
SLC35A2 Deficiency Promotes an Epithelial-to-Mesenchymal Transition-like Phenotype in Madin–Darby Canine Kidney Cells

 , , , , , and
, , , , , and {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Cell Culture Maintenance
2.2. CRISPR/Cas9-Assisted Gene Inactivation
2.3. Immunofluorescence and Fluorescence Staining
2.4. Fluorescence Lectin Staining
2.5. In Situ Proximity Ligation Assay (PLA)
2.6. Western Blotting and Lectin Blotting
2.7. SYBR Green-Based Quantitative PCR
2.8. Spontaneous Migration Assay
2.9. In Vitro Invasion Assay
2.10. Image Processing
2.11. Statistical Analysis
3. Results
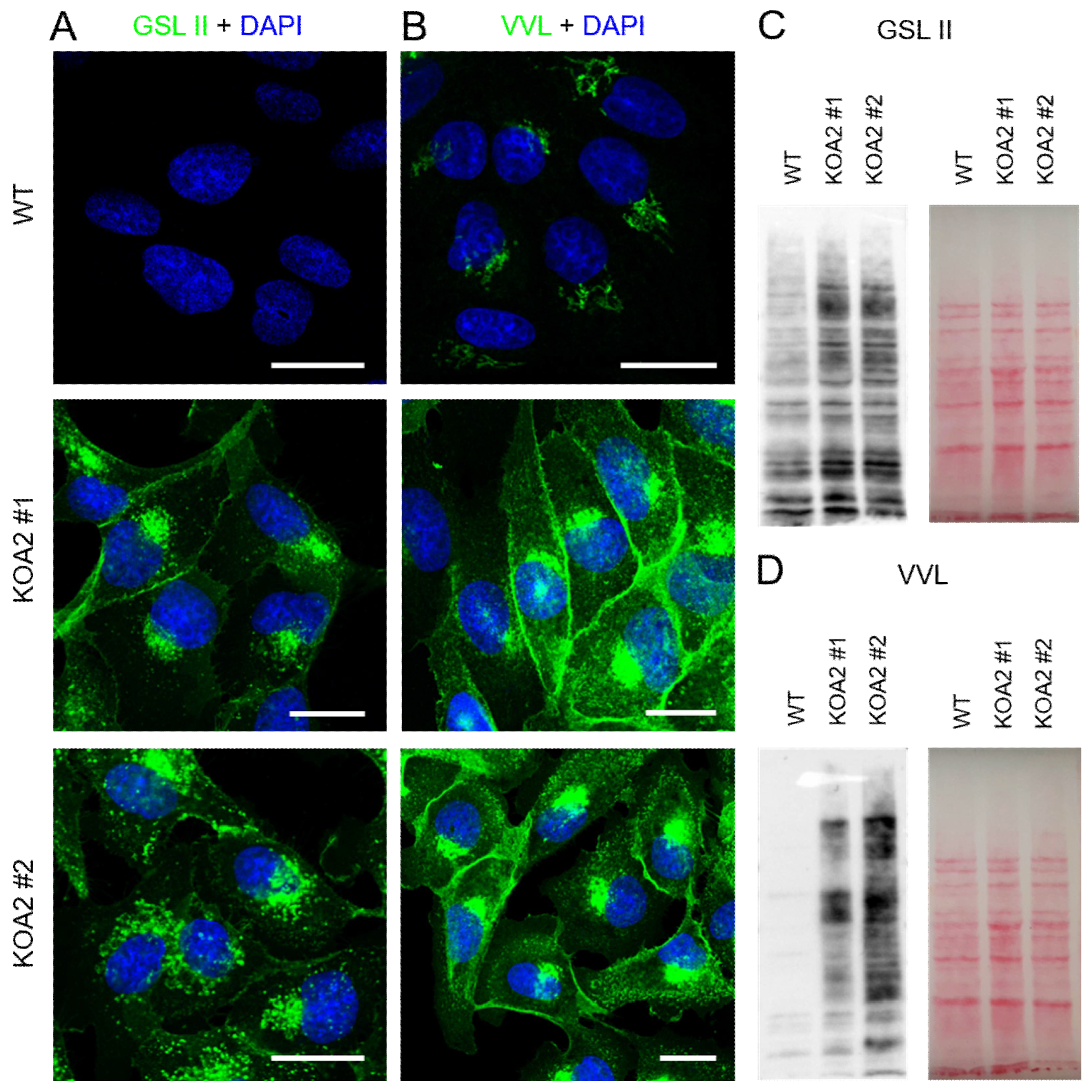
3.1. Wild-Type and SLC35A2-Deficient MDCK Cells Display Distinct Morphologies
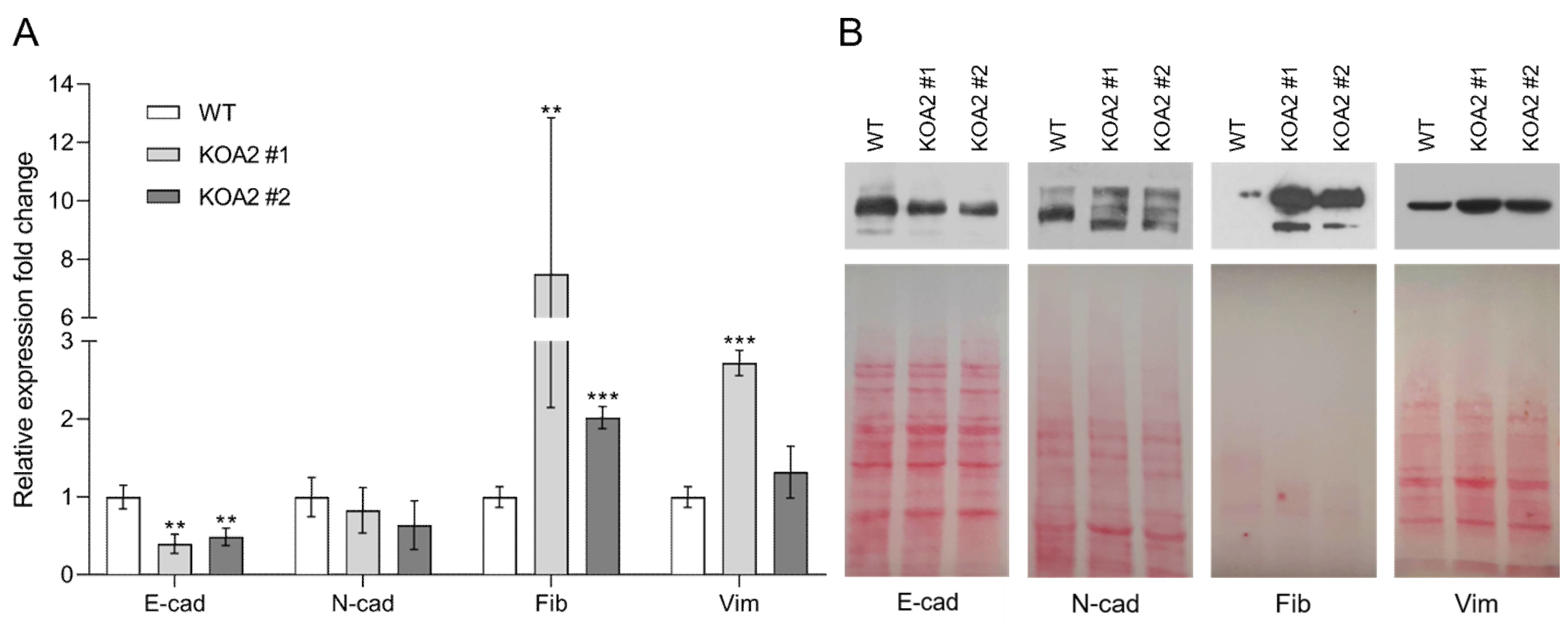
3.2. SLC35A2 Deficiency Downregulates E-Cadherin Expression
3.3. SLC35A2 Deficiency Upregulates Fibronectin Expression
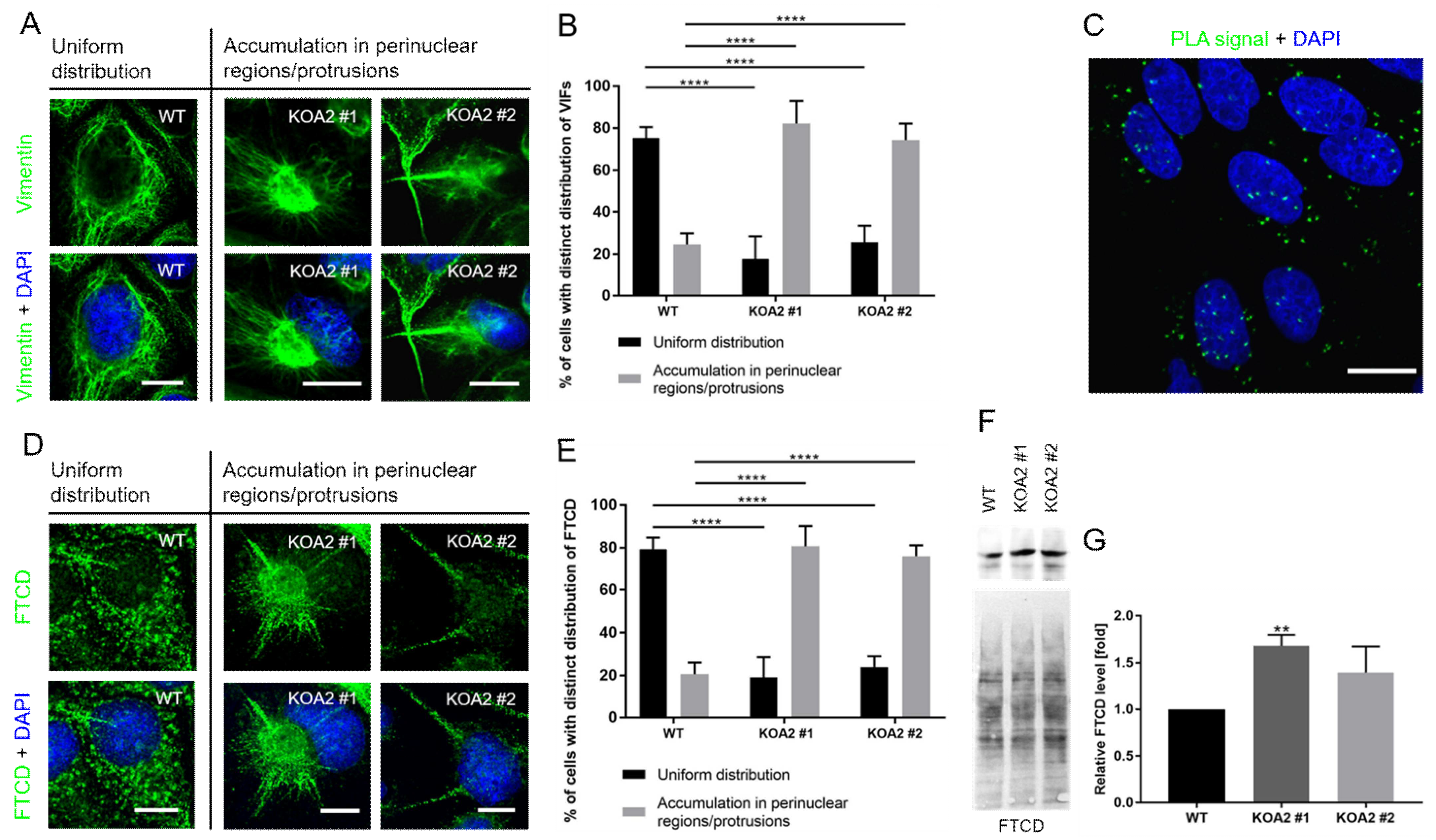
3.4. SLC35A2 Deficiency Triggers Reorganization of the Vimentin Cytoskeleton
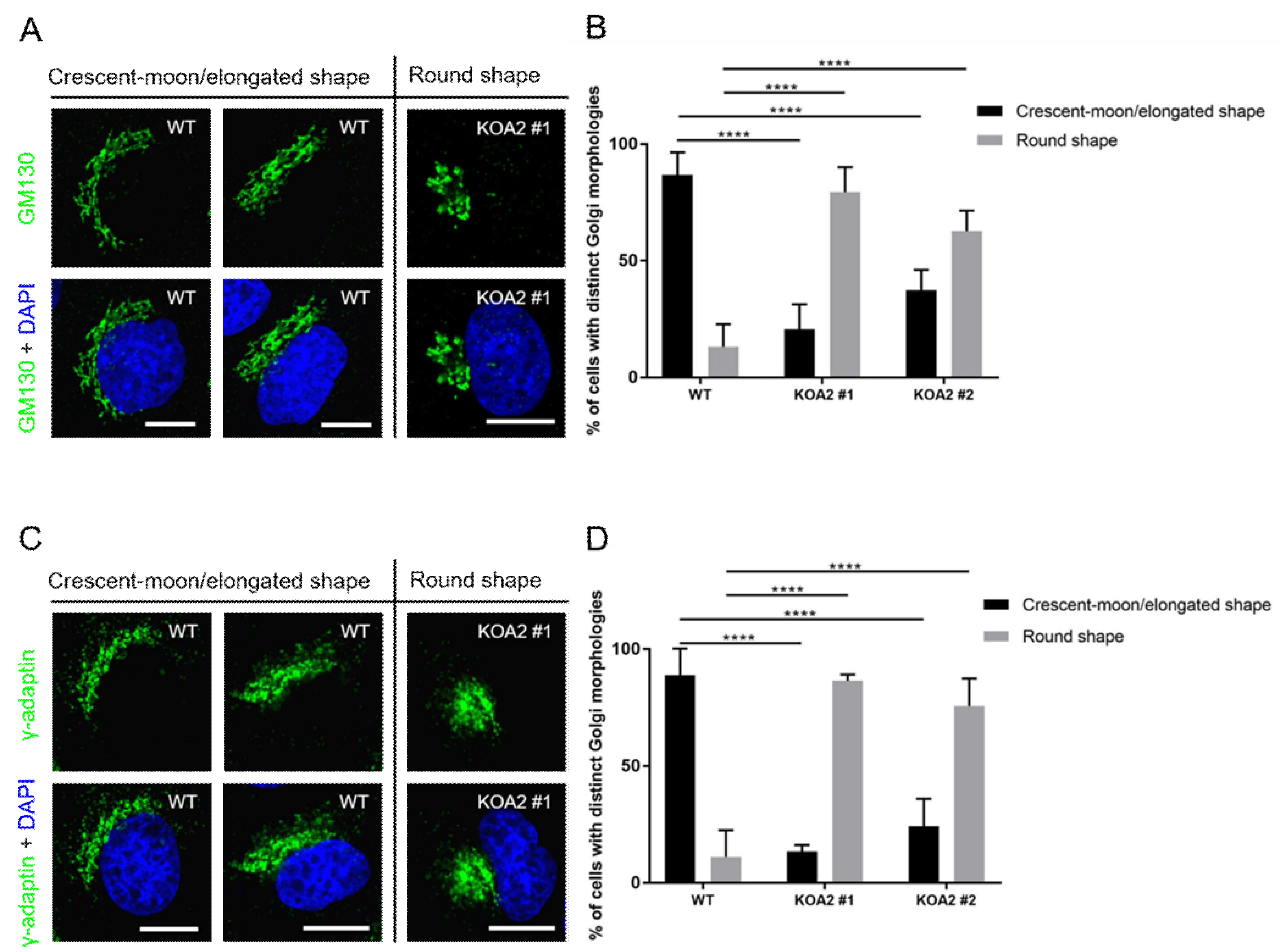
3.5. SLC35A2 Deficiency Triggers Golgi Compation
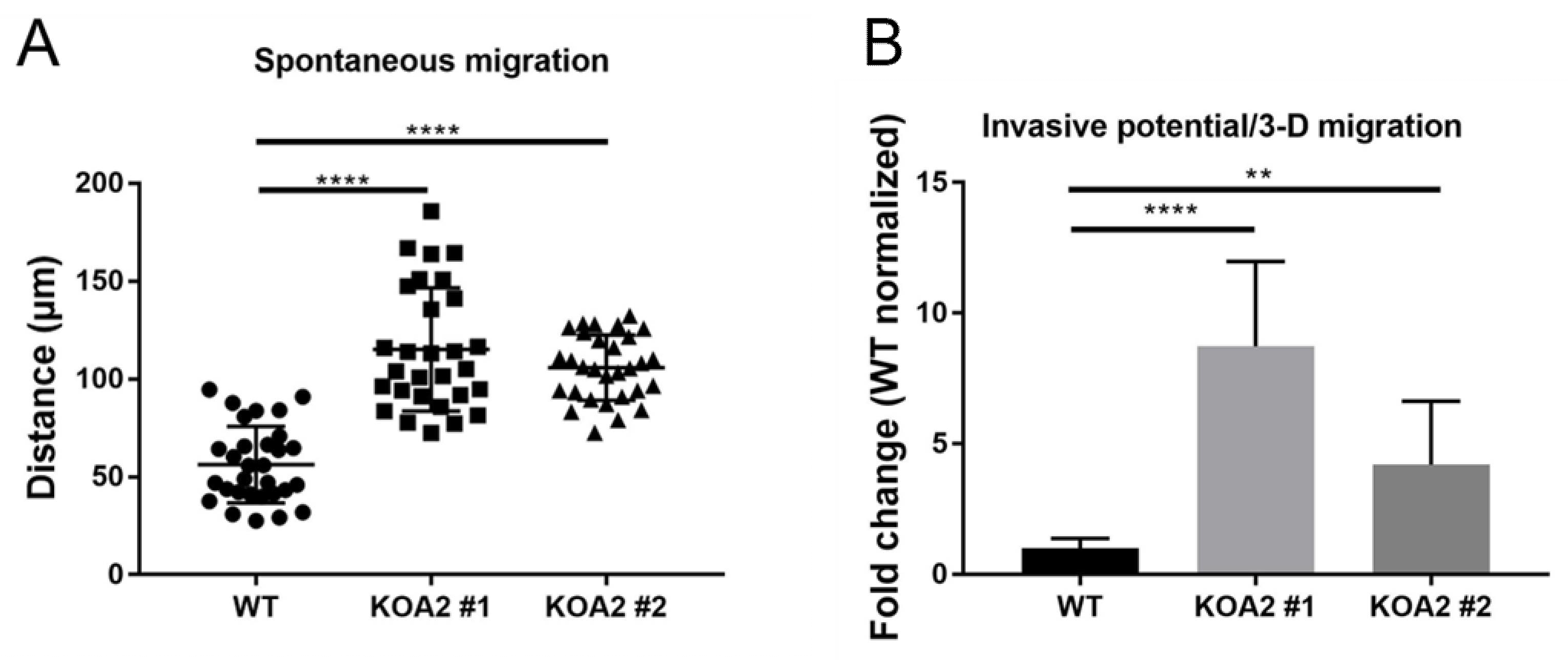
3.6. SLC35A2 Deficiency Enhances Cell Migration and Invasiveness
3.7. SLC35A2 Deficiency Upregulates Expression of Selected EMT-Activating Transcription Factors
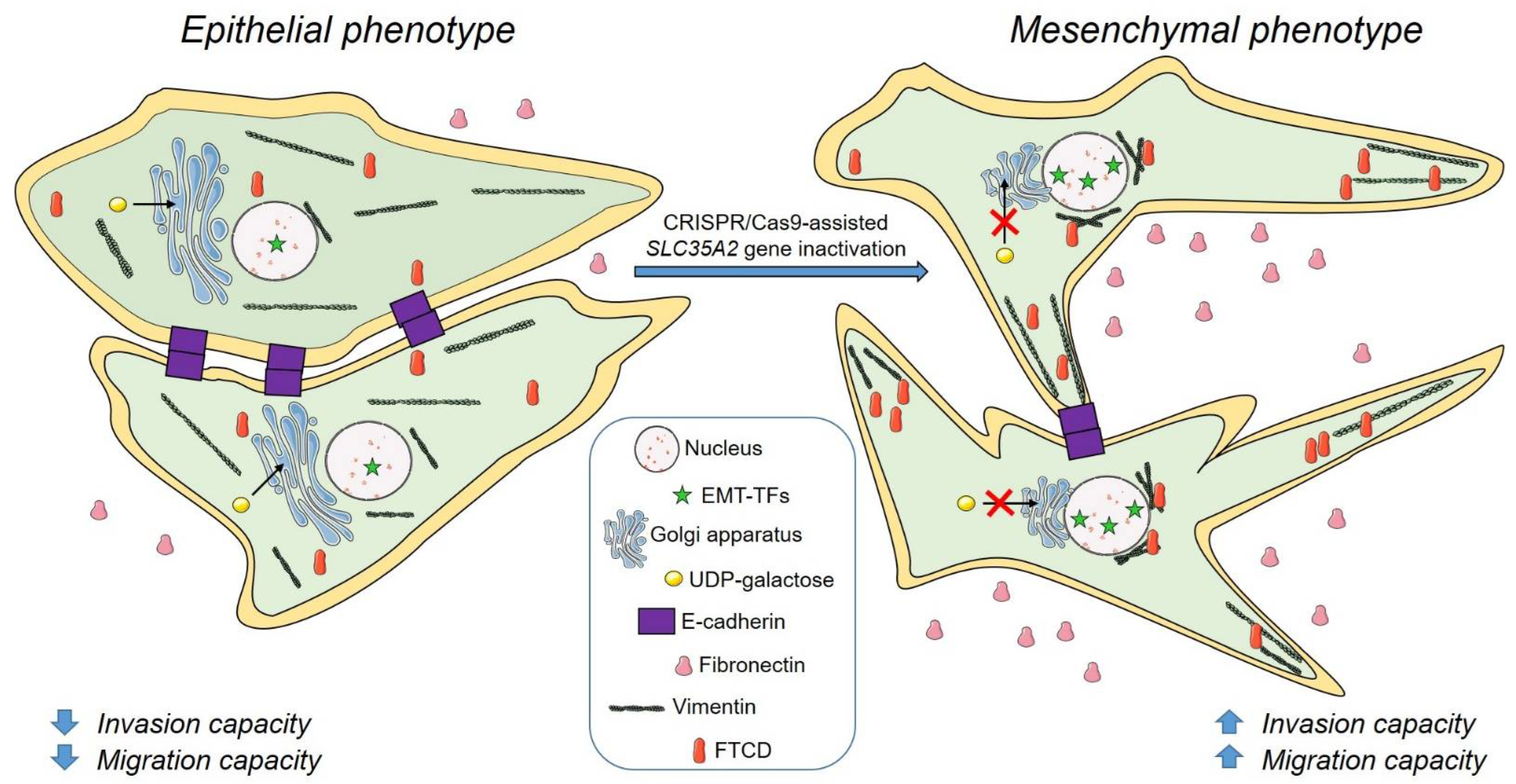
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
References
- Reily, C.; Stewart, T.J.; Renfrow, M.B.; Novak, J. Glycosylation in health and disease. Nat. Rev. Nephrol. 2019, 15, 346–366. [Google Scholar] [CrossRef] [PubMed]
- Breton, C.; Šnajdrová, L.; Jeanneau, C.; Koča, J.; Imberty, A. Structures and mechanisms of glycosyltransferases. Glycobiology 2006, 16, 29R–37R. [Google Scholar] [CrossRef] [PubMed]
- Hadley, B.; Maggioni, A.; Ashikov, A.; Day, C.J.; Haselhorst, T.; Tiralongo, J. Structure and function of nucleotide sugar transporters: Current progress. Comput. Struct. Biotechnol. J. 2014, 10, 23–32. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kabuss, R.; Ashikov, A.; Oelmann, S.; Gerardy-Schahn, R.; Bakker, H. Endoplasmic reticulum retention of the large splice variant of the UDP-galactose transporter is caused by a dilysine motif. Glycobiology 2005, 15, 905–911. [Google Scholar] [CrossRef]
