
Absence of EPAC1 Signaling to Stabilize CFTR in Intestinal Organoids



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Cell Culture and Compound Treatment
2.2. Forskolin-Induced Swelling Assay
2.3. Plasmid Transfection
2.4. Live Cell Imaging
2.5. Western Blotting
2.6. Cell Surface Biotinylation Assay
2.7. Statistical Analysis
3. Results
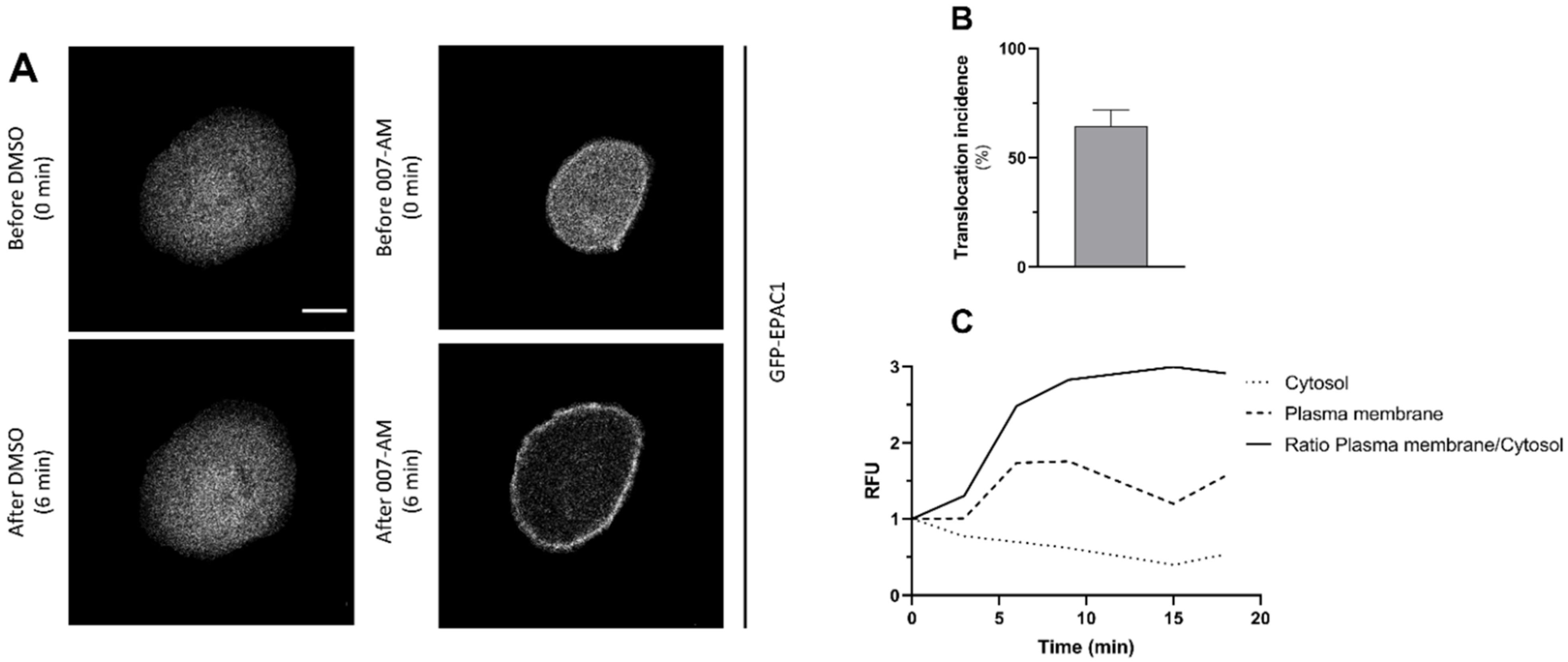
3.1. Activation of EPAC1 by AM-007 Promotes Its Translocation to the PM in CFBE
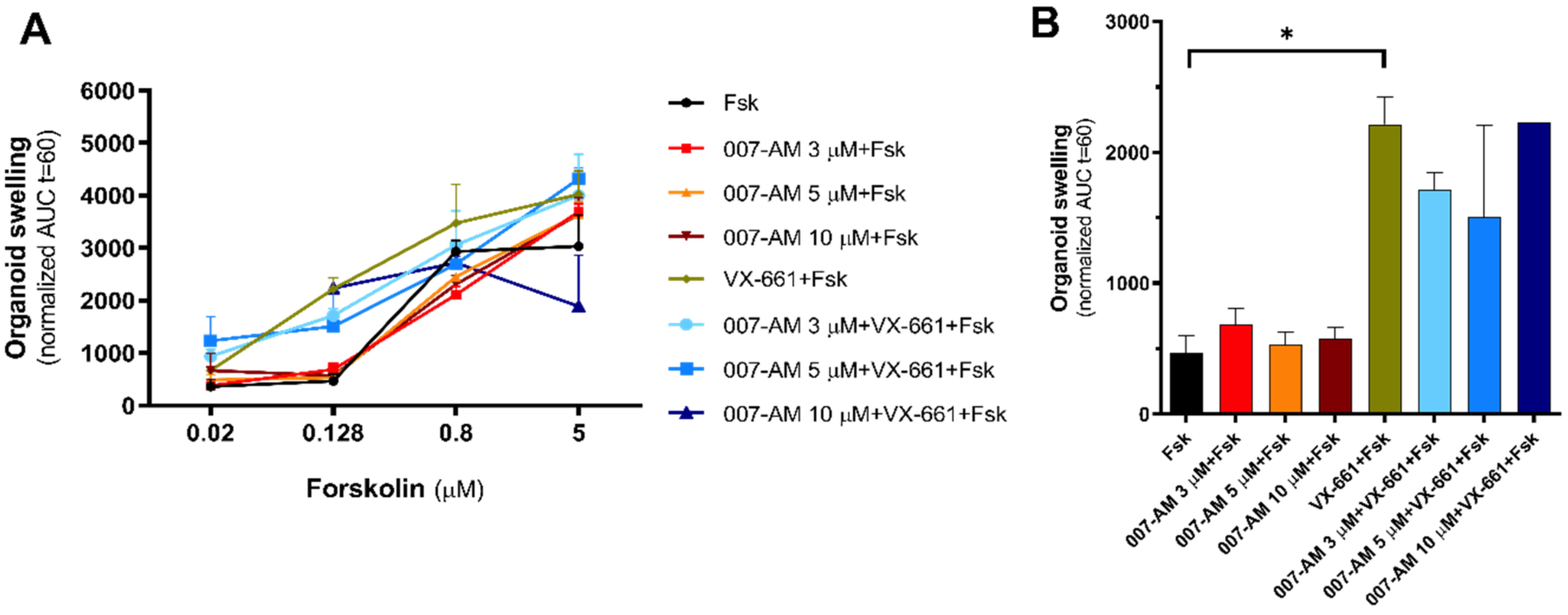
3.2. EPAC1 Activation Does Not Increase Intestinal Organoid Swelling
3.3. EPAC1 Expression Is Not Detected in Intestinal Organoids
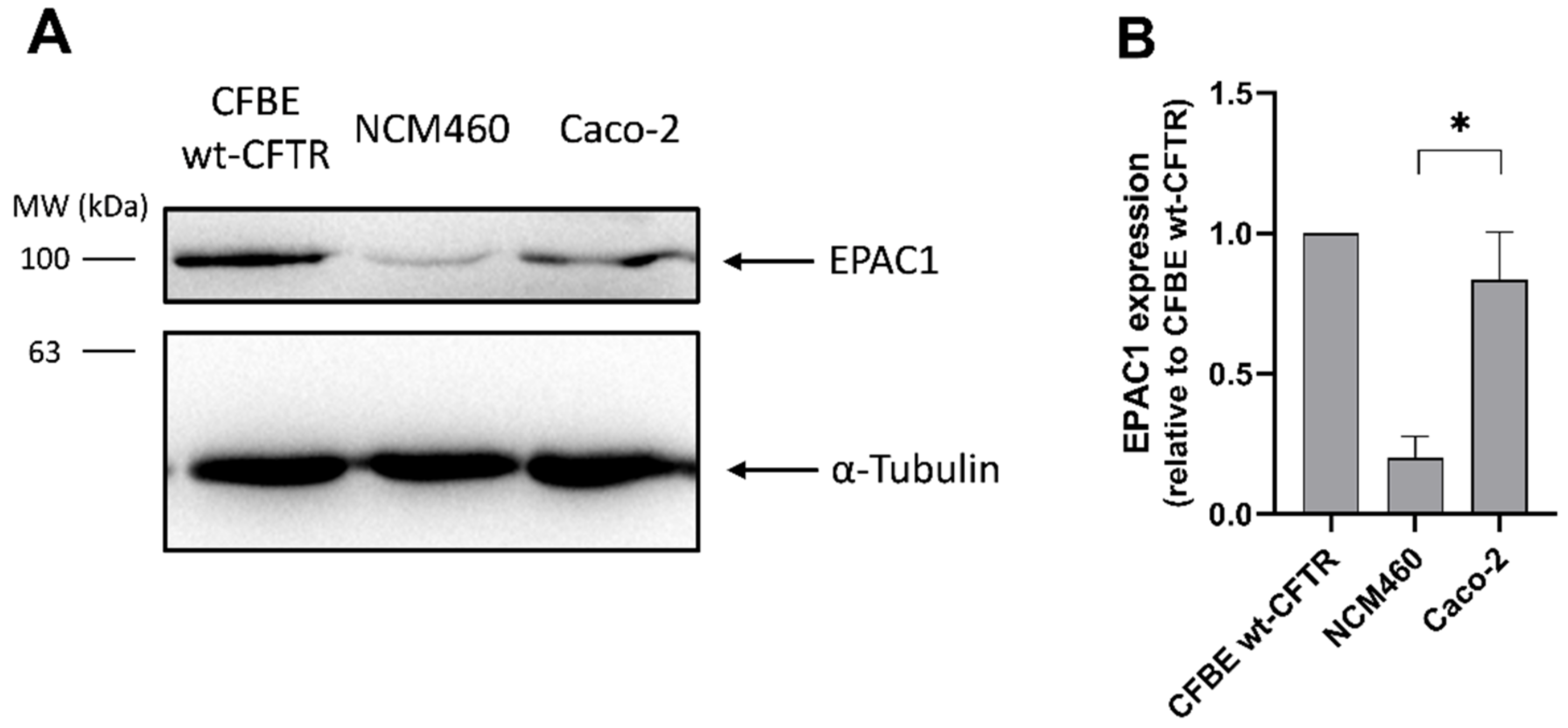
3.4. EPAC1 Is Expressed in Intestinal Cell Lines
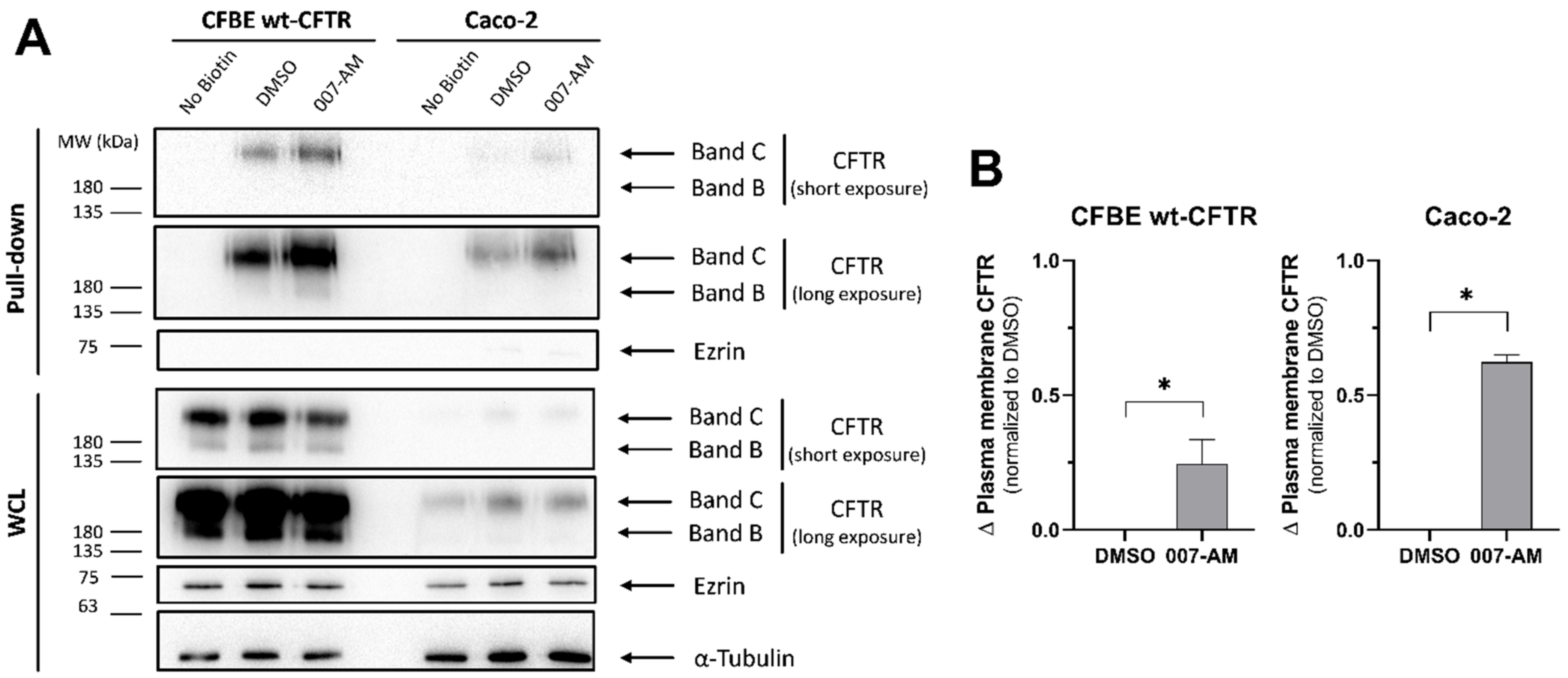
3.5. EPAC1 Activation Increases PM CFTR in Intestinal Organoids
4. Discussion
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Riordan, J.R. CFTR Function and Prospects for Therapy. Annu. Rev. Biochem. 2008, 77, 701–726. [Google Scholar] [CrossRef] [PubMed]
- Collins, F. Cystic Fibrosis: Molecular Biology and Therapeutic Implications. Science 1992, 256, 774–779. [Google Scholar] [CrossRef] [PubMed]
- Rowe, S.M.; Miller, S.; Sorscher, E.J. Cystic Fibrosis. N. Engl. J. Med. 2005, 352, 1992–2001. [Google Scholar] [CrossRef] [PubMed]
- Okiyoneda, T.; Barrière, H.; Bagdány, M.; Rabeh, W.M.; Du, K.; Höhfeld, J.; Young, J.C.; Lukacs, G.L. Peripheral Protein Quality Control Removes Unfolded CFTR from the Plasma Membrane. Science 2010, 329, 805–810. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Farinha, C.M.; Canato, S. From the Endoplasmic Reticulum to the Plasma Membrane: Mechanisms of CFTR Folding and Trafficking. Cell. Mol. Life Sci. 2017, 74, 39–55. [Google Scholar] [CrossRef]
