
Mitochondrial Damage-Associated Molecular Patterns Content in Extracellular Vesicles Promotes Early Inflammation in Neurodegenerative Disorders





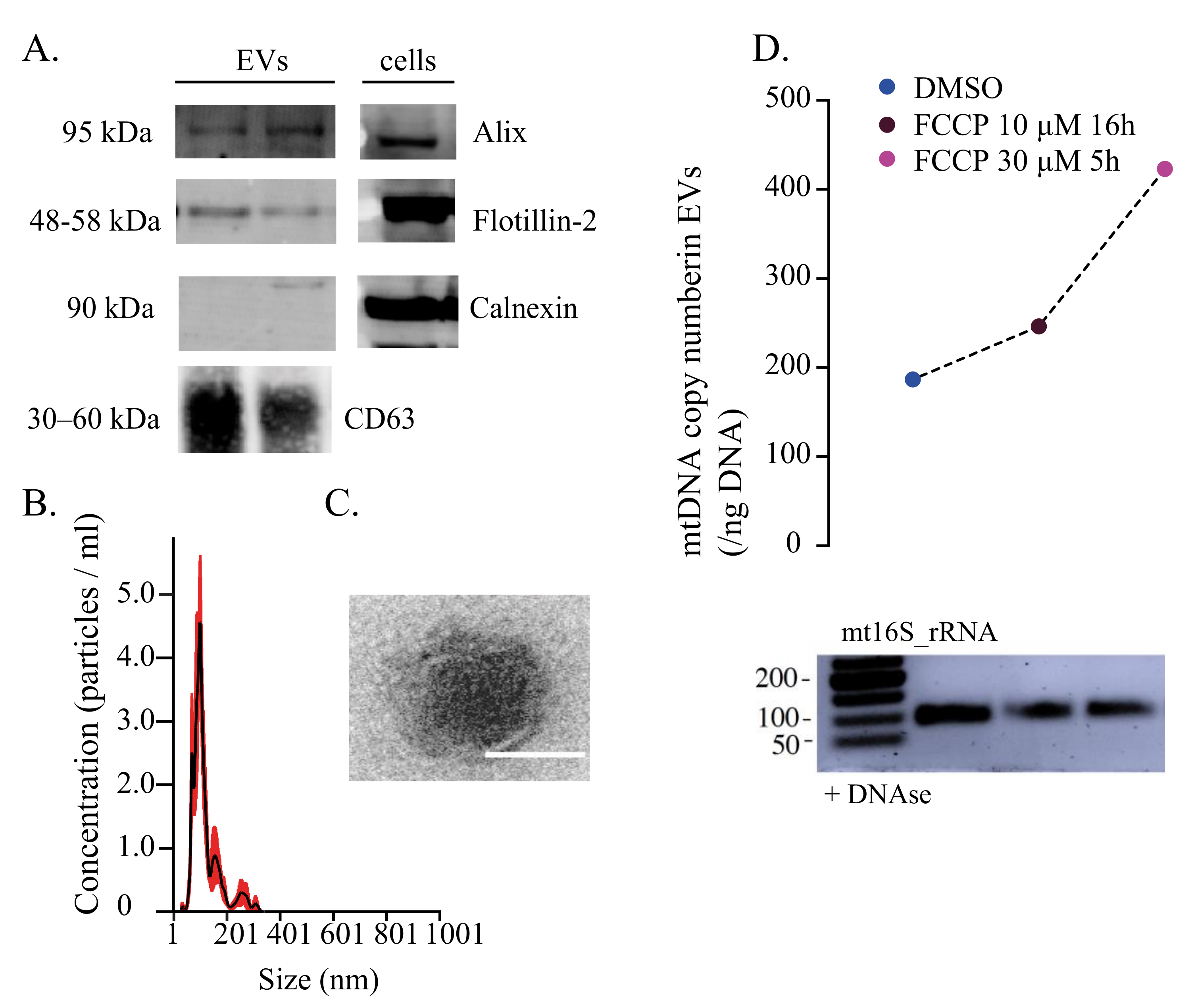
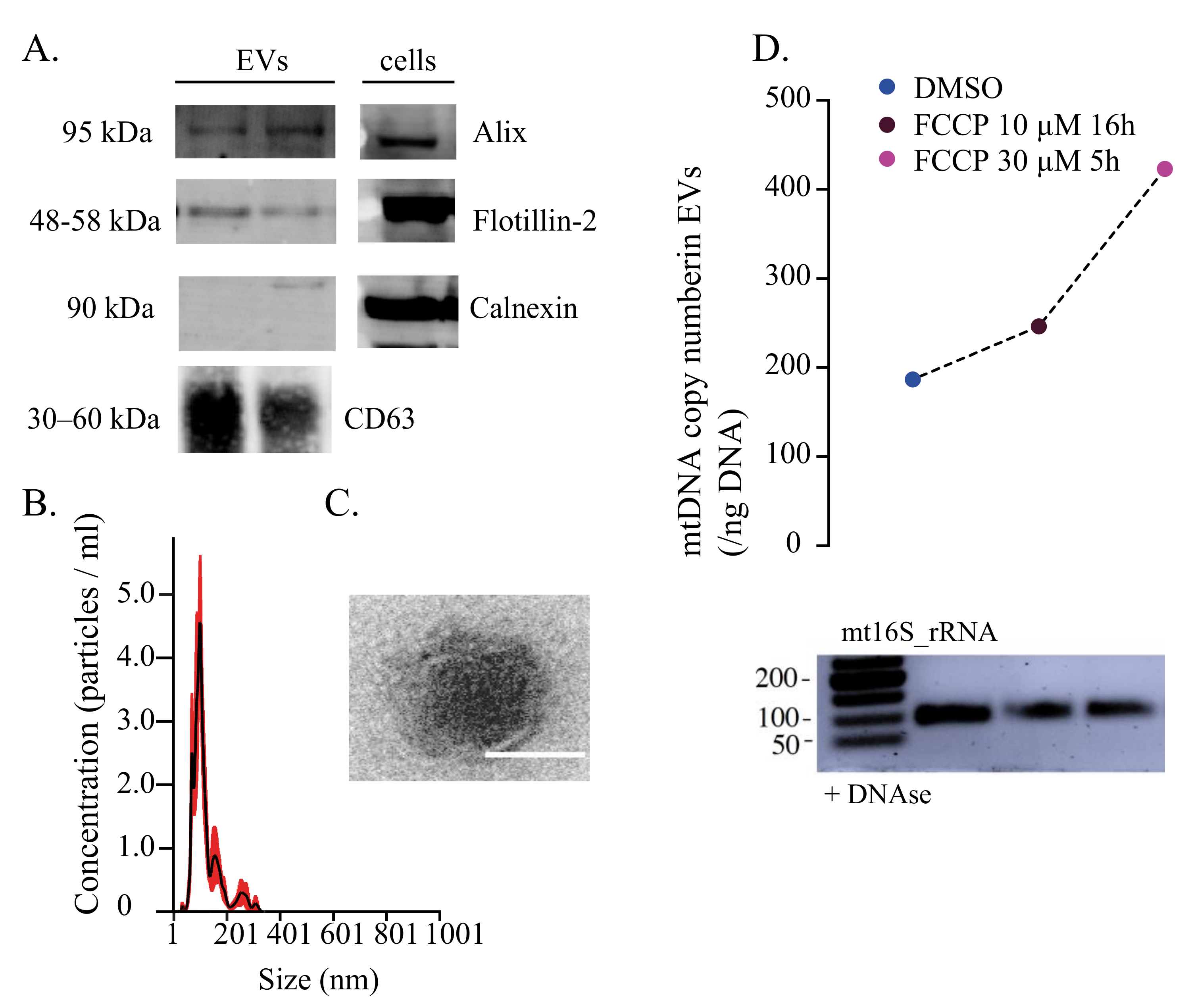{kind=link}
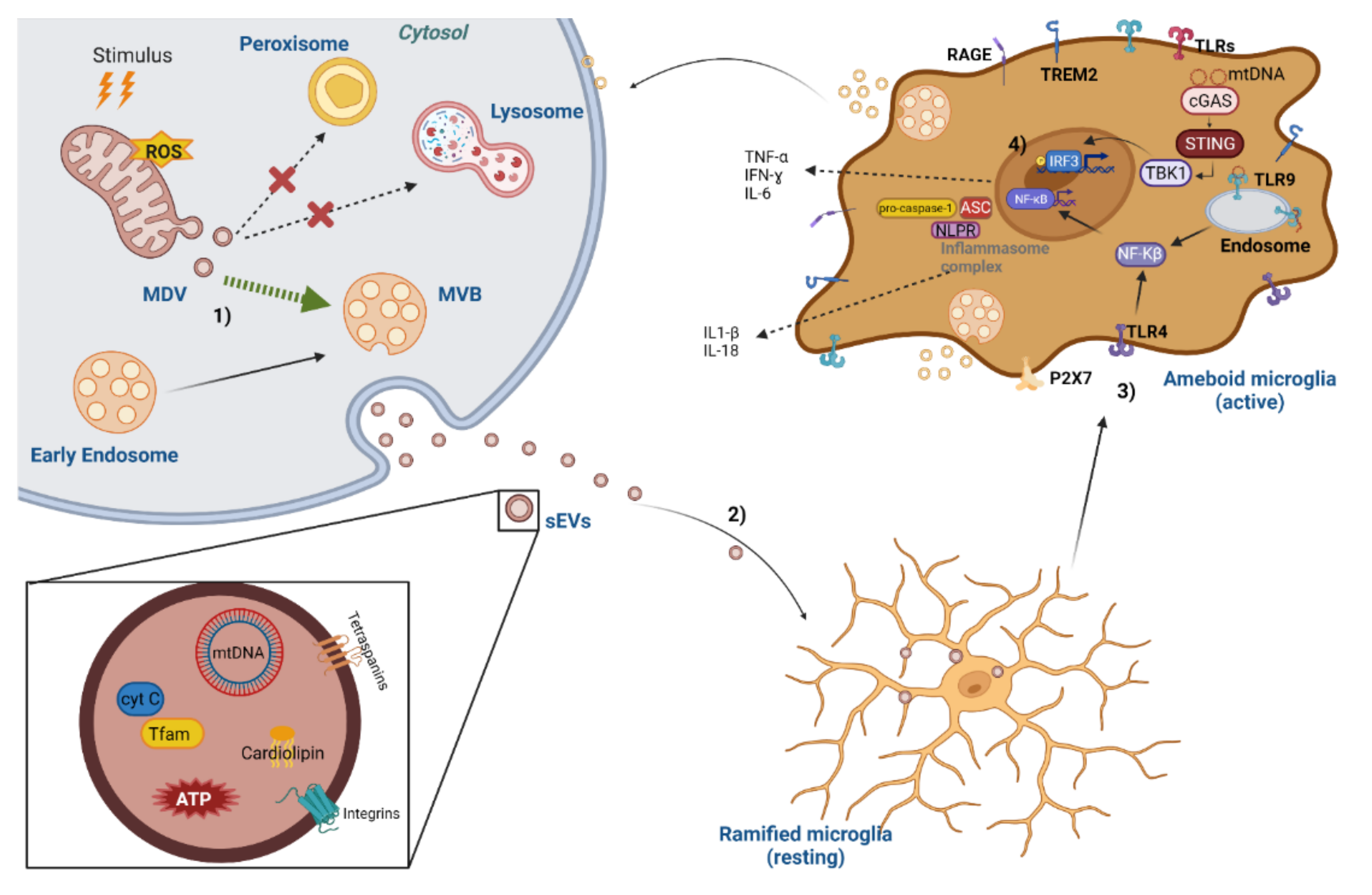{kind=link}
Abstract
:1. Introduction
2. The Role of Microglia in Brain Homeostasis and Neuroinflammation
3. Damage-Associated Molecular Patterns Released from Mitochondria: Role in Neurodegenerative Disorders
3.1. Mitochondrial DNA
3.2. ATP
3.3. Cytochrome C
3.4. TFAM
3.5. Cardiolipin
4. Mitochondrial DAMPs in Extracellular Vesicles as Inflammasome Activators
5. Mitochondrial-Derived Vesicles and Mitovesicles
6. Concluding Remarks
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Kwon, H.S.; Koh, S.H. Neuroinflammation in neurodegenerative disorders: The roles of microglia and astrocytes. Transl. Neurodegener. 2020, 9, 42. [Google Scholar] [CrossRef] [PubMed]
- Lyman, M.; Lloyd, D.G.; Ji, X.; Vizcaychipi, M.P.; Ma, D. Neuroinflammation: The role and consequences. Neurosci. Res. 2014, 79, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Kempuraj, D.; Thangavel, R.; Yang, E.; Pattani, S.; Zaheer, S.; Santillan, D.A.; Santillan, M.K.; Zaheer, A. Dopaminergic Toxin 1-Methyl-4-Phenylpyridinium, Proteins alpha-Synuclein and Glia Maturation Factor Activate Mast Cells and Release Inflammatory Mediators. PLoS ONE 2015, 10, e0135776. [Google Scholar] [CrossRef] [PubMed]
- Takeda, S.; Sato, N.; Ikimura, K.; Nishino, H.; Rakugi, H.; Morishita, R. Increased blood-brain barrier vulnerability to systemic inflammation in an Alzheimer disease mouse model. Neurobiol. Aging 2013, 34, 2064–2070. [Google Scholar] [CrossRef]
- Li, W.W.; Guo, T.Z.; Shi, X.; Sun, Y.; Wei, T.; Clark, D.J.; Kingery, W.S. Substance P spinal signaling induces glial activation and nociceptive sensitization after fracture. Neuroscience 2015, 310, 73–90. [Google Scholar] [CrossRef] [Green Version]
- Leng, F.; Edison, P. Neuroinflammation and microglial activation in Alzheimer disease: Where do we go from here? Nat. Rev. Neurol. 2021, 17, 157–172. [Google Scholar] [CrossRef]
- Nakahira, K.; Haspel, J.A.; Rathinam, V.A.; Lee, S.J.; Dolinay, T.; Lam, H.C.; Englert, J.A.; Rabinovitch, M.; Cernadas, M.; Kim, H.P.; et al. Autophagy proteins regulate innate immune responses by inhibiting the release of mitochondrial DNA mediated by the NALP3 inflammasome. Nat. Immunol. 2011, 12, 222–230. [Google Scholar] [CrossRef] [Green Version]
- De Biase, L.M.; Schuebel, K.E.; Fusfeld, Z.H.; Jair, K.; Hawes, I.A.; Cimbro, R.; Zhang, H.Y.; Liu, Q.R.; Shen, H.; Xi, Z.X.; et al. Local Cues Establish and Maintain Region-Specific Phenotypes of Basal Ganglia Microglia. Neuron 2017, 95, 341–356.e346. [Google Scholar] [CrossRef] [Green Version]
- Henkel, J.S.; Engelhardt, J.I.; Siklos, L.; Simpson, E.P.; Kim, S.H.; Pan, T.; Goodman, J.C.; Siddique, T.; Beers, D.R.; Appel, S.H. Presence of dendritic cells, MCP-1, and activated microglia/macrophages in amyotrophic lateral sclerosis spinal cord tissue. Ann. Neurol. 2004, 55, 221–235. [Google Scholar] [CrossRef]
- Jesudasan, S.J.B.; Gupta, S.J.; Churchward, M.A.; Todd, K.G.; Winship, I.R. Inflammatory Cytokine Profile and Plasticity of Brain and Spinal Microglia in Response to ATP and Glutamate. Front. Cell. Neurosci. 2021, 15, 634020. [Google Scholar] [CrossRef]
- Shaked, I.; Porat, Z.; Gersner, R.; Kipnis, J.; Schwartz, M. Early activation of microglia as antigen-presenting cells correlates with T cell-mediated protection and repair of the injured central nervous system. J. Neuroimmunol. 2004, 146, 84–93. [Google Scholar] [CrossRef]
- Conde, J.R.; Streit, W.J. Effect of aging on the microglial response to peripheral nerve injury. Neurobiol. Aging 2006, 27, 1451–1461. [Google Scholar] [CrossRef]
- Battista, D.; Ferrari, C.C.; Gage, F.H.; Pitossi, F.J. Neurogenic niche modulation by activated microglia: Transforming growth factor beta increases neurogenesis in the adult dentate gyrus. Eur. J. Neurosci. 2006, 23, 83–93. [Google Scholar] [CrossRef]
- Venkatesan, C.; Chrzaszcz, M.; Choi, N.; Wainwright, M.S. Chronic upregulation of activated microglia immunoreactive for galectin-3/Mac-2 and nerve growth factor following diffuse axonal injury. J. Neuroinflamm. 2010, 7, 32. [Google Scholar] [CrossRef] [Green Version]
- Bido, S.; Muggeo, S.; Massimino, L.; Marzi, M.J.; Giannelli, S.G.; Melacini, E.; Nannoni, M.; Gambare, D.; Bellini, E.; Ordazzo, G.; et al. Microglia-specific overexpression of alpha-synuclein leads to severe dopaminergic neurodegeneration by phagocytic exhaustion and oxidative toxicity. Nat. Commun. 2021, 12, 6237. [Google Scholar] [CrossRef]
- Zrzavy, T.; Hoftberger, R.; Berger, T.; Rauschka, H.; Butovsky, O.; Weiner, H.; Lassmann, H. Pro-inflammatory activation of microglia in the brain of patients with sepsis. Neuropathol. Appl. Neurobiol. 2019, 45, 278–290. [Google Scholar] [CrossRef] [Green Version]
- Moss, D.W.; Bates, T.E. Activation of murine microglial cell lines by lipopolysaccharide and interferon-gamma causes NO-mediated decreases in mitochondrial and cellular function. Eur. J. Neurosci. 2001, 13, 529–538. [Google Scholar] [CrossRef]
- Huo, Y.; Rangarajan, P.; Ling, E.A.; Dheen, S.T. Dexamethasone inhibits the Nox-dependent ROS production via suppression of MKP-1-dependent MAPK pathways in activated microglia. BMC Neurosci. 2011, 12, 49. [Google Scholar] [CrossRef] [Green Version]
- Mander, P.; Brown, G.C. Activation of microglial NADPH oxidase is synergistic with glial iNOS expression in inducing neuronal death: A dual-key mechanism of inflammatory neurodegeneration. J. Neuroinflamm. 2005, 2, 20. [Google Scholar] [CrossRef] [Green Version]
- Zhang, G.; He, J.L.; Xie, X.Y.; Yu, C. LPS-induced iNOS expression in N9 microglial cells is suppressed by geniposide via ERK, p38 and nuclear factor-kappaB signaling pathways. Int. J. Mol. Med. 2012, 30, 561–568. [Google Scholar] [CrossRef] [Green Version]
- Wen, X.; Xiao, L.; Zhong, Z.; Wang, L.; Li, Z.; Pan, X.; Liu, Z. Astaxanthin acts via LRP-1 to inhibit inflammation and reverse lipopolysaccharide-induced M1/M2 polarization of microglial cells. Oncotarget 2017, 8, 69370–69385. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cui, W.; Sun, C.; Ma, Y.; Wang, S.; Wang, X.; Zhang, Y. Inhibition of TLR4 Induces M2 Microglial Polarization and Provides Neuroprotection via the NLRP3 Inflammasome in Alzheimer’s Disease. Front. Neurosci. 2020, 14, 444. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Li, Y.; Yu, J.; Guo, M.; Meng, J.; Liu, C.; Xie, Y.; Feng, L.; Xiao, B.; Ma, C. Rho kinase inhibitor fasudil regulates microglia polarization and function. Neuroimmunomodulation 2013, 20, 313–322. [Google Scholar] [CrossRef] [PubMed]