- Oelmann, S.; Stanley, P.; Gerardy-Schahn, R. Point mutations identified in Lec8 Chinese hamster ovary glycosylation mutants that inactivate both the UDP-galactose and CMP-sialic acid transporters. J. Biol. Chem. 2001, 276, 26291–26300. [Google Scholar] [CrossRef] [Green Version]
- Olczak, M.; Guillen, E. Characterization of a mutation and an alternative splicing of UDP-galactose transporter in MDCK-RCAr cell line. Biochim. Biophys. Acta 2006, 1763, 82–92. [Google Scholar] [CrossRef] [Green Version]
- Maszczak-Seneczko, D.; Olczak, T.; Wunderlich, L.; Olczak, M. Comparative analysis of involvement of UGT1 and UGT2 splice variants of UDP-galactose transporter in glycosylation of macromolecules in MDCK and CHO cell lines. Glycoconj. J. 2011, 28, 481–492. [Google Scholar] [CrossRef] [Green Version]
- Deutscher, S.L.; Hirschberg, C.B. Mechanism of galactosylation in the Golgi apparatus. A Chinese hamster ovary cell mutant deficient in translocation of UDP-galactose across Golgi vesicle membranes. J. Biol. Chem. 1986, 261, 96–100. [Google Scholar] [CrossRef]
- Brändli, A.W.; Hansson, G.C.; Rodriguez-Boulan, E.; Simons, K. A polarized epithelial cell mutant deficient in translocation of UDP-galactose into the Golgi complex. J. Biol. Chem. 1988, 263, 16283–16290. [Google Scholar] [CrossRef]
- Taki, T.; Ogura, K.; Rokukawa, C.; Hara, T.; Kawakita, M.; Endo, T.; Kobata, A.; Handa, S. Had-1, a uridine 5’-diphosphogalactose transport-defective mutant of mouse mammary tumor cell FM3A: Composition of glycolipids, cell growth inhibition by lactosylceramide, and loss of tumorigenicity. Cancer Res. 1991, 51, 1701–7107. [Google Scholar]
- Szulc, B.; Sosicka, P.; Maszczak-Seneczko, D.; Skurska, E.; Shauchuk, A.; Olczak, T.; Freeze, H.H.; Olczak, M. Biosynthesis of GlcNAc-rich N- and O-glycans in the Golgi apparatus does not require the nucleotide sugar transporter SLC35A3. J. Biol. Chem. 2020, 295, 16445–16463. [Google Scholar] [CrossRef] [PubMed]