- Farinha, C.M.; Matos, P.; Amaral, M.D. Control of Cystic Fibrosis Transmembrane Conductance Regulator Membrane Trafficking: Not Just from the Endoplasmic Reticulum to the Golgi. FEBS J. 2013, 280, 4396–4406. [Google Scholar] [CrossRef]
- Cantiello, H.F. Role of the Actin Cytoskeleton in the Regulation of the Cystic Fibrosis Transmembrane Conductance Regulator. Exp. Physiol. 1996, 81, 505–514. [Google Scholar] [CrossRef]
- Schmidt, M.; Dekker, F.J.; Maarsingh, H. Exchange Protein Directly Activated by CAMP (Epac): A Multidomain CAMP Mediator in the Regulation of Diverse Biological Functions. Pharmacol. Rev. 2013, 65, 670–709. [Google Scholar] [CrossRef] [Green Version]
- Lobo, M.J.; Amaral, M.D.; Zaccolo, M.; Farinha, C.M. EPAC1 Activation by CAMP Stabilizes CFTR at the Membrane by Promoting Its Interaction with NHERF1. J. Cell Sci. 2016, 129, 2599–2612. [Google Scholar] [CrossRef] [Green Version]
- Santos, J.D.; Pinto, F.R.; Ferreira, J.F.; Amaral, M.D.; Zaccolo, M.; Farinha, C.M. Cytoskeleton Regulators CAPZA2 and INF2 Associate with CFTR to Control Its Plasma Membrane Levels under EPAC1 Activation. Biochem. J. 2020, 477, 2561–2580. [Google Scholar] [CrossRef]
- Zaccolo, M.; Zerio, A.; Lobo, M.J. Subcellular Organization of the CAMP Signaling Pathway. Pharmacol. Rev. 2021, 73, 278–309. [Google Scholar] [CrossRef] [PubMed]
- De Rooij, J.; Rehmann, H.; Van Triest, M.; Cool, R.H.; Wittinghofer, A.; Bos, J.L. Mechanism of Regulation of the Epac Family of CAMP-Dependent RapGEFs. J. Biol. Chem. 2000, 275, 20829–20836. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gloerich, M.; Bos, J.L. Epac: Defining a New Mechanism for CAMP Action. Annu. Rev. Pharmacol. Toxicol. 