
Modeling MEN1 with Patient-Origin iPSCs Reveals GLP-1R Mediated Hypersecretion of Insulin

 , ,
, , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Cell Culture
2.2. Cell Differentiation
2.3. Immunofluorescence Microscopy
2.4. Cell Cycle and Cell Proliferation Assay
2.5. Insulin Secretion
2.6. Animal Model and Cell Transplantation
2.7. Histological Analysis
2.8. Immunohistochemistry
2.9. Western Blot Analysis
2.10. Statistical Analysis
2.11. Study Approval
3. Results
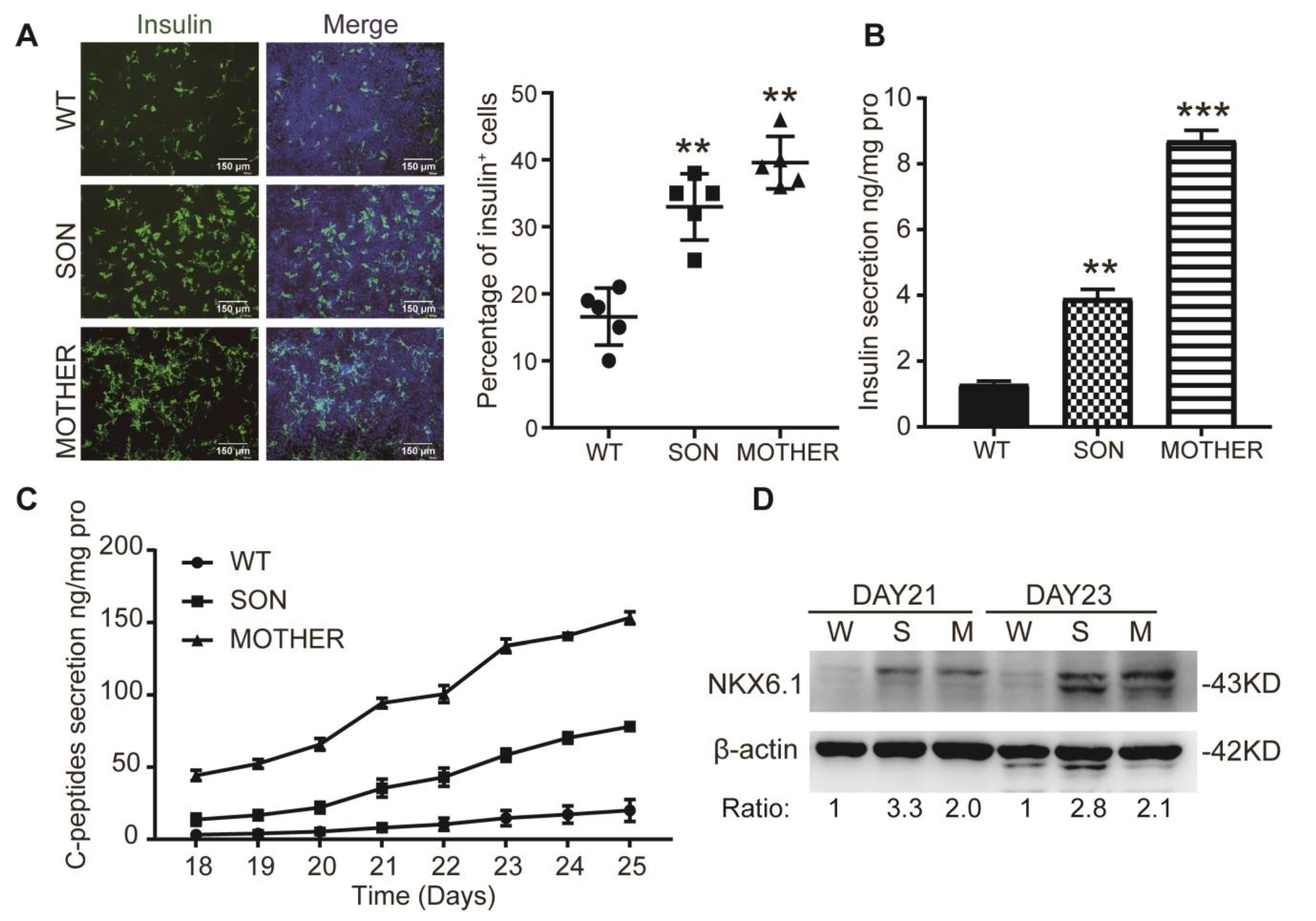
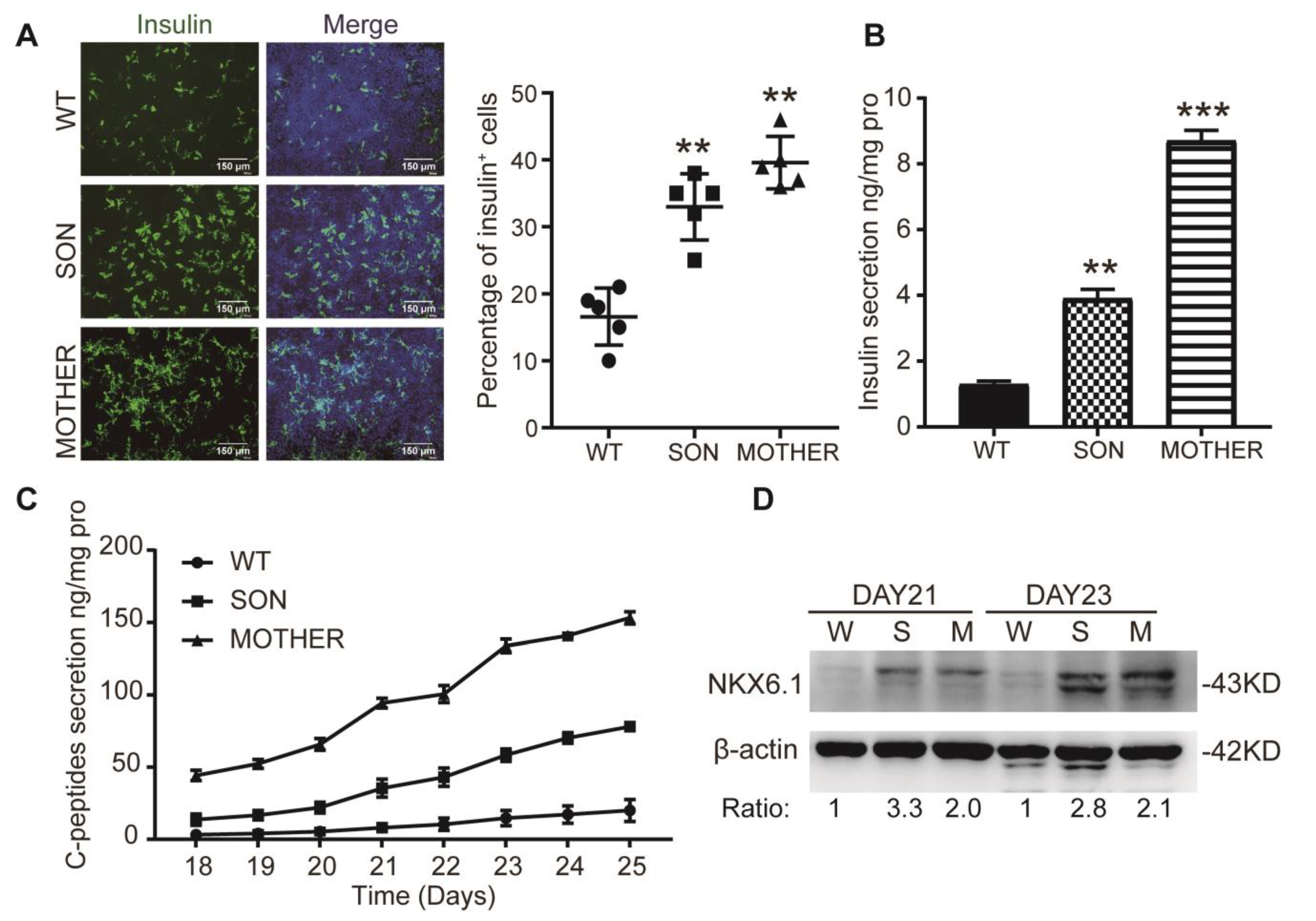
3.1. MEN1-iPSCs Obtained More Insulin-Positive Cells and Increased Insulin Production
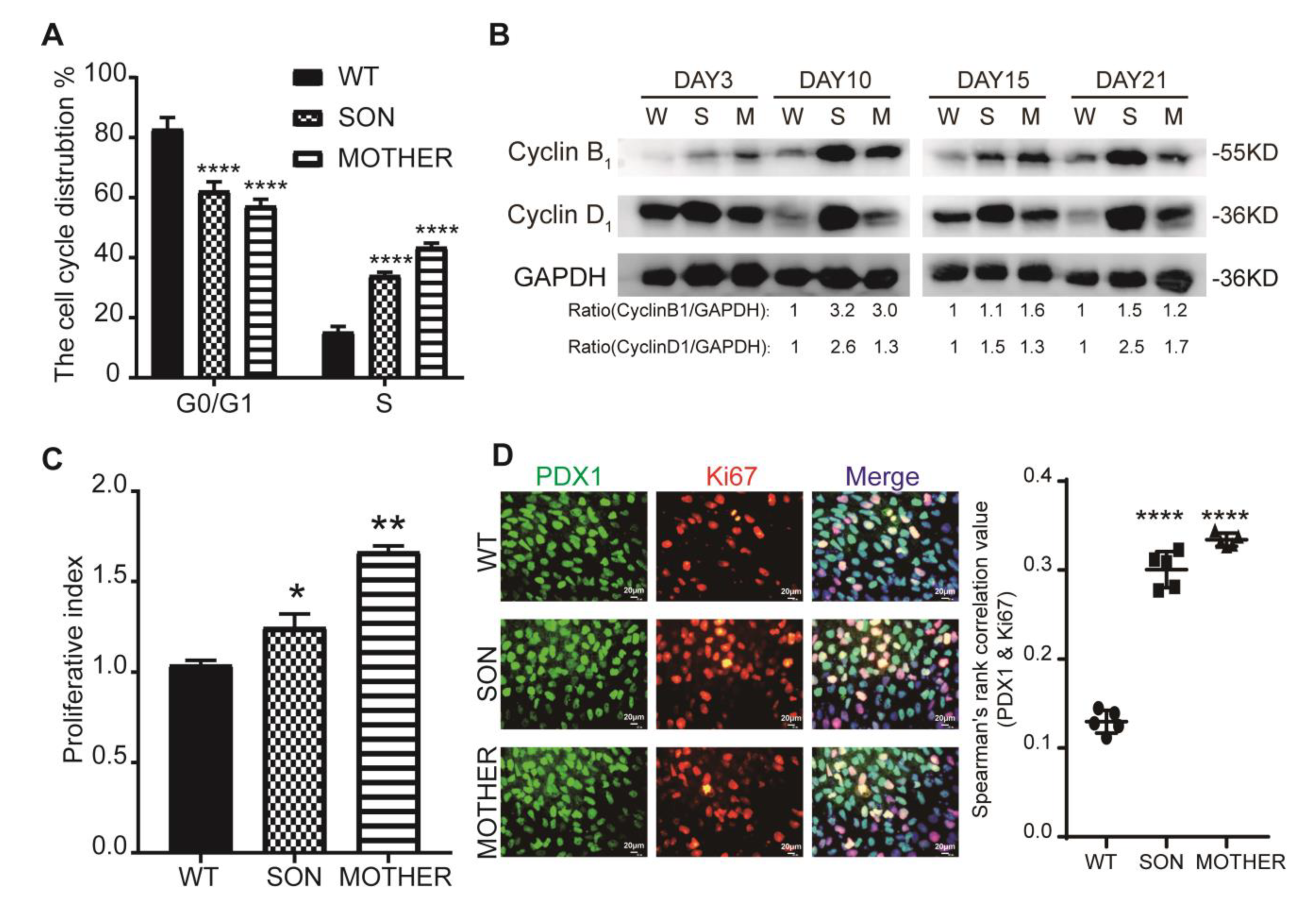
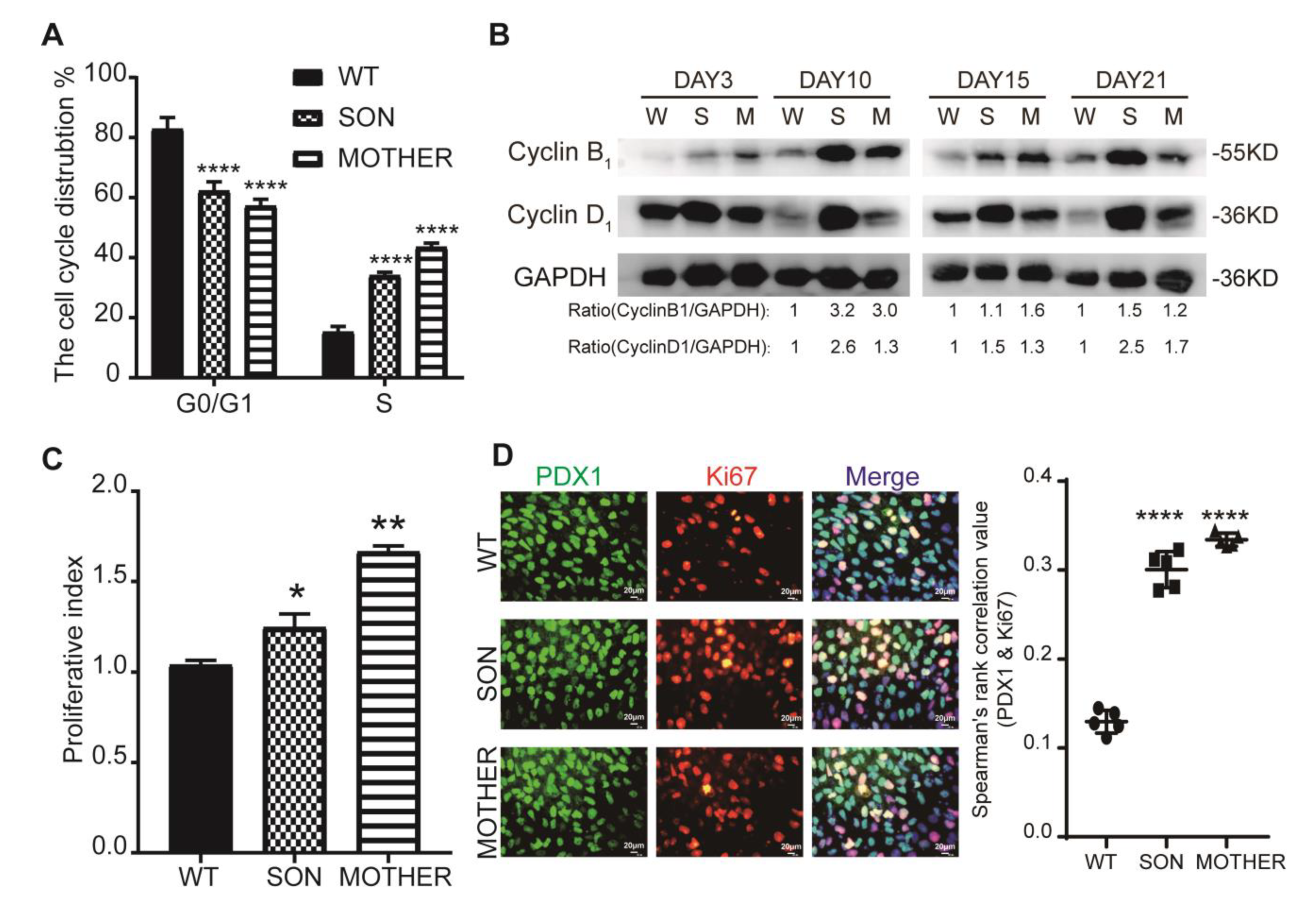
3.2. MEN1-Derived Cells Presented Neoplastic Features with Stepped up Proliferation Ability
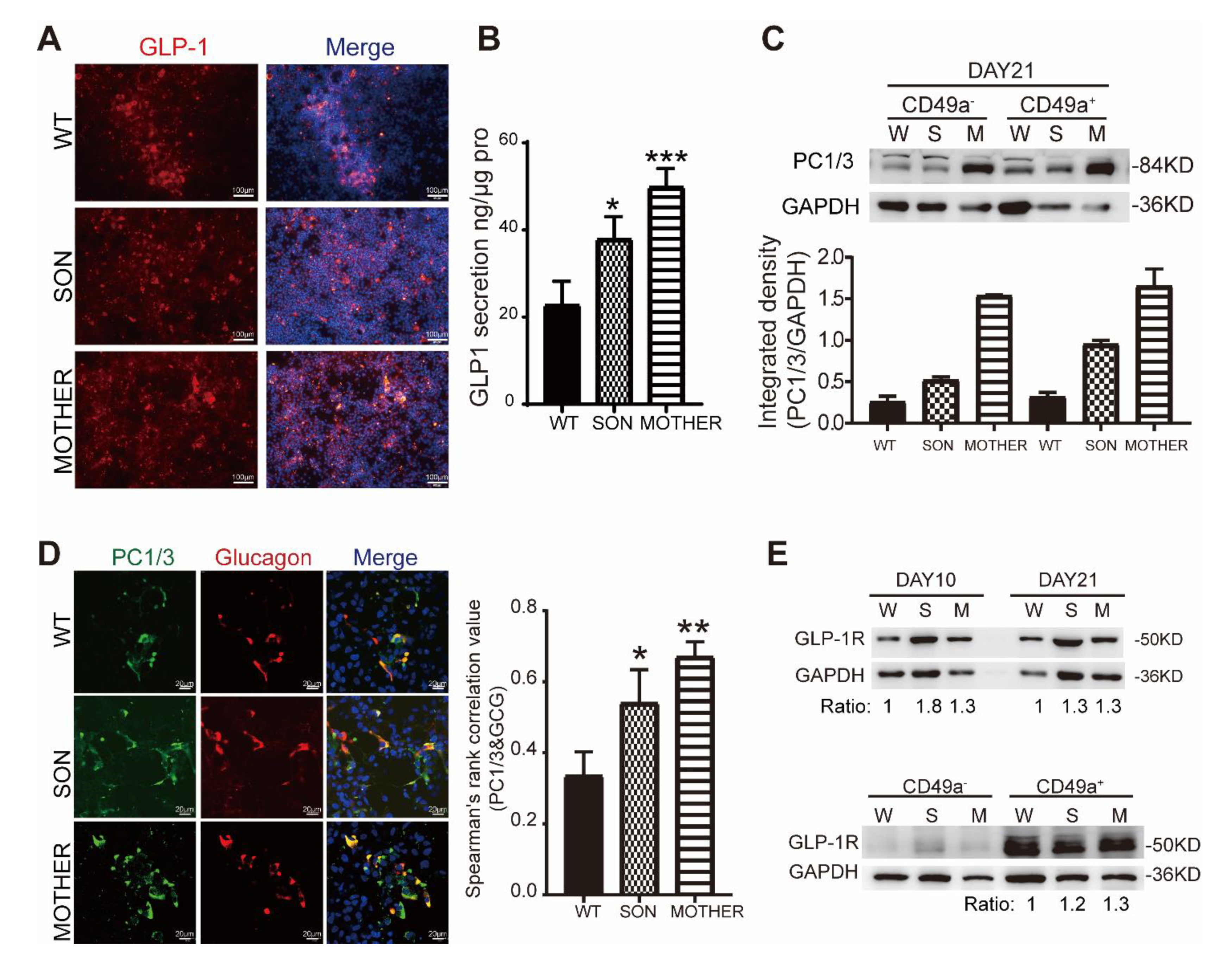
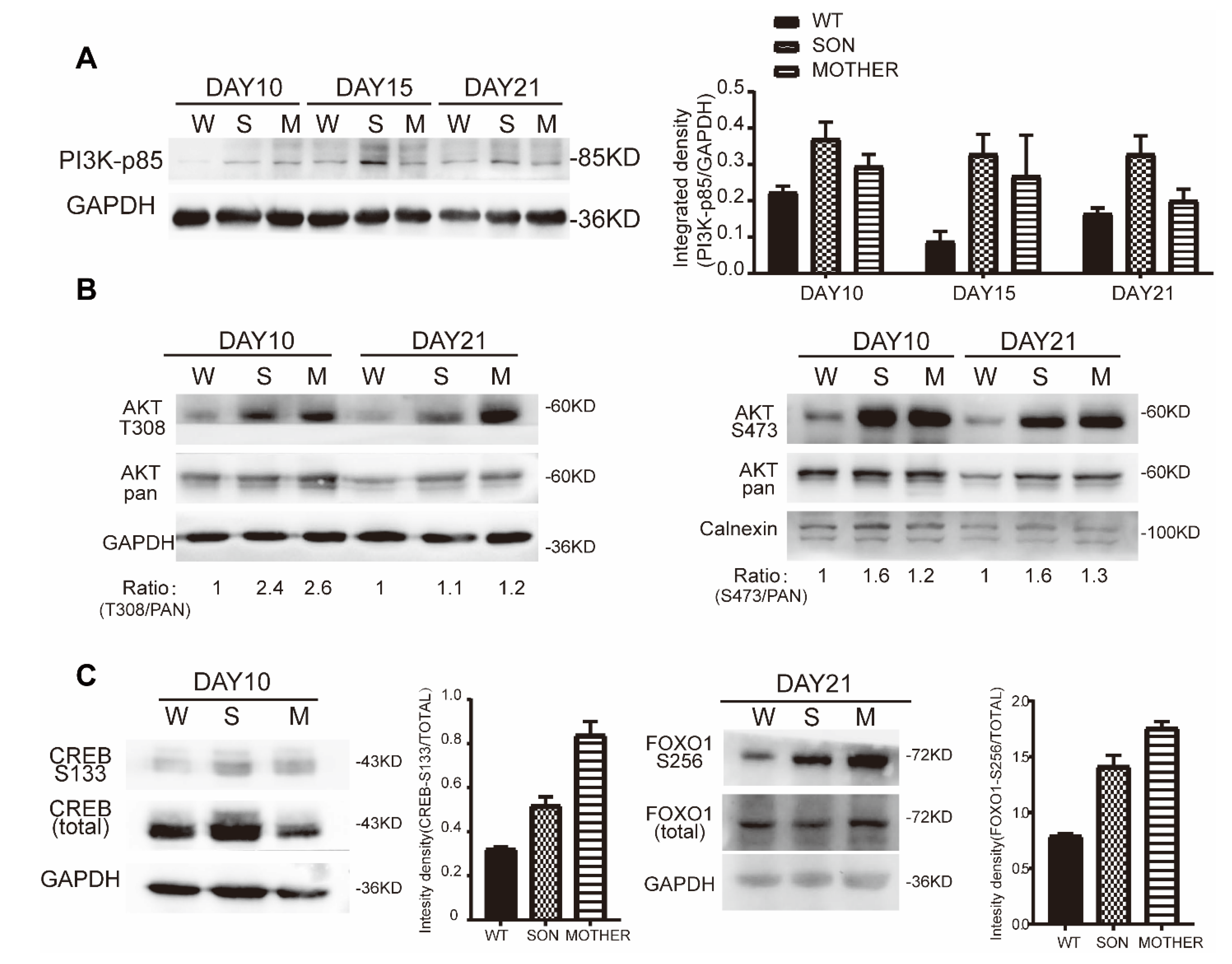
3.3. MEN1-Derived Cells Presented Increased GLP-1 Production and PI3K/AKT Signaling
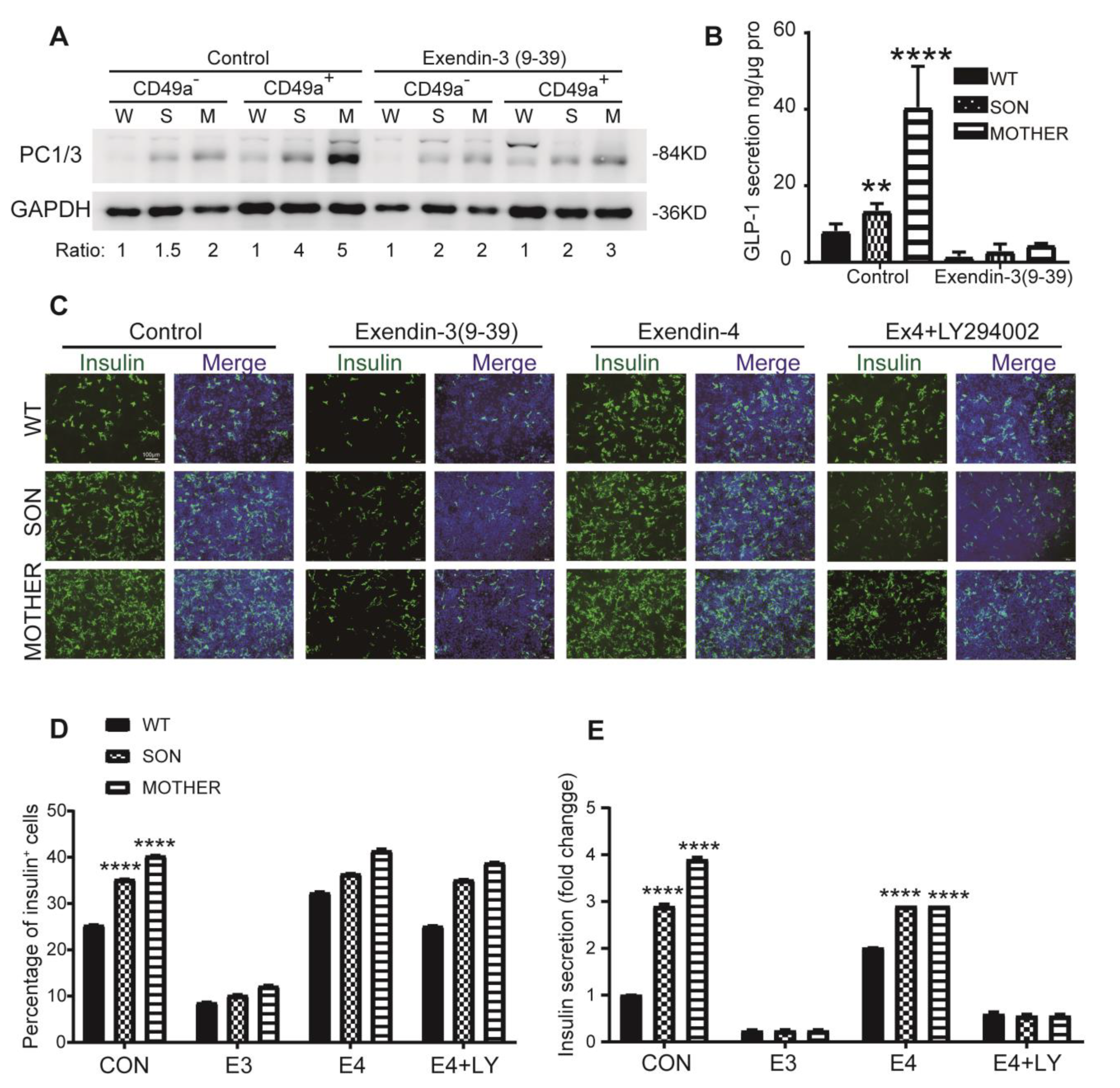
3.4. Inhibition of GLP-1R Signaling Rescued the MEN1-IPCs Phenotypes
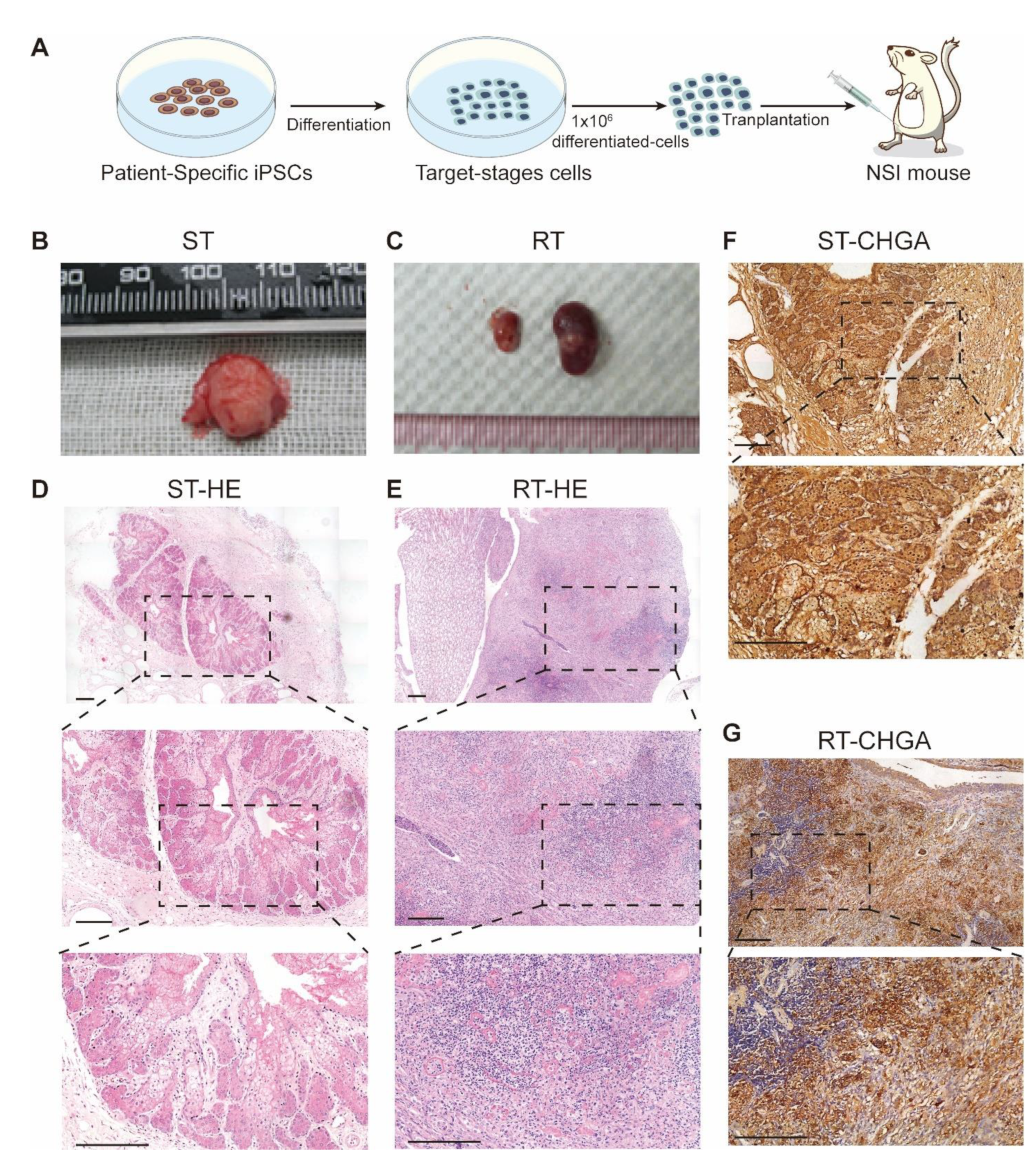
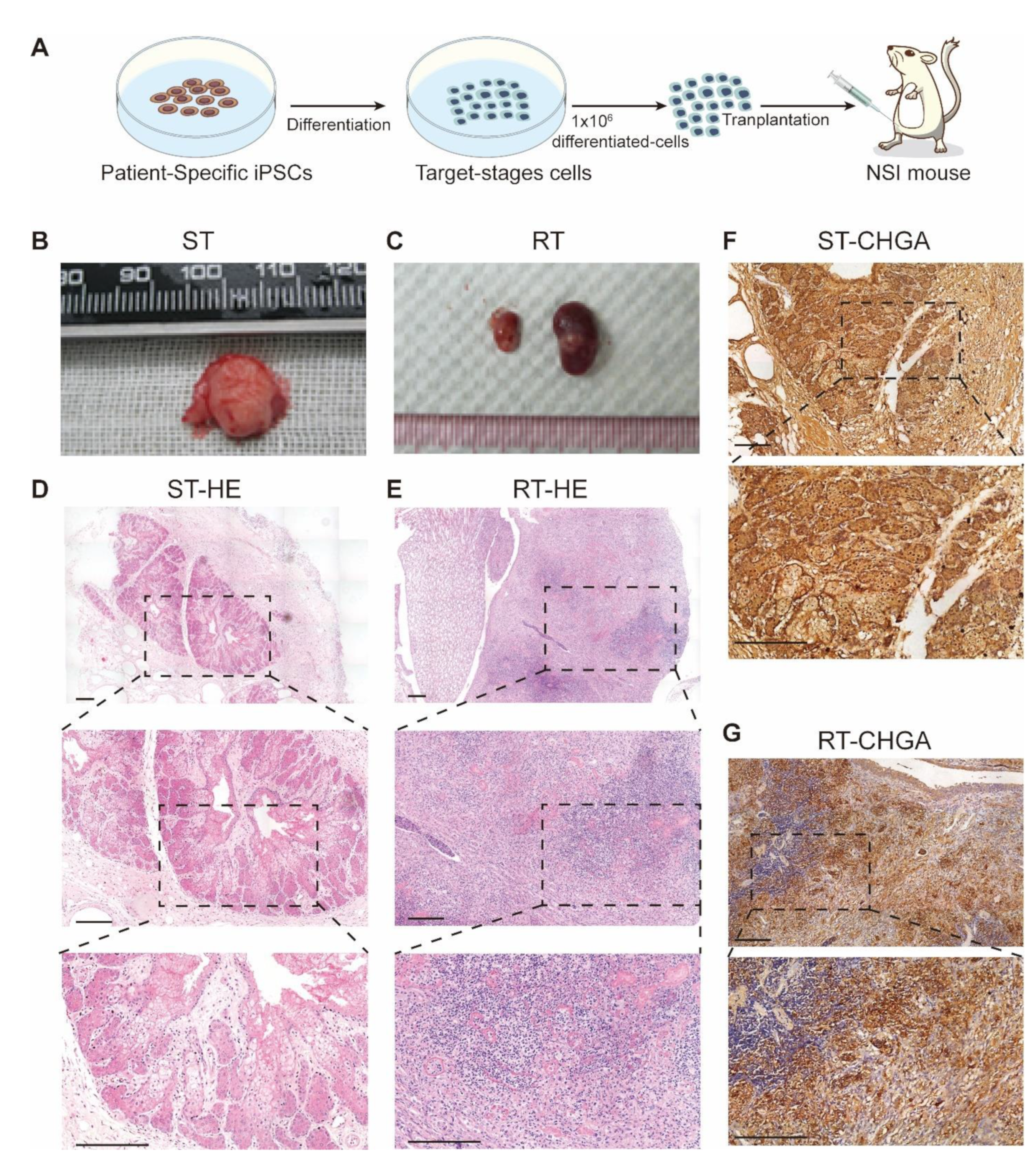
3.5. Transplantation of MEN1-IPCs into NSI Mice Partially Recapitulated some Features of the Original Tumor in Patient
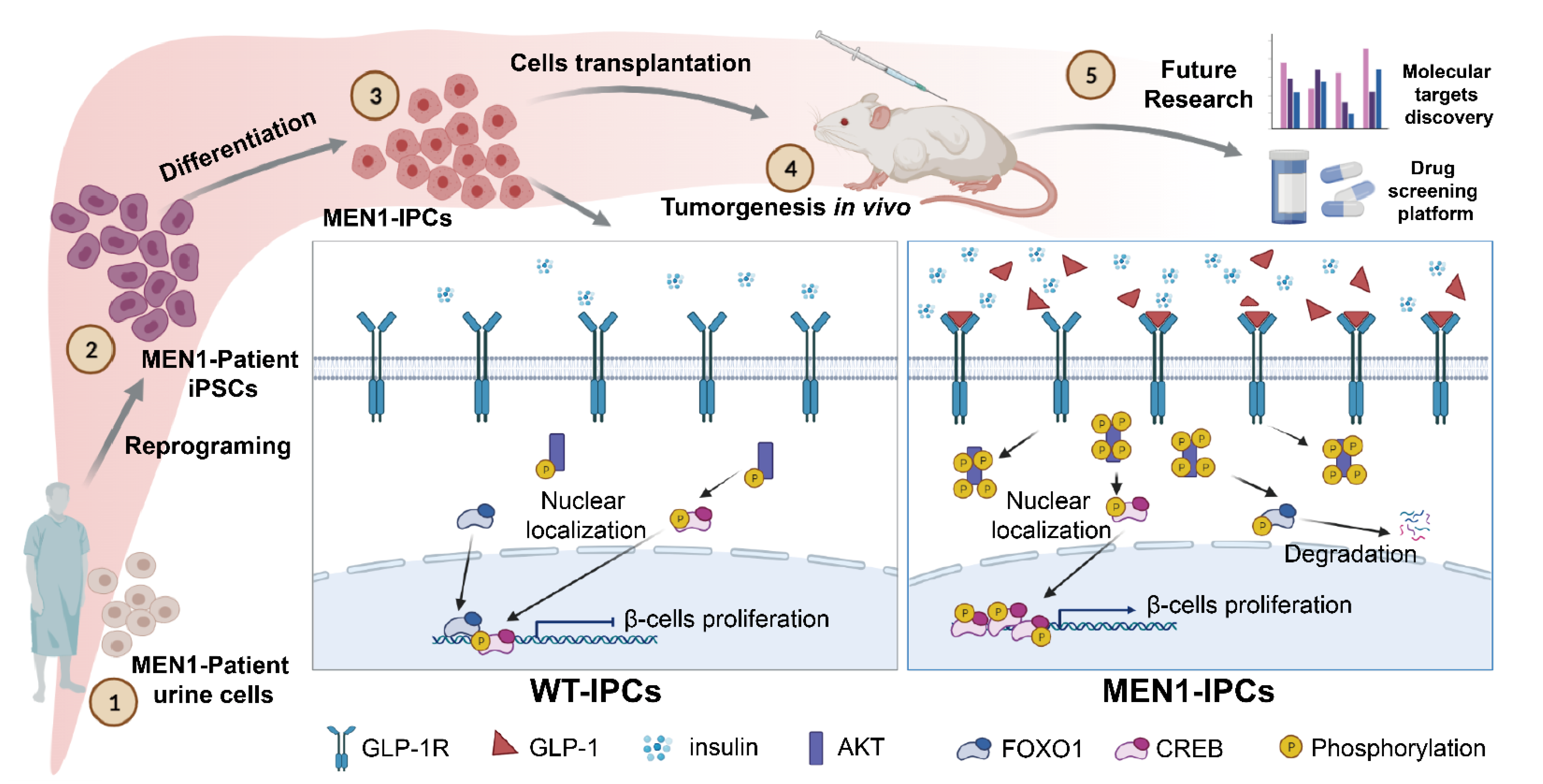
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Acknowledgments
Conflicts of Interest
References
- Marini, F.; Falchetti, A.; Del Monte, F.; Sala, S.C.; Gozzini, A.; Luzi, E.; Brandi, M.L. Multiple endocrine neoplasia type 1. Orphanet J. Rare Dis. 2006, 1, 38. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Effraimidis, G.; Knigge, U.; Rossing, M.; Oturai, P.; Rasmussen, A.K.; Feldt-Rasmussen, U. Multiple endocrine neoplasia type 1 (MEN-1) and neuroendocrine neoplasms (NENs). In Seminars in Cancer Biology; Academic Press: Cambridge, MA, USA, 2021. [Google Scholar] [CrossRef]
- Weiss, R.E.; Refetoff, S. Genetic Diagnosis of Endocrine Disorders, 2nd ed.; Academic Press: Amsterdam, The Netherlands; Boston, MA, USA, 2016; p. 444. [Google Scholar]
- Lemmens, I.; Van de Ven, W.J.; Kas, K.; Zhang, C.X.; Giraud, S.; Wautot, V.; Buisson, N.; De Witte, K.; Salandre, J.; Lenoir, G.; et al. Identification of the multiple endocrine neoplasia type 1 (MEN1) gene. The European Consortium on MEN1. Hum. Mol. Genet. 1997, 6, 1177–1183. [Google Scholar] [CrossRef] [PubMed]