- Maszczak-Seneczko, D.; Sosicka, P.; Majkowski, M.; Olczak, T.; Olczak, M. UDP-N-acetylglucosamine transporter and UDP-galactose transporter form heterologous complexes in the Golgi membrane. FEBS Lett. 2012, 586, 4082–4087. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wiertelak, W.; Olczak, M.; Maszczak-Seneczko, D. Demonstration of Heterologous Complexes formed by Golgi-Resident Type III Membrane Proteins using Split Luciferase Complementation Assay. J. Vis. Exp. 2020, 10, e61669. [Google Scholar] [CrossRef] [PubMed]
- Sosicka, P.; Bazan, B.; Maszczak-Seneczko, D.; Shauchuk, Y.; Olczak, T.; Olczak, M. SLC35A5 Protein-A Golgi Complex Member with Putative Nucleotide Sugar Transport Activity. Int. J. Mol. Sci. 2019, 20, 276. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sprong, H.; Degroote, S.; Nilsson, T.; Kawakita, M.; Ishida, N.; van der Sluijs, P.; van Meer, G. Association of the Golgi UDP-galactose transporter with UDP-galactose:ceramide galactosyltransferase allows UDP-galactose import in the endoplasmic reticulum. Mol. Biol. Cell 2003, 14, 3482–3493. [Google Scholar] [CrossRef]
- Maszczak-Seneczko, D.; Sosicka, P.; Kaczmarek, B.; Majkowski, M.; Luzarowski, M.; Olczak, T.; Olczak, M. UDP-galactose (SLC35A2) and UDP-N-acetylglucosamine (SLC35A3) Transporters Form Glycosylation-related Complexes with Mannoside Acetylglucosaminyltransferases (Mgats). J. Biol. Chem. 2015, 290, 15475–15486. [Google Scholar] [CrossRef] [Green Version]
- Wiertelak, W.; Sosicka, P.; Olczak, M.; Maszczak-Seneczko, D. Analysis of homologous and heterologous interactions between UDP-galactose transporter and beta-1,4-galactosyltransferase 1 using NanoBiT. Anal. Biochem. 2020, 593, 113599. [Google Scholar] [CrossRef]
- Shauchuk, A.; Szulc, B.; Maszczak-Seneczko, D.; Wiertelak, W.; Skurska, E.; Olczak, M. N-glycosylation of the human β1,4-galactosyltransferase 4 is crucial for its activity and Golgi localization. Glycoconj. J. 2020, 37, 577–588. [Google Scholar] [CrossRef]
- Wiktor, M.; Wiertelak, W.; Maszczak-Seneczko, D.; Balwierz, P.J.; Szulc, B.; Olczak, M. Identification of novel potential interaction partners of UDP-galactose (SLC35A2), UDP-N-acetylglucosamine (SLC35A3) and an orphan (SLC35A4) nucleotide sugar transporters. J. Proteom. 2021, 249, 104321. [Google Scholar] [CrossRef]