2010, 50, 355–375. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ehrhardt, C.; Collnot, E.M.; Baldes, C.; Becker, U.; Laue, M.; Kim, K.J.; Lehr, C.M. Towards an in Vitro Model of Cystic Fibrosis Small Airway Epithelium: Characterisation of the Human Bronchial Epithelial Cell Line CFBE41o. Cell Tissue Res. 2006, 323, 405–415. [Google Scholar] [CrossRef]
- Bebok, Z.; Collawn, J.F.; Wakefield, J.; Parker, W.; Li, Y.; Varga, K.; Sorscher, E.J.; Clancy, J.P. Failure of CAMP Agonists to Activate Rescued ΔF508 CFTR in CFBE41o– Airway Epithelial Monolayers. J. Physiol. 2005, 569, 601–615. [Google Scholar] [CrossRef]
- Awatade, N.T.; Ramalho, S.; Silva, I.A.L.; Felício, V.; Botelho, H.M.; de Poel, E.; Vonk, A.; Beekman, J.M.; Farinha, C.M.; Amaral, M.D. R560S: A Class II CFTR Mutation That Is Not Rescued by Current Modulators. J. Cyst. Fibros. 2019, 18, 182–189. [Google Scholar] [CrossRef]
- Vonk, A.M.; van Mourik, P.; Ramalho, A.S.; Silva, I.A.L.; Statia, M.; Kruisselbrink, E.; Suen, S.W.F.; Dekkers, J.F.; Vleggaar, F.P.; Houwen, R.H.J.; et al. Protocol for Application, Standardization and Validation of the Forskolin-Induced Swelling Assay in Cystic Fibrosis Human Colon Organoids. STAR Protoc. 2020, 1, 100019. [Google Scholar] [CrossRef]
- Silva, I.A.L.; Doušová, T.; Ramalho, S.; Centeio, R.; Clarke, L.A.; Railean, V.; Botelho, H.M.; Holubová, A.; Valášková, I.; Yeh, J.T.; et al. Organoids as a Personalized Medicine Tool for Ultra-Rare Mutations in Cystic Fibrosis: The Case of S955P and 1717-2A>G. Biochim. Biophys. Acta-Mol. Basis Dis. 2020, 1866, 165905. [Google Scholar] [CrossRef]
