
ATP-Sensitive Potassium Channels in Migraine: Translational Findings and Therapeutic Potential


Abstract
:1. Introduction
2. Molecular Basis and Physiological Function of KATP Channels
2.1. Molecular Structure and Regulation of Channel Activity
2.2. Tissue Distribution
2.3. Physiological Functions of KATP Channels
2.3.1. Vascular System
2.3.2. Neuronal Function
2.3.3. Analgesia, Antinociception, and Opioid Signaling
2.3.4. Insulin Secretion and Glucose Metabolism
3. Pharmacological Tools Targeting KATP Channels
3.1. KATP Channel Openers
3.2. KATP Channel Blockers
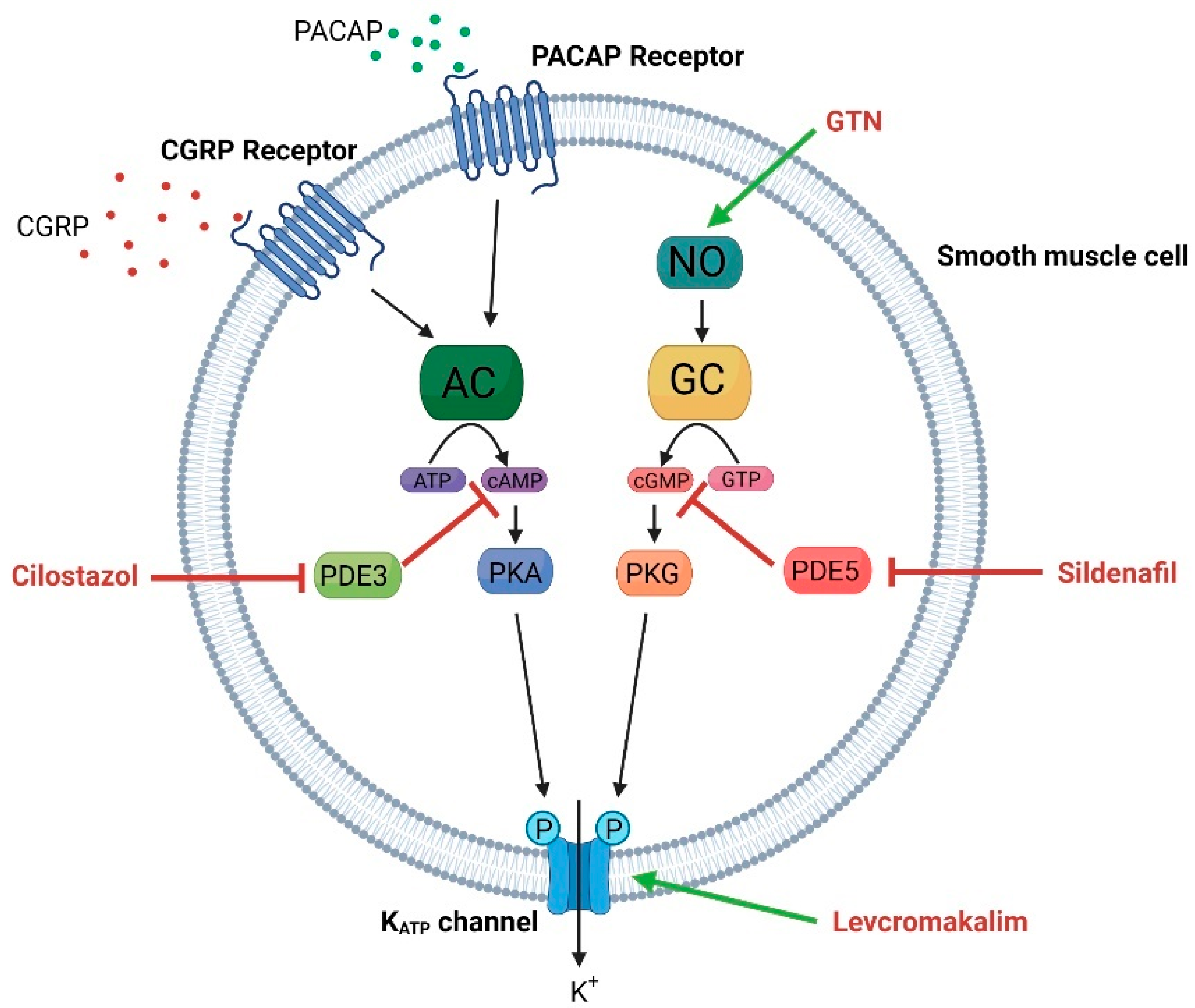
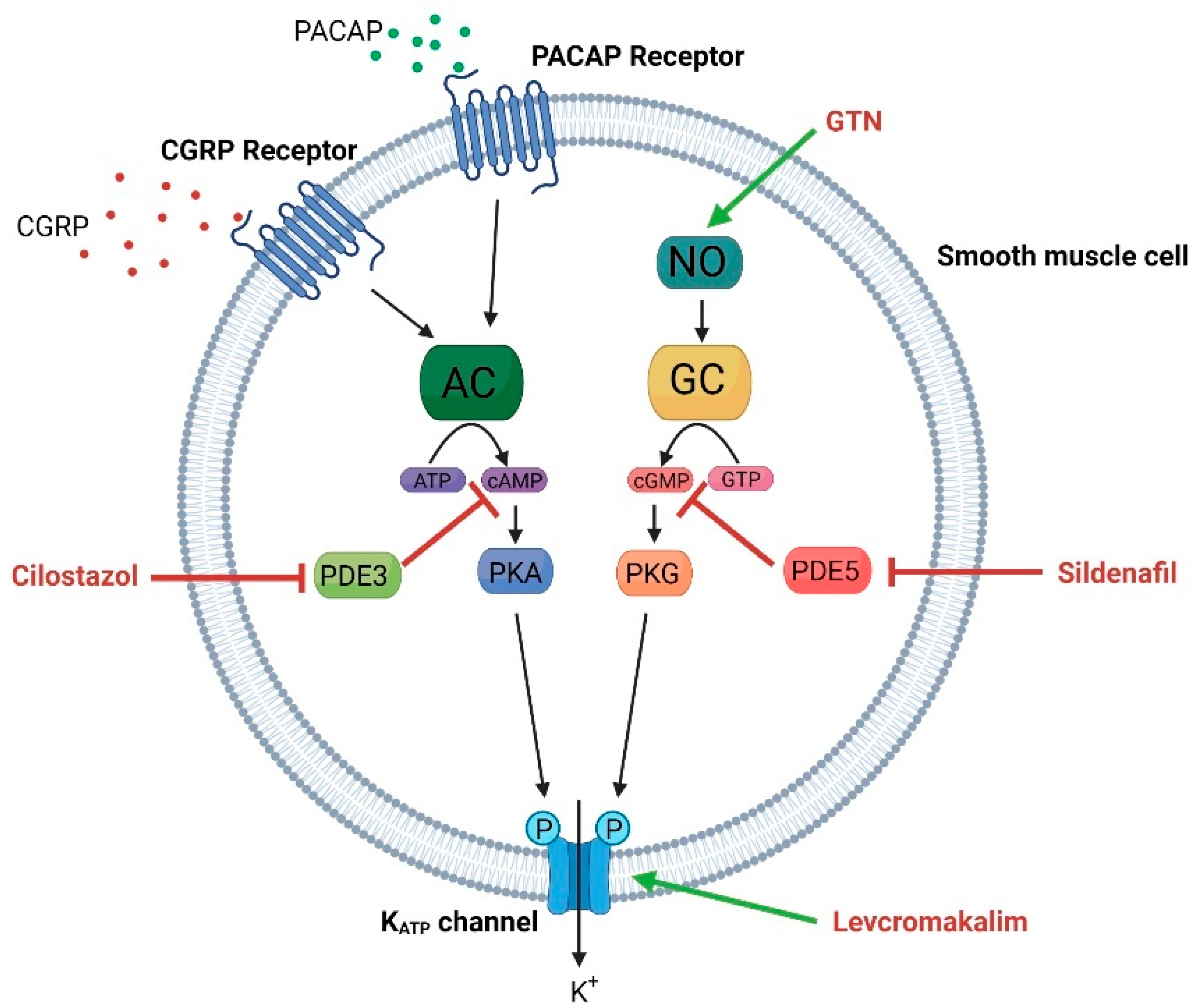
4. KATP Channels and Headache
4.1. Levcromakalim Is a Potent Trigger of Experimental Headache and Migraine
4.2. KATP Channel Opening in Preclinical Migraine Models
4.2.1. Dilatory Effects on Cranial Arteries
4.2.2. Stimulation of CGRP Release
4.2.3. Mast Cell Degranulation
4.2.4. In Vivo Mouse Model
4.3. KATP Channel Blockage as Therapeutic Target in Migraine
4.3.1. Effect of KATP Channel Blockers in Preclinical Models
4.3.2. Clinical Effect of KATP Channel Inhibition in Human Migraine Models
5. Discussion
5.1. KATP Channel Opening Has Similar Effect in Preclinical and Clinical Studies
5.2. Discrepant Results on KATP Channel Inhibition in Preclinical and Clinical Studies
5.2.1. Discrepant Effect of Glibenclamide on Cranial Arteries
5.2.2. Discrepant Effect of Glibenclamide on Headache Measures
5.2.3. Target Engagement
5.3. Possible Mechanism of Headache Induction and Prevention
5.3.1. Dilation of Meningeal Arteries
5.3.2. Effect on CGRP Signaling
5.3.3. Hyperpolarization-Activated Cyclic Nucleotide-Gated (HCN) Channels
5.4. Clinical Therapeutic Perspectives
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Steiner, T.J.; Stovner, L.J.; Jensen, R.; Uluduz, D.; Katsarava, Z. Migraine Remains Second among the World’s Causes of Disability, and First among Young Women: Findings from GBD2019. J. Headache Pain 2020, 21, 4–7. [Google Scholar] [CrossRef] [PubMed]
- James, S.L.; Abate, D.; Abate, K.H.; Abay, S.M.; Abbafati, C.; Abbasi, N.; Abbastabar, H.; Abd-Allah, F.; Abdela, J.; Abdelalim, A.; et al. Global, Regional, and National Incidence, Prevalence, and Years Lived with Disability for 354 Diseases and Injuries for 195 Countries and Territories, 1990–2017: A Systematic Analysis for the Global Burden of Disease Study 2017. Lancet 2018, 392, 1789–1858. [Google Scholar] [CrossRef] [Green Version]
- Olesen, J. Headache Classification Committee of the International Headache Society (IHS) The International Classification of Headache Disorders, 3rd Edition. Cephalalgia 2018, 38, 1–211. [Google Scholar] [CrossRef]
- Vgontzas, A.; Pavlović, J.M. Sleep Disorders and Migraine: Review of Literature and Potential Pathophysiology Mechanisms. Headache 2018, 58, 1030–1039. [Google Scholar] [CrossRef]
- Vuralli, D.; Ayata, C.; Bolay, H. Cognitive Dysfunction and Migraine 17 Psychology and Cognitive Sciences 1701 Psychology 11 Medical and Health Sciences 1103 Clinical Sciences 11 Medical and Health Sciences 1109 Neurosciences. J. Headache Pain 2018, 19, 109. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ashina, M.; Katsarava, Z.; Do, T.P.; Buse, D.C.; Pozo-Rosich, P.; Özge, A.; Krymchantowski, A.V.; Lebedeva, E.R.; Ravishankar, K.; Yu, S.; et al. Migraine: Epidemiology and Systems of Care. Lancet 2021, 397, 1485–1495. [Google Scholar] [CrossRef]
- Ashina, M. Migraine. N. Engl. J. Med. 2020, 383, 1866–1876. [Google Scholar] [CrossRef]
- Ashina, M.; Hansen, J.M.; Do, T.P.; Melo-Carrillo, A.; Burstein, R.; Moskowitz, M.A. Migraine and the Trigeminovascular System-40 Years and Counting. Lancet. Neurol. 2019, 18, 795–804. [Google Scholar] [CrossRef]
- Peng, K.P.; May, A. Migraine Understood as a Sensory Threshold Disease. Pain 2019, 160, 1494–1501. [Google Scholar] [CrossRef]
- Edvinsson, L.; Haanes, K.A.; Warfvinge, K.; Krause, D.N. CGRP as the Target of New Migraine Therapies—Successful Translation from Bench to Clinic. Nat. Rev. Neurol. 2018, 14, 338–350. [Google Scholar] [CrossRef]
- Stauffer, V.L.; Dodick, D.W.; Zhang, Q.; Carter, J.N.; Ailani, J.; Conley, R.R. Evaluation of Galcanezumab for the Prevention of Episodic Migraine: The EVOLVE-1 Randomized Clinical Trial. JAMA Neurol. 2018, 75, 1080–1088. [Google Scholar] [CrossRef] [PubMed]
- Förderreuther, S.; Zhang, Q.; Stauffer, V.L.; Aurora, S.K.; Láinez, M.J.A. Preventive Effects of Galcanezumab in Adult Patients with Episodic or Chronic Migraine Are Persistent: Data from the Phase 3, Randomized, Double-Blind, Placebo-Controlled EVOLVE-1, EVOLVE-2, and REGAIN Studies. J. Headache Pain 2018, 19, 121. [Google Scholar] [CrossRef] [PubMed]
- Ashina, M.; Goadsby, P.J.; Reuter, U.; Silberstein, S.; Dodick, D.W.; Xue, F.; Zhang, F.; Paiva da Silva Lima, G.; Cheng, S.; Mikol, D.D. Long-Term Efficacy and Safety of Erenumab in Migraine Prevention: Results from a 5-Year, Open-Label Treatment Phase of a Randomized Clinical Trial. Eur. J. Neurol. 2021, 28, 1716–1725. [Google Scholar] [CrossRef] [PubMed]
- Silberstein, S.D.; Dodick, D.W.; Bigal, M.E.; Yeung, P.P.; Goadsby, P.J.; Blankenbiller, T.; Grozinski-Wolff, M.; Yang, R.; Ma, Y.; Aycardi, E. Fremanezumab for the Preventive Treatment of Chronic Migraine. N. Engl. J. Med. 2017, 377, 2113–2122. [Google Scholar] [CrossRef] [PubMed]
- Ashina, M.; Saper, J.; Cady, R.; Schaeffler, B.A.; Biondi, D.M.; Hirman, J.; Pederson, S.; Allan, B.; Smith, J. Eptinezumab in Episodic Migraine: A Randomized, Double-Blind, Placebo-Controlled Study (PROMISE-1). Cephalalgia 2020, 40, 241–254. [Google Scholar] [CrossRef] [Green Version]
- Lassen, L.H.; Haderslev, P.A.; Jacobsen, V.B.; Iversen, H.K.; Sperling, B.; Olesen, J. CGRP May Play a Causative Role in Migraine. Cephalalgia 2002, 22, 54–61. [Google Scholar] [CrossRef]
- Schytz, H.W.; Birk, S.; Wienecke, T.; Kruuse, C.; Olesen, J.; Ashina, M. PACAP38 Induces Migraine-like Attacks in Patients with Migraine without Aura. Brain 2009, 132, 16–25. [Google Scholar] [CrossRef] [Green Version]
