef] [Green Version]
- Watanabe, Y.; Kobayashi, T.; Yamamoto, H.; Hoshida, H.; Akada, R.; Inagaki, F.; Ohsumi, Y.; Noda, N.N. Structure-based analyses reveal distinct binding sites for Atg2 and phosphoinositides in Atg18. J. Biol. Chem. 2012, 287, 31681–31690. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Krick, R.; Henke, S.; Tolstrup, J.; Thumm, M. Dissecting the localization and function of Atg18, Atg21 and Ygr223c. Autophagy 2008, 4, 896–910. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Obara, K.; Sekito, T.; Niimi, K.; Ohsumi, Y. The Atg18-Atg2 complex is recruited to autophagic membranes via phosphatidylinositol 3-phosphate and exerts an essential function. J. Biol. Chem. 2008, 283, 23972–23980. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hervas, J.H.; Landajuela, A.; Anton, Z.; Shnyrova, A.V.; Goni, F.M.; Alonso, A. Human ATG3 binding to lipid bilayers: Role of lipid geometry, and electric charge. Sci. Rep. 2017, 7, 15614. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yamada, Y.; Suzuki, N.N.; Hanada, T.; Ichimura, Y.; Kumeta, H.; Fujioka, Y.; Ohsumi, Y.; Inagaki, F. The crystal structure of Atg3, an autophagy-related ubiquitin carrier protein (E2) enzyme that mediates Atg8 lipidation. J. Biol. Chem. 2007, 282, 8036–8043. [Google Scholar] [CrossRef] [Green Version]
- Zhang, L.; Li, J.; Ouyang, L.; Liu, B.; Cheng, Y. Unraveling the roles of Atg4 proteases from autophagy modulation to targeted cancer therapy. Cancer Lett. 2016, 373, 19–26. [Google Scholar] [CrossRef]
- Kirisako, T.; Ichimura, Y.; Okada, H.; Kabeya, Y.; Mizushima, N.; Yoshimori, T.; Ohsumi, M.; Takao, T.; Noda, T.; Ohsumi, Y. The reversible modification regulates the membrane-binding state of Apg8/Aut7 essential for autophagy and the cytoplasm to vacuole targeting pathway. J. Cell Biol. 2000, 151, 263–276. [Google Scholar] [CrossRef]
- Tanida, I.; Sou, Y.S.; Ezaki, J.; Minematsu-Ikeguchi, N.; Ueno, T.; Kominami, E. HsAtg4B/HsApg4B/autophagin-1 cleaves the carboxyl termini of three human Atg8 homologues and delipidates microtubule-associated protein light chain 3- and GABAA receptor-associated protein-phospholipid conjugates. J. Biol. Chem. 2004, 279, 36268–36276. [Google Scholar] [CrossRef] [Green Version]
- Sakoh-Nakatogawa, M.; Matoba, K.; Asai, E.; Kirisako, H.; Ishii, J.; Noda, N.N.; Inagaki, F.; Nakatogawa, H.; Ohsumi, Y. Atg12-Atg5 conjugate enhances E2 activity of Atg3 by rearranging its catalytic site. Nat. Struct. Mol. Biol. 2013, 20, 433–439. [Google Scholar] [CrossRef]
- Tanida, I. Autophagosome formation and molecular mechanism of autophagy. Antioxid. Redox. Signal. 2011, 14, 2201–2214. [Google Scholar] [CrossRef]
- Feng, Y.; Yao, Z.; Klionsky, D.J. How to control self-digestion: Transcriptional, post-transcriptional, and post-translational regulation of autophagy. Trends Cell Biol. 2015, 25, 354–363. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cao, Y.; Cheong, H.; Song, H.; Klionsky, D.J. In vivo reconstitution of autophagy in Saccharomyces cerevisiae. J. Cell Biol. 2008, 182, 703–713. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Komatsu, M.; Waguri, S.; Ueno, T.; Iwata, J.; Murata, S.; Tanida, I.; Ezaki, J.; Mizushima, N.; Ohsumi, Y.; Uchiyama, Y.; et al. Impairment of starvation-induced and constitutive autophagy in Atg7-deficient mice. J. Cell Biol. 2005, 169, 425–434. [Google Scholar] [CrossRef]
- Ichimura, Y.; Kirisako, T.; Takao, T.; Satomi, Y.; Shimonishi, Y.; Ishihara, N.; Mizushima, N.; Tanida, I.; Kominami, E.; Ohsumi, M.; et al. A ubiquitin-like system mediates protein lipidation. Nature 2000, 408, 488–492. [Google Scholar] [CrossRef]
- Wild, P.; McEwan, D.G.; Dikic, I. The LC3 interactome at a glance. J. Cell Sci. 2014, 127, 3–9. [Google Scholar] [CrossRef] [Green Version]
- Lamb, C.A.; Yoshimori, T.; Tooze, S.A. The autophagosome: Origins unknown, biogenesis complex. Nat. Rev. Mol. Cell Biol. 2013, 14, 759–774. [Google Scholar] [CrossRef]
- Hong, S.B.; Kim, B.W.; Kim, J.H.; Song, H.K. Structure of the autophagic E2 enzyme Atg10. Acta Cryst. D Biol. Cryst. 2012, 68, 1409–1417. [Google Scholar] [CrossRef] [PubMed]
- Tanida, I.; Mizushima, N.; Kiyooka, M.; Ohsumi, M.; Ueno, T.; Ohsumi, Y.; Kominami, E. Apg7p/Cvt2p: A novel protein-activating enzyme essential for autophagy. Mol. Biol. Cell 1999, 10, 1367–1379. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Radoshevich, L.; Murrow, L.; Chen, N.; Fernandez, E.; Roy, S.; Fung, C.; Debnath, J. ATG12 conjugation to ATG3 regulates mitochondrial homeostasis and cell death. Cell 2010, 142, 590–600. [Google Scholar] [CrossRef] [Green Version]
- Murrow, L.; Malhotra, R.; Debnath, J. ATG12-ATG3 interacts with Alix to promote basal autophagic flux and late endosome function. Nat. Cell Biol. 2015, 17, 300–310. [Google Scholar] [CrossRef] [Green Version]
- Fujioka, Y.; Noda, N.N.; Matsushita, M.; Ohsumi, Y.; Inagaki, F. Crystallization of the coiled-coil domain of Atg16 essential for autophagy. Acta Cryst. Sect. F Struct. Biol. Cryst. Commun. 2008, 64, 1046–1048. [Google Scholar] [CrossRef] [PubMed]
- Nishimura, T.; Kaizuka, T.; Cadwell, K.; Sahani, M.H.; Saitoh, T.; Akira, S.; Virgin, H.W.; Mizushima, N. FIP200 regulates targeting of Atg16L1 to the isolation membrane. EMBO Rep. 2013, 14, 284–291. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kuma, A.; Mizushima, N.; Ishihara, N.; Ohsumi, Y. Formation of the approximately 350-kDa Apg12-Apg5.Apg16 multimeric complex, mediated by Apg16 oligomerization, is essential for autophagy in yeast. J. Biol. Chem. 2002, 277, 18619–18625. [Google Scholar] [CrossRef] [Green Version]
- Boada-Romero, E.; Letek, M.; Fleischer, A.; Pallauf, K.; Ramon-Barros, C.; Pimentel-Muinos, F.X. TMEM59 defines a novel ATG16L1-binding motif that promotes local activation of LC3. EMBO J. 2013, 32, 566–582. [Google Scholar] [CrossRef] [Green Version]
- Travassos, L.H.; Carneiro, L.A.; Ramjeet, M.; Hussey, S.; Kim, Y.G.; Magalhaes, J.G.; Yuan, L.; Soares, F.; Chea, E.; Le Bourhis, L.; et al. Nod1 and Nod2 direct autophagy by recruiting ATG16L1 to the plasma membrane at the site of bacterial entry. Nat. Immunol. 2010, 11, 55–62. [Google Scholar] [CrossRef] [PubMed]
- Xiong, Q.; Li, W.; Li, P.; Yang, M.; Wu, C.; Eichinger, L. The Role of ATG16 in Autophagy and The Ubiquitin Proteasome System. Cells 2018, 8, 2. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gonzalez Porras, M.A.; Sieck, G.C.; Mantilla, C.B. Impaired Autophagy in Motor Neurons: A Final Common Mechanism of Injury and Death. Physiol. Bethesda 2018, 33, 211–224. [Google Scholar] [CrossRef]
- Itakura, E.; Kishi-Itakura, C.; Mizushima, N. The hairpin-type tail-anchored SNARE syntaxin 17 targets to autophagosomes for fusion with endosomes/lysosomes. Cell 2012, 151, 1256–1269. [Google Scholar] [CrossRef] [Green Version]