- Thakker, R.V.; Newey, P.J.; Walls, G.V.; Bilezikian, J.; Dralle, H.; Ebeling, P.R.; Melmed, S.; Sakurai, A.; Tonelli, F.; Brandi, M.L.; et al. Clinical practice guidelines for multiple endocrine neoplasia type 1 (MEN1). J. Clin. Endocrinol. Metab. 2012, 97, 2990–3011. [Google Scholar] [CrossRef] [PubMed]
- Kottemann, M.C.; Bale, A.E. Characterization of DNA damage-dependent cell cycle checkpoints in a menin-deficient model. DNA Repair. 2009, 8, 944–952. [Google Scholar] [CrossRef] [Green Version]
- Concolino, P.; Costella, A.; Capoluongo, E. Multiple endocrine neoplasia type 1 (MEN1): An update of 208 new germline variants reported in the last nine years. Cancer Genet. 2016, 209, 36–41. [Google Scholar] [CrossRef]
- Lemos, M.C.; Thakker, R.V. Multiple endocrine neoplasia type 1 (MEN1): Analysis of 1336 mutations reported in the first decade following identification of the gene. Hum. Mutat. 2008, 29, 22–32. [Google Scholar] [CrossRef]
- Wu, J.; Yang, X.; Chen, B.; Xu, X. Pancreas beta cell regeneration and type 1 diabetes (Review). Exp. Ther. Med. 2015, 9, 653–657. [Google Scholar] [CrossRef] [Green Version]
- Karnik, S.K.; Chen, H.; McLean, G.W.; Heit, J.J.; Gu, X.; Zhang, A.Y.; Fontaine, M.; Yen, M.H.; Kim, S.K. Menin controls growth of pancreatic beta-cells in pregnant mice and promotes gestational diabetes mellitus. Science 2007, 318, 806–809. [Google Scholar] [CrossRef] [Green Version]
- Crabtree, J.S.; Scacheri, P.C.; Ward, J.M.; McNally, S.R.; Swain, G.P.; Montagna, C.; Hager, J.H.; Hanahan, D.; Edlund, H.; Magnuson, M.A.; et al. Of mice and MEN1: Insulinomas in a conditional mouse knockout. Mol. Cell. Biol. 2003, 23, 6075–6085. [Google Scholar] [CrossRef] [Green Version]
- Chamberlain, C.E.; Scheel, D.W.; McGlynn, K.; Kim, H.; Miyatsuka, T.; Wang, J.H.; Nguyen, V.; Zhao, S.H.; Mavropoulos, A.; Abraham, A.G.; et al. Menin determines K-RAS proliferative outputs in endocrine cells. J. Clin. Investig. 2014, 124, 4093–4101. [Google Scholar] [CrossRef] [Green Version]