- Thomsen, L.L. Investigations into the Role of Nitric Oxide and the Large Intracranial Arteries in Migraine Headache. Cephalalgia 1997, 17, 873–895. [Google Scholar] [CrossRef]
- Pellesi, L.; Al-Karagholi, M.A.M.; Chaudhry, B.A.; Lopez, C.L.; Snellman, J.; Hannibal, J.; Amin, F.M.; Ashina, M. Two-Hour Infusion of Vasoactive Intestinal Polypeptide Induces Delayed Headache and Extracranial Vasodilation in Healthy Volunteers. Cephalalgia 2020, 40, 1212–1223. [Google Scholar] [CrossRef]
- Kruuse, C.; Thomsen, L.L.; Birk, S.; Olesen, J. Migraine Can Be Induced by Sildenafil without Changes in Middle Cerebral Artery Diameter. Brain 2003, 126, 241–247. [Google Scholar] [CrossRef]
- Guo, S.; Olesen, J.; Ashina, M. Phosphodiesterase 3 Inhibitor Cilostazol Induces Migraine-like Attacks via Cyclic AMP Increase. Brain 2014, 137, 2951–2959. [Google Scholar] [CrossRef] [Green Version]
- Al-Karagholi, M.A.M.; Hansen, J.M.; Guo, S.; Olesen, J.; Ashina, M. Opening of ATP-Sensitive Potassium Channels Causes Migraine Attacks: A New Target for the Treatment of Migraine. Brain 2019, 142, 2644–2654. [Google Scholar] [CrossRef]
- Al-Karagholi, M.A.M.; Ghanizada, H.; Nielsen, C.A.W.; Hougaard, A.; Ashina, M. Opening of ATP Sensitive Potassium Channels Causes Migraine Attacks with Aura. Brain 2021, 144, 2322–2332. [Google Scholar] [CrossRef]
- Noma, A. ATP-Regulated K+ Channels in Cardiac Muscle. Nature 1983, 305, 147–148. [Google Scholar] [CrossRef] [PubMed]
- Miki, T.; Nagashima, K.; Seino, S. The Structure and Function of the ATP-Sensitive K+ Channel in Insulin-Secreting Pancreatic Beta-Cells. J. Mol. Endocrinol. 1999, 22, 113–123. [Google Scholar] [CrossRef]
- Proks, P.; Ashcroft, F.M. Modeling KATP Channel Gating and Its Regulation. Prog. Biophys. Mol. Biol. 2009, 99, 7–19. [Google Scholar] [CrossRef]
- Hibino, H.; Inanobe, A.; Furutani, K.; Murakami, S.; Findlay, I.; Kurachi, Y. Inwardly Rectifying Potassium Channels: Their Structure, Function, and Physiological Roles. Physiol. Rev. 2010, 90, 291–366. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Choma, K.; Bednarczyk, P.; Koszela-Piotrowska, I.; Kulawiak, B.; Kudin, A.; Kunz, W.S.; Dołowy, K.; Szewczyk, A. Single Channel Studies of the ATP-Regulated Potassium Channel in Brain Mitochondria. J. Bioenerg. Biomembr. 2009, 41, 323–334. [Google Scholar] [CrossRef]
- Babenko, A.P.; Aguilar-Bryan, L.; Bryan, J. A View of Sur/Kir6.X, KATP Channels. Annu. Rev. Physiol 1998, 60, 667–687. [Google Scholar] [CrossRef]
- Seino, S.; Miki, T. Physiological and Pathophysiological Roles of ATP-Sensitive K+ Channels. Prog. Biophys. Mol. Biol. 2003, 81, 133–176. [Google Scholar] [CrossRef]
- Syed, A.U.; Koide, M.; Brayden, J.E.; Wellman, G.C. Tonic Regulation of Middle Meningeal Artery Diameter by ATP-Sensitive Potassium Channels. J. Cereb. Blood Flow Metab. 2019, 39, 670–679. [Google Scholar] [CrossRef]
- Standen, N.B.; Quayle, J.M.; Davies, N.W.; Brayden, J.E.; Huang, Y.; Nelson, M.T. Hyperpolarizing Vasodilators Activate ATP-Sensitive K+ Channels in Arterial Smooth Muscle. Science 1989, 245, 177–180. [Google Scholar] [CrossRef]
- Sung, M.W.; Yang, Z.; Driggers, C.M.; Patton, B.L.; Mostofian, B.; Russo, J.D.; Zuckerman, D.M.; Shyng, S.L. Vascular KATP Channel Structural Dynamics Reveal Regulatory Mechanism by Mg-Nucleotides. Proc. Natl. Acad. Sci. USA 2021, 118. [Google Scholar] [CrossRef]
- Ribalet, B.; John, S.A.; Xie, L.H.; Weiss, J.N. Regulation of the ATP-Sensitive K Channel Kir6.2 by ATP and PIP(2). J. Mol. Cell. Cardiol. 2005, 39, 71–77. [Google Scholar] [CrossRef]
- Baukrowitz, T.; Schulte, U.; Oliver, D.; Herlitze, S.; Krauter, T.; Tucker, S.J.; Ruppersberg, J.P.; Fakler, B. PIP2 and PIP as Determinants for ATP Inhibition of KATP Channels. Science 1998, 282, 1141–1144. [Google Scholar] [CrossRef]
- Larsson, O.; Barker, C.J.; Berggren, P.O. Phosphatidylinositol 4,5-Bisphosphate and ATP-Sensitive Potassium Channel Regulation: A Word of Caution. Diabetes 2000, 49, 1409–1412. [Google Scholar] [CrossRef]
- Shi, Y.; Cui, N.; Shi, W.; Jiang, C. A Short Motif in Kir6.1 Consisting of Four Phosphorylation Repeats Underlies the Vascular KATP Channel Inhibition by Protein Kinase C. J. Biol. Chem. 2008, 283, 2488–2494. [Google Scholar] [CrossRef] [Green Version]
- Shi, W.-W.; Yang, Y.; Shi, Y.; Chun, J. KATP Channel Action in Vascular Tone Regulation: From Genetics to Diseases. Sheng Li Xue Bao 2012, 64, 1–13. [Google Scholar] [PubMed]
- Quinn, K.V.; Giblin, J.P.; Tinker, A. Multisite Phosphorylation Mechanism for Protein Kinase A Activation of the Smooth Muscle ATP-Sensitive K+ Channel. Circ. Res. 2004, 94, 1359–1366. [Google Scholar] [CrossRef] [Green Version]
- Shi, Y.; Chen, X.; Wu, Z.; Shi, W.; Yang, Y.; Cui, N.; Jiang, C.; Harrison, R.W. CAMP-Dependent Protein Kinase Phosphorylation Produces Interdomain Movement in SUR2B Leading to Activation of the Vascular KATP Channel. J. Biol. Chem. 2008, 283, 7523–7530. [Google Scholar] [CrossRef] [Green Version]
- Kokoti, L.; Al-Karagholi, M.A.M.; Ashina, M. Latest Insights into the Pathophysiology of Migraine: The ATP-Sensitive Potassium Channels. Curr. Pain Headache Rep. 2020, 24, 77. [Google Scholar] [CrossRef]
- Schytz, H.W.; Schoonman, G.G.; Ashina, M. What Have We Learnt from Triggering Migraine? Curr. Opin. Neurol. 2010, 23, 259–265. [Google Scholar] [CrossRef] [PubMed]
- Yamada, M.; Isomoto, S.; Matsumoto, S.; Kondo, C.; Shindo, T.; Horio, Y.; Kurachi, Y. Sulphonylurea Receptor 2B and Kir6.1 Form a Sulphonylurea-Sensitive but ATP-Insensitive K+ Channel. J. Physiol. 1997, 499, 715–720. [Google Scholar] [CrossRef]
- Teramoto, N.; Zhu, H.-L.; Shibata, A.; Aishima, M.; Walsh, E.J.; Nagao, M.; Cole, W.C. ATP-Sensitive K+ Channels in Pig Urethral Smooth Muscle Cells Are Heteromultimers of Kir6.1 and Kir6.2. Am. J. Physiol Ren. Physiol 2009, 296, 107–117. [Google Scholar] [CrossRef]
- Christensen, S.L.; Rasmussen, R.H.; Cour, S.L.; Ernstsen, C.; Hansen, T.F.; Kogelman, L.J.; Lauritzen, S.P.; Guzaite, G.; Styrishave, B.; Janfelt, C.; et al. Smooth Muscle ATP-Sensitive Potassium Channels Mediate Migraine-Relevant Hypersensitivity in Mouse Models. Cephalalgia 2022, 42, 93–107. [Google Scholar] [CrossRef] [PubMed]