- Silver, G.; Bahl, S.; Cordeiro, D.; Thakral, A.; Athey, T.; Mercimek-Andrews, S. Prevalence of Congenital Disorders of Glycosylation in Childhood Epilepsy and Effects of Anti-Epileptic Drugs on the Transferrin Isoelectric Focusing Test. Genes 2021, 12, 1227. [Google Scholar] [CrossRef]
- Chang, I.J.; He, M.; Lam, C.T. Congenital disorders of glycosylation. Ann. Transl. Med. 2018, 6, 477. [Google Scholar] [CrossRef] [PubMed]
- Lübke, T.; Marquardt, T.; von Figura, K.; Körner, C. A new type of carbohydrate-deficient glycoprotein syndrome due to a decreased import of GDP-fucose into the golgi. J. Biol. Chem. 1999, 274, 25986–25989. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ng, B.G.; Buckingham, K.J.; Raymond, K.; Kircher, M.; Turner, E.H.; He, M.; Smith, J.D.; Eroshkin, A.; Szybowska, M.; Losfeld, M.E.; et al. Mosaicism of the UDP-galactose transporter SLC35A2 causes a congenital disorder of glycosylation. Am. J. Hum. Genet. 2013, 92, 632–636. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kodera, H.; Nakamura, K.; Osaka, H.; Maegaki, Y.; Haginoya, K.; Mizumoto, S.; Kato, M.; Okamoto, N.; Iai, M.; Kondo, Y.; et al. De novo mutations in SLC35A2 encoding a UDP-galactose transporter cause early-onset epileptic encephalopathy. Hum. Mutat. 2013, 34, 1708–1714. [Google Scholar] [CrossRef] [PubMed]
- Dörre, K.; Olczak, M.; Wada, Y.; Sosicka, P.; Grüneberg, M.; Reunert, J.; Kurlemann, G.; Fiedler, B.; Biskup, S.; Hörtnagel, K.; et al. A new case of UDP-galactose transporter deficiency (SLC35A2-CDG): Molecular basis, clinical phenotype, and therapeutic approach. J. Inherit. Metab. Dis. 2015, 38, 931–940. [Google Scholar] [CrossRef] [PubMed]
- Yates, T.M.; Suri, M.; Desurkar, A.; Lesca, G.; Wallgren-Pettersson, C.; Hammer, T.B.; Raghavan, A.; Poulat, A.L.; Møller, R.S.; Thuresson, A.C.; et al. SLC35A2-related congenital disorder of glycosylation: Defining the phenotype. Eur. J. Paediatr. Neurol. 2018, 22, 1095–1102. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ng, B.G.; Sosicka, P.; Agadi, S.; Almannai, M.; Bacino, C.A.; Barone, R.; Botto, L.D.; Burton, J.E.; Carlston, C.; Chung, B.H.; et al. SLC35A2-CDG: Functional characterization, expanded molecular, clinical, and biochemical phenotypes of 30 unreported Individuals. Hum. Mutat. 2019, 40, 908–925. [Google Scholar] [CrossRef]