- Milne, T.A.; Hughes, C.M.; Lloyd, R.; Yang, Z.; Rozenblatt-Rosen, O.; Dou, Y.; Schnepp, R.W.; Krankel, C.; Livolsi, V.A.; Gibbs, D.; et al. Menin and MLL cooperatively regulate expression of cyclin-dependent kinase inhibitors. Proc. Natl. Acad. Sci. USA 2005, 102, 749–754. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Karnik, S.K.; Hughes, C.M.; Gu, X.; Rozenblatt-Rosen, O.; McLean, G.W.; Xiong, Y.; Meyerson, M.; Kim, S.K. Menin regulates pancreatic islet growth by promoting histone methylation and expression of genes encoding p27Kip1 and p18INK4c. Proc. Natl. Acad. Sci. USA 2005, 102, 14659–14664. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schnepp, R.W.; Chen, Y.X.; Wang, H.; Cash, T.; Silva, A.; Diehl, J.A.; Brown, E.; Hua, X. Mutation of tumor suppressor gene Men1 acutely enhances proliferation of pancreatic islet cells. Cancer Res. 2006, 66, 5707–5715. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jhala, U.S.; Canettieri, G.; Screaton, R.A.; Kulkarni, R.N.; Krajewski, S.; Reed, J.; Walker, J.; Lin, X.; White, M.; Montminy, M. cAMP promotes pancreatic beta-cell survival via CREB-mediated induction of IRS2. Genes Dev. 2003, 17, 1575–1580. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Heit, J.J.; Karnik, S.K.; Kim, S.K. Intrinsic regulators of pancreatic beta-cell proliferation. Annu. Rev. Cell Dev. Biol. 2006, 22, 311–338. [Google Scholar] [CrossRef]
- Zhang, H.; Li, W.; Wang, Q.; Wang, X.; Li, F.; Zhang, C.; Wu, L.; Long, H.; Liu, Y.; Li, X.; et al. Glucose-mediated repression of menin promotes pancreatic beta-cell proliferation. Endocrinology 2012, 153, 602–611. [Google Scholar] [CrossRef] [Green Version]
- Arumugam, R.; Fleenor, D.; Lu, D.H.; Freemark, M. Differential and Complementary Effects of Glucose and Prolactin on Islet DNA Synthesis and Gene Expression. Endocrinology 2011, 152, 856–868. [Google Scholar] [CrossRef] [Green Version]
- Perren, A.; Anlauf, M.; Henopp, T.; Rudolph, T.; Schmitt, A.; Raffel, A.; Gimm, O.; Weihe, E.; Knoefel, W.T.; Dralle, H.; et al. Multiple endocrine neoplasia type 1 (MEN1): Loss of one MEN1 allele in tumors and monohormonal endocrine cell clusters but not in islet hyperplasia of the pancreas. J. Clin. Endocr. Metab. 2007, 92, 1118–1128. [Google Scholar] [CrossRef] [Green Version]
- Habener, J.F.; Stanojevic, V. alpha-cell role in beta-cell generation and regeneration. Islets 2012, 4, 188–198. [Google Scholar] [CrossRef] [Green Version]
- Baggio, L.L.; Drucker, D.J. Biology of incretins: GLP-1 and GIP. Gastroenterology 2007, 132, 2131–2157. [Google Scholar] [CrossRef]
