
Taste Receptor Activation in Tracheal Brush Cells by Denatonium Modulates ENaC Channels via Ca2+, cAMP and ACh

 , ,
, ,  , and
, and {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Animals
2.2. Reagents
2.3. Ussing Chamber Experiments
2.4. Cloning
2.5. Calcium Imaging Experiments
2.6. Ablation of Sensory Nerves with Resiniferatoxin
2.7. Whole-Mount Staining
2.8. Experimental Design and Statistical Analysis
3. Results
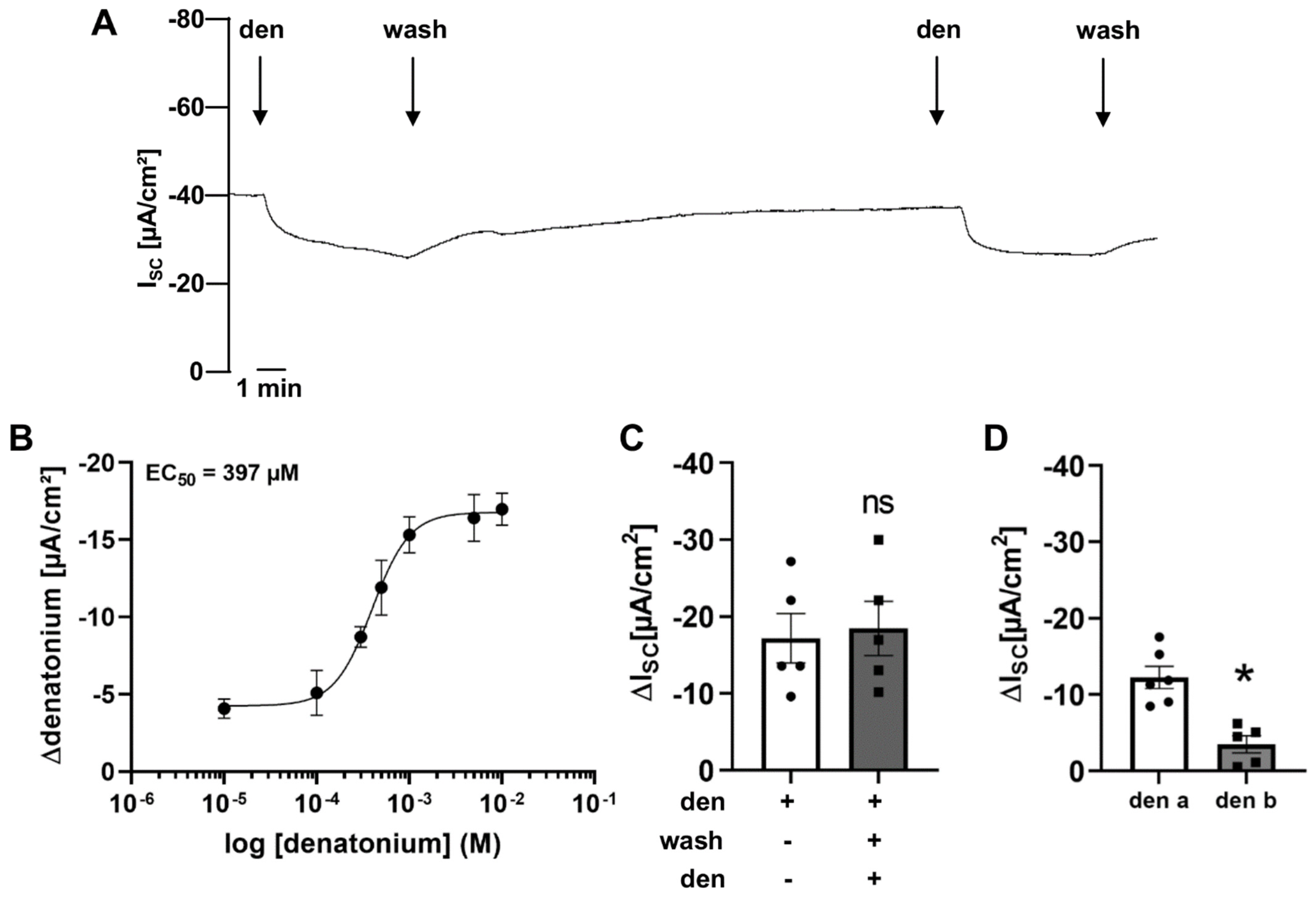
3.1. Denatonium Modulates Transepithelial Ion Transport in Wildtype Mice
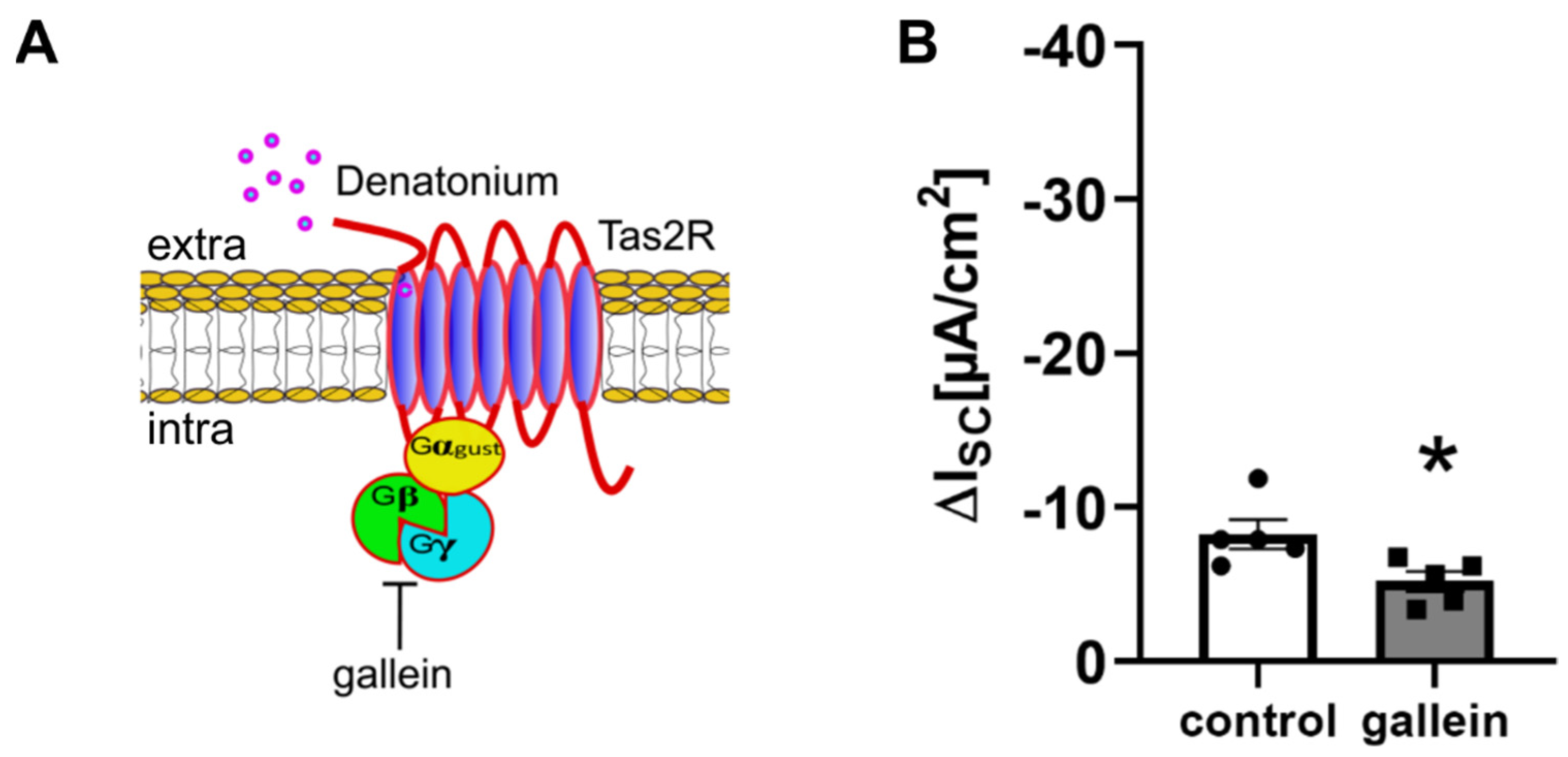
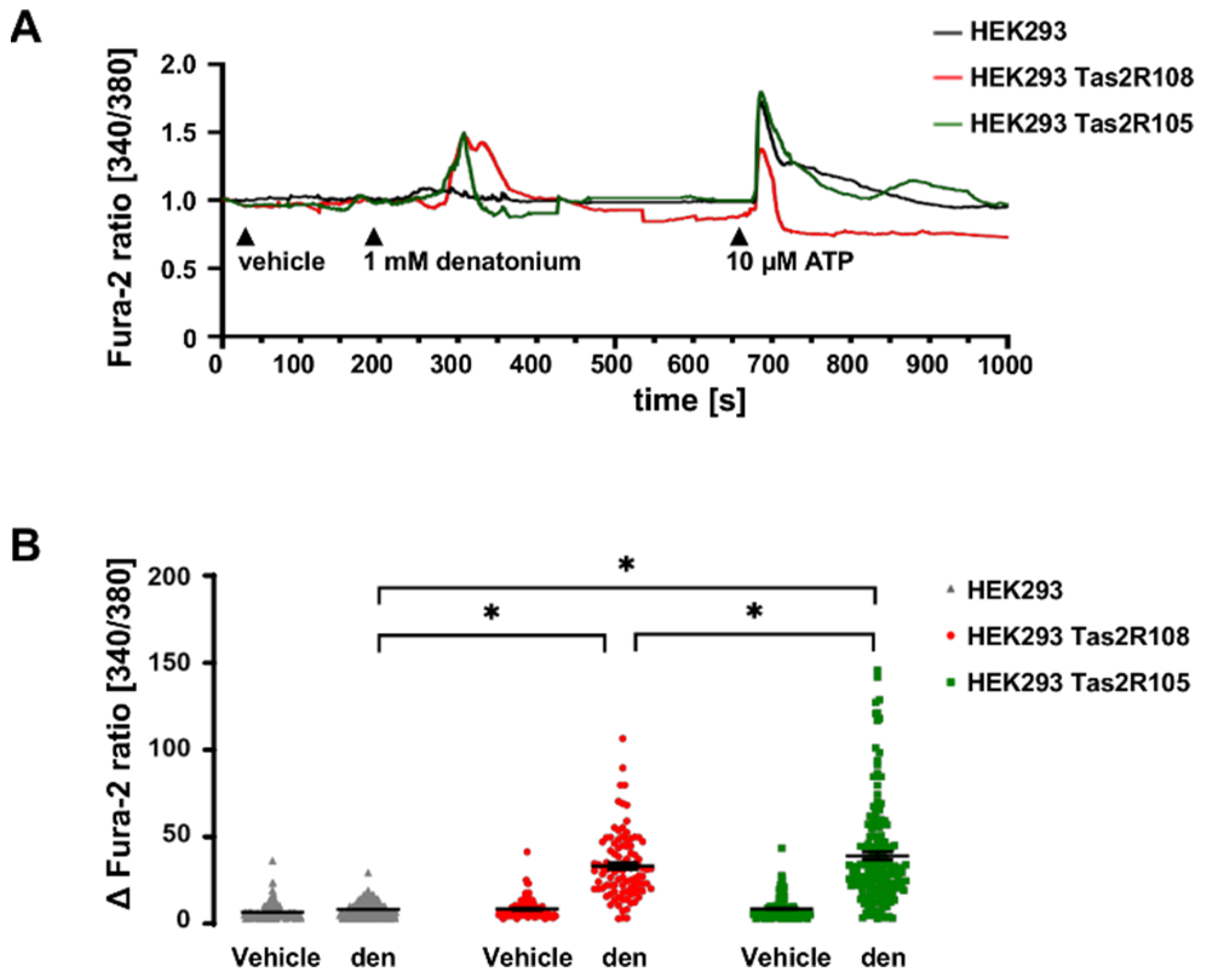
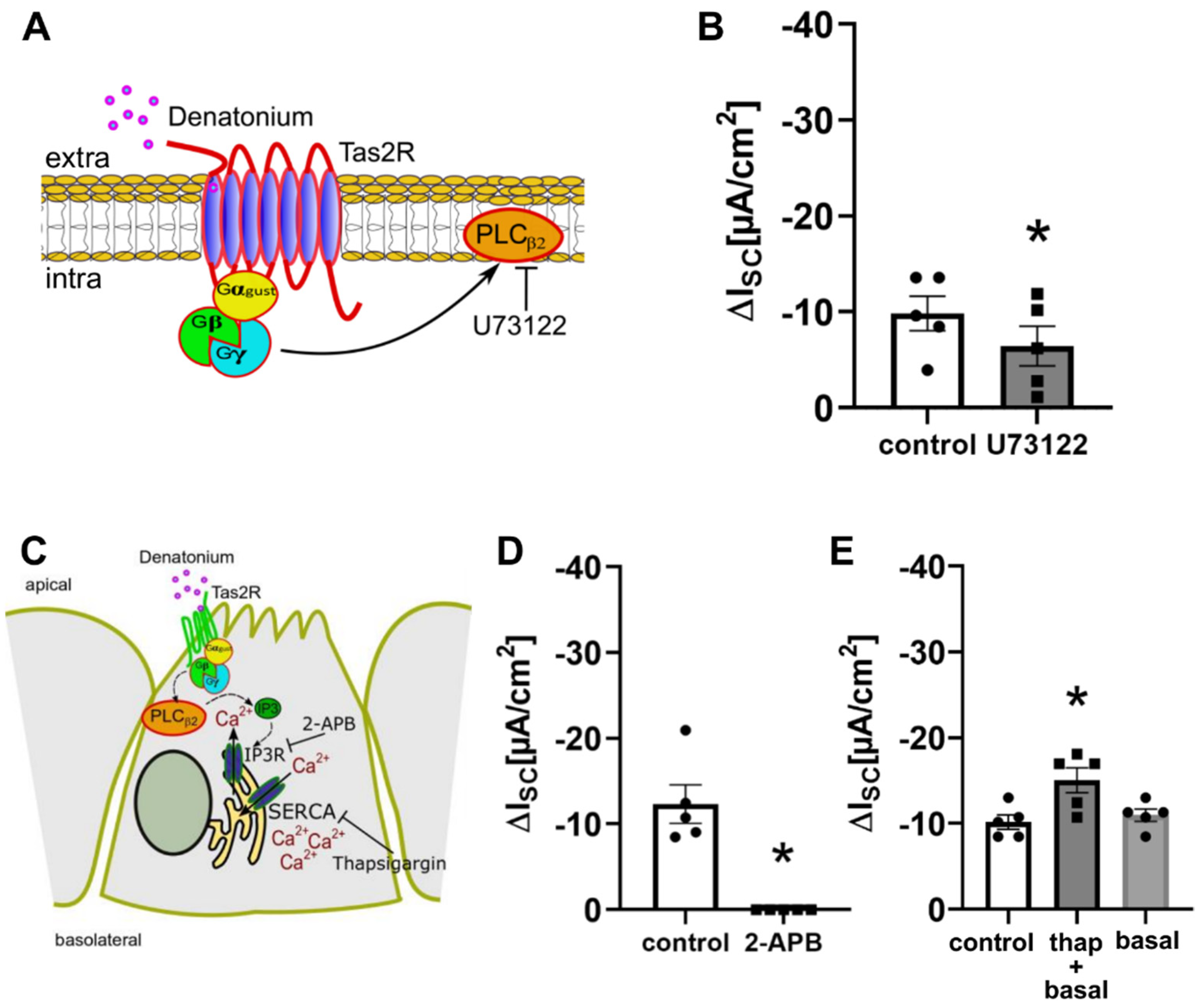
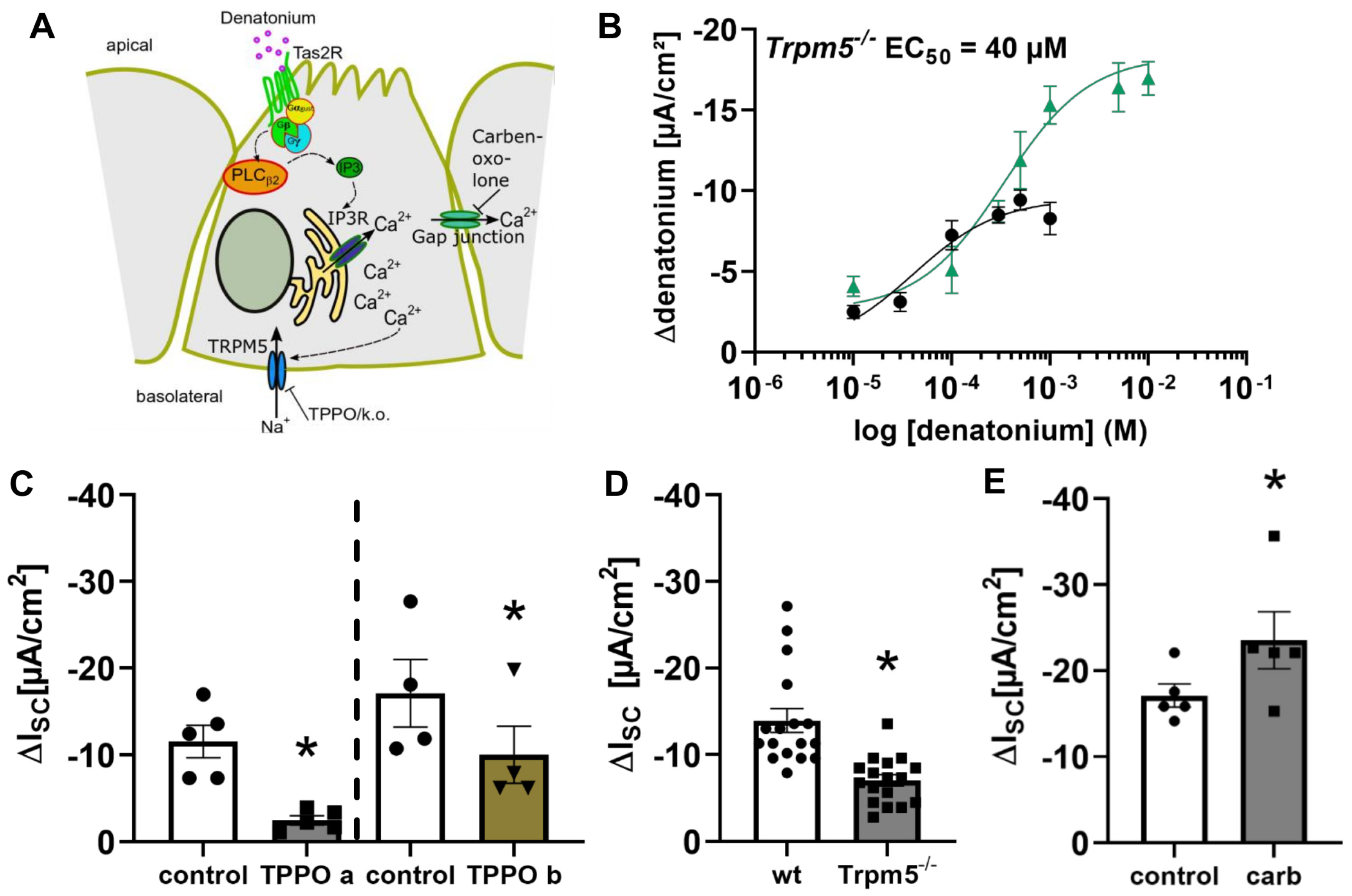
3.2. Denatonium Acts via the Canonical Bitter Taste Signalling Cascade
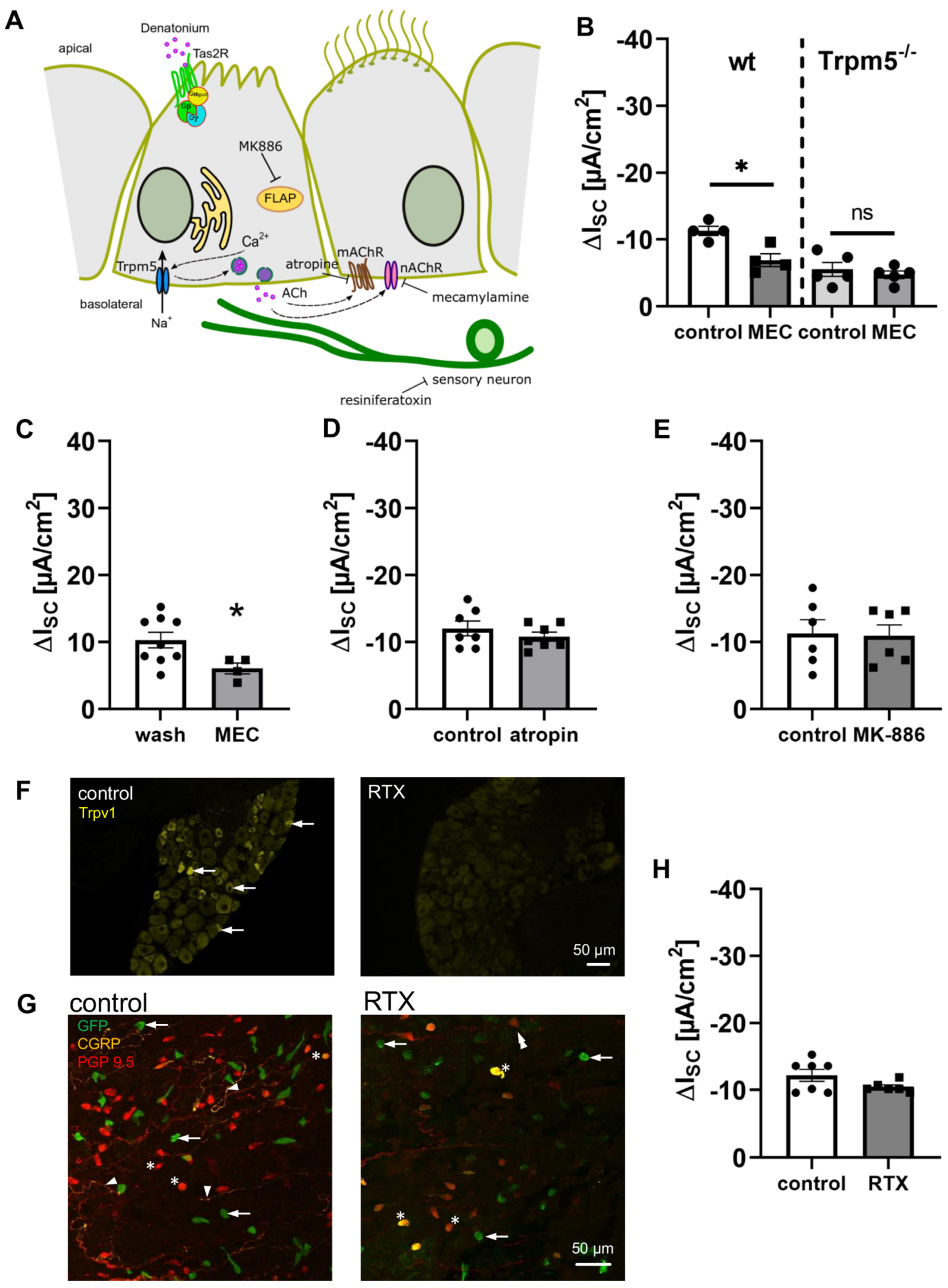
3.3. Activation of Brush Cells Leads to Cholinergic Paracrine Signalling
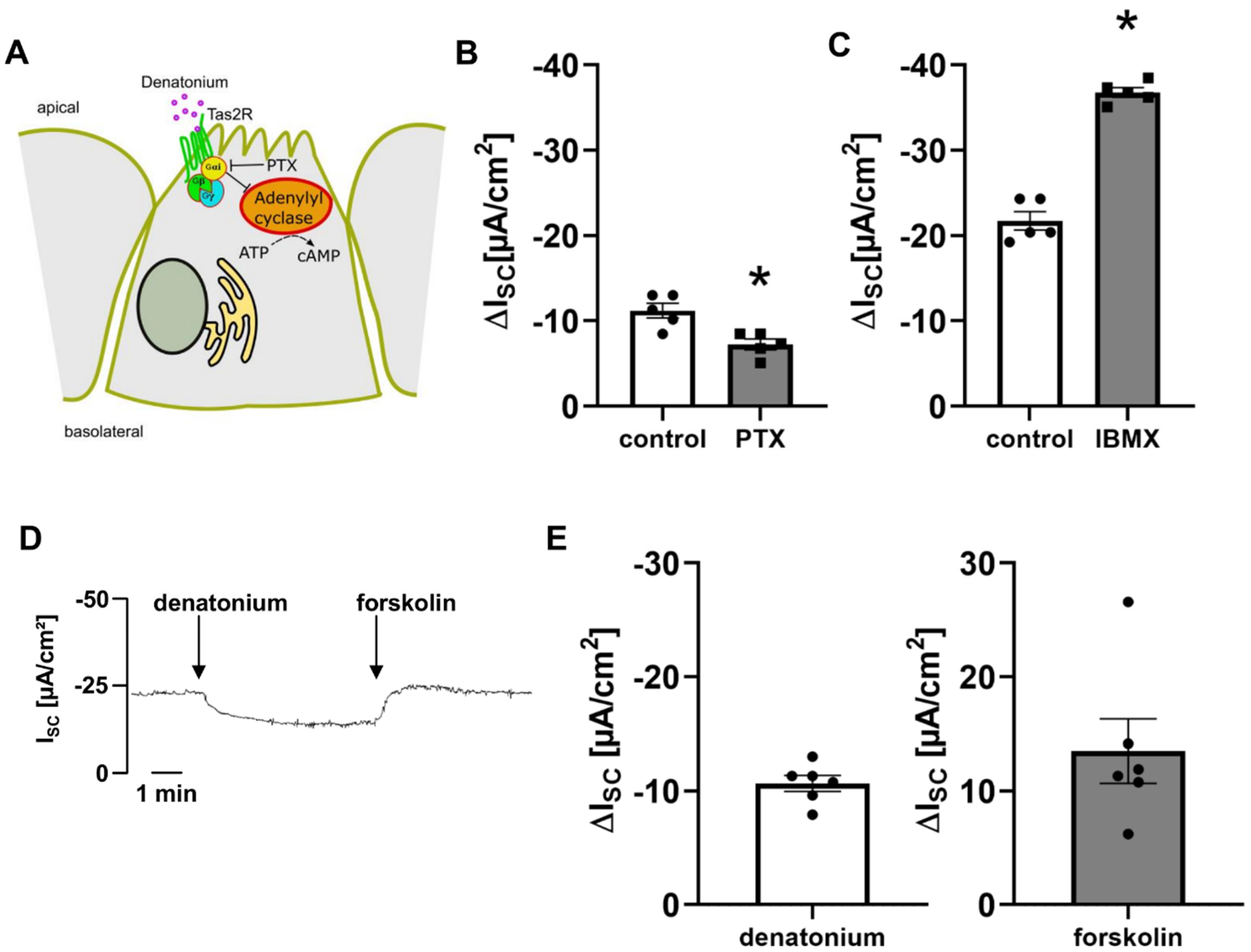
3.4. Bitter Taste Signalling Leads to an Inhibition of cAMP Signalling Pathways
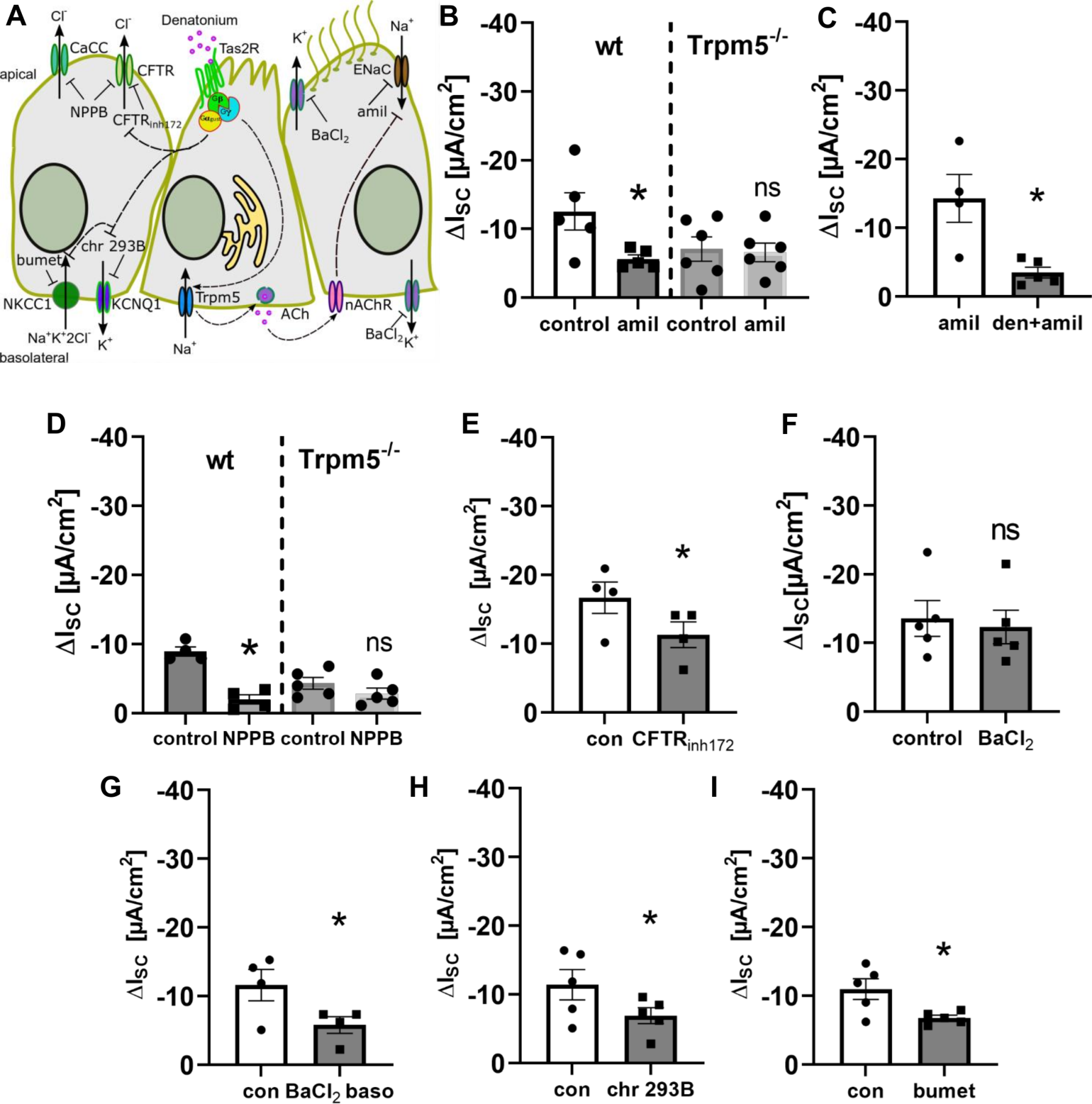
3.5. Bitter Taste Signalling Inhibits the Epithelial Sodium Channel
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Hollenhorst, M.I.; Richter, K.; Fronius, M. Ion transport by pulmonary epithelia. J. Biomed. Biotechnol. 2011, 2011, 174306. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Song, Y.; Namkung, W.; Nielson, D.W.; Lee, J.W.; Finkbeiner, W.E.; Verkman, A.S. Airway surface liquid depth measured in ex vivo fragments of pig and human trachea: Dependence on Na+ and Cl− channel function. Am. J. Physiol. Lung Cell. Mol. Physiol. 2009, 297, 1131–1140. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bartoszewski, R.; Matalon, S.; Collawn, J.F. Ion channels of the lung and their role in disease pathogenesis. Am. J. Physiol. Lung Cell. Mol. Physiol. 2017, 313, L859–L872. [Google Scholar] [CrossRef] [PubMed]
- Flagella, M.; Clarke, L.L.; Miller, M.L.; Erway, L.C.; Giannella, R.A.; Andringa, A.; Gawenis, L.R.; Kramer, J.; Duffy, J.J.; Doetschman, T.; et al. Mice lacking the basolateral Na-K-2Cl cotransporter have impaired epithelial chloride secretion and are profoundly deaf. J. Biol. Chem. 1999, 274, 26946–26955. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mall, M.; Wissner, A.; Schreiber, R.; Kuehr, J.; Seydewitz, H.H.; Brandis, M.; Greger, R.; Kunzelmann, K. Role of K(v)LQT1 in cyclic adenosine monophosphate-mediated Cl- secretion in human airway epithelia. Am. J. Respir. Cell Mol. Biol. 2000, 23, 283–289. [Google Scholar] [CrossRef] [Green Version]
- Dransfield, M.T.; Wilhelm, A.M.; Flanagan, B.; Courville, C.; Tidwell, S.L.; Raju, S.V.; Gaggar, A.; Steele, C.; Tang, L.P.; Liu, B.; et al. Acquired cystic fibrosis transmembrane conductance regulator dysfunction in the lower airways in COPD. Chest 2013, 144, 498–506. [Google Scholar] [CrossRef] [Green Version]