- Matsui, T.; Jiang, P.; Nakano, S.; Sakamaki, Y.; Yamamoto, H.; Mizushima, N. Autophagosomal YKT6 is required for fusion with lysosomes independently of syntaxin 17. J. Cell Biol. 2018, 217, 2633–2645. [Google Scholar] [CrossRef]
- Hegedus, K.; Takats, S.; Boda, A.; Jipa, A.; Nagy, P.; Varga, K.; Kovacs, A.L.; Juhasz, G. The Ccz1-Mon1-Rab7 module and Rab5 control distinct steps of autophagy. Mol. Biol. Cell 2016, 27, 3132–3142. [Google Scholar] [CrossRef] [Green Version]
- Vaites, L.P.; Paulo, J.A.; Huttlin, E.L.; Harper, J.W. Systematic Analysis of Human Cells Lacking ATG8 Proteins Uncovers Roles for GABARAPs and the CCZ1/MON1 Regulator C18orf8/RMC1 in Macroautophagic and Selective Autophagic Flux. Mol. Cell Biol. 2018, 38. [Google Scholar] [CrossRef] [Green Version]
- Ferguson, C.J.; Lenk, G.M.; Meisler, M.H. Defective autophagy in neurons and astrocytes from mice deficient in PI(3,5)P2. Hum. Mol. Genet. 2009, 18, 4868–4878. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ravikumar, B.; Acevedo-Arozena, A.; Imarisio, S.; Berger, Z.; Vacher, C.; O’Kane, C.J.; Brown, S.D.; Rubinsztein, D.C. Dynein mutations impair autophagic clearance of aggregate-prone proteins. Nat. Genet. 2005, 37, 771–776. [Google Scholar] [CrossRef] [PubMed]
- Pena-Llopis, S.; Vega-Rubin-de-Celis, S.; Schwartz, J.C.; Wolff, N.C.; Tran, T.A.; Zou, L.; Xie, X.J.; Corey, D.R.; Brugarolas, J. Regulation of TFEB and V-ATPases by mTORC1. EMBO J. 2011, 30, 3242–3258. [Google Scholar] [CrossRef] [Green Version]
- Yu, L.; McPhee, C.K.; Zheng, L.; Mardones, G.A.; Rong, Y.; Peng, J.; Mi, N.; Zhao, Y.; Liu, Z.; Wan, F.; et al. Termination of autophagy and reformation of lysosomes regulated by mTOR. Nature 2010, 465, 942–946. [Google Scholar] [CrossRef] [PubMed]
- Offei, E.B.; Yang, X.; Brand-Saberi, B. The role of autophagy in morphogenesis and stem cell maintenance. Histochem. Cell Biol. 2018, 150, 721–732. [Google Scholar] [CrossRef] [PubMed]
- Cecconi, F.; Levine, B. The role of autophagy in mammalian development: Cell makeover rather than cell death. Dev. Cell 2008, 15, 344–357. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Levine, B.; Klionsky, D.J. Development by Self-Digestion. Dev. Cell 2004, 6, 463–477. [Google Scholar] [CrossRef]
- Yue, Z.; Jin, S.; Yang, C.; Levine, A.J.; Heintz, N. Beclin 1, an autophagy gene essential for early embryonic development, is a haploinsufficient tumor suppressor. Proc. Natl. Acad. Sci. USA 2003, 100, 15077–15082. [Google Scholar] [CrossRef] [Green Version]
- Lorda-Diez, C.I.; Montero, J.A.; Garcia-Porrero, J.A.; Hurle, J.M. Interdigital tissue regression in the developing limb of vertebrates. Int. J. Dev. Biol. 2015, 59, 55–62. [Google Scholar] [CrossRef] [Green Version]
- Elmore, S. Apoptosis: A review of programmed cell death. Toxicol. Pathol. 2007, 35, 495–516. [Google Scholar] [CrossRef] [PubMed]
- Chen, Q.; Kang, J.; Fu, C. The independence of and associations among apoptosis, autophagy, and necrosis. Signal Transduct. Target. Ther. 2018, 3, 18. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kroemer, G.; Marino, G.; Levine, B. Autophagy and the integrated stress response. Mol. Cell 2010, 40, 280–293. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pattingre, S.; Tassa, A.; Qu, X.; Garuti, R.; Liang, X.H.; Mizushima, N.; Packer, M.; Schneider, M.D.; Levine, B. Bcl-2 antiapoptotic proteins inhibit Beclin 1-dependent autophagy. Cell 2005, 122, 927–939. [Google Scholar] [CrossRef] [Green Version]
- Lockshin, R.A.; Zakeri, Z. Apoptosis, autophagy, and more. Int. J. Biochem. Cell Biol. 2004, 36, 2405–2419. [Google Scholar] [CrossRef]
- Nikoletopoulou, V.; Markaki, M.; Palikaras, K.; Tavernarakis, N. Crosstalk between apoptosis, necrosis and autophagy. Biochim. Biophys. Acta BBA Mol. Cell Res. 2013, 1833, 3448–3459. [Google Scholar] [CrossRef] [Green Version]
- Reichrath, J.; Reichrath, S. Notch Signaling and Embryonic Development: An Ancient Friend, Revisited. Adv. Exp. Med. Biol. 2020, 1218, 9–37. [Google Scholar] [CrossRef]
- Balasubramanian, R.; Zhang, X. Mechanisms of FGF gradient formation during embryogenesis. Semin. Cell Dev. Biol. 2016, 53, 94–100. [Google Scholar] [CrossRef] [Green Version]
- Sagai, T.; Amano, T.; Maeno, A.; Ajima, R.; Shiroishi, T. SHH signaling mediated by a prechordal and brain enhancer controls forebrain organization. Proc. Natl. Acad. Sci. USA 2019, 116, 23636–23642. [Google Scholar] [CrossRef] [Green Version]
- Wu, M.Y.; Hill, C.S. Tgf-beta superfamily signaling in embryonic development and homeostasis. Dev. Cell 2009, 16, 329–343. [Google Scholar] [CrossRef] [Green Version]
- Steinhart, Z.; Angers, S. Wnt signaling in development and tissue homeostasis. Development 2018, 145, dev146589. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kuma, A.; Hatano, M.; Matsui, M.; Yamamoto, A.; Nakaya, H.; Yoshimori, T.; Ohsumi, Y.; Tokuhisa, T.; Mizushima, N. The role of autophagy during the early neonatal starvation period. Nature 2004, 432, 1032–1036. [Google Scholar] [CrossRef] [PubMed]
- Sou, Y.S.; Waguri, S.; Iwata, J.; Ueno, T.; Fujimura, T.; Hara, T.; Sawada, N.; Yamada, A.; Mizushima, N.; Uchiyama, Y.; et al. The Atg8 conjugation system is indispensable for proper development of autophagic isolation membranes in mice. Mol. Biol. Cell 2008, 19, 4762–4775. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fimia, G.M.; Stoykova, A.; Romagnoli, A.; Giunta, L.; Di Bartolomeo, S.; Nardacci, R.; Corazzari, M.; Fuoco, C.; Ucar, A.; Schwartz, P.; et al. Ambra1 regulates autophagy and development of the nervous system. Nature 2007, 447, 1121–1125. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jimenez-Sanchez, M.; Menzies, F.M.; Chang, Y.Y.; Simecek, N.; Neufeld, T.P.; Rubinsztein, D.C. The Hedgehog signalling pathway regulates autophagy. Nat. Commun. 2012, 3, 1200. [Google Scholar] [CrossRef] [Green Version]
- Gao, C.; Cao, W.; Bao, L.; Zuo, W.; Xie, G.; Cai, T.; Fu, W.; Zhang, J.; Wu, W.; Zhang, X.; et al. Autophagy negatively regulates Wnt signalling by promoting Dishevelled degradation. Nat. Cell Biol. 2010, 12, 781–790. [Google Scholar] [CrossRef]
- Suzuki, H.I.; Kiyono, K.; Miyazono, K. Regulation of autophagy by transforming growth factor-beta (TGF-beta) signaling. Autophagy 2010, 6, 645–647. [Google Scholar] [CrossRef] [Green Version]
- Zhang, J.; Liu, J.; Liu, L.; McKeehan, W.L.; Wang, F. The fibroblast growth factor signaling axis controls cardiac stem cell differentiation through regulating autophagy. Autophagy 2012, 8, 690–691. [Google Scholar] [CrossRef] [Green Version]
- Barth, J.M.; Hafen, E.; Kohler, K. The lack of autophagy triggers precocious activation of Notch signaling during Drosophila oogenesis. BMC Dev. Biol. 2012, 12, 35. [Google Scholar] [CrossRef] [Green Version]