- Hagemeijer, M.C.; Vonk, A.M.; Awatade, N.T.; Silva, I.A.L.; Tischer, C.; Hilsenstein, V.; Beekman, J.M.; Amaral, M.D.; Botelho, H.M. An Open-Source High-Content Analysis Workflow for CFTR Function Measurements Using the Forskolin-Induced Swelling Assay. Bioinformatics 2021, 36, 5686–5694. [Google Scholar] [CrossRef]
- Jones, T.R.; Kang, I.H.; Wheeler, D.B.; Lindquist, R.A.; Papallo, A.; Sabatini, D.M.; Golland, P.; Carpenter, A.E. CellProfiler Analyst: Data Exploration and Analysis Software for Complex Image-Based Screens. BMC Bioinform. 2008, 9, 482. [Google Scholar] [CrossRef] [Green Version]
- Luz, S.; Cihil, K.M.; Brautigan, D.L.; Amaral, M.D.; Farinha, C.M.; Swiatecka-Urban, A. LMTK2-Mediated Phosphorylation Regulates CFTR Endocytosis in Human Airway Epithelial Cells. J. Biol. Chem. 2014, 289, 15080–15093. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Enserink, J.M.; Christensen, A.E.; De Rooij, J.; Van Triest, M.; Schwede, F.; Genieser, H.G.; Døskeland, S.O.; Blank, J.L.; Bos, J.L. A Novel Epac-Specific CAMP Analogue Demonstrates Independent Regulation of Rap1 and ERK. Nat. Cell Biol. 2002, 4, 901–906. [Google Scholar] [CrossRef] [PubMed]
- Rehmann, H.; Schwede, F.; Doøskeland, S.O.; Wittinghofer, A.; Bos, J.L. Ligand-Mediated Activation of the CAMP-Responsive Guanine Nucleotide Exchange Factor Epac. J. Biol. Chem. 2003, 278, 38548–38556. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vliem, M.J.; Ponsioen, B.; Schwede, F.; Pannekoek, W.J.; Riedl, J.; Kooistra, M.R.H.; Jalink, K.; Genieser, H.G.; Bos, J.L.; Rehmann, H. 8-PCPT-2′-O-Me-CAMP-AM: An Improved Epac-Selective CAMP Analogue. ChemBioChem 2008, 9, 2052–2054. [Google Scholar] [CrossRef]