- Knowles, M.R.; Boucher, R.C. Mucus clearance as a primary innate defense mechanism for mammalian airways. J. Clin. Investig. 2002, 109, 571–577. [Google Scholar] [CrossRef]
- O’Sullivan, B.P.; Freedman, S.D. Cystic fibrosis. Lancet 2009, 373, 1891–1904. [Google Scholar] [CrossRef]
- Perniss, A.; Liu, S.; Boonen, B.; Keshavarz, M.; Ruppert, A.L.; Timm, T.; Pfeil, U.; Soultanova, A.; Kusumakshi, S.; Delventhal, L.; et al. Chemosensory Cell-Derived Acetylcholine Drives Tracheal Mucociliary Clearance in Response to Virulence-Associated Formyl Peptides. Immunity 2020, 52, 683–699.e11. [Google Scholar] [CrossRef]
- Hollenhorst, M.I.; Jurastow, I.; Nandigama, R.; Appenzeller, S.; Li, L.; Vogel, J.; Wiederhold, S.; Althaus, M.; Empting, M.; Altmüller, J.; et al. Tracheal brush cells release acetylcholine in response to bitter tastants for paracrine and autocrine signaling. FASEB J. 2020, 34, 316–332. [Google Scholar] [CrossRef] [Green Version]
- Lee, R.J.; Xiong, G.; Kofonow, J.M.; Chen, B.; Lysenko, A.; Jiang, P.; Abraham, V.; Doghramji, L.; Adappa, N.D.; Palmer, J.N.; et al. T2R38 taste receptor polymorphisms underlie susceptibility to upper respiratory infection. J. Clin. Investig. 2012, 122, 4145–4159. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, R.J.; Kofonow, J.M.; Rosen, P.L.; Siebert, A.P.; Chen, B.; Doghramji, L.; Xiong, G.; Adappa, N.D.; Palmer, J.N.; Kennedy, D.W.; et al. Bitter and sweet taste receptors regulate human upper respiratory innate immunity. J. Clin. Investig. 2014, 124, 1393–1405. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Saunders, C.J.; Christensen, M.; Finger, T.E.; Tizzano, M. Cholinergic neurotransmission links solitary chemosensory cells to nasal inflammation. Proc. Natl. Acad. Sci. USA 2014, 111, 6075–6080. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hollenhorst, M.I.; Nandigama, R.; Evers, S.B.; Gamayun, I.; Abdel Wadood, N.; Salah, A.; Pieper, M.; Wyatt, A.; Stukalov, A.; Gebhardt, A.; et al. Bitter taste signaling in tracheal epithelial brush cells elicits innate immune responses to bacterial infection. J. Clin. Investig. 2022, 132, e150951. [Google Scholar] [CrossRef]
- Hofmann, T.; Chubanov, V.; Gudermann, T.; Montell, C. TRPM5 Is a Voltage-Modulated and Ca2+-Activated Monovalent Selective Cation Channel Thomas. Curr. Biol. 2003, 13, 1153–1158. [Google Scholar] [CrossRef] [Green Version]
- Perniss, A.; Latz, A.; Boseva, I.; Papadakis, T.; Dames, C.; Meisel, C.; Meisel, A.; Scholze, P.; Kummer, W.; Krasteva-Christ, G. Acute nicotine administration stimulates ciliary activity via α3β4 nAChR in the mouse trachea. Int. Immunopharmacol. 2020, 84, 106496. [Google Scholar] [CrossRef]