- Al-Karagholi, M.A.-M.; Hansen, J.M.; Severinsen, J.; Jansen-Olesen, I.; Ashina, M. The KATP Channel in Migraine Pathophysiology: A Novel Therapeutic Target for Migraine. J. Headache Pain 2017, 18, 90. [Google Scholar] [CrossRef]
- Christensen, S.L.; Munro, G.; Petersen, S.; Shabir, A.; Jansen-Olesen, I.; Kristensen, D.M.; Olesen, J. ATP Sensitive Potassium (KATP) Channel Inhibition: A Promising New Drug Target for Migraine. Cephalalgia 2020, 40, 650–664. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Aziz, Q.; Tinker, A. The Pharmacology of ATP-Sensitive K + Channels (K ATP). Handb. Exp. Pharmacol. 2021, 267, 357–378. [Google Scholar] [CrossRef]
- Skatchkov, S.N.; Rojas, L.; Eaton, M.J.; Orkand, R.K.; Biedermann, B.; Bringmann, A.; Pannicke, T.; Veh, R.W.; Reichenbach, A. Functional Expression of Kir 6.1/SUR1-KATP Channels in Frog Retinal Müller Glial Cells. Glia 2002, 38, 256–267. [Google Scholar] [CrossRef]
- Ploug, K.B.; Sørensen, M.A.; Strøbech, L.; Klaerke, D.A.; Hay-Schmidt, A.; Sheykhzade, M.; Olesen, J.; Jansen-Olesen, I. KATP Channels in Pig and Human Intracranial Arteries. Eur. J. Pharmacol. 2008, 601, 43–49. [Google Scholar] [CrossRef]
- Aziz, Q.; Li, Y.; Tinker, A. ATP-Sensitive Potassium Channels and Vascular Function. Channels 2015, 9, 3–4. [Google Scholar] [CrossRef] [PubMed]
- Ploug, K.B.; Baun, M.; Hay-Schmidt, A.; Olesen, J.; Jansen-Olesen, I. Presence and Vascular Pharmacology of KATP Channel Subtypes in Rat Central and Peripheral Tissues. Eur. J. Pharmacol. 2010, 637, 109–117. [Google Scholar] [CrossRef] [PubMed]
- Rodrigo, G.; Standen, N. ATP-Sensitive Potassium Channels. Curr. Pharm. Des. 2005, 11, 1915–1940. [Google Scholar] [CrossRef] [PubMed]
- Bao, L.; Kefaloyianni, E.; Lader, J.; Hong, M.; Morley, G.; Fishman, G.I.; Sobie, E.A.; Coetzee, W.A. Unique Properties of the ATP-Sensitive K+ Channel in the Mouse Ventricular Cardiac Conduction System. Circ. Arrhythm. Electrophysiol. 2011, 4, 926–935. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aguilar-Bryan, L.; Nichols, C.G.; Wechsler, S.W.; Clement IV, J.P.; Boyd, A.E.; González, G.; Herrera-Sosa, H.; Nguy, K.; Bryan, J.; Nelson, D.A. Cloning of the Beta Cell High-Affinity Sulfonylurea Receptor: A Regulator of Insulin Secretion. Science 1995, 268, 423–426. [Google Scholar] [CrossRef]
- Flagg, T.P.; Kurata, H.T.; Masia, R.; Caputa, G.; Magnuson, M.A.; Lefer, D.J.; Coetzee, W.A.; Nichols, C.G. Differential Structure of Atrial and Ventricular KATP: Atrial KATP Channels Require SUR1. Circ. Res. 2008, 103, 1458–1465. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zoga, V.; Kawano, T.; Liang, M.Y.; Bienengraeber, M.; Weihrauch, D.; McCallum, B.; Gemes, G.; Hogan, Q.; Sarantopoulos, C. KATPchannel Subunits in Rat Dorsal Root Ganglia: Alterations by Painful Axotomy. Mol. Pain 2010, 6. [Google Scholar] [CrossRef] [Green Version]
- Kawano, T.; Zoga, V.; McCallum, J.B.; Wu, H.E.; Gemes, G.; Liang, M.Y.; Abram, S.; Kwok, W.M.; Hogan, Q.H.; Sarantopoulos, C.D. ATP-Sensitive Potassium Currents in Rat Primary Afferent Neurons: Biophysical, Pharmacological Properties, and Alterations by Painful Nerve Injury. Neuroscience 2009, 162, 431–443. [Google Scholar] [CrossRef] [PubMed]
- Tricarico, D.; Mele, A.; Lundquist, A.L.; Desai, R.R.; George, A.L.; Conte Camerino, D. Hybrid Assemblies of ATP-Sensitive K+ Channels Determine Their Muscle-Type-Dependent Biophysical and Pharmacological Properties. Proc. Natl. Acad. Sci. USA 2006, 103, 1118–1123. [Google Scholar] [CrossRef] [Green Version]
- Inagaki, N.; Gonoi, T.; Clement IV, J.P.; Wang, C.Z.; Aguilar-Bryan, L.; Bryan, J.; Seino, S. A Family of Sulfonylurea Receptors Determines the Pharmacological Properties of ATP-Sensitive K+ Channels. Neuron 1996, 16, 1011–1017. [Google Scholar] [CrossRef] [Green Version]
- Sun, H.S.; Feng, Z.P.; Barber, P.A.; Buchan, A.M.; French, R.J. Kir6.2-Containing ATP-Sensitive Potassium Channels Protect Cortical Neurons from Ischemic/Anoxic Injury in Vitro and in Vivo. Neuroscience 2007, 144, 1509–1515. [Google Scholar] [CrossRef] [PubMed]
- Jovanović, S.; Ballantyne, T.; Du, Q.; Blagojević, M.; Jovanović, A. Phenylephrine Preconditioning in Embryonic Heart H9c2 Cells Is Mediated by Up-Regulation of SUR2B/Kir6.2: A First Evidence for Functional Role of SUR2B in Sarcolemmal KATP Channels and Cardioprotection. Int. J. Biochem. Cell Biol. 2016, 70, 23–28. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jahangir, A.; Terzic, A. K(ATP) Channel Therapeutics at the Bedside. J. Mol. Cell. Cardiol. 2005, 39, 99–112. [Google Scholar] [CrossRef] [Green Version]
- Aziz, Q.; Thomas, A.M.; Gomes, J.; Ang, R.; Sones, W.R.; Li, Y.; Ng, K.E.; Gee, L.; Tinker, A. The ATP-Sensitive Potassium Channel Subunit, Kir6.1, in Vascular Smooth Muscle Plays a Major Role in Blood Pressure Control. Hypertension 2014, 64, 523–529. [Google Scholar] [CrossRef] [Green Version]
- Gozalov, A.; Jansen-Olesen, I.; Klaerke, D.; Olesen, J. Role of KATP Channels in Cephalic Vasodilatation Induced by Calcitonin Gene-Related Peptide, Nitric Oxide, and Transcranial Electrical Stimulation in the Rat. Headache 2008, 48, 1202–1213. [Google Scholar] [CrossRef]
- Nelson, M.T.; Quayle, J.M. Physiological Roles and Properties of Potassium Channels in Arterial Smooth Muscle. Am. J. Physiol. Cell Physiol. 1995, 268, 799–822. [Google Scholar] [CrossRef]
- Tinker, A.; Aziz, Q.; Thomas, A. The Role of ATP-Sensitive Potassium Channels in Cellular Function and Protection in the Cardiovascular System. Br. J. Pharmacol. 2014, 171, 12–23. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Murphy, M.E.; Brayden, J.E. Nitric Oxide Hyperpolarizes Rabbit Mesenteric Arteries via ATP-Sensitive Potassium Channels. J. Physiol. 1995, 486, 58. [Google Scholar] [CrossRef] [PubMed]
- Olesen, J. The Role of Nitric Oxide (NO) in Migraine, Tension-Type Headache and Cluster Headache. Pharmacol. Ther. 2008, 120, 157–171. [Google Scholar] [CrossRef]