- Terlizzi, V.; Carnovale, V.; Castaldo, G.; Castellani, C.; Cirilli, N.; Colombo, C.; Corti, F.; Cresta, F.; D’Adda, A.; Lucarelli, M.; et al. Clinical Expression of Patients with the D1152H CFTR Mutation. J. Cyst. Fibros. 2015, 14, 447–452. [Google Scholar] [CrossRef] [Green Version]
- Dekkers, J.F.; Berkers, G.; Kruisselbrink, E.; Vonk, A.; De Jonge, H.R.; Janssens, H.M.; Bronsveld, I.; Van De Graaf, E.A.; Nieuwenhuis, E.E.S.; Houwen, R.H.J.; et al. Characterizing Responses to CFTR-Modulating Drugs Using Rectal Organoids Derived from Subjects with Cystic Fibrosis. Sci. Transl. Med. 2016, 8, 344ra84. [Google Scholar] [CrossRef]
- Berkers, G.; van Mourik, P.; Vonk, A.M.; Kruisselbrink, E.; Dekkers, J.F.; de Winter-de Groot, K.M.; Arets, H.G.M.; Marck-van der Wilt, R.E.P.; Dijkema, J.S.; Vanderschuren, M.M.; et al. Rectal Organoids Enable Personalized Treatment of Cystic Fibrosis. Cell Rep. 2019, 26, 1701–1708.e3. [Google Scholar] [CrossRef] [Green Version]
- Silva, I.A.L.; Railean, V.; Duarte, A.; Amaral, M.D. Personalized Medicine Based on Nasal Epithelial Cells: Comparative Studies with Rectal Biopsies and Intestinal Organoids. J. Pers. Med. 2021, 11, 421. [Google Scholar] [CrossRef]
- Shanthikumar, S.; Massie, J. A Review of Treatments That Improve Cystic Fibrosis Transmembrane Conductance Regulator Function. Clin. Med. Insights Ther. 2017, 9, 1–7. [Google Scholar] [CrossRef]
- Monterisi, S.; Favia, M.; Guerra, L.; Cardone, R.A.; Marzulli, D.; Reshkin, S.J.; Casavola, V.; Zaccolo, M. CFTR Regulation in Human Airway Epithelial Cells Requires Integrity of the Actin Cytoskeleton and Compartmentalized CAMP and PKA Activity. J. Cell Sci. 2012, 125, 1106–1117. [Google Scholar] [CrossRef] [Green Version]