- Song, W.J.; Schreiber, W.E.; Zhong, E.; Liu, F.F.; Kornfeld, B.D.; Wondisford, F.E.; Hussain, M.A. Exendin-4 stimulation of cyclin A2 in beta-cell proliferation. Diabetes 2008, 57, 2371–2381. [Google Scholar] [CrossRef] [Green Version]
- Perfetti, R.; Merkel, P. Glucagon-like peptide-1: A major regulator of pancreatic beta-cell function. Eur. J. Endocrinol. 2000, 143, 717–725. [Google Scholar] [CrossRef] [PubMed]
- Muhammad, A.B.; Xing, B.; Liu, C.; Naji, A.; Ma, X.; Simmons, R.A.; Hua, X. Menin and PRMT5 suppress GLP1 receptor transcript and PKA-mediated phosphorylation of FOXO1 and CREB. Am. J. Physiol. Endocrinol. Metab. 2017, 313, E148–E166. [Google Scholar] [CrossRef] [PubMed]
- Xing, B.; Ma, J.; Jiang, Z.; Feng, Z.; Ling, S.; Szigety, K.; Su, W.; Zhang, L.; Jia, R.; Sun, Y.; et al. GLP-1 signaling suppresses menin’s transcriptional block by phosphorylation in beta cells. J. Cell Biol. 2019, 218, 855–870. [Google Scholar] [CrossRef]
- Lu, J.; Herrera, P.L.; Carreira, C.; Bonnavion, R.; Seigne, C.; Calender, A.; Bertolino, P.; Zhang, C.X. Alpha cell-specific Men1 ablation triggers the transdifferentiation of glucagon-expressing cells and insulinoma development. Gastroenterology 2010, 138, 1954–1965. [Google Scholar] [CrossRef] [PubMed]
- Mohr, H.; Pellegata, N.S. Animal models of MEN1. Endocr. Relat. Cancer 2017, 24, T161–T177. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Loffler, K.A.; Biondi, C.A.; Gartside, M.; Waring, P.; Stark, M.; Serewko-Auret, M.M.; Muller, H.K.; Hayward, N.K.; Kay, G.F. Broad tumor spectrum in a mouse model of multiple endocrine neoplasia type 1. Int. J. Cancer 2007, 120, 259–267. [Google Scholar] [CrossRef]
- Zhou, Q.; Melton, D.A. Pancreas regeneration. Nature 2018, 557, 351–358. [Google Scholar] [CrossRef]
- Lehv, M.; Fitzgerald, P.J. Pancreatic acinar cell regeneration. IV. Regeneration after resection. Am. J. Pathol. 1968, 53, 513–535. [Google Scholar]
- Bonner-Weir, S.; Trent, D.F.; Weir, G.C. Partial pancreatectomy in the rat and subsequent defect in glucose-induced insulin release. J. Clin. Investig. 1983, 71, 1544–1553. [Google Scholar] [CrossRef]
- Watanabe, H.; Saito, H.; Rychahou, P.G.; Uchida, T.; Evers, B.M. Aging is associated with decreased pancreatic acinar cell regeneration and phosphatidylinositol 3-kinase/Akt activation. Gastroenterology 2005, 128, 1391–1404. [Google Scholar] [CrossRef]