- Hollenhorst, M.I.; Lips, K.S.; Wolff, M.; Wess, J.; Gerbig, S.; Takats, Z.; Kummer, W.; Fronius, M. Luminal cholinergic signalling in airway lining fluid: A novel mechanism for activating chloride secretion via Ca2+-dependent Cl− and K+ channels. Br. J. Pharmacol. 2012, 166, 1388–1402. [Google Scholar] [CrossRef] [Green Version]
- Kumar, P.; Scholze, P.; Fronius, M.; Krasteva-Christ, G.; Hollenhorst, M.I. Nicotine stimulates ion transport via metabotropic β4 subunit containing nicotinic acetylcholine receptors. Br. J. Pharmacol. 2020, 177, 5595–5608. [Google Scholar] [CrossRef]
- Kohanski, M.A.; Brown, L.; Orr, M.; Tan, L.H.; Adappa, N.D.; Palmer, J.N.; Rubenstein, R.C.; Cohen, N.A. Bitter taste receptor agonists regulate epithelial two-pore potassium channels via cAMP signaling. Respir. Res. 2021, 22, 31. [Google Scholar] [CrossRef]
- Damak, S.; Rong, M.; Yasumatsu, K.; Kokrashvili, Z.; Pérez, C.A.; Shigemura, N.; Yoshida, R.; Mosinger, B., Jr.; Glendinning, J.I.; Ninomiya, Y.; et al. Trpm5 Null Mice Respond to Bitter, Sweet, and Umami Compounds. Chem. Senses 2006, 31, 253–264. [Google Scholar] [CrossRef] [Green Version]
- Wyatt, A.; Wartenberg, P.; Candlish, M.; Krasteva-Christ, G.; Flockerzi, V.; Boehm, U. Genetic strategies to analyze primary TRP channel-expressing cells in mice. Cell Calcium 2017, 67, 91–104. [Google Scholar] [CrossRef] [PubMed]