- Swiatecka-Urban, A.; Brown, A.; Moreau-Marquis, S.; Renuka, J.; Coutermarsh, B.; Barnaby, R.; Karlson, K.H.; Flotte, T.R.; Fukuda, M.; Langford, G.M.; et al. The Short Apical Membrane Half-Life of Rescued ΔF508-Cystic Fibrosis Transmembrane Conductance Regulator (CFTR) Results from Accelerated Endocytosis of ΔF508-CFTR in Polarized Human Airway Epithelial Cells. J. Biol. Chem. 2005, 280, 36762–36772. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Farinha, C.M.; King-Underwood, J.; Sousa, M.; Correia, A.R.; Henriques, B.J.; Roxo-Rosa, M.; Da Paula, A.C.; Williams, J.; Hirst, S.; Gomes, C.M.; et al. Revertants, Low Temperature, and Correctors Reveal the Mechanism of F508del-CFTR Rescue by VX-809 and Suggest Multiple Agents for Full Correction. Chem. Biol. 2013, 20, 943–955. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Farinha, C.M.; Matos, P. Repairing the Basic Defect in Cystic Fibrosis—One Approach Is Not Enough. FEBS J. 2016, 283, 246–264. [Google Scholar] [CrossRef] [PubMed]
- Van Mourik, P.; Beekman, J.M.; Van Der Ent, C.K. Intestinal Organoids to Model Cystic Fibrosis. Eur. Respir. J. 2019, 54, 1802379. [Google Scholar] [CrossRef] [PubMed]
- Saint-Criq, V.; Gray, M.A. Role of CFTR in Epithelial Physiology. Cell. Mol. Life Sci. 2017, 74, 93–115. [Google Scholar] [CrossRef] [Green Version]
- Kawasaki, H.; Springett, G.M.; Mochizuki, N.; Toki, S.; Nakaya, M.; Matsuda, M.; Housman, D.E.; Graybiel, A.M. A Family of CAMP-Binding Proteins That Directly Activate Rap1. Science 1998, 282, 2275–2279. [Google Scholar] [CrossRef] [Green Version]