- Ghosh, M.; Xu, Y.; Pearse, D.D. Cyclic AMP is a key regulator of M1 to M2a phenotypic conversion of microglia in the presence of Th2 cytokines. J. Neuroinflamm. 2016, 13, 9. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Casella, G.; Garzetti, L.; Gatta, A.T.; Finardi, A.; Maiorino, C.; Ruffini, F.; Martino, G.; Muzio, L.; Furlan, R. IL4 induces IL6-producing M2 macrophages associated to inhibition of neuroinflammation in vitro and in vivo. J. Neuroinflamm. 2016, 13, 139. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xie, Y.; Guo, H.; Wang, L.; Xu, L.; Zhang, X.; Yu, L.; Liu, Q.; Li, Y.; Zhao, N.; Zhao, N.; et al. Human albumin attenuates excessive innate immunity via inhibition of microglial Mincle/Syk signaling in subarachnoid hemorrhage. Brain Behav. Immun. 2017, 60, 346–360. [Google Scholar] [CrossRef]
- You, W.; Wang, Z.; Li, H.; Shen, H.; Xu, X.; Jia, G.; Chen, G. Inhibition of mammalian target of rapamycin attenuates early brain injury through modulating microglial polarization after experimental subarachnoid hemorrhage in rats. J. Neurol. Sci. 2016, 367, 224–231. [Google Scholar] [CrossRef]
- Li, R.; Liu, W.; Yin, J.; Chen, Y.; Guo, S.; Fan, H.; Li, X.; Zhang, X.; He, X.; Duan, C. TSG-6 attenuates inflammation-induced brain injury via modulation of microglial polarization in SAH rats through the SOCS3/STAT3 pathway. J. Neuroinflamm. 2018, 15, 231. [Google Scholar] [CrossRef] [PubMed]
- Morganti, J.M.; Riparip, L.K.; Rosi, S. Call Off the Dog(ma): M1/M2 Polarization Is Concurrent following Traumatic Brain Injury. PLoS ONE 2016, 11, e0148001. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chiu, I.M.; Morimoto, E.T.; Goodarzi, H.; Liao, J.T.; O’Keeffe, S.; Phatnani, H.P.; Muratet, M.; Carroll, M.C.; Levy, S.; Tavazoie, S.; et al. A neurodegeneration-specific gene-expression signature of acutely isolated microglia from an amyotrophic lateral sclerosis mouse model. Cell Rep. 2013, 4, 385–401. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xue, J.; Schmidt, S.V.; Sander, J.; Draffehn, A.; Krebs, W.; Quester, I.; De Nardo, D.; Gohel, T.D.; Emde, M.; Schmidleithner, L.; et al. Transcriptome-based network analysis reveals a spectrum model of human macrophage activation. Immunity 2014, 40, 274–288. [Google Scholar] [CrossRef] [Green Version]
- Stratoulias, V.; Venero, J.L.; Tremblay, M.E.; Joseph, B. Microglial subtypes: Diversity within the microglial community. EMBO J. 2019, 38, e101997. [Google Scholar] [CrossRef]
- Ajami, B.; Samusik, N.; Wieghofer, P.; Ho, P.P.; Crotti, A.; Bjornson, Z.; Prinz, M.; Fantl, W.J.; Nolan, G.P.; Steinman, L. Single-cell mass cytometry reveals distinct populations of brain myeloid cells in mouse neuroinflammation and neurodegeneration models. Nat. Neurosci. 2018, 21, 541–551. [Google Scholar] [CrossRef]
- Du, R.H.; Sun, H.B.; Hu, Z.L.; Lu, M.; Ding, J.H.; Hu, G. Kir6.1/K-ATP channel modulates microglia phenotypes: Implication in Parkinson’s disease. Cell Death Dis. 2018, 9, 404. [Google Scholar] [CrossRef] [PubMed]
- Furube, E.; Kawai, S.; Inagaki, H.; Takagi, S.; Miyata, S. Brain Region-dependent Heterogeneity and Dose-dependent Difference in Transient Microglia Population Increase during Lipopolysaccharide-induced Inflammation. Sci. Rep. 2018, 8, 2203. [Google Scholar] [CrossRef] [Green Version]
- Tan, Y.L.; Yuan, Y.; Tian, L. Microglial regional heterogeneity and its role in the brain. Mol. Psychiatry 2020, 25, 351–367. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Askew, K.; Li, K.; Olmos-Alonso, A.; Garcia-Moreno, F.; Liang, Y.; Richardson, P.; Tipton, T.; Chapman, M.A.; Riecken, K.; Beccari, S.; et al. Coupled Proliferation and Apoptosis Maintain the Rapid Turnover of Microglia in the Adult Brain. Cell Rep. 2017, 18, 391–405. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tay, T.L.; Mai, D.; Dautzenberg, J.; Fernandez-Klett, F.; Lin, G.; Sagar; Datta, M.; Drougard, A.; Stempfl, T.; Ardura-Fabregat, A.; et al. A new fate mapping system reveals context-dependent random or clonal expansion of microglia. Nat. Neurosci. 2017, 20, 793–803. [Google Scholar] [CrossRef] [PubMed]
- Savchenko, V.L.; Nikonenko, I.R.; Skibo, G.G.; McKanna, J.A. Distribution of microglia and astrocytes in different regions of the normal adult rat brain. Neurophysiology 1997, 29, 343–351. [Google Scholar] [CrossRef]
- Stowell, R.D.; Wong, E.L.; Batchelor, H.N.; Mendes, M.S.; Lamantia, C.E.; Whitelaw, B.S.; Majewska, A.K. Cerebellar microglia are dynamically unique and survey Purkinje neurons in vivo. Dev. Neurobiol. 2018, 78, 627–644. [Google Scholar] [CrossRef] [PubMed]
- Takagi, S.; Furube, E.; Nakano, Y.; Morita, M.; Miyata, S. Microglia are continuously activated in the circumventricular organs of mouse brain. J. Neuroimmunol. 2019, 331, 74–86. [Google Scholar] [CrossRef] [PubMed]
- Vela, J.M.; Dalmau, I.; Gonzalez, B.; Castellano, B. Morphology and distribution of microglial cells in the young and adult mouse cerebellum. J. Comp. Neurol. 1995, 361, 602–616. [Google Scholar] [CrossRef] [PubMed]
- Keren-Shaul, H.; Spinrad, A.; Weiner, A.; Matcovitch-Natan, O.; Dvir-Szternfeld, R.; Ulland, T.K.; David, E.; Baruch, K.; Lara-Astaiso, D.; Toth, B.; et al. A Unique Microglia Type Associated with Restricting Development of Alzheimer’s Disease. Cell 2017, 169, 1276–1290.e1217. [Google Scholar] [CrossRef] [PubMed]
- Lopes, K.P.; Snijders, G.J.L.; Humphrey, J.; Allan, A.; Sneeboer, M.A.M.; Navarro, E.; Schilder, B.M.; Vialle, R.A.; Parks, M.; Missall, R.; et al. Genetic analysis of the human microglial transcriptome across brain regions, aging and disease pathologies. Nat. Genet. 2022, 54, 4–17. [Google Scholar] [CrossRef]
- Andersen, M.S.; Bandres-Ciga, S.; Reynolds, R.H.; Hardy, J.; Ryten, M.; Krohn, L.; Gan-Or, Z.; Holtman, I.R.; Pihlstrom, L. International Parkinson’s Disease Genomics. C. Heritability Enrichment Implicates Microglia in Parkinson’s Disease Pathogenesis. Ann. Neurol. 2021, 89, 942–951. [Google Scholar] [CrossRef]
- International Multiple Sclerosis Genetics Consortium. Multiple sclerosis genomic map implicates peripheral immune cells and microglia in susceptibility. Science 2019, 365, aav7188. [Google Scholar] [CrossRef] [Green Version]
- Lambert, J.C.; Ibrahim-Verbaas, C.A.; Harold, D.; Naj, A.C.; Sims, R.; Bellenguez, C.; DeStafano, A.L.; Bis, J.C.; Beecham, G.W.; Grenier-Boley, B.; et al. Meta-analysis of 74,046 individuals identifies 11 new susceptibility loci for Alzheimer’s disease. Nat. Genet. 2013, 45, 1452–1458. [Google Scholar] [CrossRef] [Green Version]
- Lee, C.Y.D.; Daggett, A.; Gu, X.; Jiang, L.L.; Langfelder, P.; Li, X.; Wang, N.; Zhao, Y.; Park, C.S.; Cooper, Y.; et al. Elevated TREM2 Gene Dosage Reprograms Microglia Responsivity and Ameliorates Pathological Phenotypes in Alzheimer’s Disease Models. Neuron 2018, 97, 1032–1048.e1035. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Borroni, B.; Ferrari, F.; Galimberti, D.; Nacmias, B.; Barone, C.; Bagnoli, S.; Fenoglio, C.; Piaceri, I.; Archetti, S.; Bonvicini, C.; et al. Heterozygous TREM2 mutations in frontotemporal dementia. Neurobiol. Aging 2014, 35, 934.e7–934.e10. [Google Scholar] [CrossRef] [PubMed]