- Duarte, I.D.G.; dos Santos, I.R.; Lorenzetti, B.B.; Ferreira, S.H. Analgesia by Direct Antagonism of Nociceptor Sensitization Involves the Arginine-Nitric Oxide-CGMP Pathway. Eur. J. Pharmacol. 1992, 217, 225–227. [Google Scholar] [CrossRef]
- Aziz, Q.; Li, Y.; Anderson, N.; Ojake, L.; Tsisanova, E.; Tinker, A. Molecular and Functional Characterization of the Endothelial ATP-Sensitive Potassium Channel. J. Biol. Chem. 2017, 292, 17587–17597. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De Logu, F.; Landini, L.; Janal, M.N.; Li Puma, S.; De Cesaris, F.; Geppetti, P.; Nassini, R. Migraine-Provoking Substances Evoke Periorbital Allodynia in Mice. J. Headache Pain 2019, 20, 18. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schytz, H.W.; Olesen, J.; Ashina, M. The PACAP Receptor: A Novel Target for Migraine Treatment. Neurotherapeutics 2010, 7, 191–196. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brown, T.; Forster, R.B.; Cleanthis, M.; Mikhailidis, D.P.; Stansby, G.; Stewart, M. Cilostazol for Intermittent Claudication. Cochrane Database Syst. Rev. 2021, 2021. [Google Scholar] [CrossRef]
- Moore, R.A.; Edwards, J.E.; McQuay, H.J. Sildenafil (Viagra) for Male Erectile Dysfunction: A Meta-Analysis of Clinical Trial Reports. BMC Urol. 2002, 2, 1–12. [Google Scholar] [CrossRef] [Green Version]
- Trainor, D.C.; Jones, R.C. Headaches in Explosive Magazine Workers. Arch. Environ. Health 1966, 12, 231–234. [Google Scholar] [CrossRef]
- Miki, T.; Suzuki, M.; Shibasaki, T.; Uemura, H.; Sato, T.; Yamaguchi, K.; Koseki, H.; Iwanaga, T.; Nakaya, H.; Seino, S. Mouse Model of Prinzmetal Angina by Disruption of the Inward Rectifier Kir6.1. Nat. Med. 2002, 8, 466–472. [Google Scholar] [CrossRef] [PubMed]
- Porter, A. Controlling Your Losses: Conditional Gene Silencing in Mammals. Trends Genet. 1998, 14, 73–79. [Google Scholar] [CrossRef]
- Sauer, B. Inducible Gene Targeting in Mice Using the Cre/Lox System. Methods 1998, 14, 381–392. [Google Scholar] [CrossRef] [Green Version]
- Utomo, A.R.H.; Nikitin, A.Y.; Lee, W.H. Temporal, Spatial, and Cell Type-Specified Control of Cre-Mediated DNA Recombination in Transgenic Mice. Nat. Biotechnol. 1999, 17, 1091–1096. [Google Scholar] [CrossRef]
- Soundarapandian, M.M.; Zhong, X.; Peng, L.; Wu, D.; Lu, Y. Role of K ATP Channels in Protection against Neuronal Excitatory Insults. J. Neurochem 2007, 103, 1721–1729. [Google Scholar] [CrossRef] [PubMed]
- Yildirim, C.; Özkaya, B.; Bal, R. KATP and TRPM2-like Channels Couple Metabolic Status to Resting Membrane Potential of Octopus Neurons in the Mouse Ventral Cochlear Nucleus. Brain Res. Bull. 2021, 170, 115–128. [Google Scholar] [CrossRef] [PubMed]
- Kase, D.; Imoto, K. The Role of HCN Channels on Membrane Excitability in the Nervous System. J. Signal. Transduct. 2012, 2012, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Country, M.W.; Jonz, M.G. Mitochondrial KATP Channels Stabilize Intracellular Ca2+ during Hypoxia in Retinal Horizontal Cells of Goldfish (Carassius Auratus). J. Exp. Biol. 2021, 224. [Google Scholar] [CrossRef]
- Guo, W.; Tang, Z.Y.; Cai, Z.Y.; Zhao, W.E.; Yang, J.; Wang, X.P.; Ji, J.; Huang, X.X.; Sun, X.L. Iptakalim Alleviates Synaptic Damages via Targeting Mitochondrial ATP-Sensitive Potassium Channel in Depression. FASEB J. 2021, 35. [Google Scholar] [CrossRef]
- Toulorge, D.; Guerreiro, S.; Hirsch, E.C.; Michel, P.P. KATP Channel Blockade Protects Midbrain Dopamine Neurons by Repressing a Glia-to-Neuron Signaling Cascade That Ultimately Disrupts Mitochondrial Calcium Homeostasis. J. Neurochem. 2010, 114, 553–564. [Google Scholar] [CrossRef]
- Khanna, N.; Malhotra, R.S.; Mehta, A.K.; Garg, G.R.; Halder, S.; Sharma, K.K. Interaction of Morphine and Potassium Channel Openers on Experimental Models of Pain in Mice. Fundam. Clin. Pharmacol. 2011, 25, 479–484. [Google Scholar] [CrossRef]
- Tsantoulas, C.; McMahon, S.B. Opening Paths to Novel Analgesics: The Role of Potassium Channels in Chronic Pain. Trends Neurosci. 2014, 37, 146–158. [Google Scholar] [CrossRef] [Green Version]
- Cunha, T.M.; Roman-Campos, D.; Lotufo, C.M.; Duarte, H.L.; Souza, G.R.; Verri, W.A.; Funez, M.I.; Dias, Q.M.; Schivo, I.R.; Domingues, A.C.; et al. Morphine Peripheral Analgesia Depends on Activation of the PI3Kγ/AKT/NNOS/NO/KATP Signaling Pathway. Proc. Natl. Acad. Sci. USA 2010, 107, 4442–4447. [Google Scholar] [CrossRef] [Green Version]
- Kusuda, R.; Carreira, E.U.; Ulloa, L.; Cunha, F.Q.; Kanashiro, A.; Cunha, T.M. Choline Attenuates Inflammatory Hyperalgesia Activating Nitric Oxide/CGMP/ATP-Sensitive Potassium Channels Pathway. Brain Res. 2020, 1727, 146567. [Google Scholar] [CrossRef]
- Fakhri, S.; Ahmadpour, Y.; Rezaei, H.; Kooshki, L.; Moradi, S.Z.; Iranpanah, A.; Gravandi, M.M.; Abbaszadeh, F.; Ghanbarveisi, F. The Antinociceptive Mechanisms of Melatonin: Role of L-Arginine/Nitric Oxide/Cyclic GMP/KATP Channel Signaling Pathway. Behav. Pharmacol. 2020, 31, 728–737. [Google Scholar] [CrossRef] [PubMed]
- Gong, L.; Gao, F.; Li, J.; Li, J.; Yu, X.; Ma, X.; Zheng, W.; Cui, S.; Liu, K.; Zhang, M.; et al. Oxytocin-Induced Membrane Hyperpolarization in Pain-Sensitive Dorsal Root Ganglia Neurons Mediated by Ca(2+)/NNOS/NO/KATP Pathway. Neuroscience 2015, 289, 417–428. [Google Scholar] [CrossRef] [PubMed]
- Sarantopoulos, C.; McCallum, B.; Sapunar, D.; Kwok, W.M.; Hogan, Q. ATP-Sensitive Potassium Channels in Rat Primary Afferent Neurons: The Effect of Neuropathic Injury and Gabapentin. Neurosci. Lett. 2003, 343, 185–189. [Google Scholar] [CrossRef]
- Wu, X.F.; Liu, W.T.; Liu, Y.P.; Huang, Z.J.; Zhang, Y.K.; Song, X.J. Reopening of ATP-Sensitive Potassium Channels Reduces Neuropathic Pain and Regulates Astroglial Gap Junctions in the Rat Spinal Cord. Pain 2011, 152, 2605–2615. [Google Scholar] [CrossRef]
- Sakamaki, G.; Johnson, K.; Mensinger, M.; Hmu, E.; Klein, A.H. Loss of SUR1 Subtype KATP Channels Alters Antinociception and Locomotor Activity after Opioid Administration. Behav. Brain Res. 2021, 414. [Google Scholar] [CrossRef]
- Fisher, C.; Johnson, K.; Okerman, T.; Jurgenson, T.; Nickell, A.; Salo, E.; Moore, M.; Doucette, A.; Bjork, J.; Klein, A.H. Morphine Efficacy, Tolerance, and Hypersensitivity Are Altered After Modulation of SUR1 Subtype KATP Channel Activity in Mice. Front. Neurosci. 2019, 13. [Google Scholar] [CrossRef]