- Abuduxikuer, K.; Wang, J.S. Four New Cases of SLC35A2-CDG with Novel Mutations and Clinical Features. Front. Genet. 2021, 12, 658786. [Google Scholar] [CrossRef]
- Witters, P.; Tahata, S.; Barone, R.; Õunap, K.; Salvarinova, R.; Grønborg, S.; Hoganson, G.; Scaglia, F.; Lewis, A.M.; Mori, M.; et al. Clinical and biochemical improvement with galactose supplementation in SLC35A2-CDG. Genet. Med. 2020, 22, 1102–1107. [Google Scholar] [CrossRef]
- Kalluri, R.; Weinberg, R.A. The basics of epithelial-mesenchymal transition. J. Clin. Investig. 2009, 119, 1420–1428. [Google Scholar] [CrossRef] [Green Version]
- Hay, E.D. An overview of epithelio-mesenchymal transformation. Acta Anat. 1995, 154, 8–20. [Google Scholar] [CrossRef] [PubMed]
- Kim, Y.S.; Yi, B.R.; Kim, N.H.; Choi, K.C. Role of the epithelial-mesenchymal transition and its effects on embryonic stem cells. Exp. Mol. Med. 2014, 46, e108. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pucci, M.; Malagolini, N.; Dall’Olio, F. Glycobiology of the Epithelial to Mesenchymal Transition. Biomedicines 2021, 9, 770. [Google Scholar] [CrossRef] [PubMed]
- Bazan, B.; Wiktor, M.; Maszczak-Seneczko, D.; Olczak, T.; Kaczmarek, B.; Olczak, M. Lysine at position 329 within a C-terminal dilysine motif is crucial for the ER localization of human SLC35B4. PLoS ONE 2018, 13, e0207521. [Google Scholar] [CrossRef]
- Mrówczyńska, E.; Mazurkiewicz, E.; Mazur, A.J. Gelatin Zymography to Detect Gelatinase Activity in Melanoma Cells. J. Vis. Exp. 2022, 9, 181. [Google Scholar] [CrossRef] [PubMed]
- Szulc, B.; Zadorozhna, Y.; Olczak, M.; Wiertelak, W.; Maszczak-Seneczko, D. Novel Insights into Selected Disease-Causing Mutations within the SLC35A1 Gene Encoding the CMP-Sialic Acid Transporter. Int. J. Mol. Sci. 2020, 22, 304. [Google Scholar] [CrossRef] [PubMed]
- Jung, Y.S.; Liu, X.W.; Chirco, R.; Warner, R.B.; Fridman, R.; Kim, H.R. TIMP-1 induces an EMT-like phenotypic conversion in MDCK cells independent of its MMP-inhibitory domain. PLoS ONE 2012, 7, e38773. [Google Scholar] [CrossRef] [Green Version]