- Rayaprolu, S.; Mullen, B.; Baker, M.; Lynch, T.; Finger, E.; Seeley, W.W.; Hatanpaa, K.J.; Lomen-Hoerth, C.; Kertesz, A.; Bigio, E.H.; et al. TREM2 in neurodegeneration: Evidence for association of the p.R47H variant with frontotemporal dementia and Parkinson’s disease. Mol. Neurodegener. 2013, 8, 19. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Y.; Cella, M.; Mallinson, K.; Ulrich, J.D.; Young, K.L.; Robinette, M.L.; Gilfillan, S.; Krishnan, G.M.; Sudhakar, S.; Zinselmeyer, B.H.; et al. TREM2 lipid sensing sustains the microglial response in an Alzheimer’s disease model. Cell 2015, 160, 1061–1071. [Google Scholar] [CrossRef] [Green Version]
- Cady, J.; Koval, E.D.; Benitez, B.A.; Zaidman, C.; Jockel-Balsarotti, J.; Allred, P.; Baloh, R.H.; Ravits, J.; Simpson, E.; Appel, S.H.; et al. TREM2 variant p.R47H as a risk factor for sporadic amyotrophic lateral sclerosis. JAMA Neurol. 2014, 71, 449–453. [Google Scholar] [CrossRef] [Green Version]
- Walker, D.G.; Lue, L.F.; Serrano, G.; Adler, C.H.; Caviness, J.N.; Sue, L.I.; Beach, T.G. Altered Expression Patterns of Inflammation-Associated and Trophic Molecules in Substantia Nigra and Striatum Brain Samples from Parkinson’s Disease, Incidental Lewy Body Disease and Normal Control Cases. Front. Neurosci. 2015, 9, 507. [Google Scholar] [CrossRef] [Green Version]
- Sawada, M.; Imamura, K.; Nagatsu, T. Role of cytokines in inflammatory process in Parkinson’s disease. J. Neural Transm. Suppl. 2006, 70, 373–381. [Google Scholar]
- Na, S.J.; DiLella, A.G.; Lis, E.V.; Jones, K.; Levine, D.M.; Stone, D.J.; Hess, J.F. Molecular profiling of a 6-hydroxydopamine model of Parkinson’s disease. Neurochem. Res. 2010, 35, 761–772. [Google Scholar] [CrossRef]
- Mogi, M.; Togari, A.; Kondo, T.; Mizuno, Y.; Komure, O.; Kuno, S.; Ichinose, H.; Nagatsu, T. Caspase activities and tumor necrosis factor receptor R1 (p55) level are elevated in the substantia nigra from parkinsonian brain. J. Neural Transm. 2000, 107, 335–341. [Google Scholar] [CrossRef]
- Mishra, A.; Kim, H.J.; Shin, A.H.; Thayer, S.A. Synapse loss induced by interleukin-1beta requires pre- and post-synaptic mechanisms. J. Neuroimmune Pharmacol. 2012, 7, 571–578. [Google Scholar] [CrossRef] [Green Version]
- Sobue, A.; Komine, O.; Hara, Y.; Endo, F.; Mizoguchi, H.; Watanabe, S.; Murayama, S.; Saito, T.; Saido, T.C.; Sahara, N.; et al. Microglial gene signature reveals loss of homeostatic microglia associated with neurodegeneration of Alzheimer’s disease. Acta Neuropathol. Commun. 2021, 9, 1. [Google Scholar] [CrossRef]
- Grammas, P.; Ovase, R. Inflammatory factors are elevated in brain microvessels in Alzheimer’s disease. Neurobiol. Aging 2001, 22, 837–842. [Google Scholar] [CrossRef]
- Vogelzangs, N.; Duivis, H.E.; Beekman, A.T.; Kluft, C.; Neuteboom, J.; Hoogendijk, W.; Smit, J.H.; de Jonge, P.; Penninx, B.W. Association of depressive disorders, depression characteristics and antidepressant medication with inflammation. Transl. Psychiatry 2012, 2, e79. [Google Scholar] [CrossRef] [Green Version]
- MacDowell, K.S.; Garcia-Bueno, B.; Madrigal, J.L.; Parellada, M.; Arango, C.; Mico, J.A.; Leza, J.C. Risperidone normalizes increased inflammatory parameters and restores anti-inflammatory pathways in a model of neuroinflammation. Int. J. Neuropsychopharmacol. 2013, 16, 121–135. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Galatro, T.F.; Holtman, I.R.; Lerario, A.M.; Vainchtein, I.D.; Brouwer, N.; Sola, P.R.; Veras, M.M.; Pereira, T.F.; Leite, R.E.P.; Moller, T.; et al. Transcriptomic analysis of purified human cortical microglia reveals age-associated changes. Nat. Neurosci. 2017, 20, 1162–1171. [Google Scholar] [CrossRef] [PubMed]
- Taipa, R.; das Neves, S.P.; Sousa, A.L.; Fernandes, J.; Pinto, C.; Correia, A.P.; Santos, E.; Pinto, P.S.; Carneiro, P.; Costa, P.; et al. Proinflammatory and anti-inflammatory cytokines in the CSF of patients with Alzheimer’s disease and their correlation with cognitive decline. Neurobiol. Aging 2019, 76, 125–132. [Google Scholar] [CrossRef] [PubMed]
- Heneka, M.T.; Kummer, M.P.; Stutz, A.; Delekate, A.; Schwartz, S.; Vieira-Saecker, A.; Griep, A.; Axt, D.; Remus, A.; Tzeng, T.C.; et al. NLRP3 is activated in Alzheimer’s disease and contributes to pathology in APP/PS1 mice. Nature 2013, 493, 674–678. [Google Scholar] [CrossRef]
- Zrzavy, T.; Hametner, S.; Wimmer, I.; Butovsky, O.; Weiner, H.L.; Lassmann, H. Loss of ‘homeostatic’ microglia and patterns of their activation in active multiple sclerosis. Brain 2017, 140, 1900–1913. [Google Scholar] [CrossRef]
- Matzinger, P. Tolerance, danger, and the extended family. Annu. Rev. Immunol. 1994, 12, 991–1045. [Google Scholar] [CrossRef]
- Schroder, K.; Tschopp, J. The inflammasomes. Cell 2010, 140, 821–832. [Google Scholar] [CrossRef] [Green Version]
- Santoni, G.; Cardinali, C.; Morelli, M.B.; Santoni, M.; Nabissi, M.; Amantini, C. Danger- and pathogen-associated molecular patterns recognition by pattern-recognition receptors and ion channels of the transient receptor potential family triggers the inflammasome activation in immune cells and sensory neurons. J. Neuroinflamm. 2015, 12, 21. [Google Scholar] [CrossRef] [Green Version]
- Roh, J.S.; Sohn, D.H. Damage-Associated Molecular Patterns in Inflammatory Diseases. Immune Netw. 2018, 18, e27. [Google Scholar] [CrossRef]
- Jimenez-Dalmaroni, M.J.; Gerswhin, M.E.; Adamopoulos, I.E. The critical role of toll-like receptors--From microbial recognition to autoimmunity: A comprehensive review. Autoimmun. Rev. 2016, 15, 1–8. [Google Scholar] [CrossRef] [Green Version]
- Kufer, T.A.; Banks, D.J.; Philpott, D.J. Innate immune sensing of microbes by Nod proteins. Ann. N. Y. Acad. Sci. 2006, 1072, 19–27. [Google Scholar] [CrossRef] [Green Version]
- Rock, K.L.; Latz, E.; Ontiveros, F.; Kono, H. The sterile inflammatory response. Annu. Rev. Immunol. 2010, 28, 321–342. [Google Scholar] [CrossRef] [Green Version]
- Schaefer, L. Complexity of danger: The diverse nature of damage-associated molecular patterns. J. Biol. Chem. 2014, 289, 35237–35245. [Google Scholar] [CrossRef] [Green Version]
- Rock, K.L.; Kono, H. The inflammatory response to cell death. Annu. Rev. Pathol. 2008, 3, 99–126. [Google Scholar] [CrossRef]
- Murao, A.; Aziz, M.; Wang, H.; Brenner, M.; Wang, P. Release mechanisms of major DAMPs. Apoptosis 2021, 26, 152–162. [Google Scholar] [CrossRef]
- Wang, H.; Liu, C.; Zhao, Y.; Gao, G. Mitochondria regulation in ferroptosis. Eur. J. Cell Biol. 2020, 99, 151058. [Google Scholar] [CrossRef]
- Wen, Q.; Liu, J.; Kang, R.; Zhou, B.; Tang, D. The release and activity of HMGB1 in ferroptosis. Biochem. Biophys. Res. Commun. 2019, 510, 278–283. [Google Scholar] [CrossRef]