- Jia, Z.; Wang, J.; Wang, W.; Tian, Y.; XiangWei, W.; Chen, P.; Ma, K.; Zhou, C. Autophagy eliminates cytoplasmic beta-catenin and NICD to promote the cardiac differentiation of P19CL6 cells. Cell Signal. 2014, 26, 2299–2305. [Google Scholar] [CrossRef] [Green Version]
- Hu, Z.; Chen, B.; Zhao, Q. Hedgehog signaling regulates osteoblast differentiation in zebrafish larvae through modulation of autophagy. Biol. Open 2019, 8, bio040840. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pan, Y.; Yan, L.; Chen, Q.; Wei, C.; Dai, Y.; Tong, X.; Zhu, H.; Lu, M.; Zhang, Y.; Jin, X.; et al. Dysfunction of Shh signaling activates autophagy to inhibit trophoblast motility in recurrent miscarriage. Exp. Mol. Med. 2021, 53, 52–66. [Google Scholar] [CrossRef] [PubMed]
- Sobolewska, A.; Motyl, T.; Gajewska, M. Role and regulation of autophagy in the development of acinar structures formed by bovine BME-UV1 mammary epithelial cells. Eur. J. Cell Biol. 2011, 90, 854–864. [Google Scholar] [CrossRef] [PubMed]
- Gajewska, M.; Gajkowska, B.; Motyl, T. Apoptosis and autophagy induced by TGF-B1 in bovine mammary epithelial BME-UV1 cells. J. Physiol. Pharm. 2005, 56 (Suppl. S3), 143–157. [Google Scholar]
- Cinque, L.; Forrester, A.; Bartolomeo, R.; Svelto, M.; Venditti, R.; Montefusco, S.; Polishchuk, E.; Nusco, E.; Rossi, A.; Medina, D.L.; et al. FGF signalling regulates bone growth through autophagy. Nature 2015, 528, 272–275. [Google Scholar] [CrossRef]
- Zhang, J.; Liu, J.; Huang, Y.; Chang, J.Y.; Liu, L.; McKeehan, W.L.; Martin, J.F.; Wang, F. FRS2alpha-mediated FGF signals suppress premature differentiation of cardiac stem cells through regulating autophagy activity. Circ. Res. 2012, 110, e29–e39. [Google Scholar] [CrossRef] [Green Version]
- Boya, P.; Mellen, M.A.; de la Rosa, E.J. How autophagy is related to programmed cell death during the development of the nervous system. Biochem. Soc. Trans. 2008, 36, 813–817. [Google Scholar] [CrossRef] [Green Version]
- Kim, W.R.; Sun, W. Programmed cell death during postnatal development of the rodent nervous system. Dev. Growth Differ. 2011, 53, 225–235. [Google Scholar] [CrossRef]
- Siebel, C.; Lendahl, U. Notch Signaling in Development, Tissue Homeostasis, and Disease. Physiol. Rev 2017, 97, 1235–1294. [Google Scholar] [CrossRef] [Green Version]
- Casares-Crespo, L.; Calatayud-Baselga, I.; Garcia-Corzo, L.; Mira, H. On the Role of Basal Autophagy in Adult Neural Stem Cells and Neurogenesis. Front. Cell Neurosci. 2018, 12, 339. [Google Scholar] [CrossRef]
- Vazquez, P.; Arroba, A.I.; Cecconi, F.; de la Rosa, E.J.; Boya, P.; de Pablo, F. Atg5 and Ambra1 differentially modulate neurogenesis in neural stem cells. Autophagy 2012, 8, 187–199. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Boya, P.; Codogno, P.; Rodriguez-Muela, N. Autophagy in stem cells: Repair, remodelling and metabolic reprogramming. Development 2018, 145, dev146506. [Google Scholar] [CrossRef] [Green Version]
- Gao, Y.; Tan, L.; Yu, J.T.; Tan, L. Tau in Alzheimer’s Disease: Mechanisms and Therapeutic Strategies. Curr. Alzheimer Res. 2018, 15, 283–300. [Google Scholar] [CrossRef]
- Kulkarni, A.; Chen, J.; Maday, S. Neuronal autophagy and intercellular regulation of homeostasis in the brain. Curr. Opin. Neurobiol. 2018, 51, 29–36. [Google Scholar] [CrossRef] [PubMed]
- Yin, Y.; Sun, G.; Li, E.; Kiselyov, K.; Sun, D. ER stress and impaired autophagy flux in neuronal degeneration and brain injury. Ageing Res. Rev. 2017, 34, 3–14. [Google Scholar] [CrossRef] [Green Version]
- Iván, J.C.; Will, T.; Erin, M.S. Protein homeostasis and synaptic plasticity. EMBO J. 2010, 29, 2746–2752. [Google Scholar] [CrossRef]
- Melentijevic, I.; Toth, M.L.; Arnold, M.L.; Guasp, R.J.; Harinath, G.; Nguyen, K.C.; Taub, D.; Parker, J.A.; Neri, C.; Gabel, C.V.; et al. C. elegans neurons jettison protein aggregates and mitochondria under neurotoxic stress. Nature 2017, 542, 367–371. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tydlacka, S.; Wang, C.E.; Wang, X.; Li, S.; Li, X.J. Differential activities of the ubiquitin-proteasome system in neurons versus glia may account for the preferential accumulation of misfolded proteins in neurons. J. Neurosci. 2008, 28, 13285–13295. [Google Scholar] [CrossRef] [Green Version]
- Bostanciklioglu, M. An update on the interactions between Alzheimer’s disease, autophagy and inflammation. Gene 2019, 705, 157–166. [Google Scholar] [CrossRef]
- Cardenas-Aguayo Mdel, C.; Kazim, S.F.; Grundke-Iqbal, I.; Iqbal, K. Neurogenic and neurotrophic effects of BDNF peptides in mouse hippocampal primary neuronal cell cultures. PLoS ONE 2013, 8, e53596. [Google Scholar] [CrossRef]
- Skaper, S.D. Neurotrophic Factors: An Overview. Methods Mol. Biol. 2018, 1727, 1–17. [Google Scholar] [CrossRef] [PubMed]
- Popova, N.K.; Ilchibaeva, T.V.; Naumenko, V.S. Neurotrophic Factors (BDNF and GDNF) and the Serotonergic System of the Brain. Biochem. Mosc. 2017, 82, 308–317. [Google Scholar] [CrossRef] [PubMed]
- Simonato, M. Neurotrophic factors and status epilepticus. Epilepsia 2018, 59 (Suppl. S2), 87–91. [Google Scholar] [CrossRef] [PubMed]
- Nuria, G.-B. Mecanismos de Señalización Intracelular de Supervivencia y Muerte Neuronal Regulados por Factores Tróficos; Universitat de Barcelona: Barcelona, Spain, 2005. [Google Scholar]
- Liu, S. Neurotrophic factors in enteric physiology and pathophysiology. Neurogastroenterol. Motil. 2018, 30, e13446. [Google Scholar] [CrossRef]
- Lanni, C.; Stanga, S.; Govoni, S. The Expanding Universe of Neurotrophic Factors: Therapeutic Potential in Aging and Age-Associated Disorders. Curr. Pharm. Des. 2010, 16, 698–717. [Google Scholar] [CrossRef] [Green Version]
- Kalinowska-Lyszczarz, A.; Losy, J. The role of neurotrophins in multiple sclerosis-pathological and clinical implications. Int. J. Mol. Sci. 2012, 13, 13713–13725. [Google Scholar] [CrossRef] [Green Version]
- Diaz-Galindo, M.D.C.; Calderon-Vallejo, D.; Olvera-Sandoval, C.; Quintanar, J.L. Therapeutic approaches of trophic factors in animal models and in patients with spinal cord injury. Growth Factors 2020, 38, 1–15. [Google Scholar] [CrossRef]
- Weiss, A.; Attisano, L. The TGFbeta superfamily signaling pathway. Wiley Interdiscip. Rev. Dev. Biol. 2013, 2, 47–63. [Google Scholar] [CrossRef]
- Hanna, A.; Frangogiannis, N.G. The Role of the TGF-beta Superfamily in Myocardial Infarction. Front. Cardiovasc. Med. 2019, 6, 140. [Google Scholar] [CrossRef]
- Xiao, N.; Le, Q.T. Neurotrophic Factors and Their Potential Applications in Tissue Regeneration. Arch. Immunol. Exp. Warsz. 2016, 64, 89–99. [Google Scholar] [CrossRef]