- Taylor, S.C.; Nadeau, K.; Abbasi, M.; Lachance, C.; Nguyen, M.; Fenrich, J. The Ultimate qPCR Experiment: Producing Publication Quality, Reproducible Data the First Time. Trends Biotechnol. 2019, 37, 761–774. [Google Scholar] [CrossRef] [Green Version]
- Malek, N.; Michrowska, A.; Mazurkiewicz, E.; Mrówczyńska, E.; Mackiewicz, P.; Mazur, A.J. The origin of the expressed retrotransposed gene ACTBL2 and its influence on human melanoma cells’ motility and focal adhesion formation. Sci. Rep. 2021, 11, 3329. [Google Scholar] [CrossRef]
- Mazurkiewicz, E.; Makowiecka, A.; Mrówczyńska, E.; Kopernyk, I.; Nowak, D.; Mazur, A.J. Gelsolin Contributes to the Motility of A375 Melanoma Cells and This Activity Is Mediated by the Fibrous Extracellular Matrix Protein Profile. Cells 2021, 10, 1848. [Google Scholar] [CrossRef]
- Oike, T.; Uchihara, Y.; Permata, T.B.M.; Gondhowiardjo, S.; Ohno, T.; Shibata, A. Quantitative volumetric analysis of the Golgi apparatus following X-ray irradiation by super-resolution 3D-SIM microscopy. Med. Mol. Morphol. 2021, 54, 166–172. [Google Scholar] [CrossRef] [PubMed]
- Gao, Y.; Sztul, E. A novel interaction of the Golgi complex with the vimentin intermediate filament cytoskeleton. J. Cell Biol. 2001, 152, 877–894. [Google Scholar] [CrossRef] [PubMed]
- Petrosyan, A. Onco-Golgi: Is Fragmentation a Gate to Cancer Progression? Biochem. Mol. Biol. J. 2015, 1, 16. [Google Scholar] [CrossRef] [Green Version]
- Tan, X.; Banerjee, P.; Guo, H.F.; Ireland, S.; Pankova, D.; Ahn, Y.H.; Nikolaidis, I.M.; Liu, X.; Zhao, Y.; Xue, Y.; et al. Epithelial-to-mesenchymal transition drives a pro-metastatic Golgi compaction process through scaffolding protein PAQR11. J. Clin. Investig. 2017, 127, 117–131. [Google Scholar] [CrossRef] [Green Version]
- Robinson, M.S. Cloning and expression of gamma-adaptin, a component of clathrin-coated vesicles associated with the Golgi apparatus. J. Cell Biol. 1990, 111, 2319–2326. [Google Scholar] [CrossRef] [Green Version]