- Wen, S.; Götze, I.N.; Mai, O.; Schauer, C.; Leinders-Zufall, T.; Boehm, U. Genetic identification of GnRH receptor neurons: A new model for studying neural circuits underlying reproductive physiology in the mouse brain. Endocrinology 2011, 152, 1515–1526. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kusumakshi, S.; Voigt, A.; Hübner, S.; Hermans-Borgmeyer, I.; Ortalli, A.; Pyrski, M.; Dörr, J.; Zufall, F.; Flockerzi, V.; Meyerhof, W.; et al. A binary genetic approach to characterize TRPM5 cells in mice. Chem. Senses 2015, 40, 413–425. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bankova, L.G.; Dwyer, D.F.; Yoshimoto, E.; Ualiyeva, S.; McGinty, J.W.; Raff, H.; von Moltke, J.; Kanaoka, Y.; Frank Austen, K.; Barrett, N.A. The cysteinyl leukotriene 3 receptor regulates expansion of IL-25–producing airway brush cells leading to type 2 inflammation. Sci. Immunol. 2018, 3, eaat9453. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Montoro, D.T.; Haber, A.L.; Biton, M.; Vinarsky, V.; Lin, B.; Birket, S.E.; Yuan, F.; Chen, S.; Leung, H.M.; Villoria, J.; et al. A revised airway epithelial hierarchy includes CFTR-expressing ionocytes. Nature 2018, 560, 319–324. [Google Scholar] [CrossRef] [PubMed]
- Plasschaert, L.W.; Žilionis, R.; Choo-wing, R.; Savova, V.; Knehr, J.; Roma, G.; Klein, A.M.; Jaffe, A.B. A single-cell atlas of the airway epithelium reveals the CFTR-rich pulmonary ionocyte. Nature 2018, 560, 377–381. [Google Scholar] [CrossRef]