- Hoque, K.M.; Woodward, O.M.; Van Rossum, D.B.; Zachos, N.C.; Chen, L.; Leung, G.P.H.; Guggino, W.B.; Guggino, S.E.; Tse, C.M. Epac1 Mediates Protein Kinase A–Independent Mechanism of Forskolin-Activated Intestinal Chloride Secretion. J. Gen. Physiol. 2010, 135, 43–58. [Google Scholar] [CrossRef] [Green Version]
- Wang, D.; Eraslan, B.; Wieland, T.; Hallström, B.; Hopf, T.; Zolg, D.P.; Zecha, J.; Asplund, A.; Li, L.; Meng, C.; et al. A Deep Proteome and Transcriptome Abundance Atlas of 29 Healthy Human Tissues. Mol. Syst. Biol. 2019, 15, e8503. [Google Scholar] [CrossRef]
- Lindeboom, R.G.; van Voorthuijsen, L.; Oost, K.C.; Rodríguez-Colman, M.J.; Luna-Velez, M.V.; Furlan, C.; Baraille, F.; Jansen, P.W.; Ribeiro, A.; Burgering, B.M.; et al. Integrative Multi-omics Analysis of Intestinal Organoid Differentiation. Mol. Syst. Biol. 2018, 14, 8227. [Google Scholar] [CrossRef]

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ferreira, J.F.; Silva, I.A.L.; Botelho, H.M.; Amaral, M.D.; Farinha, C.M. Absence of EPAC1 Signaling to Stabilize CFTR in Intestinal Organoids. Cells 2022, 11, 2295. https://doi.org/10.3390/cells11152295
Ferreira JF, Silva IAL, Botelho HM, Amaral MD, Farinha CM. Absence of EPAC1 Signaling to Stabilize CFTR in Intestinal Organoids. Cells. 2022; 11(15):2295. https://doi.org/10.3390/cells11152295
Chicago/Turabian StyleFerreira, João F., Iris A. L. Silva, Hugo M. Botelho, Margarida D. Amaral, and Carlos M. Farinha. 2022. "Absence of EPAC1 Signaling to Stabilize CFTR in Intestinal Organoids" Cells 11, no. 15: 2295. https://doi.org/10.3390/cells11152295
APA StyleFerreira, J. F., Silva, I. A. L., Botelho, H. M., Amaral, M. D., & Farinha, C. M. (2022). Absence of EPAC1 Signaling to Stabilize CFTR in Intestinal Organoids. Cells, 11(15), 2295. https://doi.org/10.3390/cells11152295