- Burgess, W.H.; Maciag, T. The heparin-binding (fibroblast) growth factor family of proteins. Annu. Rev. Biochem. 1989, 58, 575–606. [Google Scholar] [CrossRef] [PubMed]
- Thomas, K.A. Transforming potential of fibroblast growth factor genes. Trends Biochem. Sci. 1988, 13, 327–328. [Google Scholar] [CrossRef]
- Ornitz, D.M.; Itoh, N. Fibroblast growth factors. Genome Biol. 2001, 2, REVIEWS3005. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Blaber, M.; DiSalvo, J.; Thomas, K.A. X-ray crystal structure of human acidic fibroblast growth factor. Biochemistry 1996, 35, 2086–2094. [Google Scholar] [CrossRef] [Green Version]
- Zechel, S.; Werner, S.; Unsicker, K.; von Bohlen und Halbach, O. Expression and functions of fibroblast growth factor 2 (FGF-2) in hippocampal formation. Neuroscientist 2010, 16, 357–373. [Google Scholar] [CrossRef]
- Tang, C.; Shan, Y.; Hu, Y.; Fang, Z.; Tong, Y.; Chen, M.; Wei, X.; Fu, X.; Xu, X. FGF2 Attenuates Neural Cell Death via Suppressing Autophagy after Rat Mild Traumatic Brain Injury. Stem Cells Int. 2017, 2017, 2923182. [Google Scholar] [CrossRef] [Green Version]
- Kardami, E.; Koleini, N. The Role of FGF2 Isoforms in Cell Survival in the Heart. In Biochemistry of Apoptosis and Autophagy; Kirshenbaum, L.A., Ed.; Springer International Publishing: Cham, Switzerland, 2022; pp. 269–283. [Google Scholar]
- Domigan, C.K.; Warren, C.M.; Antanesian, V.; Happel, K.; Ziyad, S.; Lee, S.; Krall, A.; Duan, L.; Torres-Collado, A.X.; Castellani, L.W.; et al. Autocrine VEGF maintains endothelial survival through regulation of metabolism and autophagy. J. Cell Sci. 2015, 128, 2236–2248. [Google Scholar] [CrossRef] [Green Version]
- Cardenas-Aguayo Mdel, C.; Santa-Olalla, J.; Baizabal, J.M.; Salgado, L.M.; Covarrubias, L. Growth factor deprivation induces an alternative non-apoptotic death mechanism that is inhibited by Bcl2 in cells derived from neural precursor cells. J. Hematother. Stem. Cell Res. 2003, 12, 735–748. [Google Scholar] [CrossRef]
- Klionsky, D.J.; Emr, S.D. Autophagy as a regulated pathway of cellular degradation. Science 2000, 290, 1717–1721. [Google Scholar] [CrossRef]
- Reggiori, F.; Klionsky, D.J. Autophagy in the Eukaryotic Cell. Eukaryot. Cell 2002, 1, 11–21. [Google Scholar] [CrossRef] [Green Version]
- Oppenheim, R.W.; Flavell, R.A.; Vinsant, S.; Prevette, D.; Kuan, C.Y.; Rakic, P. Programmed cell death of developing mammalian neurons after genetic deletion of caspases. J. Neurosci. 2001, 21, 4752–4760. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Paglin, S.; Hollister, T.; Delohery, T.; Hackett, N.; McMahill, M.; Sphicas, E.; Domingo, D.; Yahalom, J. A novel response of cancer cells to radiation involves autophagy and formation of acidic vesicles. Cancer Res. 2001, 61, 439–444. [Google Scholar] [PubMed]
- Yao, K.C.; Komata, T.; Kondo, Y.; Kanzawa, T.; Kondo, S.; Germano, I.M. Molecular response of human glioblastoma multiforme cells to ionizing radiation: Cell cycle arrest, modulation of the expression of cyclin-dependent kinase inhibitors, and autophagy. J. Neurosurg. 2003, 98, 378–384. [Google Scholar] [CrossRef] [PubMed]
- Kanzawa, T.; Kondo, Y.; Ito, H.; Kondo, S.; Germano, I. Induction of autophagic cell death in malignant glioma cells by arsenic trioxide. Cancer Res. 2003, 63, 2103–2108. [Google Scholar]
- Gomez-Santos, C.; Ferrer, I.; Santidrian, A.F.; Barrachina, M.; Gil, J.; Ambrosio, S. Dopamine induces autophagic cell death and alpha-synuclein increase in human neuroblastoma SH-SY5Y cells. J. Neurosci. Res. 2003, 73, 341–350. [Google Scholar] [CrossRef]
- Bursch, W.; Ellinger, A.; Kienzl, H.; Torok, L.; Pandey, S.; Sikorska, M.; Walker, R.; Hermann, R.S. Active cell death induced by the anti-estrogens tamoxifen and ICI 164 384 in human mammary carcinoma cells (MCF-7) in culture: The role of autophagy. Carcinogenesis 1996, 17, 1595–1607. [Google Scholar] [CrossRef] [Green Version]
- Chau, Y.P.; Lin, S.Y.; Chen, J.H.; Tai, M.H. Endostatin induces autophagic cell death in EAhy926 human endothelial cells. Histol. Histopathol. 2003, 18, 715–726. [Google Scholar] [CrossRef]
- Bauvy, C.; Gane, P.; Arico, S.; Codogno, P.; Ogier-Denis, E. Autophagy delays sulindac sulfide-induced apoptosis in the human intestinal colon cancer cell line HT-29. Exp. Cell Res. 2001, 268, 139–149. [Google Scholar] [CrossRef]
- Levine, B.; Kroemer, G. Autophagy in the pathogenesis of disease. Cell 2008, 132, 27–42. [Google Scholar] [CrossRef] [Green Version]
- Levine, B.; Yuan, J. Autophagy in cell death: An innocent convict? J. Clin. Investig. 2005, 115, 2679–2688. [Google Scholar] [CrossRef]
- Menzies, F.M.; Fleming, A.; Caricasole, A.; Bento, C.F.; Andrews, S.P.; Ashkenazi, A.; Fullgrabe, J.; Jackson, A.; Jimenez Sanchez, M.; Karabiyik, C.; et al. Autophagy and Neurodegeneration: Pathogenic Mechanisms and Therapeutic Opportunities. Neuron 2017, 93, 1015–1034. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Leidal, A.M.; Levine, B.; Debnath, J. Autophagy and the cell biology of age-related disease. Nat. Cell Biol. 2018, 20, 1338–1348. [Google Scholar] [CrossRef] [PubMed]
- Cuervo, A.M.; Dice, J.F. How do intracellular proteolytic systems change with age? Front. Biosci. 1998, 3, d25-43. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, Z.; Klionsky, D. Mammalian autophagy: Core molecular machinery and signaling regulation. Curr. Opin. Cell Biol. 2010, 22, 124–131. [Google Scholar] [CrossRef] [Green Version]
- Yang, X.; Pan, X.; Zhao, X.; Luo, J.; Xu, M.; Bai, D.; Hu, Y.; Liu, X.; Yu, Q.; Gao, D. Autophagy and Age-Related Eye Diseases. Biomed. Res. Int. 2019, 2019, 5763658. [Google Scholar] [CrossRef] [PubMed]
- Sancak, Y.; Bar-Peled, L.; Zoncu, R.; Markhard, A.L.; Nada, S.; Sabatini, D.M. Ragulator-Rag complex targets mTORC1 to the lysosomal surface and is necessary for its activation by amino acids. Cell 2010, 141, 290–303. [Google Scholar] [CrossRef] [Green Version]
- Sies, H.; Jones, D. Oxidative Stress. In Encyclopedia of Stress, 2nd ed.; Fink, G., Ed.; Academic Press: New York, NY, USA, 2007; pp. 45–48. [Google Scholar]
- Laurindo, F.R.M. Chapter 10—Redox Cellular Signaling Pathways in Endothelial Dysfunction and Vascular Disease. In Endothelium and Cardiovascular Diseases; Da Luz, P.L., Libby, P., Chagas, A.C.P., Laurindo, F.R.M., Eds.; Academic Press: Cambridge, MA, USA, 2018; pp. 127–145. [Google Scholar]
- Navarro-Yepes, J.; Burns, M.; Anandhan, A.; Khalimonchuk, O.; del Razo, L.M.; Quintanilla-Vega, B.; Pappa, A.; Panayiotidis, M.I.; Franco, R. Oxidative stress, redox signaling, and autophagy: Cell death versus survival. Antioxid. Redox. Signal. 2014, 21, 66–85. [Google Scholar] [CrossRef] [Green Version]