- Zhou, T.; Benda, C.; Duzinger, S.; Huang, Y.; Li, X.; Li, Y.; Guo, X.; Cao, G.; Chen, S.; Hao, L.; et al. Generation of induced pluripotent stem cells from urine. J. Am. Soc. Nephrol. 2011, 22, 1221–1228. [Google Scholar] [CrossRef] [PubMed]
- Zhou, T.; Benda, C.; Dunzinger, S.; Huang, Y.; Ho, J.C.; Yang, J.; Wang, Y.; Zhang, Y.; Zhuang, Q.; Li, Y.; et al. Generation of human induced pluripotent stem cells from urine samples. Nat. Protoc. 2012, 7, 2080–2089. [Google Scholar] [CrossRef] [PubMed]
- Guo, D.; Wu, F.; Liu, H.; Gao, G.; Kou, S.; Yang, F.; Abbas, N.; Zhou, T.; Cai, X.; Zhang, H.; et al. Generation of non-integrated induced pluripotent stem cells from a 59-year-old female with multiple endocrine neoplasia type 1 syndrome. Stem. Cell Res. 2017, 18, 64–66. [Google Scholar] [CrossRef]
- Guo, D.; Wu, F.; Liu, H.; Gao, G.; Kou, S.; Yang, F.; Abbas, N.; Zhou, T.; Cai, X.; Zhang, H.; et al. Generation of non-integrated induced pluripotent stem cells from a 23-year-old male with multiple endocrine neoplasia type 1 syndrome. Stem. Cell Res. 2017, 18, 70–72. [Google Scholar] [CrossRef]
- Xue, Y.; Cai, X.; Wang, L.; Liao, B.; Zhang, H.; Shan, Y.; Chen, Q.; Zhou, T.; Li, X.; Hou, J.; et al. Generating a non-integrating human induced pluripotent stem cell bank from urine-derived cells. PLoS ONE 2013, 8, e70573. [Google Scholar] [CrossRef]
- Kunisada, Y.; Tsubooka-Yamazoe, N.; Shoji, M.; Hosoya, M. Small molecules induce efficient differentiation into insulin-producing cells from human induced pluripotent stem cells. Stem. Cell Res. 2012, 8, 274–284. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ye, W.; Jiang, Z.W.; Li, G.X.; Xiao, Y.R.; Lin, S.M.; Lai, Y.X.; Wang, S.N.; Li, B.H.; Jia, B.; Li, Y.; et al. Quantitative evaluation of the immunodeficiency of a mouse strain by tumor engraftments. J. Hematol. Oncol. 2015, 8, 59. [Google Scholar] [CrossRef] [Green Version]
- Xu, Y.; Tao, J.; Yu, X.; Wu, Y.; Chen, Y.; You, K.; Zhang, J.; Getachew, A.; Pan, T.; Zhuang, Y.; et al. Hypomorphic ASGR1 modulates lipid homeostasis via INSIG1-mediated SREBP signaling suppression. JCI Insight 2021, 6, e147038. [Google Scholar] [CrossRef]
- Jialiang, L.; Changming, Z.; Jibing, C.; Fei, Y.; Jianying, Z.; Liwen, H.; Xiuli, Y.; Weiqun, L.; Feng, C.; Keqiang, X. Two case reports of pilot percutaneous cryosurgery in familial multiple endocrine neoplasia type 1. Pancreas 2013, 42, 353. [Google Scholar]