- Shi, W.; Cui, N.; Wu, Z.; Yang, Y.; Zhang, S.; Gai, H.; Zhu, D.; Jiang, C. Lipopolysaccharides Up-Regulate Kir6.1/SUR2B Channel Expression and Enhance Vascular KATP Channel Activity via NF-KappaB-Dependent Signaling. J. Biol. Chem. 2010, 285, 3021–3029. [Google Scholar] [CrossRef] [Green Version]
- Cao, Z.; Dai, W.; Zhang, R.; Chen, L.; Yang, X.; Hu, L.; Chiang, L.Y.; Liu, W. Opening of the Adenosine Triphosphate-Sensitive Potassium Channel Attenuates Morphine Tolerance by Inhibiting JNK and Astrocyte Activation in the Spinal Cord. Clin. J. Pain 2016, 32, 617–623. [Google Scholar] [CrossRef]
- Christensen, S.L.; Rasmussen, R.H.; Ernstsen, C.; La Cour, S.; David, A.; Chaker, J.; Haanes, K.A.; Christensen, S.T.; Olesen, J.; Kristensen, D.M. CGRP-Dependent Signalling Pathways Involved in Mouse Models of GTN- Cilostazol- and Levcromakalim-Induced Migraine. Cephalalgia 2021, 41, 1413–1426. [Google Scholar] [CrossRef]
- Narita, M.; Suzuki, T.; Misawa, M.; Nagase, H.; Nabeshima, A.; Ashizawa, T.; Ozawa, H.; Saito, T.; Takahata, N. Role of Central ATP-Sensitive Potassium Channels in the Analgesic Effect and Spinal Noradrenaline Turnover-Enhancing Effect of Intracerebroventricularly Injected Morphine in Mice. Brain Res. 1992, 596, 209–214. [Google Scholar] [CrossRef]
- Saint-Martin, C.; Arnoux, J.B.; de Lonlay, P.; Bellanné-Chantelot, C. KATP Channel Mutations in Congenital Hyperinsulinism. Semin. Pediatr. Surg. 2011, 20, 18–22. [Google Scholar] [CrossRef] [PubMed]
- De Franco, E.; Saint-Martin, C.; Brusgaard, K.; Knight Johnson, A.E.; Aguilar-Bryan, L.; Bowman, P.; Arnoux, J.B.; Larsen, A.R.; May, S.; Greeley, S.A.W.; et al. Update of Variants Identified in the Pancreatic β-Cell KATP Channel Genes KCNJ11 and ABCC8 in Individuals with Congenital Hyperinsulinism and Diabetes. Hum. Mutat. 2020, 41, 884–905. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kane, C.; Shepherd, R.M.; Squires, P.E.; Johnson, P.R.V.; James, R.F.L.; Milla, P.J.; Aynsley-Green, A.; Lindley, K.J.; Dunne, M.J. Loss of Functional KATP Channels in Pancreatic Beta-Cells Causes Persistent Hyperinsulinemic Hypoglycemia of Infancy. Nat. Med. 1996, 2, 1344–1347. [Google Scholar] [CrossRef] [PubMed]
- Li, C.; Ackermann, A.M.; Boodhansingh, K.E.; Bhatti, T.R.; Liu, C.; Schug, J.; Doliba, N.; Han, B.; Cosgrove, K.E.; Banerjee, I.; et al. Functional and Metabolomic Consequences of K ATP Channel Inactivation in Human Islets. Diabetes 2017, 66, 1901–1913. [Google Scholar] [CrossRef] [Green Version]
- Lontchi-Yimagou, E.; Sobngwi, E.; Matsha, T.E.; Kengne, A.P. Diabetes Mellitus and Inflammation. Curr. Diab. Rep. 2013, 13, 435–444. [Google Scholar] [CrossRef]
- Duncan, B.B.; Schmidt, M.I. The Epidemiology of Low-Grade Chronic Systemic Inflammation and Type 2 Diabetes. Diabetes Technol. Ther. 2006, 8, 7–17. [Google Scholar] [CrossRef]
- Gier, B.; Krippeit-Drews, P.; Sheiko, T.; Aguilar-Bryan, L.; Bryan, J.; Düfer, M.; Drews, G. Suppression of KATP Channel Activity Protects Murine Pancreatic Beta Cells against Oxidative Stress. J. Clin. Investig. 2009, 119, 3246–3256. [Google Scholar] [CrossRef] [Green Version]
- Krippeit-Drews, P.; Lang, F.; Häussinger, D.; Drews, G. H2O2 Induced Hyperpolarization of Pancreatic B-Cells. Pflug. Arch. 1994, 426, 552–554. [Google Scholar] [CrossRef]
- Maechler, P.; Jornot, L.; Wollheim, C.B. Hydrogen Peroxide Alters Mitochondrial Activation and Insulin Secretion in Pancreatic Beta Cells. J. Biol. Chem. 1999, 274, 27905–27913. [Google Scholar] [CrossRef] [Green Version]
- Nakazaki, M.; Kakei, M.; Koriyama, N.; Tanaka, H. Involvement of ATP-Sensitive K+ Channels in Free Radical-Mediated Inhibition of Insulin Secretion in Rat Pancreatic Beta-Cells. Diabetes 1995, 44, 878–883. [Google Scholar] [CrossRef]
- Edalat, A.; Schulte-Mecklenbeck, P.; Bauer, C.; Undank, S.; Krippeit-Drews, P.; Drews, G.; Düfer, M. Mitochondrial Succinate Dehydrogenase Is Involved in Stimulus-Secretion Coupling and Endogenous ROS Formation in Murine Beta Cells. Diabetologia 2015, 58, 1532–1541. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Benemei, S.; Fusi, C.; Trevisan, G.; Geppetti, P. The TRPA1 Channel in Migraine Mechanism and Treatment. Br. J. Pharmacol. 2014, 171, 2552–2567. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Akerman, S.; Salvemini, D.; Romero-Reyes, M. Targeting Reactive Nitroxidative Species in Preclinical Models of Migraine. Cephalalgia 2021, 41, 1187–1200. [Google Scholar] [CrossRef] [PubMed]
- Hambrock, A.; Löffler-Walz, C.; Kloor, D.; Delabar, U.; Horio, Y.; Kurachi, Y.; Quast, U. ATP-Sensitive K+ Channel Modulator Binding to Sulfonylurea Receptors SUR2A and SUR2B: Opposite Effects of MgADP. Mol. Pharmacol. 1999, 55, 832–840. [Google Scholar] [PubMed]
- Petersson, J.; Ryman, T.; Högestätt, E.D. Vasodilator Effects of KRN2391, Levcromakalim and 3-Morpholino- Sydnonimin in Human Pial and Omental Arteries. Naunyn. Schmiedebergs. Arch. Pharmacol. 2000, 362, 68–73. [Google Scholar] [CrossRef]
- Jansen-Olesen, I.; Mortensen, C.H.; El-Bariaki, N.; Ploug, K.B. Characterization of KATP-Channels in Rat Basilar and Middle Cerebral Arteries: Studies of Vasomotor Responses and MRNA Expression. Eur. J. Pharmacol. 2005, 523, 109–118. [Google Scholar] [CrossRef]
- Ploug, K.B.; Boni, L.J.; Baun, M.; Hay-Schmidt, A.; Olesen, J.; Jansen-Olesen, I. KATP Channel Expression and Pharmacological in Vivo and in Vitro Studies of the KATP Channel Blocker PNU-37883A in Rat Middle Meningeal Arteries. Br. J. Pharmacol. 2008, 154, 72–81. [Google Scholar] [CrossRef] [Green Version]
- Carlsen, J.E.; Kardel, T.; Jensen, H.; Tangø, M.; Trap-Jensen, J. Pinacidil, a New Vasodilator: Pharmacokinetics and Pharmacodynamics of a New Retarded Release Tablet in Essential Hypertension. Eur. J. Clin. Pharmacol. 1983, 25, 557–561. [Google Scholar] [CrossRef] [PubMed]
- Ward, J.W.; McBurney, A.; Farrow, P.R.; Sharp, P. Pharmacokinetics and Hypotensive Effect in Healthy Volunteers of Pinacidil, a New Potent Vasodilator. Eur. J. Clin. Pharmacol. 1984, 26, 603–608. [Google Scholar] [CrossRef]