- Jung, S.; Jeong, H.; Yu, S.W. Autophagy as a decisive process for cell death. Exp. Mol. Med. 2020, 52, 921–930. [Google Scholar] [CrossRef]
- Bensaad, K.; Cheung, E.C.; Vousden, K.H. Modulation of intracellular ROS levels by TIGAR controls autophagy. EMBO J. 2009, 28, 3015–3026. [Google Scholar] [CrossRef] [Green Version]
- Zhang, H.; Bosch-Marce, M.; Shimoda, L.A.; Tan, Y.S.; Baek, J.H.; Wesley, J.B.; Gonzalez, F.J.; Semenza, G.L. Mitochondrial autophagy is an HIF-1-dependent adaptive metabolic response to hypoxia. J. Biol. Chem. 2008, 283, 10892–10903. [Google Scholar] [CrossRef] [Green Version]
- Bellot, G.; Garcia-Medina, R.; Gounon, P.; Chiche, J.; Roux, D.; Pouyssegur, J.; Mazure, N.M. Hypoxia-induced autophagy is mediated through hypoxia-inducible factor induction of BNIP3 and BNIP3L via their BH3 domains. Mol. Cell Biol. 2009, 29, 2570–2581. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rubio, N.; Verrax, J.; Dewaele, M.; Verfaillie, T.; Johansen, T.; Piette, J.; Agostinis, P. p38(MAPK)-regulated induction of p62 and NBR1 after photodynamic therapy promotes autophagic clearance of ubiquitin aggregates and reduces reactive oxygen species levels by supporting Nrf2-antioxidant signaling. Free Radic. Biol. Med. 2014, 67, 292–303. [Google Scholar] [CrossRef] [PubMed]
- Aucello, M.; Dobrowolny, G.; Musaro, A. Localized accumulation of oxidative stress causes muscle atrophy through activation of an autophagic pathway. Autophagy 2009, 5, 527–529. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jain, A.; Lamark, T.; Sjottem, E.; Larsen, K.B.; Awuh, J.A.; Overvatn, A.; McMahon, M.; Hayes, J.D.; Johansen, T. p62/SQSTM1 is a target gene for transcription factor NRF2 and creates a positive feedback loop by inducing antioxidant response element-driven gene transcription. J. Biol. Chem. 2010, 285, 22576–22591. [Google Scholar] [CrossRef] [Green Version]
- Kim, I.; Rodriguez-Enriquez, S.; Lemasters, J.J. Selective degradation of mitochondria by mitophagy. Arch. Biochem. Biophys. 2007, 462, 245–253. [Google Scholar] [CrossRef] [Green Version]
- Levine, B. Eating oneself and uninvited guests: Autophagy-related pathways in cellular defense. Cell 2005, 120, 159–162. [Google Scholar] [CrossRef] [Green Version]
- Thurston, T.L.; Ryzhakov, G.; Bloor, S.; von Muhlinen, N.; Randow, F. The TBK1 adaptor and autophagy receptor NDP52 restricts the proliferation of ubiquitin-coated bacteria. Nat. Immunol. 2009, 10, 1215–1221. [Google Scholar] [CrossRef]
- Deretic, V. Autophagy as an innate immunity paradigm: Expanding the scope and repertoire of pattern recognition receptors. Curr. Opin. Immunol. 2012, 24, 21–31. [Google Scholar] [CrossRef] [Green Version]
- Nakagawa, I.; Amano, A.; Mizushima, N.; Yamamoto, A.; Yamaguchi, H.; Kamimoto, T.; Nara, A.; Funao, J.; Nakata, M.; Tsuda, K.; et al. Autophagy defends cells against invading group A Streptococcus. Science 2004, 306, 1037–1040. [Google Scholar] [CrossRef]
- Birmingham, C.L.; Smith, A.C.; Bakowski, M.A.; Yoshimori, T.; Brumell, J.H. Autophagy controls Salmonella infection in response to damage to the Salmonella-containing vacuole. J. Biol. Chem. 2006, 281, 11374–11383. [Google Scholar] [CrossRef] [Green Version]
- Yu, X.; Li, C.; Hong, W.; Pan, W.; Xie, J. Autophagy during Mycobacterium tuberculosis infection and implications for future tuberculosis medications. Cell Signal 2013, 25, 1272–1278. [Google Scholar] [CrossRef] [PubMed]
- Amer, A.O.; Swanson, M.S. Autophagy is an immediate macrophage response to Legionella pneumophila. Cell Microbiol. 2005, 7, 765–778. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ogawa, M.; Yoshimori, T.; Suzuki, T.; Sagara, H.; Mizushima, N.; Sasakawa, C. Escape of intracellular Shigella from autophagy. Science 2005, 307, 727–731. [Google Scholar] [CrossRef] [PubMed]
- Ogawa, M.; Yoshikawa, Y.; Kobayashi, T.; Mimuro, H.; Fukumatsu, M.; Kiga, K.; Piao, Z.; Ashida, H.; Yoshida, M.; Kakuta, S.; et al. A Tecpr1-dependent selective autophagy pathway targets bacterial pathogens. Cell Host Microbe 2011, 9, 376–389. [Google Scholar] [CrossRef] [Green Version]
- Dorn, B.R.; Dunn, W.A., Jr.; Progulske-Fox, A. Porphyromonas gingivalis traffics to autophagosomes in human coronary artery endothelial cells. Infect. Immun. 2001, 69, 5698–5708. [Google Scholar] [CrossRef] [Green Version]
- Ling, Y.M.; Shaw, M.H.; Ayala, C.; Coppens, I.; Taylor, G.A.; Ferguson, D.J.; Yap, G.S. Vacuolar and plasma membrane stripping and autophagic elimination of Toxoplasma gondii in primed effector macrophages. J. Exp. Med. 2006, 203, 2063–2071. [Google Scholar] [CrossRef]
- Salassa, B.N.; Romano, P.S. Autophagy: A necessary process during the Trypanosoma cruzi life-cycle. Virulence 2019, 10, 460–469. [Google Scholar] [CrossRef] [Green Version]
- Nicolao, M.C.; Loos, J.A.; Rodriguez Rodrigues, C.; Beas, V.; Cumino, A.C. Bortezomib initiates endoplasmic reticulum stress, elicits autophagy and death in Echinococcus granulosus larval stage. PLoS ONE 2017, 12, e0181528. [Google Scholar] [CrossRef] [Green Version]
- Kirkegaard, K. Subversion of the cellular autophagy pathway by viruses. Curr. Top. Microbiol. Immunol. 2009, 335, 323–333. [Google Scholar] [CrossRef]
- Cirone, M. EBV and KSHV Infection Dysregulates Autophagy to Optimize Viral Replication, Prevent Immune Recognition and Promote Tumorigenesis. Viruses 2018, 10, 599. [Google Scholar] [CrossRef] [Green Version]
- Blanchet, F.P.; Moris, A.; Nikolic, D.S.; Lehmann, M.; Cardinaud, S.; Stalder, R.; Garcia, E.; Dinkins, C.; Leuba, F.; Wu, L.; et al. Human immunodeficiency virus-1 inhibition of immunoamphisomes in dendritic cells impairs early innate and adaptive immune responses. Immunity 2010, 32, 654–669. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Orvedahl, A.; Alexander, D.; Talloczy, Z.; Sun, Q.; Wei, Y.; Zhang, W.; Burns, D.; Leib, D.A.; Levine, B. HSV-1 ICP34.5 confers neurovirulence by targeting the Beclin 1 autophagy protein. Cell Host Microbe 2007, 1, 23–35. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liang, Q.; Chang, B.; Brulois, K.F.; Castro, K.; Min, C.K.; Rodgers, M.A.; Shi, M.; Ge, J.; Feng, P.; Oh, B.H.; et al. Kaposi’s sarcoma-associated herpesvirus K7 modulates Rubicon-mediated inhibition of autophagosome maturation. J. Virol. 2013, 87, 12499–12503. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gassen, N.C.; Papies, J.; Bajaj, T.; Emanuel, J.; Dethloff, F.; Chua, R.L.; Trimpert, J.; Heinemann, N.; Niemeyer, C.; Weege, F.; et al. SARS-CoV-2-mediated dysregulation of metabolism and autophagy uncovers host-targeting antivirals. Nat. Commun. 2021, 12, 3818. [Google Scholar] [CrossRef]
- Miao, G.; Zhao, H.; Li, Y.; Ji, M.; Chen, Y.; Shi, Y.; Bi, Y.; Wang, P.; Zhang, H. ORF3a of the COVID-19 virus SARS-CoV-2 blocks HOPS complex-mediated assembly of the SNARE complex required for autolysosome formation. Dev. Cell 2021, 56, 427–442.e425. [Google Scholar] [CrossRef] [PubMed]