- Rindi, G.; Terenghi, G.; Westermark, G.; Westermark, P.; Moscoso, G.; Polak, J.M. Islet Amyloid Polypeptide in Proliferating Pancreatic B-Cells during Development, Hyperplasia, and Neoplasia in Humans and Mice. Am. J. Pathol. 1991, 138, 1321–1334. [Google Scholar]
- Veres, A.; Faust, A.L.; Bushnell, H.L.; Engquist, E.N.; Kenty, J.H.; Harb, G.; Poh, Y.C.; Sintov, E.; Gurtler, M.; Pagliuca, F.W.; et al. Charting cellular identity during human in vitro beta-cell differentiation. Nature 2019, 569, 368–373. [Google Scholar] [CrossRef]
- Kim, J.; Hoffman, J.P.; Alpaugh, R.K.; Rhim, A.D.; Reichert, M.; Stanger, B.Z.; Furth, E.E.; Sepulveda, A.R.; Yuan, C.X.; Won, K.J.; et al. An iPSC line from human pancreatic ductal adenocarcinoma undergoes early to invasive stages of pancreatic cancer progression. Cell Rep. 2013, 3, 2088–2099. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cheng, H.; Liu, C.; Cai, X.C.; Lu, Y.; Xu, Y.F.; Yu, X.J. iPSCs Derived from Malignant Tumor Cells: Potential Application for Cancer Research. Curr. Stem. Cell Res. T 2016, 11, 444–450. [Google Scholar] [CrossRef] [PubMed]
- Zhou, R.J.; Xu, A.; Tu, J.; Liu, M.; Gingold, J.A.; Zhao, R.Y.; Lee, D.F. Modeling Osteosarcoma Using Li-Fraumeni Syndrome Patient-derived Induced Pluripotent Stem Cells. Jove-J. Vis. Exp. 2018, 136, e57664. [Google Scholar] [CrossRef] [PubMed]
- Portier, L.; Desterke, C.; Chaker, D.; Oudrhiri, N.; Asgarova, A.; Dkhissi, F.; Turhan, A.G.; Bennaceur-Griscelli, A.; Griscelli, F. iPSC-Derived Hereditary Breast Cancer Model Reveals the BRCA1-Deleted Tumor Niche as a New Culprit in Disease Progression. Int. J. Mol. Sci. 2021, 22, 1227. [Google Scholar] [CrossRef]
- Agarwal, S.K.; Jothi, R. Genome-Wide Characterization of Menin-Dependent H3K4me3 Reveals a Specific Role for Menin in the Regulation of Genes Implicated in MEN1-Like Tumors. PLoS ONE 2012, 7, e37952. [Google Scholar] [CrossRef] [Green Version]
- Gherardi, S.; Ripoche, D.; Mikaelian, I.; Chanal, M.; Teinturier, R.; Goehrig, D.; Cordier-Bussat, M.; Zhang, C.X.; Hennino, A.; Bertolino, P. Menin regulates Inhbb expression through an Akt/Ezh2-mediated H3K27 histone modification. BBA-Gene Regul. Mech. 2017, 1860, 427–437. [Google Scholar] [CrossRef]
- Yokoyama, A.; Cleary, M.L. Menin critically links MLL proteins with LEDGE on cancer-associated target genes. Cancer Cell 2008, 14, 36–46. [Google Scholar] [CrossRef] [Green Version]