- Clapham, J.C.; Trail, B.K.; Hamilton, T.C. K+ Channel Activators, Acute Glucose Tolerance and Glibenclamide-Induced Hypoglycaemia in the Hypertensive Rat. Eur. J. Pharmacol. 1994, 257, 79–85. [Google Scholar] [CrossRef]
- Mannhold, R. KATP Channel Openers: Structure-Activity Relationships and Therapeutic Potential. Med. Res. Rev. 2004, 24, 213–266. [Google Scholar] [CrossRef] [PubMed]
- Wolff, F. Diazoxide Hyperglycaemia and Its Continued Relief by Tolbutamide. Lancet 1964, 1, 309–310. [Google Scholar] [CrossRef]
- Rubin, A.A.; Roth, F.E.; Taylor, R.M.; Rosenkilde, H. Pharmacology of Diazoxide, an Antihypertensive, Nondiuretic Benzothiadiazine. J. Pharmacol. Exp. Ther. 1962, 136, 344–352. [Google Scholar] [PubMed]
- Lodwick, D.; Rainbow, R.D.; Rubaiy, H.N.; Al Johi, M.; Vuister, G.W.; Norman, R.I. Sulfonylurea Receptors Regulate the Channel Pore in ATP-Sensitive Potassium Channels via an Intersubunit Salt Bridge. Biochem. J. 2014, 464, 343–354. [Google Scholar] [CrossRef] [PubMed]
- Maruthur, N.M.; Tseng, E.; Hutfless, S.; Wilson, L.M.; Suarez-Cuervo, C.; Berger, Z.; Chu, Y.; Iyoha, E.; Segal, J.B.; Bolen, S. Diabetes Medications as Monotherapy or Metformin-Based Combination Therapy for Type 2 Diabetes: A Systematic Review and Meta-Analysis. Ann. Intern. Med. 2016, 164, 740–751. [Google Scholar] [CrossRef]
- Coghlan, M.J.; Carroll, W.A.; Gopalakrishnan, M. Recent Developments in the Biology and Medicinal Chemistry of Potassium Channel Modulators: Update from a Decade of Progress. J. Med. Chem. 2001, 44, 1627–1653. [Google Scholar] [CrossRef]
- Gribble, F.M.; Tucker, S.J.; Seino, S.; Ashcroft, F.M. Tissue Specificity of Sulfonylureas Studies on Cloned Cardiac and β- Cell K(ATP) Channels. Diabetes 1998, 47, 1412–1418. [Google Scholar] [CrossRef]
- Liu, S.Y.; Tian, H.M.; Liao, D.Q.; Chen, Y.F.; Gou, Z.P.; Xie, X.Y.; Li, X.J. The Effect of Gliquidone on KATP Channels in Pancreatic β-Cells, Cardiomyocytes, and Vascular Smooth Muscle Cells. Diabetes Res. Clin. Pract. 2015, 109, 334–339. [Google Scholar] [CrossRef]
- Glibenclamide|Ligand Activity Charts|IUPHAR/BPS Guide to PHARMACOLOGY. Available online: https://www.guidetopharmacology.org/GRAC/LigandActivityRangeVisForward?ligandId=2414 (accessed on 26 June 2022).
- Wang, Y.; Yu, L.; Cui, N.; Jin, X.; Zhu, D.; Jiang, C. Differential Sensitivities of the Vascular K(ATP) Channel to Various PPAR Activators. Biochem. Pharmacol. 2013, 85, 1495–1503. [Google Scholar] [CrossRef] [Green Version]
- Yu, L.; Jin, X.; Cui, N.; Wu, Y.; Shi, Z.; Zhu, D.; Jiang, C. Rosiglitazone Selectively Inhibits KATP Channels by Acting on the KIR6 Subunit. Br. J. Pharmacol. 2012, 167, 26. [Google Scholar] [CrossRef] [Green Version]
- Humphrey, S.J. Pharmacology of the K-ATP Channel Blocking Morpholinoguanidine PNU- 37883A. Cardiovasc. Drug Rev. 1999, 17, 295–328. [Google Scholar] [CrossRef]
- D’Arcy, V.; Laher, M.; McCoy, D.; Sullivan, P.; Walsh, C.H.; Hickey, M.P. Pinacidil, a New Vasodilator, in the Treatment of Mild to Moderate Essential Hypertension. Eur. J. Clin. Pharmacol. 1985, 28, 347–349. [Google Scholar] [CrossRef] [PubMed]
- Kidney, J.C.; Fuller, R.W.; Worsdell, Y.M.; Lavender, E.A.; Chung, K.F.; Barnes, P.J. Effect of an Oral Potassium Channel Activator, BRL 38227, on Airway Function and Responsiveness in Asthmatic Patients: Comparison with Oral Salbutamol. Thorax 1993, 48, 130–133. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ashina, M.; Hansen, J.M.; Olesen, J. Pearls and Pitfalls in Human Pharmacological Models of Migraine: 30 Years’ Experience. Cephalalgia 2013, 33, 540–553. [Google Scholar] [CrossRef]
- Al-Karagholi, M.A.-M.; Ghanizada, H.; Hansen, J.M.; Skovgaard, L.T.; Olesen, J.; Larsson, H.B.W.; Amin, F.M.; Ashina, M. Levcromakalim, an Adenosine Triphosphate-Sensitive Potassium Channel Opener, Dilates Extracerebral but Not Cerebral Arteries. Headache 2019, 59, 1468–1480. [Google Scholar] [CrossRef] [PubMed]
- Al-Karagholi, M.A.M.; Ghanizada, H.; Nielsen, C.A.W.; Ansari, A.; Gram, C.; Younis, S.; Vestergaard, M.B.; Larsson, H.B.W.; Skovgaard, L.T.; Amin, F.M.; et al. Cerebrovascular Effects of Glibenclamide Investigated Using High-Resolution Magnetic Resonance Imaging in Healthy Volunteers. J. Cereb. Blood Flow Metab. 2021, 41, 1328–1337. [Google Scholar] [CrossRef]
- Gozalov, A.; Petersen, K.A.; Mortensen, C.; Jansen-Olesen, I.; Klaerke, D.; Olesen, J. Role of KATP Channels in the Regulation of Rat Dura and Pia Artery Diameter. Cephalalgia 2005, 25, 249–260. [Google Scholar] [CrossRef]
- Ploug, K.B.; Edvinsson, L.; Olesen, J.; Jansen-Olesen, I. Pharmacological and Molecular Comparison of KATP Channels in Rat Basilar and Middle Cerebral Arteries. Eur. J. Pharmacol. 2006, 553, 254–262. [Google Scholar] [CrossRef]
- Rasmussen, R.H.; Jansen-Olesen, I.; Kristensen, D.M.; Christensen, S.L. Ex Vivo Release of Calcitonin Gene-Related Peptide from the Trigeminovascular System in Rodents|Protocol. J. Vis. Exp. 2022, 183. [Google Scholar] [CrossRef]
- Ploug, K.B.; Amrutkar, D.V.; Baun, M.; Ramachandran, R.; Iversen, A.; Lund, T.M.; Gupta, S.; Hay-Schmidt, A.; Olesen, J.; Jansen-Olesen, I. K ATP Channel Openers in the Trigeminovascular System. Cephalalgia 2012, 32, 55–65. [Google Scholar] [CrossRef]
- Russell, F.A.; King, R.; Smillie, S.-J.; Kodji, X.; Brain, S.D. Calcitonin Gene-Related Peptide: Physiology and Pathophysiology. Physiol. Rev. 2014, 94, 1099–1142. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sicuteri, F. Mast cells and their active substances: Their role in the pathogenesis of migraine. Headache 1963, 3, 86–92. [Google Scholar] [CrossRef] [PubMed]
- Bates, E.A.; Nikai, T.; Brennan, K.C.; Fu, Y.-H.; Charles, A.C.; Basbaum, A.I.; Ptácek, L.J.; Ahn, A.H. Sumatriptan Alleviates Nitroglycerin-Induced Mechanical and Thermal Allodynia in Mice. Cephalalgia 2010, 30, 170–178. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pradhan, A.A.; Smith, M.L.; McGuire, B.; Tarash, I.; Evans, C.J.; Charles, A. Characterization of a Novel Model of Chronic Migraine. PAIN® 2014, 155, 269–274. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ernstsen, C.; Christensen, S.L.; Rasmussen, R.H.; Nielsen, B.S.; Jansen-Olesen, I.; Olesen, J.; Kristensen, D.M. The PACAP Pathway Is Independent of CGRP in Mouse Models of Migraine: Possible New Drug Target? Brain 2022, 145, 2450–2460. [Google Scholar] [CrossRef] [PubMed]