- Miyake, S.; Murai, S.; Kakuta, S.; Uchiyama, Y.; Nakano, H. Identification of the hallmarks of necroptosis and ferroptosis by transmission electron microscopy. Biochem. Biophys. Res. Commun. 2020, 527, 839–844. [Google Scholar] [CrossRef]
- Rosin, D.L.; Okusa, M.D. Dangers within: DAMP responses to damage and cell death in kidney disease. J. Am. Soc. Nephrol. JASN 2011, 22, 416–425. [Google Scholar] [CrossRef] [Green Version]
- Yang, D.; Chen, Q.; Yang, H.; Tracey, K.J.; Bustin, M.; Oppenheim, J.J. High mobility group box-1 protein induces the migration and activation of human dendritic cells and acts as an alarmin. J. Leukoc. Biol. 2007, 81, 59–66. [Google Scholar] [CrossRef]
- Gao, H.M.; Zhou, H.; Zhang, F.; Wilson, B.C.; Kam, W.; Hong, J.S. HMGB1 acts on microglia Mac1 to mediate chronic neuroinflammation that drives progressive neurodegeneration. J. Neurosci. 2011, 31, 1081–1092. [Google Scholar] [CrossRef]
- Rubartelli, A. DAMP-Mediated Activation of NLRP3-Inflammasome in Brain Sterile Inflammation: The Fine Line between Healing and Neurodegeneration. Front. Immunol. 2014, 5, 99. [Google Scholar] [CrossRef]
- Grazioli, S.; Pugin, J. Mitochondrial Damage-Associated Molecular Patterns: From Inflammatory Signaling to Human Diseases. Front. Immunol. 2018, 9, 832. [Google Scholar] [CrossRef]
- Bajwa, E.; Pointer, C.B.; Klegeris, A. The Role of Mitochondrial Damage-Associated Molecular Patterns in Chronic Neuroinflammation. Mediat. Inflamm. 2019, 2019, 4050796. [Google Scholar] [CrossRef] [Green Version]
- Nakahira, K.; Hisata, S.; Choi, A.M. The Roles of Mitochondrial Damage-Associated Molecular Patterns in Diseases. Antioxid. Redox Signal. 2015, 23, 1329–1350. [Google Scholar] [CrossRef] [Green Version]
- Bruni, F.; Lightowlers, R.N.; Chrzanowska-Lightowlers, Z.M. Human mitochondrial nucleases. FEBS J. 2017, 284, 1767–1777. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sagan, L. On the origin of mitosing cells. J. Theor. Biol. 1967, 14, 255–274. [Google Scholar] [CrossRef]
- Krysko, D.V.; Agostinis, P.; Krysko, O.; Garg, A.D.; Bachert, C.; Lambrecht, B.N.; Vandenabeele, P. Emerging role of damage-associated molecular patterns derived from mitochondria in inflammation. Trends Immunol. 2011, 32, 157–164. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Q.; Raoof, M.; Chen, Y.; Sumi, Y.; Sursal, T.; Junger, W.; Brohi, K.; Itagaki, K.; Hauser, C.J. Circulating mitochondrial DAMPs cause inflammatory responses to injury. Nature 2010, 464, 104–107. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Golpich, M.; Amini, E.; Mohamed, Z.; Azman Ali, R.; Mohamed Ibrahim, N.; Ahmadiani, A. Mitochondrial Dysfunction and Biogenesis in Neurodegenerative diseases: Pathogenesis and Treatment. CNS Neurosci. Ther. 2017, 23, 5–22. [Google Scholar] [CrossRef]
- Jetto, C.T.; Nambiar, A.; Manjithaya, R. Mitophagy and Neurodegeneration: Between the Knowns and the Unknowns. Front. Cell Dev. Biol. 2022, 10, 837337. [Google Scholar] [CrossRef] [PubMed]
- Beatriz, M.; Vilaça, R.; Anjo, S.I.; Manadas, B.; Januário, C.; Rego, A.C.; Lopes, C. Defective mitochondrial-lysosomal axis promotes extracellular vesicles release of mitochondrial components in Huntington’s Disease. bioRxiv 2022. [Google Scholar]
- Larsson, N.G. Somatic mitochondrial DNA mutations in mammalian aging. Annu. Rev. Biochem. 2010, 79, 683–706. [Google Scholar] [CrossRef] [PubMed]
- Lee, C.; Zeng, J.; Drew, B.G.; Sallam, T.; Martin-Montalvo, A.; Wan, J.; Kim, S.J.; Mehta, H.; Hevener, A.L.; de Cabo, R.; et al. The mitochondrial-derived peptide MOTS-c promotes metabolic homeostasis and reduces obesity and insulin resistance. Cell Metab. 2015, 21, 443–454. [Google Scholar] [CrossRef] [Green Version]
- Cobb, L.J.; Lee, C.; Xiao, J.; Yen, K.; Wong, R.G.; Nakamura, H.K.; Mehta, H.H.; Gao, Q.; Ashur, C.; Huffman, D.M.; et al. Naturally occurring mitochondrial-derived peptides are age-dependent regulators of apoptosis, insulin sensitivity, and inflammatory markers. Aging 2016, 8, 796–809. [Google Scholar] [CrossRef] [Green Version]
- Lopes, A.F.C. Mitochondrial metabolism and DNA methylation: A review of the interaction between two genomes. Clin. Epigenetics 2020, 12, 182. [Google Scholar] [CrossRef]
- Nakayama, H.; Otsu, K. Mitochondrial DNA as an inflammatory mediator in cardiovascular diseases. Biochem. J. 2018, 475, 839–852. [Google Scholar] [CrossRef]
- Torregrosa-Muñumer, R.; Forslund, J.M.E.; Goffart, S.; Pfeiffer, A.; Stojkovič, G.; Carvalho, G.; Al-Furoukh, N.; Blanco, L.; Wanrooij, S.; Pohjoismäki, J.L.O. PrimPol is required for replication reinitiation after mtDNA damage. Proc. Natl. Acad. Sci. USA 2017, 114, 11398–11403. [Google Scholar] [CrossRef] [Green Version]
- Collins, L.V.; Hajizadeh, S.; Holme, E.; Jonsson, I.M.; Tarkowski, A. Endogenously oxidized mitochondrial DNA induces in vivo and in vitro inflammatory responses. J. Leukoc. Biol. 2004, 75, 995–1000. [Google Scholar] [CrossRef]
- Qiu, Y.; Huang, Y.; Chen, M.; Yang, Y.; Li, X.; Zhang, W. Mitochondrial DNA in NLRP3 inflammasome activation. Int. Immunopharmacol. 2022, 108, 108719. [Google Scholar] [CrossRef]
- Hu, H.; Zhao, R.; He, Q.; Cui, C.; Song, J.; Guo, X.; Zang, N.; Yang, M.; Zou, Y.; Yang, J.; et al. cGAS-STING mediates cytoplasmic mitochondrial-DNA-induced inflammatory signal transduction during accelerated senescence of pancreatic beta-cells induced by metabolic stress. FASEB J. 2022, 36, e22266. [Google Scholar] [CrossRef]
- Zhang, W.; Li, G.; Luo, R.; Lei, J.; Song, Y.; Wang, B.; Ma, L.; Liao, Z.; Ke, W.; Liu, H.; et al. Cytosolic escape of mitochondrial DNA triggers cGAS-STING-NLRP3 axis-dependent nucleus pulposus cell pyroptosis. Exp. Mol. Med. 2022, 54, 129–142. [Google Scholar] [CrossRef]
- Ma, X.M.; Geng, K.; Law, B.Y.; Wang, P.; Pu, Y.L.; Chen, Q.; Xu, H.W.; Tan, X.Z.; Jiang, Z.Z.; Xu, Y. Lipotoxicity-induced mtDNA release promotes diabetic cardiomyopathy by activating the cGAS-STING pathway in obesity-related diabetes. Cell Biol. Toxicol. 2022. [CrossRef]
- Zhong, F.; Liang, S.; Zhong, Z. Emerging Role of Mitochondrial DNA as a Major Driver of Inflammation and Disease Progression. Trends Immunol. 2019, 40, 1120–1133. [Google Scholar] [CrossRef]
- Jauhari, A.; Baranov, S.V.; Suofu, Y.; Kim, J.; Singh, T.; Yablonska, S.; Li, F.; Wang, X.; Oberly, P.; Minnigh, M.B.; et al. Melatonin inhibits cytosolic mitochondrial DNA-induced neuroinflammatory signaling in accelerated aging and neurodegeneration. J. Clin. Investig. 2020, 130, 3124–3136. [Google Scholar] [CrossRef] [Green Version]
- Yu, C.H.; Davidson, S.; Harapas, C.R.; Hilton, J.B.; Mlodzianoski, M.J.; Laohamonthonkul, P.; Louis, C.; Low, R.R.J.; Moecking, J.; De Nardo, D.; et al. TDP-43 Triggers Mitochondrial DNA Release via mPTP to Activate cGAS/STING in ALS. Cell 2020, 183, 636–649.e618. [Google Scholar] [CrossRef]
- Pyle, A.; Brennan, R.; Kurzawa-Akanbi, M.; Yarnall, A.; Thouin, A.; Mollenhauer, B.; Burn, D.; Chinnery, P.F.; Hudson, G. Reduced cerebrospinal fluid mitochondrial DNA is a biomarker for early-stage Parkinson’s disease. Ann. Neurol. 2015, 78, 1000–1004. [Google Scholar] [CrossRef]