- Lakis, V.; Lawlor, R.T.; Newell, F.; Patch, A.M.; Mafficini, A.; Sadanandam, A.; Koufariotis, L.T.; Johnston, R.L.; Leonard, C.; Wood, S.; et al. DNA methylation patterns identify subgroups of pancreatic neuroendocrine tumors with clinical association. Commun. Biol. 2021, 4, 155. [Google Scholar] [CrossRef]
- Slater, E.P.; Strauch, K.; Rospleszcz, S.; Ramaswamy, A.; Esposito, I.; Kloppel, G.; Matthai, E.; Heeger, K.; Fendrich, V.; Langer, P.; et al. MicroRNA-196a and-196b as Potential Biomarkers for the Early Detection of Familial Pancreatic Cancer. Transl. Oncol. 2014, 7, 464–471. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bartsch, D.K.; Slater, E.P.; Albers, M.; Knoop, R.; Chaloupka, B.; Lopez, C.L.; Fendrich, V.; Kann, P.H.; Waldmann, J. Higher Risk of Aggressive Pancreatic Neuroendocrine Tumors in MEN1 Patients With MEN1 Mutations Affecting the CHES1 Interacting MENIN Domain. J. Clin. Endocr. Metab. 2014, 99, E2387–E2391. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Raef, H.; Zou, M.J.; Baitei, E.Y.; Al-Rijjal, R.A.; Kaya, N.; Al-Hamed, M.; Monies, D.; Abu-Dheim, N.N.; Al-Hindi, H.; Al-Ghamdi, M.H.; et al. A novel deletion of the MEN1 gene in a large family of multiple endocrine neoplasia type 1 (MEN1) with aggressive phenotype. Clin. Endocrinol. 2011, 75, 791–800. [Google Scholar] [CrossRef] [PubMed]
- Qiu, W.; Christakis, I.; Silva, A.; Bassett, R.L., Jr.; Cao, L.; Meng, Q.H.; Gardner Grubbs, E.; Zhao, H.; Yao, J.C.; Lee, J.E.; et al. Utility of chromogranin A, pancreatic polypeptide, glucagon and gastrin in the diagnosis and follow-up of pancreatic neuroendocrine tumours in multiple endocrine neoplasia type 1 patients. Clin. Endocrinol. 2016, 85, 400–407. [Google Scholar] [CrossRef] [Green Version]
- Bates, S.E. Epigenetic Therapies for Cancer. N. Engl. J. Med. 2020, 383, 650–663. [Google Scholar] [CrossRef]
- Yuan, F.; Shi, M.; Ji, J.; Shi, H.; Zhou, C.; Yu, Y.; Liu, B.; Zhu, Z.; Zhang, J. KRAS and DAXX/ATRX gene mutations are correlated with the clinicopathological features, advanced diseases, and poor prognosis in Chinese patients with pancreatic neuroendocrine tumors. Int. J. Biol. Sci. 2014, 10, 957–965. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Frost, M.; Lines, K.E.; Thakker, R.V. Current and emerging therapies for PNETs in patients with or without MEN1. Nat. Rev. Endocrinol. 2018, 14, 216–227. [Google Scholar] [CrossRef] [Green Version]