- Galluzzi, L.; Bravo-San Pedro, J.M.; Levine, B.; Green, D.R.; Kroemer, G. Pharmacological modulation of autophagy: Therapeutic potential and persisting obstacles. Nat. Rev. Drug Discov. 2017, 16, 487–511. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Amaravadi, R.; Kimmelman, A.C.; White, E. Recent insights into the function of autophagy in cancer. Genes Dev. 2016, 30, 1913–1930. [Google Scholar] [CrossRef] [PubMed]
- Menzies, F.M.; Fleming, A.; Rubinsztein, D.C. Compromised autophagy and neurodegenerative diseases. Nat. Rev. Neurosci. 2015, 16, 345–357. [Google Scholar] [CrossRef]
- Fornai, F.; Longone, P.; Ferrucci, M.; Lenzi, P.; Isidoro, C.; Ruggieri, S.; Paparelli, A. Autophagy and amyotrophic lateral sclerosis: The multiple roles of lithium. Autophagy 2008, 4, 527–530. [Google Scholar] [CrossRef] [Green Version]
- Fornai, F.; Longone, P.; Cafaro, L.; Kastsiuchenka, O.; Ferrucci, M.; Manca, M.L.; Lazzeri, G.; Spalloni, A.; Bellio, N.; Lenzi, P.; et al. Lithium delays progression of amyotrophic lateral sclerosis. Proc. Natl. Acad. Sci. USA 2008, 105, 2052–2057. [Google Scholar] [CrossRef] [Green Version]
- Natale, G.; Lenzi, P.; Lazzeri, G.; Falleni, A.; Biagioni, F.; Ryskalin, L.; Fornai, F. Compartment-dependent mitochondrial alterations in experimental ALS, the effects of mitophagy and mitochondriogenesis. Front. Cell Neurosci. 2015, 9, 434. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bonaldo, P.; Sandri, M. Cellular and molecular mechanisms of muscle atrophy. Dis. Model Mech. 2013, 6, 25–39. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, H.; Li, D.; Ma, Z.; Qian, Z.; Kang, X.; Jin, X.; Li, F.; Wang, X.; Chen, Q.; Sun, H.; et al. Defective autophagy in osteoblasts induces endoplasmic reticulum stress and causes remarkable bone loss. Autophagy 2018, 14, 1726–1741. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rezaei, S.; Mahjoubin-Tehran, M.; Aghaee-Bakhtiari, S.H.; Jalili, A.; Movahedpour, A.; Khan, H.; Moghoofei, M.; Shojaei, Z.; Mirzaei, H. Autophagy-related MicroRNAs in chronic lung diseases and lung cancer. Crit. Rev. Oncol. Hematol. 2020, 153, 103063. [Google Scholar] [CrossRef]
- Ueno, T.; Komatsu, M. Autophagy in the liver: Functions in health and disease. Nat. Rev. Gastroenterol. Hepatol. 2017, 14, 170–184. [Google Scholar] [CrossRef]
- Gukovskaya, A.S.; Gukovsky, I.; Algul, H.; Habtezion, A. Autophagy, Inflammation, and Immune Dysfunction in the Pathogenesis of Pancreatitis. Gastroenterology 2017, 153, 1212–1226. [Google Scholar] [CrossRef] [Green Version]
- Biczo, G.; Vegh, E.T.; Shalbueva, N.; Mareninova, O.A.; Elperin, J.; Lotshaw, E.; Gretler, S.; Lugea, A.; Malla, S.R.; Dawson, D.; et al. Mitochondrial Dysfunction, Through Impaired Autophagy, Leads to Endoplasmic Reticulum Stress, Deregulated Lipid Metabolism, and Pancreatitis in Animal Models. Gastroenterology 2018, 154, 689–703. [Google Scholar] [CrossRef] [Green Version]
- Lassen, K.G.; Xavier, R.J. Mechanisms and function of autophagy in intestinal disease. Autophagy 2018, 14, 216–220. [Google Scholar] [CrossRef] [Green Version]
- Du, J.; Li, Y.; Zhao, W. Autophagy and Myocardial Ischemia. Adv. Exp. Med. Biol. 2020, 1207, 217–222. [Google Scholar] [CrossRef]
- Yamaguchi, O. Autophagy in the Heart. Circ. J. 2019, 83, 697–704. [Google Scholar] [CrossRef] [Green Version]
- Bello-Perez, M.; Sola, I.; Novoa, B.; Klionsky, D.J.; Falco, A. Canonical and Noncanonical Autophagy as Potential Targets for COVID-19. Cells 2020, 9, 1619. [Google Scholar] [CrossRef] [PubMed]
- Levine, B.; Kroemer, G. Biological Functions of Autophagy Genes: A Disease Perspective. Cell 2019, 176, 11–42. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- White, E.; Mehnert, J.M.; Chan, C.S. Autophagy, Metabolism, and Cancer. Clin. Cancer Res. 2015, 21, 5037–5046. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, X.; He, S.; Ma, B. Autophagy and autophagy-related proteins in cancer. Mol. Cancer 2020, 19, 12. [Google Scholar] [CrossRef]
- Reddy, P.H.; Yin, X.; Manczak, M.; Kumar, S.; Pradeepkiran, J.A.; Vijayan, M.; Reddy, A.P. Mutant APP and amyloid beta-induced defective autophagy, mitophagy, mitochondrial structural and functional changes and synaptic damage in hippocampal neurons from Alzheimer’s disease. Hum. Mol. Genet. 2018, 27, 2502–2516. [Google Scholar] [CrossRef]
- Reddy, P.H.; Oliver, D.M. Amyloid Beta and Phosphorylated Tau-Induced Defective Autophagy and Mitophagy in Alzheimer’s Disease. Cells 2019, 8, 488. [Google Scholar] [CrossRef] [Green Version]
- Jiao, J.; Demontis, F. Skeletal muscle autophagy and its role in sarcopenia and organismal aging. Curr. Opin. Pharm. 2017, 34, 1–6. [Google Scholar] [CrossRef]
- Li, Y.; Jiang, J.; Liu, W.; Wang, H.; Zhao, L.; Liu, S.; Li, P.; Zhang, S.; Sun, C.; Wu, Y.; et al. microRNA-378 promotes autophagy and inhibits apoptosis in skeletal muscle. Proc. Natl. Acad. Sci. USA 2018, 115, E10849–E10858. [Google Scholar] [CrossRef] [Green Version]
- Masiero, E.; Agatea, L.; Mammucari, C.; Blaauw, B.; Loro, E.; Komatsu, M.; Metzger, D.; Reggiani, C.; Schiaffino, S.; Sandri, M. Autophagy is required to maintain muscle mass. Cell Metab. 2009, 10, 507–515. [Google Scholar] [CrossRef]
- Wang, T.; Liu, X.; He, C. Glucocorticoid-induced autophagy and apoptosis in bone. Apoptosis 2020, 25, 157–168. [Google Scholar] [CrossRef]
- Ma, Y.; Qi, M.; An, Y.; Zhang, L.; Yang, R.; Doro, D.H.; Liu, W.; Jin, Y. Autophagy controls mesenchymal stem cell properties and senescence during bone aging. Aging Cell 2018, 17, e12709. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nollet, M.; Santucci-Darmanin, S.; Breuil, V.; Al-Sahlanee, R.; Cros, C.; Topi, M.; Momier, D.; Samson, M.; Pagnotta, S.; Cailleteau, L.; et al. Autophagy in osteoblasts is involved in mineralization and bone homeostasis. Autophagy 2014, 10, 1965–1977. [Google Scholar] [CrossRef] [PubMed]
- Racanelli, A.C.; Kikkers, S.A.; Choi, A.M.K.; Cloonan, S.M. Autophagy and inflammation in chronic respiratory disease. Autophagy 2018, 14, 221–232. [Google Scholar] [CrossRef] [PubMed]
- Liao, S.X.; Sun, P.P.; Gu, Y.H.; Rao, X.M.; Zhang, L.Y.; Ou-Yang, Y. Autophagy and pulmonary disease. Adv. Respir. Dis. 2019, 13, 1753466619890538. [Google Scholar] [CrossRef]
- Qu, L.; Chen, C.; Chen, Y.; Li, Y.; Tang, F.; Huang, H.; He, W.; Zhang, R.; Shen, L. High-Mobility Group Box 1 (HMGB1) and Autophagy in Acute Lung Injury (ALI): A Review. Med. Sci. Monit. 2019, 25, 1828–1837. [Google Scholar] [CrossRef]
- Allaire, M.; Rautou, P.E.; Codogno, P.; Lotersztajn, S. Autophagy in liver diseases: Time for translation? J. Hepatol. 2019, 70, 985–998. [Google Scholar] [CrossRef] [Green Version]