- Bruch, L.; Bychkov, R.; Kästner, A.; Bülow, T.; Ried, C.; Gollasch, M.; Baumann, G.; Luft, F.C.; Haller, H. Pituitary Adenylate-Cyclase-Activating Peptides Relax Human Coronary Arteries by Activating K(ATP) and K(Ca) Channels in Smooth Muscle Cells. J. Vasc. Res. 1997, 34, 11–18. [Google Scholar] [CrossRef] [PubMed]
- Hayabuchi, Y.; Davies, N.W.; Standen, N.B. Angiotensin II Inhibits Rat Arterial KATP Channels by Inhibiting Steady-State Protein Kinase A Activity and Activating Protein Kinase Cε. J. Physiol. 2001, 530, 193. [Google Scholar] [CrossRef]
- Meisheri, K.D.; Humphrey, S.J.; Khan, S.A.; Cipkus-Dubray, L.A.; Smith, M.P.; Jones, A.W. 4-Morpholinecarboximidine-N-1-Adamantyl-N’-Cyclohexylhydrochloride (U- 37883A): Pharmacological Characterization of a Novel Antagonist of Vascular ATP-Sensitive K+ Channel Openers. J. Pharmacol. Exp. Ther. 1993, 266, 655–665. [Google Scholar]
- Armstead, W.M. Role of Nitric Oxide, Cyclic Nucleotides, and the Activation of ATP-Sensitive K+ Channels in the Contribution of Adenosine to Hypoxia-Induced Pial Artery Dilation. J. Cereb. Blood Flow Metab. 1997, 17, 100–108. [Google Scholar] [CrossRef] [Green Version]
- Oshinsky, M.L.; Sanghvi, M.M.; Maxwell, C.R.; Gonzalez, D.; Spangenberg, R.J.; Cooper, M.; Silberstein, S.D. Spontaneous Trigeminal Allodynia in Rats: A Model of Primary Headache. Headache 2012, 52, 1336–1349. [Google Scholar] [CrossRef] [Green Version]
- Munro, G.; Petersen, S.; Jansen-Olesen, I.; Olesen, J. A Unique Inbred Rat Strain with Sustained Cephalic Hypersensitivity as a Model of Chronic Migraine-like Pain. Sci. Rep. 2018, 8, 1836. [Google Scholar] [CrossRef] [PubMed]
- Al-Karagholi, M.A.M.; Ghanizada, H.; Kokoti, L.; Paulsen, J.S.; Hansen, J.M.; Ashina, M. Effect of KATP Channel Blocker Glibenclamide on Levcromakalim-Induced Headache. Cephalalgia 2020, 40, 1045–1054. [Google Scholar] [CrossRef] [PubMed]
- Coskun, H.; Elbahi, F.A.; Al-Karagholi, M.A.M.; Ghanizada, H.; Sheykhzade, M.; Ashina, M. The Effect of KATP Channel Blocker Glibenclamide on CGRP-Induced Headache and Hemodynamic in Healthy Volunteers. Front. Physiol. 2021, 12, 652136. [Google Scholar] [CrossRef] [PubMed]
- Kokoti, L.; Al-Mahdi Al-Karagholi, M.; Elbahi, F.A.; Coskun, H.; Ghanizada, H.; Amin, F.M.; Ashina, M. Effect of KATP Channel Blocker Glibenclamide on PACAP38-Induced Headache and Hemodynamic. Cephalalgia 2022, 42, 846–858. [Google Scholar] [CrossRef]
- Home—ClinicalTrials.Gov. Available online: https://www.clinicaltrials.gov/ (accessed on 27 June 2022).
- Nair, A.; Jacob, S. A Simple Practice Guide for Dose Conversion between Animals and Human. J. Basic Clin. Pharm. 2016, 7, 27. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Reagan-Shaw, S.; Nihal, M.; Ahmad, N. Dose Translation from Animal to Human Studies Revisited. FASEB J. 2008, 22, 659–661. [Google Scholar] [CrossRef] [Green Version]
- Lukas, G.; Brindle, S.D.; Greengard, P. The Route of Absorption of Intraperitoneally Administered Compounds. J. Pharmacol. Exp. Ther. 1971, 178, 562–564. [Google Scholar] [PubMed]
- Remedi, M.S.; Nichols, C.G. Chronic Antidiabetic Sulfonylureas In Vivo: Reversible Effects on Mouse Pancreatic β-Cells. PLoS Med. 2008, 5, e206. [Google Scholar] [CrossRef] [Green Version]
- Lahmann, C.; Kramer, H.B.; Ashcroft, F.M. Systemic Administration of Glibenclamide Fails to Achieve Therapeutic Levels in the Brain and Cerebrospinal Fluid of Rodents. PLoS ONE 2015, 10, 1–18. [Google Scholar] [CrossRef] [Green Version]
- Ashcroft, F.M.; Gribble, F.M. ATP-Sensitive K+ Channels and Insulin Secretion: Their Role in Health and Disease. Diabetologia 1999, 42, 903–919. [Google Scholar] [CrossRef] [Green Version]
- Sheykhzade, M.; Berg Nyborg, N.C. Mechanism of CGRP-Induced Relaxation in Rat Intramural Coronary Arteries. Br. J. Pharmacol. 2001, 132, 1235–1246. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Haanes, K.A.; Edvinsson, L. Hyperpolarization through ATP-Sensitive Potassium Channels; Relevance to Migraine Pathology. Brain 2020, 143, e13. [Google Scholar] [CrossRef]
- Al-Mahdi Al-Karagholi, M.; Olesen, J.; Ashina, M. Reply: Hyperpolarization through ATP-Sensitive Potassium Channels; Relevance to Migraine Pathology. Brain 2020, 143, E14. [Google Scholar] [CrossRef] [PubMed]
- Tsantoulas, C.; Lainez, S.; Wong, S.; Mehta, I.; Vilar, B.; McNaughton, P.A. Hyperpolarization-Activated Cyclic Nucleotide-Gated 2 (HCN2) Ion Channels Drive Pain in Mouse Models of Diabetic Neuropathy. Sci. Transl. Med. 2017, 9. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bernard Healey, S.A.; Scholtes, I.; Abrahams, M.; McNaughton, P.A.; Menon, D.K.; Lee, M.C. Role of Hyperpolarization-Activated Cyclic Nucleotide-Gated Ion Channels in Neuropathic Pain: A Proof-of-Concept Study of Ivabradine in Patients with Chronic Peripheral Neuropathic Pain. Pain Rep. 2021, 6, e967. [Google Scholar] [CrossRef] [PubMed]
- Young, G.T.; Emery, E.C.; Mooney, E.R.; Tsantoulas, C.; McNaughton, P.A. Inflammatory and Neuropathic Pain Are Rapidly Suppressed by Peripheral Block of Hyperpolarisation-Activated Cyclic Nucleotide-Gated Ion Channels. Pain 2014, 155, 1708–1719. [Google Scholar] [CrossRef]
- Tu, H.; Deng, L.; Sun, Q.; Yao, L.; Han, J.S.; Wan, Y. Hyperpolarization-Activated, Cyclic Nucleotide-Gated Cation Channels: Roles in the Differential Electrophysiological Properties of Rat Primary Afferent Neurons. J. Neurosci. Res. 2004, 76, 713–722. [Google Scholar] [CrossRef]
- Momin, A.; Cadiou, H.; Mason, A.; Mcnaughton, P.A. Role of the Hyperpolarization-Activated Current Ih in Somatosensory Neurons. J. Physiol. 2008, 586, 5911–5929. [Google Scholar] [CrossRef] [PubMed]