- Lowes, H.; Pyle, A.; Santibanez-Koref, M.; Hudson, G. Circulating cell-free mitochondrial DNA levels in Parkinson’s disease are influenced by treatment. Mol. Neurodegener. 2020, 15, 10. [Google Scholar] [CrossRef] [Green Version]
- Grunewald, A.; Rygiel, K.A.; Hepplewhite, P.D.; Morris, C.M.; Picard, M.; Turnbull, D.M. Mitochondrial DNA Depletion in Respiratory Chain-Deficient Parkinson Disease Neurons. Ann. Neurol. 2016, 79, 366–378. [Google Scholar] [CrossRef] [Green Version]
- Picca, A.; Guerra, F.; Calvani, R.; Bucci, C.; Lo Monaco, M.R.; Bentivoglio, A.R.; Landi, F.; Bernabei, R.; Marzetti, E. Mitochondrial-Derived Vesicles as Candidate Biomarkers in Parkinson’s Disease: Rationale, Design and Methods of the EXosomes in PArkiNson Disease (EXPAND) Study. Int. J. Mol. Sci. 2019, 20, 2373. [Google Scholar] [CrossRef] [Green Version]
- Bender, A.; Krishnan, K.J.; Morris, C.M.; Taylor, G.A.; Reeve, A.K.; Perry, R.H.; Jaros, E.; Hersheson, J.S.; Betts, J.; Klopstock, T.; et al. High levels of mitochondrial DNA deletions in substantia nigra neurons in aging and Parkinson disease. Nat. Genet. 2006, 38, 515–517. [Google Scholar] [CrossRef] [PubMed]
- Sliter, D.A.; Martinez, J.; Hao, L.; Chen, X.; Sun, N.; Fischer, T.D.; Burman, J.L.; Li, Y.; Zhang, Z.; Narendra, D.P.; et al. Parkin and PINK1 mitigate STING-induced inflammation. Nature 2018, 561, 258–262. [Google Scholar] [CrossRef] [PubMed]
- Podlesniy, P.; Figueiro-Silva, J.; Llado, A.; Antonell, A.; Sanchez-Valle, R.; Alcolea, D.; Lleo, A.; Molinuevo, J.L.; Serra, N.; Trullas, R. Low cerebrospinal fluid concentration of mitochondrial DNA in preclinical Alzheimer disease. Ann. Neurol. 2013, 74, 655–668. [Google Scholar] [CrossRef]
- Ammendolia, D.A.; Bement, W.M.; Brumell, J.H. Plasma membrane integrity: Implications for health and disease. BMC Biol. 2021, 19, 71. [Google Scholar] [CrossRef] [PubMed]
- Fliegert, R.; Heeren, J.; Koch-Nolte, F.; Nikolaev, V.O.; Lohr, C.; Meier, C.; Guse, A.H. Adenine nucleotides as paracrine mediators and intracellular second messengers in immunity and inflammation. Biochem. Soc. Trans. 2019, 47, 329–337. [Google Scholar] [CrossRef] [PubMed]
- Ferrari, D.; Chiozzi, P.; Falzoni, S.; Dal Susino, M.; Melchiorri, L.; Baricordi, O.R.; Di Virgilio, F. Extracellular ATP triggers IL-1 beta release by activating the purinergic P2Z receptor of human macrophages. J. Immunol. 1997, 159, 1451–1458. [Google Scholar] [PubMed]
- Ghiringhelli, F.; Apetoh, L.; Tesniere, A.; Aymeric, L.; Ma, Y.; Ortiz, C.; Vermaelen, K.; Panaretakis, T.; Mignot, G.; Ullrich, E.; et al. Activation of the NLRP3 inflammasome in dendritic cells induces IL-1beta-dependent adaptive immunity against tumors. Nat. Med. 2009, 15, 1170–1178. [Google Scholar] [CrossRef]
- Riteau, N.; Gasse, P.; Fauconnier, L.; Gombault, A.; Couegnat, M.; Fick, L.; Kanellopoulos, J.; Quesniaux, V.F.; Marchand-Adam, S.; Crestani, B.; et al. Extracellular ATP is a danger signal activating P2X7 receptor in lung inflammation and fibrosis. Am. J. Respir. Crit. Care Med. 2010, 182, 774–783. [Google Scholar] [CrossRef]
- Liu, Y.; Dai, Y.; Li, Q.; Chen, C.; Chen, H.; Song, Y.; Hua, F.; Zhang, Z. Beta-amyloid activates NLRP3 inflammasome via TLR4 in mouse microglia. Neurosci. Lett. 2020, 736, 135279. [Google Scholar] [CrossRef]
- Saez-Orellana, F.; Godoy, P.A.; Bastidas, C.Y.; Silva-Grecchi, T.; Guzman, L.; Aguayo, L.G.; Fuentealba, J. ATP leakage induces P2XR activation and contributes to acute synaptic excitotoxicity induced by soluble oligomers of beta-amyloid peptide in hippocampal neurons. Neuropharmacology 2016, 100, 116–123. [Google Scholar] [CrossRef]
- Carmo, M.R.; Menezes, A.P.; Nunes, A.C.; Pliassova, A.; Rolo, A.P.; Palmeira, C.M.; Cunha, R.A.; Canas, P.M.; Andrade, G.M. The P2X7 receptor antagonist Brilliant Blue G attenuates contralateral rotations in a rat model of Parkinsonism through a combined control of synaptotoxicity, neurotoxicity and gliosis. Neuropharmacology 2014, 81, 142–152. [Google Scholar] [CrossRef] [PubMed]
- Oliveira-Giacomelli, A.; Albino, C.M.; de Souza, H.D.N.; Correa-Velloso, J.; de Jesus Santos, A.P.; Baranova, J.; Ulrich, H. P2Y6 and P2X7 Receptor Antagonism Exerts Neuroprotective/Neuroregenerative Effects in an Animal Model of Parkinson’s Disease. Front. Cell. Neurosci. 2019, 13, 476. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gan, M.; Moussaud, S.; Jiang, P.; McLean, P.J. Extracellular ATP induces intracellular alpha-synuclein accumulation via P2X1 receptor-mediated lysosomal dysfunction. Neurobiol. Aging 2015, 36, 1209–1220. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jiang, T.; Hoekstra, J.; Heng, X.; Kang, W.; Ding, J.; Liu, J.; Chen, S.; Zhang, J. P2X7 receptor is critical in alpha-synuclein--mediated microglial NADPH oxidase activation. Neurobiol. Aging 2015, 36, 2304–2318. [Google Scholar] [CrossRef] [PubMed]
- Wilkaniec, A.; Gassowska, M.; Czapski, G.A.; Cieslik, M.; Sulkowski, G.; Adamczyk, A. P2X7 receptor-pannexin 1 interaction mediates extracellular alpha-synuclein-induced ATP release in neuroblastoma SH-SY5Y cells. Purinergic Signal. 2017, 13, 347–361. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Diaz-Hernandez, M.; Diez-Zaera, M.; Sanchez-Nogueiro, J.; Gomez-Villafuertes, R.; Canals, J.M.; Alberch, J.; Miras-Portugal, M.T.; Lucas, J.J. Altered P2X7-receptor level and function in mouse models of Huntington’s disease and therapeutic efficacy of antagonist administration. FASEB J. 2009, 23, 1893–1906. [Google Scholar] [CrossRef] [PubMed]
- Olla, I.; Santos-Galindo, M.; Elorza, A.; Lucas, J.J. P2X7 Receptor Upregulation in Huntington’s Disease Brains. Front. Mol. Neurosci. 2020, 13, 567430. [Google Scholar] [CrossRef]
- Kao, Y.H.; Lin, M.S.; Chen, C.M.; Wu, Y.R.; Chen, H.M.; Lai, H.L.; Chern, Y.; Lin, C.J. Targeting ENT1 and adenosine tone for the treatment of Huntington’s disease. Hum. Mol. Genet. 2017, 26, 467–478. [Google Scholar] [CrossRef]
- Nukui, T.; Matsui, A.; Niimi, H.; Sugimoto, T.; Hayashi, T.; Dougu, N.; Konishi, H.; Yamamoto, M.; Anada, R.; Matsuda, N.; et al. Increased cerebrospinal fluid adenosine 5′-triphosphate in patients with amyotrophic lateral sclerosis. BMC Neurol. 2021, 21, 255. [Google Scholar] [CrossRef]
- Apolloni, S.; Fabbrizio, P.; Amadio, S.; Napoli, G.; Freschi, M.; Sironi, F.; Pevarello, P.; Tarroni, P.; Liberati, C.; Bendotti, C.; et al. Novel P2X7 Antagonist Ameliorates the Early Phase of ALS Disease and Decreases Inflammation and Autophagy in SOD1-G93A Mouse Model. Int. J. Mol. Sci. 2021, 22, 6493. [Google Scholar] [CrossRef]
- Fabbrizio, P.; Amadio, S.; Apolloni, S.; Volonte, C. P2X7 Receptor Activation Modulates Autophagy in SOD1-G93A Mouse Microglia. Front. Cell. Neurosci. 2017, 11, 249. [Google Scholar] [CrossRef] [Green Version]