- Chao, X.; Wang, H.; Jaeschke, H.; Ding, W.X. Role and mechanisms of autophagy in acetaminophen-induced liver injury. Liver Int. 2018, 38, 1363–1374. [Google Scholar] [CrossRef] [Green Version]
- Akkoc, Y.; Gozuacik, D. Autophagy and liver cancer. Turk. J. Gastroenterol. 2018, 29, 270–282. [Google Scholar] [CrossRef]
- Sudhakar, J.N.; Lu, H.H.; Chiang, H.Y.; Suen, C.S.; Hwang, M.J.; Wu, S.Y.; Shen, C.N.; Chang, Y.M.; Li, F.A.; Liu, F.T.; et al. Lumenal Galectin-9-Lamp2 interaction regulates lysosome and autophagy to prevent pathogenesis in the intestine and pancreas. Nat. Commun. 2020, 11, 4286. [Google Scholar] [CrossRef]
- Matsuzawa-Ishimoto, Y.; Shono, Y.; Gomez, L.E.; Hubbard-Lucey, V.M.; Cammer, M.; Neil, J.; Dewan, M.Z.; Lieberman, S.R.; Lazrak, A.; Marinis, J.M.; et al. Autophagy protein ATG16L1 prevents necroptosis in the intestinal epithelium. J. Exp. Med. 2017, 214, 3687–3705. [Google Scholar] [CrossRef]
- Mehto, S.; Jena, K.K.; Nath, P.; Chauhan, S.; Kolapalli, S.P.; Das, S.K.; Sahoo, P.K.; Jain, A.; Taylor, G.A.; Chauhan, S. The Crohn’s Disease Risk Factor IRGM Limits NLRP3 Inflammasome Activation by Impeding Its Assembly and by Mediating Its Selective Autophagy. Mol. Cell 2019, 73, 429–445.e427. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Larabi, A.; Barnich, N.; Nguyen, H.T.T. New insights into the interplay between autophagy, gut microbiota and inflammatory responses in IBD. Autophagy 2020, 16, 38–51. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sciarretta, S.; Maejima, Y.; Zablocki, D.; Sadoshima, J. The Role of Autophagy in the Heart. Annu. Rev. Physiol. 2018, 80, 1–26. [Google Scholar] [CrossRef] [PubMed]
- Yang, N.; Shen, H.-M. Targeting the Endocytic Pathway and Autophagy Process as a Novel Therapeutic Strategy in COVID-19. Int. J. Biol. Sci. 2020, 16, 1724–1731. [Google Scholar] [CrossRef]
- Chen, X.; Wang, K.; Xing, Y.; Tu, J.; Yang, X.; Zhao, Q.; Li, K.; Chen, Z. Coronavirus membrane-associated papain-like proteases induce autophagy through interacting with Beclin1 to negatively regulate antiviral innate immunity. Protein Cell 2014, 5, 912–927. [Google Scholar] [CrossRef] [Green Version]
- Gassen, N.C.; Niemeyer, D.; Muth, D.; Corman, V.M.; Martinelli, S.; Gassen, A.; Hafner, K.; Papies, J.; Mosbauer, K.; Zellner, A.; et al. SKP2 attenuates autophagy through Beclin1-ubiquitination and its inhibition reduces MERS-Coronavirus infection. Nat. Commun. 2019, 10, 5770. [Google Scholar] [CrossRef]
- Bursch, W. The autophagosomal-lysosomal compartment in programmed cell death. Cell Death Differ. 2001, 8, 569–581. [Google Scholar] [CrossRef]
- Cornillon, S.; Foa, C.; Davoust, J.; Buonavista, N.; Gross, J.D.; Golstein, P. Programmed cell death in Dictyostelium. J. Cell Sci. 1994, 107 Pt 10, 2691–2704. [Google Scholar] [CrossRef]
- Hall, D.H.; Gu, G.; Garcia-Anoveros, J.; Gong, L.; Chalfie, M.; Driscoll, M. Neuropathology of degenerative cell death in Caenorhabditis elegans. J. Neurosci. 1997, 17, 1033–1045. [Google Scholar] [CrossRef]
- Hinchliffe, J.R. Cell Death in Embryogenesis. In Cell Death in Biology and Pathology; Bowen, I.D., Lockshin, R.A., Eds.; Chapman and Hall: London, UK, 1981; pp. 35–77. [Google Scholar]
- Bowen, I.D.; Mullarkey, K.; Morgan, S.M. Programmed cell death during metamorphosis in the blow-fly Calliphora vomitoria. Microsc. Res. Tech. 1996, 34, 202–217. [Google Scholar] [CrossRef]
- Baehrecke, E.H. Autophagic programmed cell death in Drosophila. Cell Death Differ. 2003, 10, 940–945. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chautan, M.; Chazal, G.; Cecconi, F.; Gruss, P.; Golstein, P. Interdigital cell death can occur through a necrotic and caspase-independent pathway. Curr. Biol. 1999, 9, 967–970. [Google Scholar] [CrossRef] [Green Version]
- Yaginuma, H.; Shiraiwa, N.; Shimada, T.; Nishiyama, K.; Hong, J.; Wang, S.; Momoi, T.; Uchiyama, Y.; Oppenheim, R.W. Caspase activity is involved in, but is dispensable for, early motoneuron death in the chick embryo cervical spinal cord. Mol. Cell Neurosci. 2001, 18, 168–182. [Google Scholar] [CrossRef]
- Tanaka, K.; Chiba, T. The proteasome: A protein-destroying machine. Genes Cells 1998, 3, 499–510. [Google Scholar] [CrossRef] [PubMed]
- Assuncao Guimaraes, C.; Linden, R. Programmed cell deaths. Apoptosis and alternative deathstyles. Eur. J. Biochem. 2004, 271, 1638–1650. [Google Scholar] [CrossRef] [PubMed]
- Bera, A.; Singh, S.; Nagaraj, R.; Vaidya, T. Induction of autophagic cell death in Leishmania donovani by antimicrobial peptides. Mol. Biochem. Parasitol. 2003, 127, 23–35. [Google Scholar] [CrossRef]
- Dunn, W.A., Jr. Studies on the mechanisms of autophagy: Formation of the autophagic vacuole. J. Cell Biol. 1990, 110, 1923–1933. [Google Scholar] [CrossRef] [Green Version]
- Ravikumar, B.; Duden, R.; Rubinsztein, D.C. Aggregate-prone proteins with polyglutamine and polyalanine expansions are degraded by autophagy. Hum. Mol. Genet. 2002, 11, 1107–1117. [Google Scholar] [CrossRef] [Green Version]
- Nixon, R.A.; Cataldo, A.M.; Mathews, P.M. The endosomal-lysosomal system of neurons in Alzheimer’s disease pathogenesis: A review. Neurochem. Res. 2000, 25, 1161–1172. [Google Scholar] [CrossRef]
- Qin, Z.H.; Wang, Y.; Kegel, K.B.; Kazantsev, A.; Apostol, B.L.; Thompson, L.M.; Yoder, J.; Aronin, N.; DiFiglia, M. Autophagy regulates the processing of amino terminal huntingtin fragments. Hum. Mol. Genet. 2003, 12, 3231–3244. [Google Scholar] [CrossRef] [Green Version]
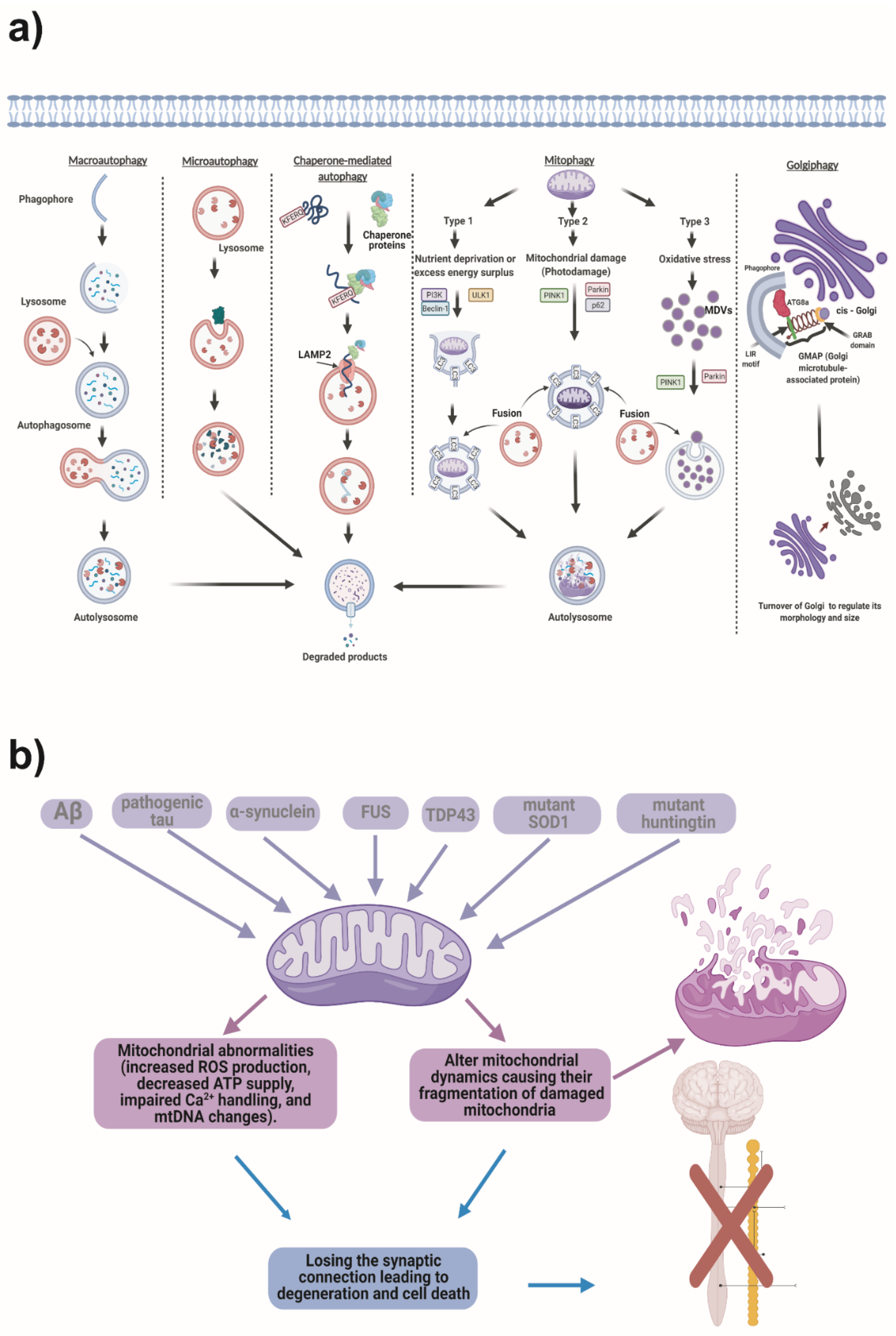

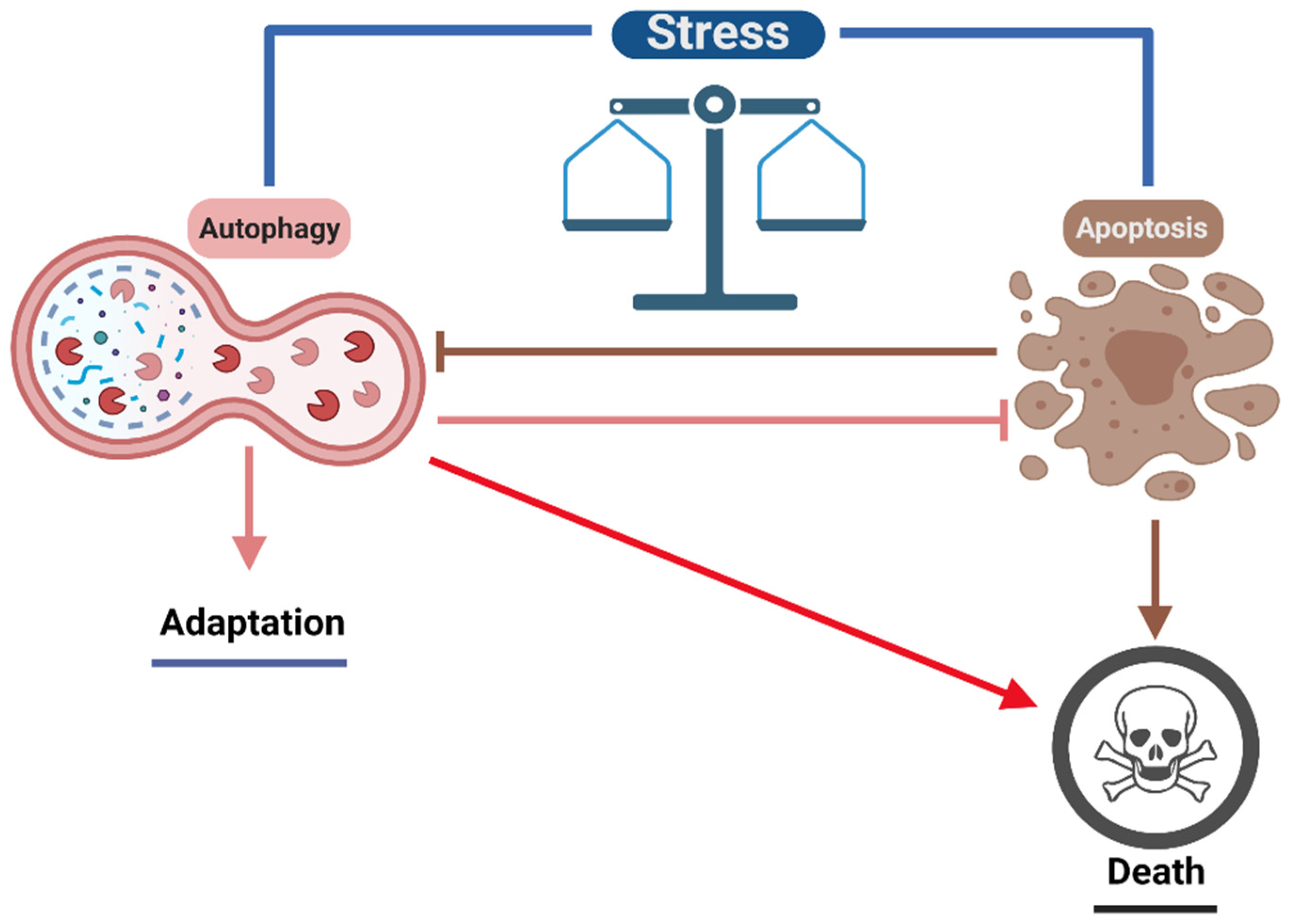
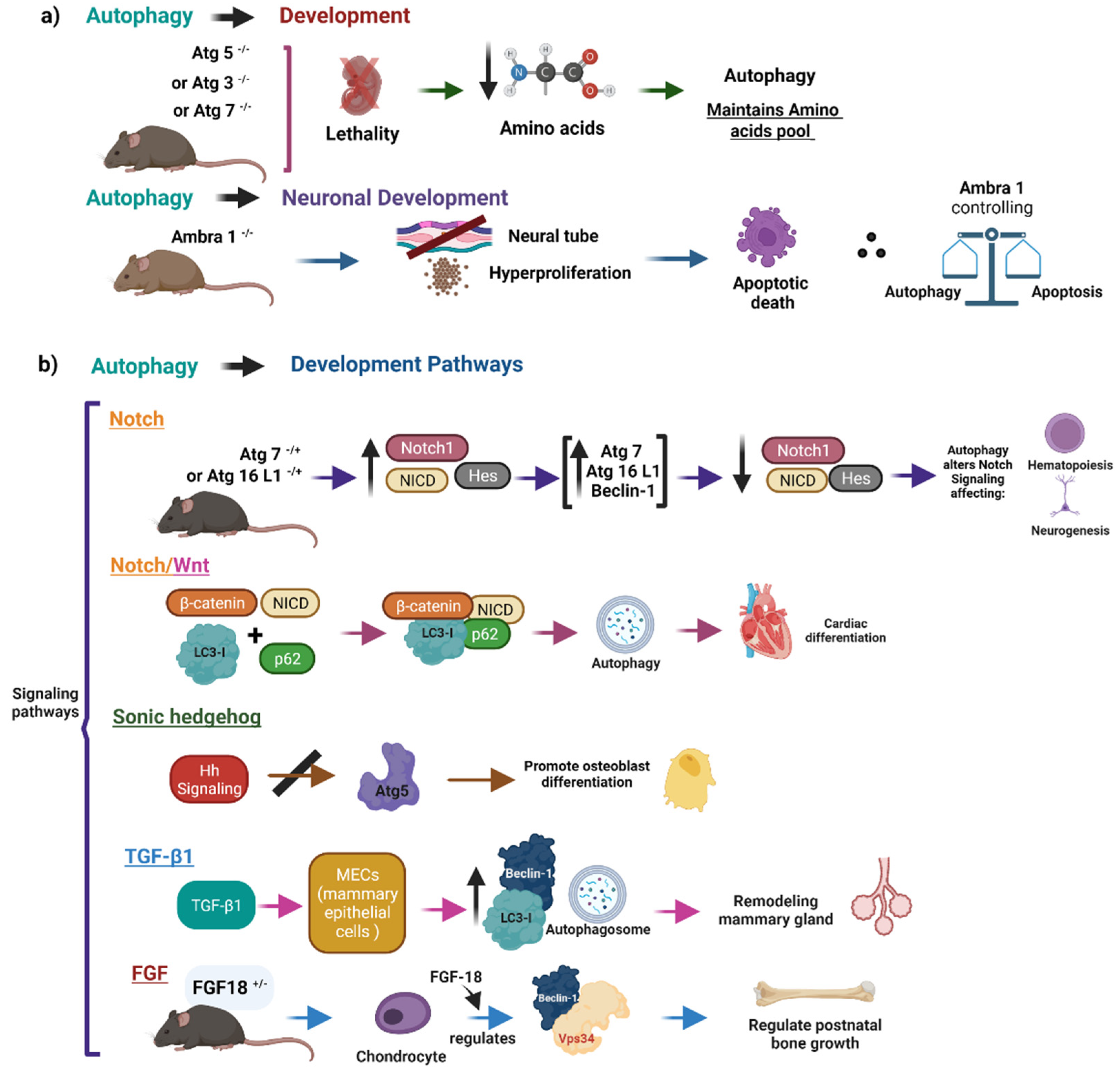
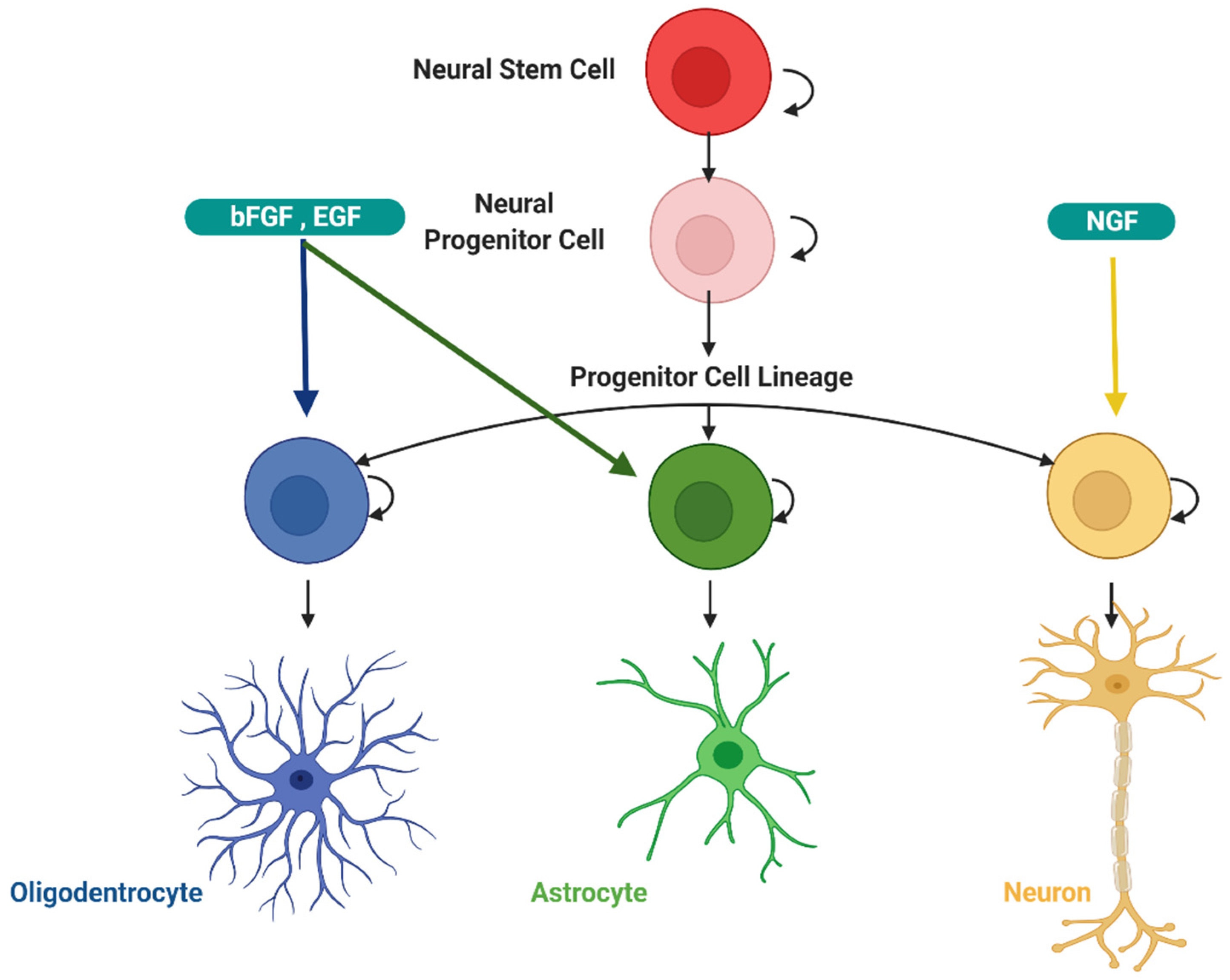


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Physiological Functions of Autophagy | Description | References |
|---|---|---|
| Energy Production | Autophagy can contribute to the mobilization of diverse energy molecular storages, regulating glucose metabolism, mobilization of neutral lipids, or is responsible for intracellular protein breakdown. | [5,6,7] |
| Intracellular Maintenance | Autophagy can contribute to the quality control and replacement of cellular components. | [8,9,10] |
| Signaling | Autophagy could, in some cases, distinguish its cargos, leaving the other cellular components unaffected because they are labeled with poly-Ub chains for degradation, a level of regulation associated with signaling. | [11,12,13] |
| Development and Differentiation | Autophagy can drive the rapid response of cells necessary for the development and play an indispensable role in differentiation. It provides the cells with the energy needed for differentiation and participates in tissue remodeling. | [14,15,16] |