- Ansieau, S.; Collin, G.; Hill, L. EMT or EMT-Promoting Transcription Factors, Where to Focus the Light? Front. Oncol. 2014, 4, 353. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ju, T.; Brewer, K.; D’Souza, A.; Cummings, R.D.; Canfield, W.M. Cloning and expression of human core 1 beta1,3-galactosyltransferase. J. Biol. Chem. 2002, 277, 178–186. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dong, X.; Jiang, Y.; Liu, J.; Liu, Z.; Gao, T.; An, G.; Wen, T. T-Synthase Deficiency Enhances Oncogenic Features in Human Colorectal Cancer Cells via Activation of Epithelial-Mesenchymal Transition. Biomed. Res. Int. 2018, 2018, 9532389. [Google Scholar] [CrossRef] [Green Version]
- Demaegd, D.; Foulquier, F.; Colinet, A.S.; Gremillon, L.; Legrand, D.; Mariot, P.; Peiter, E.; Van Schaftingen, E.; Matthijs, G.; Morsomme, P. Newly characterized Golgi-localized family of proteins is involved in calcium and pH homeostasis in yeast and human cells. Proc. Natl. Acad. Sci. USA 2013, 110, 6859–6864. [Google Scholar] [CrossRef] [Green Version]
- Foulquier, F.; Amyere, M.; Jaeken, J.; Zeevaert, R.; Schollen, E.; Race, V.; Bammens, R.; Morelle, W.; Rosnoblet, C.; Legrand, D.; et al. TMEM165 deficiency causes a congenital disorder of glycosylation. Am. J. Hum. Genet. 2012, 91, 15–26. [Google Scholar] [CrossRef] [Green Version]
- Murali, P.; Johnson, B.P.; Lu, Z.; Climer, L.; Scott, D.A.; Foulquier, F.; Oprea-Ilies, G.; Lupashin, V.; Drake, R.R.; Abbott, K.L. Novel role for the Golgi membrane protein TMEM165 in control of migration and invasion for breast carcinoma. Oncotarget 2020, 11, 2747–2762. [Google Scholar] [CrossRef] [PubMed]
- Ishida, N.; Yoshioka, S.; Chiba, Y.; Takeuchi, M.; Kawakita, M. Molecular cloning and functional expression of the human Golgi UDP-N-acetylglucosamine transporter. J. Biochem. 1999, 126, 68–77. [Google Scholar] [CrossRef] [PubMed]
- Song, W.; Isaji, T.; Nakano, M.; Liang, C.; Fukuda, T.; Gu, J. O-GlcNAcylation regulates β1,4-GlcNAc-branched N-glycan biosynthesis via the OGT/SLC35A3/GnT-IV axis. FASEB J. 2022, 36, e22149. [Google Scholar] [CrossRef]