- Apolloni, S.; Parisi, C.; Pesaresi, M.G.; Rossi, S.; Carri, M.T.; Cozzolino, M.; Volonte, C.; D’Ambrosi, N. The NADPH oxidase pathway is dysregulated by the P2X7 receptor in the SOD1-G93A microglia model of amyotrophic lateral sclerosis. J. Immunol. 2013, 190, 5187–5195. [Google Scholar] [CrossRef] [Green Version]
- D’Ambrosi, N.; Finocchi, P.; Apolloni, S.; Cozzolino, M.; Ferri, A.; Padovano, V.; Pietrini, G.; Carri, M.T.; Volonte, C. The proinflammatory action of microglial P2 receptors is enhanced in SOD1 models for amyotrophic lateral sclerosis. J. Immunol. 2009, 183, 4648–4656. [Google Scholar] [CrossRef] [Green Version]
- Atlante, A.; Calissano, P.; Bobba, A.; Azzariti, A.; Marra, E.; Passarella, S. Cytochrome c is released from mitochondria in a reactive oxygen species (ROS)-dependent fashion and can operate as a ROS scavenger and as a respiratory substrate in cerebellar neurons undergoing excitotoxic death. J. Biol. Chem. 2000, 275, 37159–37166. [Google Scholar] [CrossRef] [Green Version]
- Kagan, V.E.; Tyurin, V.A.; Jiang, J.; Tyurina, Y.Y.; Ritov, V.B.; Amoscato, A.A.; Osipov, A.N.; Belikova, N.A.; Kapralov, A.A.; Kini, V.; et al. Cytochrome c acts as a cardiolipin oxygenase required for release of proapoptotic factors. Nat. Chem. Biol. 2005, 1, 223–232. [Google Scholar] [CrossRef]
- Garrido, C.; Galluzzi, L.; Brunet, M.; Puig, P.E.; Didelot, C.; Kroemer, G. Mechanisms of cytochrome c release from mitochondria. Cell Death Differ. 2006, 13, 1423–1433. [Google Scholar] [CrossRef] [Green Version]
- Renz, A.; Berdel, W.E.; Kreuter, M.; Belka, C.; Schulze-Osthoff, K.; Los, M. Rapid extracellular release of cytochrome c is specific for apoptosis and marks cell death in vivo. Blood 2001, 98, 1542–1548. [Google Scholar] [CrossRef] [Green Version]
- Pullerits, R.; Bokarewa, M.; Jonsson, I.-M.; Verdrengh, M.; Tarkowski, A. Extracellular cytochrome c, a mitochondrial apoptosis-related protein, induces arthritis. Rheumatology 2004, 44, 32–39. [Google Scholar] [CrossRef] [Green Version]
- Ahlemeyer, B.; Klumpp, S.; Krieglstein, J. Release of cytochrome c into the extracellular space contributes to neuronal apoptosis induced by staurosporine. Brain Res. 2002, 934, 107–116. [Google Scholar] [CrossRef]
- Wenzel, T.J.; Bajwa, E.; Klegeris, A. Cytochrome c can be released into extracellular space and modulate functions of human astrocytes in a toll-like receptor 4-dependent manner. Biochim. Biophys. Acta Gen. Subj. 2019, 1863, 129400. [Google Scholar] [CrossRef]
- Lopes, C.; Tang, Y.; Anjo, S.I.; Manadas, B.; Onofre, I.; de Almeida, L.P.; Daley, G.Q.; Schlaeger, T.M.; Rego, A.C.C. Mitochondrial and Redox Modifications in Huntington Disease Induced Pluripotent Stem Cells Rescued by CRISPR/Cas9 CAGs Targeting. Front. Cell Dev. Biol. 2020, 8, 576592. [Google Scholar] [CrossRef] [PubMed]
- Chaung, W.W.; Wu, R.; Ji, Y.; Dong, W.; Wang, P. Mitochondrial transcription factor A is a proinflammatory mediator in hemorrhagic shock. Int. J. Mol. Med. 2012, 30, 199–203. [Google Scholar] [PubMed]
- Schindler, S.M.; Frank, M.G.; Annis, J.L.; Maier, S.F.; Klegeris, A. Pattern recognition receptors mediate pro-inflammatory effects of extracellular mitochondrial transcription factor A (TFAM). Mol. Cell. Neurosci. 2018, 89, 71–79. [Google Scholar] [CrossRef] [PubMed]
- Julian, M.W.; Shao, G.; Vangundy, Z.C.; Papenfuss, T.L.; Crouser, E.D. Mitochondrial transcription factor A, an endogenous danger signal, promotes TNFalpha release via RAGE- and TLR9-responsive plasmacytoid dendritic cells. PLoS ONE 2013, 8, e72354. [Google Scholar] [CrossRef] [PubMed]
- Verdier, Y.; Zarandi, M.; Penke, B. Amyloid beta-peptide interactions with neuronal and glial cell plasma membrane: Binding sites and implications for Alzheimer’s disease. J. Pept. Sci. 2004, 10, 229–248. [Google Scholar] [CrossRef]
- Lue, L.F.; Walker, D.G.; Brachova, L.; Beach, T.G.; Rogers, J.; Schmidt, A.M.; Stern, D.M.; Yan, S.D. Involvement of microglial receptor for advanced glycation endproducts (RAGE) in Alzheimer’s disease: Identification of a cellular activation mechanism. Exp. Neurol. 2001, 171, 29–45. [Google Scholar] [CrossRef]
- Jiang, X.; Wang, X.; Tuo, M.; Ma, J.; Xie, A. RAGE and its emerging role in the pathogenesis of Parkinson’s disease. Neurosci. Lett. 2018, 672, 65–69. [Google Scholar] [CrossRef]
- Huang, M.; Guo, M.; Wang, K.; Wu, K.; Li, Y.; Tian, T.; Wang, Y.; Yan, W.; Zhou, Z.; Yang, H. HMGB1 Mediates Paraquat-Induced Neuroinflammatory Responses via Activating RAGE Signaling Pathway. Neurotox. Res. 2020, 37, 913–925. [Google Scholar] [CrossRef]
- Paudel, Y.N.; Angelopoulou, E.; Piperi, C.; Othman, I.; Shaikh, M.F. Implication of HMGB1 signaling pathways in Amyotrophic lateral sclerosis (ALS): From molecular mechanisms to pre-clinical results. Pharmacol. Res. 2020, 156, 104792. [Google Scholar] [CrossRef]
- Anzilotti, S.; Giampa, C.; Laurenti, D.; Perrone, L.; Bernardi, G.; Melone, M.A.; Fusco, F.R. Immunohistochemical localization of receptor for advanced glycation end (RAGE) products in the R6/2 mouse model of Huntington’s disease. Brain Res. Bull. 2012, 87, 350–358. [Google Scholar] [CrossRef]
- Kim, J.; Waldvogel, H.J.; Faull, R.L.; Curtis, M.A.; Nicholson, L.F. The RAGE receptor and its ligands are highly expressed in astrocytes in a grade-dependant manner in the striatum and subependymal layer in Huntington’s disease. J. Neurochem. 2015, 134, 927–942. [Google Scholar] [CrossRef] [PubMed]
- Paradies, G.; Paradies, V.; Ruggiero, F.M.; Petrosillo, G. Role of Cardiolipin in Mitochondrial Function and Dynamics in Health and Disease: Molecular and Pharmacological Aspects. Cells 2019, 8, 728. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, X.X.; Tsoi, B.; Li, Y.F.; Kurihara, H.; He, R.R. Cardiolipin and its different properties in mitophagy and apoptosis. J. Histochem. Cytochem. 2015, 63, 301–311. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Esposti, M.D.; Cristea, I.M.; Gaskell, S.J.; Nakao, Y.; Dive, C. Proapoptotic Bid binds to monolysocardiolipin, a new molecular connection between mitochondrial membranes and cell death. Cell Death Differ. 2003, 10, 1300–1309. [Google Scholar] [CrossRef] [Green Version]
- Pointer, C.B.; Wenzel, T.J.; Klegeris, A. Extracellular cardiolipin regulates select immune functions of microglia and microglia-like cells. Brain Res. Bull. 2019, 146, 153–163. [Google Scholar] [CrossRef]
- Wenzel, T.J.; Ranger, A.L.; McRae, S.A.; Klegeris, A. Extracellular cardiolipin modulates microglial phagocytosis and cytokine secretion in a toll-like receptor (TLR) 4-dependent manner. J. Neuroimmunol. 2021, 353, 577496. [Google Scholar] [CrossRef]
- Iyer, S.S.; He, Q.; Janczy, J.R.; Elliott, E.I.; Zhong, Z.; Olivier, A.K.; Sadler, J.J.; Knepper-Adrian, V.; Han, R.; Qiao, L.; et al. Mitochondrial cardiolipin is required for Nlrp3 inflammasome activation. Immunity 2013, 39, 311–323. [Google Scholar] [CrossRef] [Green Version]