- Cao, Y.A.; Liu, R.X.; Jiang, X.L.; Lu, J.L.; Jiang, J.J.; Zhang, C.X.; Li, X.Y.; Ning, G. Nuclear-Cytoplasmic Shuttling of Menin Regulates Nuclear Translocation of beta-Catenin. Mol. Cell. Biol. 2009, 29, 5477–5487. [Google Scholar] [CrossRef] [Green Version]
- Klaus, A.; Birchmeier, W. Wnt signalling and its impact on development and cancer. Nat. Rev. Cancer 2008, 8, 387–398. [Google Scholar] [CrossRef]
- Mitry, E.; Walter, T.; Baudin, E.; Kurtz, J.E.; Ruszniewski, P.; Dominguez-Tinajero, S.; Bengrine-Lefevre, L.; Cadiot, G.; Dromain, C.; Farace, F.; et al. Bevacizumab plus capecitabine in patients with progressive advanced well-differentiated neuroendocrine tumors of the gastro-intestinal (GI-NETs) tract (BETTER trial)-A phase II non-randomised trial. Eur. J. Cancer 2014, 50, 3107–3115. [Google Scholar] [CrossRef]
- Moin, A.M.; Cory, M.; Gurlo, T.; Saisho, Y.; Rizza, R.A.; Butler, P.C.; Butler, A.E. Pancreatic alpha-cell mass across adult human lifespan. Eur. J. Endocrinol. 2020, 182, 219–231. [Google Scholar] [CrossRef] [PubMed]
- Hrvatin, S.; O’Donnell, C.W.; Deng, F.; Millman, J.R.; Pagliuca, F.W.; DiIorio, P.; Rezania, A.; Gifford, D.K.; Melton, D.A. Differentiated human stem cells resemble fetal, not adult, beta cells. Proc. Natl. Acad. Sci. USA 2014, 111, 3038–3043. [Google Scholar] [CrossRef] [PubMed] [Green Version]




Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cheng, Z.; Guo, D.; Ruzi, A.; Pan, T.; You, K.; Chen, Y.; Huang, X.; Zhang, J.; Yang, F.; Niu, L.; et al. Modeling MEN1 with Patient-Origin iPSCs Reveals GLP-1R Mediated Hypersecretion of Insulin. Cells 2022, 11, 2387. https://doi.org/10.3390/cells11152387
Cheng Z, Guo D, Ruzi A, Pan T, You K, Chen Y, Huang X, Zhang J, Yang F, Niu L, et al. Modeling MEN1 with Patient-Origin iPSCs Reveals GLP-1R Mediated Hypersecretion of Insulin. Cells. 2022; 11(15):2387. https://doi.org/10.3390/cells11152387
Chicago/Turabian StyleCheng, Ziqi, Dongsheng Guo, Aynisahan Ruzi, Tingcai Pan, Kai You, Yan Chen, Xinping Huang, Jiaye Zhang, Fan Yang, Lizhi Niu, and et al. 2022. "Modeling MEN1 with Patient-Origin iPSCs Reveals GLP-1R Mediated Hypersecretion of Insulin" Cells 11, no. 15: 2387. https://doi.org/10.3390/cells11152387