| Autophagy Types | Mechanism | References |
|---|---|---|
| Macroautophagy | The isolation membrane sequesters cytosolic material to generate the autophagosome, which binds to the lysosome giving rise to the autolysosome, where cargo is degraded. | [24,76,77] |
| Microautophagy | The cytoplasmic material directly enters the lysosomal through invaginations of the lysosomal membrane. | [22,38,78] |
| Chaperone Mediated Autophagy | Labeled proteins with the pentapeptide KFERQ are recognized by Hsc70 and other chaperones that unfold them and then translocate them to the lysosomal lumen throw LAMP2. | [42,79,80] |
| Mitophagy | Dysfunctional or damaged mitochondria are degraded through the Parkin-dependent and independent pathways. | [81,82,83] |
| Golgiphagy | Golgiphagy is a selective type of autophagy, that regulates Golgi Complex Turnover. The GMAP (Golgi microtubule-associated protein) protein interacts with Atg8a and the LIR motif at position 320P-325, which is important for docking between the phagophore and the Golgi complex. | [75] |
| Stage | Yeast Proteins | Mammalian Homologous | Regulatory Function in Autophagy | Interaction |
|---|---|---|---|---|
| Induction | ATG1 | ULK1, ULK2 | Interacts with mTORC1; necessary initiation of autophagy regulation | ATG13, ATG1, ATG17 |
| ATG13 | ATG13 | Control autophagy induction modulating enzyme activity and cellular localization of ULK1 | ULK1, ULK2, FIP200 | |
| ATG17 | FIP-200 | Autophagy initiator | ULK1, ATG13, ATG101 | |
| Nucleation | ATG6 | Beclin 1 | Bcl-2 binding protein creates a regulatory complex with PI3K class III (VPS34). | Vps34, Pl3K, UVRAG |
| ATG9 | ATG9A | Associates to the pre-autophagosome structure. In yeast, it helps assemble the autophagosome. | ATG2 | |
| ATG9B | ||||
| ATG14 | ATG14L | Autophagy specific subunit from the complex of PI3K Class III and Beclin-1 | LC3 | |
| ATG18 | WIPI1-4 | It binds to PI3P, possible participation in the nucleation stage. | DFCP1 | |
| Elongation | ATG3 | ATG3 | Ubiquitin E2 like enzyme acts as ligase of ATG8 and ATG12 and catalyzes the conjugation of ATG8 similar proteins to phosphatidylethanolamine (PE). | ATG7, ATG8, ATG12 |
| ATG4 | ATG4A-D | ATG8 cysteine peptidase converts pro-LC3 (ATG8) into LC3-I and delipidated autophagosomal LC3-II. | ATG8 | |
| ATG5 | ATG5 | ATG5 makes a complex with ATG12 and helps with the autophagosome elongation. | ATG12, ATG16L | |
| ATG7 | ATG7 | Ubiquitin E1 conjugase-like enzyme helps conjugate ATG8 to PE and acts as E1 enzyme for the conjugation of ATG12 to ATG5 and ATG3. | ATG8, ATG3, ATG12 | |
| ATG8 | LC3A, | Ubiquitin-like modifier; it associates with a stable component for the autophagosomal membrane | ATG3, ATG4, ATG7 | |
| LC3B, | ||||
| LC3C | ||||
| ATG10 | ATG10 | Ubiquitin E2 type enzyme catalyzes the conjugation of ATG5 and ATG12. | ATG12, ATG16L | |
| ATG12 | ATG12 | Complex with ATG5 and helps in the autophagosomal elongation. | ATG3, ATG5, ATG7, ATG10, ATG16 | |
| ATG16 | ATG16L1/L2 | Associated with the isolation membrane, making a complex with Atg5-Atg12, helps in autophagosomal elongation. | ATG5, ATG12 |
| Role of Autophagy in Diseases | |||
|---|---|---|---|
| Organ | Diseases | Autophagy Function | References |
| whole body (general function) | Tumor suppression and progression. | Selective degradation of p62, damaged mitochondria, and microbes; starvation-induced amino acid production; recycling of cytoplasmic content. | [4,301,302,303] |
| Brain | AD, amyotrophic lateral sclerosis (ALS), frontotemporal dementia, HD, and PD. | Prevention in the formation of aggregates, Parkin-dependent mitophagy, nutrient regulation, and energy balance. | [30,287,301,304,305] |
| Muscle | Danon disease, X-linked myopathy (XMEA), Ullrich congenital muscular dystrophy, Bethlem myopathy, sarcopenia. | Maintains muscle mass. | [291,306,307,308] |
| Bone | Paget’s disease, osteopetrosis, and osteopenia. | Bone metabolism. Terminal differentiation of osteoblasts. Maintains bone mass. | [292,309,310,311] |
| Lung | Cystic fibrosis, pulmonary tuberculosis, pulmonary arterial hypertension, and lung cancer. | Regulation of the responsiveness of the airways. Drive and regulate inflammatory responses in chronic lung diseases. | [293,312,313,314] |
| Liver | Hepatocellular carcinoma, alcoholic ad nonalcoholic fatty liver disease, α-antitrypsin deficiency. Viral hepatitis. | Adaptation to starvation through induction of glycogenolysis, lipolysis and protein catabolism, prevention of hepatocellular degeneration, and suppression of liver tumors. | [294,315,316,317] |
| Pancreas | Pancreatitis, and diabetes. | Adaptation of B cells to a diet rich in fat; prevention of trypsin autoactivation. | [295,296,301,318] |
| Gut | Crohn’s disease. | Maintenance of Paneth cell function. | [297,319,320,321] |
| Heart | Heart failure and atherosclerosis. | Adaptation to hemodynamic stress; prevention of age-dependent dysfunction. | [4,298,299,322] |
| Multi-Organ | COVID-19 | Inhibition of the Autophagy flux by SARS-CoV-2. | [300,323,324,325] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gómez-Virgilio, L.; Silva-Lucero, M.-d.-C.; Flores-Morelos, D.-S.; Gallardo-Nieto, J.; Lopez-Toledo, G.; Abarca-Fernandez, A.-M.; Zacapala-Gómez, A.-E.; Luna-Muñoz, J.; Montiel-Sosa, F.; Soto-Rojas, L.O.; et al. Autophagy: A Key Regulator of Homeostasis and Disease: An Overview of Molecular Mechanisms and Modulators. Cells 2022, 11, 2262. https://doi.org/10.3390/cells11152262
Gómez-Virgilio L, Silva-Lucero M-d-C, Flores-Morelos D-S, Gallardo-Nieto J, Lopez-Toledo G, Abarca-Fernandez A-M, Zacapala-Gómez A-E, Luna-Muñoz J, Montiel-Sosa F, Soto-Rojas LO, et al. Autophagy: A Key Regulator of Homeostasis and Disease: An Overview of Molecular Mechanisms and Modulators. Cells. 2022; 11(15):2262. https://doi.org/10.3390/cells11152262
Chicago/Turabian StyleGómez-Virgilio, Laura, Maria-del-Carmen Silva-Lucero, Diego-Salvador Flores-Morelos, Jazmin Gallardo-Nieto, Gustavo Lopez-Toledo, Arminda-Mercedes Abarca-Fernandez, Ana-Elvira Zacapala-Gómez, José Luna-Muñoz, Francisco Montiel-Sosa, Luis O. Soto-Rojas, and et al. 2022. "Autophagy: A Key Regulator of Homeostasis and Disease: An Overview of Molecular Mechanisms and Modulators" Cells 11, no. 15: 2262. https://doi.org/10.3390/cells11152262
APA StyleGómez-Virgilio, L., Silva-Lucero, M.-d.-C., Flores-Morelos, D.-S., Gallardo-Nieto, J., Lopez-Toledo, G., Abarca-Fernandez, A.-M., Zacapala-Gómez, A.-E., Luna-Muñoz, J., Montiel-Sosa, F., Soto-Rojas, L. O., Pacheco-Herrero, M., & Cardenas-Aguayo, M.-d.-C. (2022). Autophagy: A Key Regulator of Homeostasis and Disease: An Overview of Molecular Mechanisms and Modulators. Cells, 11(15), 2262. https://doi.org/10.3390/cells11152262