- Liu, Q.; Zhang, D.; Hu, D.; Zhou, X.; Zhou, Y. The role of mitochondria in NLRP3 inflammasome activation. Mol. Immunol. 2018, 103, 115–124. [Google Scholar] [CrossRef]
- Merighi, S.; Nigro, M.; Travagli, A.; Pasquini, S.; Borea, P.A.; Varani, K.; Vincenzi, F.; Gessi, S. A2A Adenosine Receptor: A Possible Therapeutic Target for Alzheimer’s Disease by Regulating NLRP3 Inflammasome Activity? Int. J. Mol. Sci. 2022, 23, 5056. [Google Scholar] [CrossRef]
- Islam, J.; Cho, J.A.; Kim, J.Y.; Park, K.S.; Koh, Y.J.; Chung, C.Y.; Lee, E.J.; Nam, S.J.; Lee, K.; Kim, S.H.; et al. GPCR19 Regulates P2X7R-Mediated NLRP3 Inflammasomal Activation of Microglia by Amyloid beta in a Mouse Model of Alzheimer’s Disease. Front. Immunol. 2022, 13, 766919. [Google Scholar] [CrossRef]
- Ahmed, S.; Panda, S.R.; Kwatra, M.; Sahu, B.D.; Naidu, V. Perillyl Alcohol Attenuates NLRP3 Inflammasome Activation and Rescues Dopaminergic Neurons in Experimental In Vitro and In Vivo Models of Parkinson’s Disease. ACS Chem. Neurosci. 2022, 13, 53–68. [Google Scholar] [CrossRef]
- Chen, K.P.; Hua, K.F.; Tsai, F.T.; Lin, T.Y.; Cheng, C.Y.; Yang, D.I.; Hsu, H.T.; Ju, T.C. A selective inhibitor of the NLRP3 inflammasome as a potential therapeutic approach for neuroprotection in a transgenic mouse model of Huntington’s disease. J. Neuroinflamm. 2022, 19, 56. [Google Scholar] [CrossRef]
- Banerjee, P.; Elliott, E.; Rifai, O.M.; O’Shaughnessy, J.; McDade, K.; Abrahams, S.; Chandran, S.; Smith, C.; Gregory, J.M. NLRP3 inflammasome as a key molecular target underlying cognitive resilience in amyotrophic lateral sclerosis. J. Pathol. 2022, 256, 262–268. [Google Scholar] [CrossRef]
- Beatriz, M.; Vilaça, R.; Lopes, C. Exosomes: Innocent bystanders or critical culprits in neurodegenerative diseases. Front. Cell Dev. Biol. 2021, 9, 1047. [Google Scholar] [CrossRef]
- Théry, C.; Witwer, K.W.; Aikawa, E.; Alcaraz, M.J.; Anderson, J.D.; Andriantsitohaina, R.; Antoniou, A.; Arab, T.; Archer, F.; Atkin-Smith, G.K.; et al. Minimal information for studies of extracellular vesicles 2018 (MISEV2018): A position statement of the International Society for Extracellular Vesicles and update of the MISEV2014 guidelines. J. Extracell. Vesicles 2018, 7, 1535750. [Google Scholar] [CrossRef] [Green Version]
- Théry, C.; Ostrowski, M.; Segura, E. Membrane vesicles as conveyors of immune responses. Nat. Rev. Immunol. 2009, 9, 581–593. [Google Scholar] [CrossRef]
- Joshi, A.U.; Ebert, A.E.; Haileselassie, B.; Mochly-Rosen, D. Drp1/Fis1-mediated mitochondrial fragmentation leads to lysosomal dysfunction in cardiac models of Huntington’s disease. J. Mol. Cell Cardiol. 2019, 127, 125–133. [Google Scholar] [CrossRef]
- Peruzzotti-Jametti, L.; Bernstock, J.D.; Willis, C.M.; Manferrari, G.; Rogall, R.; Fernandez-Vizarra, E.; Williamson, J.C.; Braga, A.; van den Bosch, A.; Leonardi, T.; et al. Neural stem cells traffic functional mitochondria via extracellular vesicles. PLoS Biol 2021, 19, e3001166. [Google Scholar] [CrossRef]
- Boudreau, L.H.; Duchez, A.C.; Cloutier, N.; Soulet, D.; Martin, N.; Bollinger, J.; Paré, A.; Rousseau, M.; Naika, G.S.; Lévesque, T.; et al. Platelets release mitochondria serving as substrate for bactericidal group IIA-secreted phospholipase A2 to promote inflammation. Blood 2014, 124, 2173–2183. [Google Scholar] [CrossRef] [Green Version]
- Todkar, K.; Chikhi, L.; Desjardins, V.; El-Mortada, F.; Pépin, G.; Germain, M. Selective packaging of mitochondrial proteins into extracellular vesicles prevents the release of mitochondrial DAMPs. Nat. Commun. 2021, 12, 1971. [Google Scholar] [CrossRef]
- Sansone, P.; Savini, C.; Kurelac, I.; Chang, Q.; Amato, L.B.; Strillacci, A.; Stepanova, A.; Iommarini, L.; Mastroleo, C.; Daly, L.; et al. Packaging and transfer of mitochondrial DNA via exosomes regulate escape from dormancy in hormonal therapy-resistant breast cancer. Proc. Natl. Acad. Sci. USA 2017, 114, E9066–E9075. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, X.; Weidling, I.; Koppel, S.; Menta, B.; Ortiz, J.P.; Kalani, A.; Wilkins, H.M.; Swerdlow, R.H. Detection of mitochondria-pertinent components in exosomes. Mitochondrion 2020, 55, 100–110. [Google Scholar] [CrossRef] [PubMed]
- Soubannier, V.; McLelland, G.L.; Zunino, R.; Braschi, E.; Rippstein, P.; Fon, E.A.; McBride, H.M. A vesicular transport pathway shuttles cargo from mitochondria to lysosomes. Curr. Biol. CB 2012, 22, 135–141. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Paik, S.; Kim, J.K.; Silwal, P.; Sasakawa, C.; Jo, E.K. An update on the regulatory mechanisms of NLRP3 inflammasome activation. Cell Mol. Immunol. 2021, 18, 1141–1160. [Google Scholar] [CrossRef]
- Nam, B.Y.; Jhee, J.H.; Park, J.; Kim, S.; Kim, G.; Park, J.T.; Yoo, T.H.; Kang, S.W.; Yu, J.W.; Han, S.H. PGC-1α inhibits the NLRP3 inflammasome via preserving mitochondrial viability to protect kidney fibrosis. Cell Death Dis. 2022, 13, 31. [Google Scholar] [CrossRef]
- Groß, C.J.; Mishra, R.; Schneider, K.S.; Médard, G.; Wettmarshausen, J.; Dittlein, D.C.; Shi, H.; Gorka, O.; Koenig, P.A.; Fromm, S.; et al. K + Efflux-Independent NLRP3 Inflammasome Activation by Small Molecules Targeting Mitochondria K + Efflux-Independent NLRP3 Inflammasome Activation by Small Molecules Targeting Mitochondria. Immunity 2016, 45, 761–773. [Google Scholar] [CrossRef] [Green Version]
- Billingham, L.K.; Stoolman, J.S.; Vasan, K.; Rodriguez, A.E.; Poor, T.A.; Szibor, M.; Jacobs, H.T.; Reczek, C.R.; Rashidi, A.; Zhang, P.; et al. Mitochondrial electron transport chain is necessary for NLRP3 inflammasome activation. Nat. Immunol. 2022, 23, 692–704. [Google Scholar] [CrossRef]
- Sadatomi, D.; Nakashioya, K.; Mamiya, S.; Honda, S.; Kameyama, Y.; Yamamura, Y.; Tanimura, S.; Takeda, K. Mitochondrial function is required for extracellular ATP-induced NLRP3 inflammasome activation. J. Biochem. 2017, 161, 503–512. [Google Scholar] [CrossRef]
- West, A.P.; Shadel, G.S. Mitochondrial DNA in innate immune responses and inflammatory pathology. Nat. Rev. Immunol. 2017, 17, 363–375. [Google Scholar] [CrossRef]
- Budden, C.F.; Gearing, L.J.; Kaiser, R.; Standke, L.; Hertzog, P.J.; Latz, E. Inflammasome-induced extracellular vesicles harbour distinct RNA signatures and alter bystander macrophage responses. J. Extracell. Vesicles 2021, 10, e12127. [Google Scholar] [CrossRef]
- Xia, C.; Zeng, Z.; Fang, B.; Tao, M.; Gu, C.; Zheng, L.; Wang, Y.; Shi, Y.; Fang, C.; Mei, S.; et al. Mesenchymal stem cell-derived exosomes ameliorate intervertebral disc degeneration via anti-oxidant and anti-inflammatory effects. Free Radic. Biol. Med. 2019, 143, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Koniusz, S.; Andrzejewska, A.; Muraca, M.; Srivastava, A.K.; Janowski, M.; Lukomska, B. Extracellular Vesicles in Physiology, Pathology, and Therapy of the Immune and Central Nervous System, with Focus on Extracellular Vesicles Derived from Mesenchymal Stem Cells as Therapeutic Tools. Front. Cell Neurosci. 2016, 10, 109. [Google Scholar] [CrossRef] [PubMed]