- Al-Karagholi, M.A.M.; Ghanizada, H.; Hansen, J.M.; Aghazadeh, S.; Skovgaard, L.T.; Olesen, J.; Ashina, M. Extracranial Activation of ATP-Sensitive Potassium Channels Induces Vasodilation without Nociceptive Effects. Cephalalgia 2019, 39, 1789–1797. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Yao, J.; Rong, M. Editorial: Role of Ion Channels in Pain. Front. Pharmacol. 2022, 13. [Google Scholar] [CrossRef] [PubMed]
- Perricone, S.C.; Humphrey, S.J.; Skaletzky, L.L.; Graham, B.E.; Zandt, R.A.; Zins, G.R. Synthesis and Diuretic Activity of Alkyl- and Arylguanidine Analogs of N,N’-Dicyclohexyl-4-Morpholinecarboxamidine in Rats and Dogs. J. Med. Chem. 1994, 37, 3693–3700. [Google Scholar] [CrossRef] [PubMed]
- Humphrey, S.J.; Smith, M.P.; Cimini, M.G.; Buchanan, L.V.; Gibson, J.K.; Khan, S.A.; Meisheri, K.D. Cardiovascular Effects of the K-ATP Channel Blocker U-37883A and Structurally Related Morpholinoguanidines. Methods Find. Exp. Clin. Pharmacol. 1996, 18, 247–260. [Google Scholar] [PubMed]
- Chutkow, W.A.; Pu, J.; Wheeler, M.T.; Wada, T.; Makielski, J.C.; Burant, C.F.; McNally, E.M. Episodic Coronary Artery Vasospasm and Hypertension Develop in the Absence of Sur2 K(ATP) Channels. J. Clin. Investig. 2002, 110, 203–208. [Google Scholar] [CrossRef]
- Kakkar, R.; Ye, B.; Stoller, D.A.; Smelley, M.; Shi, N.Q.; Galles, K.; Hadhazy, M.; Makielski, J.C.; McNally, E.M. Spontaneous Coronary Vasospasm in KATP Mutant Mice Arises from a Smooth Muscle-Extrinsic Process. Circ. Res. 2006, 98, 682–689. [Google Scholar] [CrossRef] [Green Version]
- Kalia, J.; Milescu, M.; Salvatierra, J.; Wagner, J.; Klint, J.K.; King, G.F.; Olivera, B.M.; Bosmans, F. From Foe to Friend: Using Animal Toxins to Investigate Ion Channel Function. J. Mol. Biol. 2015, 427, 158–175. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hutchings, C.J.; Colussi, P.; Clark, T.G. Ion Channels as Therapeutic Antibody Targets. MAbs 2019, 11, 265–296. [Google Scholar] [CrossRef]
- Gross, G.J.; Peart, J.N. KATP Channels and Myocardial Preconditioning: An Update. Am. J. Physiol. Hear. Circ. Physiol. 2003, 285, 921–930. [Google Scholar] [CrossRef] [Green Version]
- Antzelevitch, C.; Di Diego, J.M. Role of K+ Channel Activators in Cardiac Electrophysiology and Arrhythmias. Circulation 1992, 85, 1627–1629. [Google Scholar] [CrossRef] [Green Version]
- King, D.R.; Hardin, K.M.; Hoeker, G.S.; Poelzing, S. Re-Evaluating Methods Reporting Practices to Improve Reproducibility: An Analysis of Methodological Rigor for the Langendorff Whole-Heart Technique. Am. J. Physiol. Circ. Physiol. 2022. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
| Channel Subunit Composition | Tissue | References |
|---|---|---|
| Kir6.1/SUR1 | Retina | [49] |
| Nervous system | [46,48] | |
| Kir6.1/SUR2B | Vascular smooth muscle | [43,45,50,51,52] |
| Non-vascular smooth muscle | [48,53] | |
| Conduction system of the heart | [48,54] | |
| Kir6.2/SUR1 | Pancreatic β-cells | [52,55] |
| Arterial cardiac myocytes | [52,56] | |
| Nervous system | [48,52,57,58] | |
| Skeletal muscle | [48,59] | |
| Kir6.2/SUR2A | Ventricular myocytes | [54,60] |
| Skeletal muscle | [48,59] | |
| Kir6.2/SUR2B | Non-vascular smooth muscle | [53] |
| Nervous system | [48,57,61] | |
| Conduction system of the heart | [54,62] | |
| Skeletal muscle | [59] |
| Species | Endpoint | Headache Trigger mg/kg | Headache Trigger, umol/kg | Blocker mg/kg | Blocker, umol/kg | Ratio (Blocker/Trigger) | Effective Y/N/P |
|---|---|---|---|---|---|---|---|
| Rat | MMA diameter | Levcromakalim 0.025 mg/kg iv over 10 min | 0.087 | PNU-37883A 0.5 mg/kg i.v. over 10 min | 1.3 | 15 | P |
| Rat | MMA diameter | Levcromakalim 0.1 mg/kg iv over 20 min | 0.35 | Glibenclamide 20 mg/kg iv over 20 min | 40.5 | 116 | P |
| Rat | MMA diameter | Levcromakalim 0.1 mg/kg iv over 20 min | 0.35 | Glibenclamide 30 mg/kg iv over 20 min | 60.7 | 174 | Y |
| Human | MMA, STA, MCA circumference | Levcromakalim 0.014 mg/kg iv over 20 min | 0.049 | Glibenclamide 0.14 mg/kg p.o. | 0.3 | 5.8 | N |
| Rat | MMA diameter | CGRP 0.3 ug/kg iv bolus | 0.000079 | Glibenclamide 7 mg/kg iv over 20 min | 14.2 | 178,968 | P |
| Rat | MMA diameter | CGRP 0.3 ug/kg iv bolus | 0.000079 | Glibenclamide 30 mg/kg iv over 20 min | 60.7 | 767,004 | Y |
| Human | STA and RA diameter | CGRP 0.43 ug/kg iv over 20 min | 0.000011 | Glibenclamide 0.14 mg/kg p.o. | 0.3 | 24,972 # | N |
| Human | STA and RA diameter | CGRP 0.02 ug/kg/min i.v. | 0.0000053 | Glibenclamide 0.14 mg/kg p.o. | 0.3 | 53,690 | N |
| Human | MMA circumference | PACAP 200 picomol/kg over 20 min | 0.2 | Glibenclamide 0.14 mg/kg p.o. * | 0.3 | 1.4 | N |
| Human | Headache | Levcromakalim 0.014 mg/kg iv over 20 min | 0.049 | Glibenclamide 0.14 mg/kg p.o. | 0.3 | 5.8 | N/P |
| Mouse | Tactile hypersensitivity | Levcromakalim 1 mg/kg i.p $ | 3.5 | Glibenclamide 1 mg/kg i.p. | 2 | 0.6 | Y |
| Human | Headache | PACAP 200 picomol/kg over 20 min | 0.2 | Glibenclamide 0.14 mg/kg p.o. | 0.3 | 1.4 | N |
| Mouse | Tactile hypersensitivity | PACAP 0.2 ug/kg s.c. & | 0.000044 | Glibenclamide 1 mg/kg i.p. | 2 | 45,891 | P |
| Human | Headache | CGRP 0.43 ug/kg iv over 20 min | 0.000011 | Glibenclamide 0.14 mg/kg p.o. | 0.3 | 24,972 | N/P |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Clement, A.; Guo, S.; Jansen-Olesen, I.; Christensen, S.L. ATP-Sensitive Potassium Channels in Migraine: Translational Findings and Therapeutic Potential. Cells 2022, 11, 2406. https://doi.org/10.3390/cells11152406
Clement A, Guo S, Jansen-Olesen I, Christensen SL. ATP-Sensitive Potassium Channels in Migraine: Translational Findings and Therapeutic Potential. Cells. 2022; 11(15):2406. https://doi.org/10.3390/cells11152406
Chicago/Turabian StyleClement, Amalie, Song Guo, Inger Jansen-Olesen, and Sarah Louise Christensen. 2022. "ATP-Sensitive Potassium Channels in Migraine: Translational Findings and Therapeutic Potential" Cells 11, no. 15: 2406. https://doi.org/10.3390/cells11152406
APA StyleClement, A., Guo, S., Jansen-Olesen, I., & Christensen, S. L. (2022). ATP-Sensitive Potassium Channels in Migraine: Translational Findings and Therapeutic Potential. Cells, 11(15), 2406. https://doi.org/10.3390/cells11152406