- Moreno-Bueno, G.; Cubillo, E.; Sarrió, D.; Peinado, H.; Rodríguez-Pinilla, S.M.; Villa, S.; Bolós, V.; Jordá, M.; Fabra, A.; Portillo, F.; et al. Genetic profiling of epithelial cells expressing E-cadherin repressors reveals a distinct role for Snail, Slug, and E47 factors in epithelial-mesenchymal transition. Cancer Res. 2006, 66, 9543–9556. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lucena, M.C.; Carvalho-Cruz, P.; Donadio, J.L.; Oliveira, I.A.; de Queiroz, R.M.; Marinho-Carvalho, M.M.; Sola-Penna, M.; de Paula, I.F.; Gondim, K.C.; McComb, M.E.; et al. Epithelial Mesenchymal Transition Induces Aberrant Glycosylation through Hexosamine Biosynthetic Pathway Activation. J. Biol. Chem. 2016, 291, 12917–12929. [Google Scholar] [CrossRef] [Green Version]
- Priglinger, C.S.; Obermann, J.; Szober, C.M.; Merl-Pham, J.; Ohmayer, U.; Behler, J.; Gruhn, F.; Kreutzer, T.C.; Wertheimer, C.; Geerlof, A.; et al. Epithelial-to-Mesenchymal Transition of RPE Cells In Vitro Confers Increased β1,6-N-Glycosylation and Increased Susceptibility to Galectin-3 Binding. PLoS ONE 2016, 11, e0146887. [Google Scholar] [CrossRef] [Green Version]
- Lecca, M.R.; Maag, C.; Berger, E.G.; Hennet, T. Fibrotic response in fibroblasts from congenital disorders of glycosylation. J. Cell. Mol. Med. 2011, 15, 1788–1796. [Google Scholar] [CrossRef] [Green Version]



Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kot, M.; Mazurkiewicz, E.; Wiktor, M.; Wiertelak, W.; Mazur, A.J.; Rahalevich, A.; Olczak, M.; Maszczak-Seneczko, D. SLC35A2 Deficiency Promotes an Epithelial-to-Mesenchymal Transition-like Phenotype in Madin–Darby Canine Kidney Cells. Cells 2022, 11, 2273. https://doi.org/10.3390/cells11152273
Kot M, Mazurkiewicz E, Wiktor M, Wiertelak W, Mazur AJ, Rahalevich A, Olczak M, Maszczak-Seneczko D. SLC35A2 Deficiency Promotes an Epithelial-to-Mesenchymal Transition-like Phenotype in Madin–Darby Canine Kidney Cells. Cells. 2022; 11(15):2273. https://doi.org/10.3390/cells11152273
Chicago/Turabian StyleKot, Magdalena, Ewa Mazurkiewicz, Maciej Wiktor, Wojciech Wiertelak, Antonina Joanna Mazur, Andrei Rahalevich, Mariusz Olczak, and Dorota Maszczak-Seneczko. 2022. "SLC35A2 Deficiency Promotes an Epithelial-to-Mesenchymal Transition-like Phenotype in Madin–Darby Canine Kidney Cells" Cells 11, no. 15: 2273. https://doi.org/10.3390/cells11152273