- Neuspiel, M.; Schauss, A.C.; Braschi, E.; Zunino, R.; Rippstein, P.; Rachubinski, R.A.; Andrade-Navarro, M.A.; McBride, H.M. Cargo-selected transport from the mitochondria to peroxisomes is mediated by vesicular carriers. Curr. Biol. CB 2008, 18, 102–108. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Soubannier, V.; Rippstein, P.; Kaufman, B.A.; Shoubridge, E.A.; McBride, H.M. Reconstitution of mitochondria derived vesicle formation demonstrates selective enrichment of oxidized cargo. PLoS ONE 2012, 7, e52830. [Google Scholar] [CrossRef] [Green Version]
- McLelland, G.L.; Soubannier, V.; Chen, C.X.; McBride, H.M.; Fon, E.A. Parkin and PINK1 function in a vesicular trafficking pathway regulating mitochondrial quality control. EMBO J. 2014, 33, 282–295. [Google Scholar] [CrossRef]
- Towers, C.G.; Wodetzki, D.K.; Thorburn, J.; Smith, K.R.; Caino, M.C.; Thorburn, A. Mitochondrial-derived vesicles compensate for loss of LC3-mediated mitophagy. Dev. Cell 2021, 56, 2029–2042.e2025. [Google Scholar] [CrossRef]
- Audano, P.A.; Sulovari, A.; Graves-Lindsay, T.A.; Cantsilieris, S.; Sorensen, M.; Welch, A.E.; Dougherty, M.L.; Nelson, B.J.; Shah, A.; Dutcher, S.K.; et al. Characterizing the Major Structural Variant Alleles of the Human Genome. Cell 2019, 176, 663–675.e619. [Google Scholar] [CrossRef] [Green Version]
- Vasam, G.; Nadeau, R.; Cadete, V.J.J.; Lavallee-Adam, M.; Menzies, K.J.; Burelle, Y. Proteomics characterization of mitochondrial-derived vesicles under oxidative stress. FASEB J. 2021, 35, e21278. [Google Scholar] [CrossRef]
- Ryan, T.A.; Phillips, E.O.; Collier, C.L.; Jb Robinson, A.; Routledge, D.; Wood, R.E.; Assar, E.A.; Tumbarello, D.A. Tollip coordinates Parkin-dependent trafficking of mitochondrial-derived vesicles. EMBO J. 2020, 39, e102539. [Google Scholar] [CrossRef]
- Katoh, Y.; Shiba, Y.; Mitsuhashi, H.; Yanagida, Y.; Takatsu, H.; Nakayama, K. Tollip and Tom1 form a complex and recruit ubiquitin-conjugated proteins onto early endosomes. J. Biol. Chem. 2004, 279, 24435–24443. [Google Scholar] [CrossRef] [Green Version]
- Sugiura, A.; McLelland, G.L.; Fon, E.A.; McBride, H.M. A new pathway for mitochondrial quality control: Mitochondrial-derived vesicles. EMBO J. 2014, 33, 2142–2156. [Google Scholar] [CrossRef] [Green Version]
- Konig, T.; Nolte, H.; Aaltonen, M.J.; Tatsuta, T.; Krols, M.; Stroh, T.; Langer, T.; McBride, H.M. MIROs and DRP1 drive mitochondrial-derived vesicle biogenesis and promote quality control. Nat. Cell Biol. 2021, 23, 1271–1286. [Google Scholar] [CrossRef]
- Mohanty, A.; Zunino, R.; Soubannier, V.; Dilipkumar, S. A new functional role of mitochondria-anchored protein ligase in peroxisome morphology in mammalian cells. J. Cell. Biochem. 2021, 122, 1686–1700. [Google Scholar] [CrossRef]
- Braschi, E.; Goyon, V.; Zunino, R.; Mohanty, A.; Xu, L.; McBride, H.M. Vps35 mediates vesicle transport between the mitochondria and peroxisomes. Curr. Biol. CB 2010, 20, 1310–1315. [Google Scholar] [CrossRef] [Green Version]
- Thomas, M.A.; Miller, J.L.; Delco, M.L. Human mesenchymal stem cells release functional mitochondria in extracellular vesicles. Osteoarthr. Cartil. 2021, 29, S42–S43. [Google Scholar] [CrossRef]
- Puhm, F.; Afonyushkin, T.; Resch, U.; Obermayer, G.; Rohde, M.; Penz, T.; Schuster, M.; Wagner, G.; Rendeiro, A.F.; Melki, I.; et al. Mitochondria Are a Subset of Extracellular Vesicles Released by Activated Monocytes and Induce Type I IFN and TNF Responses in Endothelial Cells. Circ. Res. 2019, 125, 43–52. [Google Scholar] [CrossRef]
- Wang, W.; Wang, X.; Fujioka, H.; Hoppel, C.; Whone, A.L.; Caldwell, M.A.; Cullen, P.J.; Liu, J.; Zhu, X. Parkinson’s disease-associated mutant VPS35 causes mitochondrial dysfunction by recycling DLP1 complexes. Nat. Med. 2016, 22, 54–63. [Google Scholar] [CrossRef] [Green Version]
- Picca, A.; Guerra, F.; Calvani, R.; Marini, F.; Biancolillo, A.; Landi, G.; Beli, R.; Landi, F.; Bernabei, R.; Bentivoglio, A.R.; et al. Mitochondrial Signatures in Circulating Extracellular Vesicles of Older Adults with Parkinson’s Disease: Results from the EXosomes in PArkiNson’s Disease (EXPAND) Study. J. Clin. Med. 2020, 9, 504. [Google Scholar] [CrossRef] [Green Version]
- Kim, K.M.; Meng, Q.; Perez de Acha, O.; Mustapic, M.; Cheng, A.; Eren, E.; Kundu, G.; Piao, Y.; Munk, R.; Wood, W.H., 3rd; et al. Mitochondrial RNA in Alzheimer’s Disease Circulating Extracellular Vesicles. Front. Cell Dev. Biol. 2020, 8, 581882. [Google Scholar] [CrossRef]
- Yao, P.J.; Eren, E.; Goetzl, E.J.; Kapogiannis, D. Mitochondrial Electron Transport Chain Protein Abnormalities Detected in Plasma Extracellular Vesicles in Alzheimer’s Disease. Biomedicines 2021, 9, 1587. [Google Scholar] [CrossRef]
- Muraoka, S.; DeLeo, A.M.; Sethi, M.K.; Yukawa-Takamatsu, K.; Yang, Z.; Ko, J.; Hogan, J.D.; Ruan, Z.; You, Y.; Wang, Y.; et al. Proteomic and biological profiling of extracellular vesicles from Alzheimer’s disease human brain tissues. Alzheimers Dement. 2020, 16, 896–907. [Google Scholar] [CrossRef] [PubMed]
- Marcoux, G.; Magron, A.; Sut, C.; Laroche, A.; Laradi, S.; Hamzeh-Cognasse, H.; Allaeys, I.; Cabon, O.; Julien, A.S.; Garraud, O.; et al. Platelet-derived extracellular vesicles convey mitochondrial DAMPs in platelet concentrates and their levels are associated with adverse reactions. Transfusion 2019, 59, 2403–2414. [Google Scholar] [CrossRef] [PubMed]
- D’Acunzo, P.; Perez-Gonzalez, R.; Kim, Y.; Hargash, T.; Miller, C.; Alldred, M.J.; Erdjument-Bromage, H.; Penikalapati, S.C.; Pawlik, M.; Saito, M.; et al. Mitovesicles are a novel population of extracellular vesicles of mitochondrial origin altered in Down syndrome. Sci. Adv. 2021, 7, abe5085. [Google Scholar] [CrossRef] [PubMed]
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Deus, C.M.; Tavares, H.; Beatriz, M.; Mota, S.; Lopes, C. Mitochondrial Damage-Associated Molecular Patterns Content in Extracellular Vesicles Promotes Early Inflammation in Neurodegenerative Disorders. Cells 2022, 11, 2364. https://doi.org/10.3390/cells11152364
Deus CM, Tavares H, Beatriz M, Mota S, Lopes C. Mitochondrial Damage-Associated Molecular Patterns Content in Extracellular Vesicles Promotes Early Inflammation in Neurodegenerative Disorders. Cells. 2022; 11(15):2364. https://doi.org/10.3390/cells11152364
Chicago/Turabian StyleDeus, Cláudia M., Henrique Tavares, Margarida Beatriz, Sandra Mota, and Carla Lopes. 2022. "Mitochondrial Damage-Associated Molecular Patterns Content in Extracellular Vesicles Promotes Early Inflammation in Neurodegenerative Disorders" Cells 11, no. 15: 2364. https://doi.org/10.3390/cells11152364
APA StyleDeus, C. M., Tavares, H., Beatriz, M., Mota, S., & Lopes, C. (2022). Mitochondrial Damage-Associated Molecular Patterns Content in Extracellular Vesicles Promotes Early Inflammation in Neurodegenerative Disorders. Cells, 11(15), 2364. https://doi.org/10.3390/cells11152364