- Lehmann, D.M.; Seneviratne, A.M.P.B.; Smrcka, A. V Small molecule disruption of G protein bg subunit signaling inhibits neutrophil chemotaxis and inflammation. Mol. Pharmacol. 2008, 73, 410–418. [Google Scholar] [CrossRef] [Green Version]
- Hamada, T.; Liou, S.Y.; Fukushima, T.; Maruyama, T.; Watanabe, S.; Mikoshiba, K.; Ishida, N. The role of inositol trisphosphate-induced Ca2+ release from IP3- receptor in the rat suprachiasmatic nucleus on circadian entrainment mechanism. Neurosci. Lett. 1999, 263, 125–128. [Google Scholar] [CrossRef]
- Lievremont, J.P.; Bird, G.S.; Putney, J.W. Mechanism of inhibition of TRPC cation channels by 2- aminoethoxydiphenylborane. Mol. Pharmacol. 2005, 68, 758–762. [Google Scholar] [CrossRef]
- DeHaven, W.I.; Smyth, J.T.; Boyles, R.R.; Bird, G.S.; Putney, J.W. Complex actions of 2-aminoethyldiphenyl borate on store-operated calcium entry. J. Biol. Chem. 2008, 283, 19265–19273. [Google Scholar] [CrossRef] [Green Version]
- Thastrup, O.; Cullen, P.J.; Drobak, B.K.; Hanley, M.R.; Dawson, A.P. Thapsigargin, a tumor promoter, discharges intracellular Ca2+ stores by specific inhibition of the endoplasmic reticulum Ca2+-ATPase. Proc. Natl. Acad. Sci. USA 1990, 87, 2466–2470. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Antigny, F.; Jousset, H.; König, S.; Frieden, M. Thapsigargin activates Ca2+ entry both by store-dependent, STIM1/Orai1-mediated, and store-independent, TRPC3/PLC/PKC-mediated pathways in human endothelial cells. Cell Calcium 2011, 49, 115–127. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hodeify, R.; Selvaraj, S.; Wen, J.; Arredouani, A.; Hubrack, S.; Dib, M.; Al-Thani, S.N.; McGraw, T.; Machaca, K. A STIM1-dependent “trafficking trap” mechanism regulates Orai1 plasma membrane residence and Ca2+ influx levels. J. Cell Sci. 2015, 128, 3143–3154. [Google Scholar] [CrossRef] [Green Version]
- Baumann, K. Signalling: Stop refilling (Ca2+ stores). Nat. Rev. Mol. Cell Biol. 2012, 13, 277. [Google Scholar] [CrossRef]
- Palmer, R.K.; Atwal, K.; Bakaj, I.; Carlucci-Derbyshire, S.; Buber, M.T.; Cerne, R.; Cortés, R.Y.; Devantier, H.R.; Jorgensen, V.; Pawlyk, A.; et al. Triphenylphosphine Oxide Is a Potent and Selective Inhibitor of the Transient Receptor Potential Melastatin-5 Ion Channel. Assay Drug Dev. Technol. 2010, 8, 703–713. [Google Scholar] [CrossRef]
- Ualiyeva, S.; Hallen, N.; Kanaoka, Y.; Ledderose, C.; Matsumoto, I.; Junger, W.G.; Barrett, N.A.; Bankova, L.G. Airway brush cells generate cysteinyl leukotrienes through the ATP sensor P2Y2. Sci. Immunol. 2020, 5, eaax7224. [Google Scholar] [CrossRef]
- Krasteva, G.; Canning, B.J.; Hartmann, P.; Veres, T.Z.; Papadakis, T.; Muhlfeld, C.; Schliecker, K.; Tallini, Y.N.; Braun, A.; Hackstein, H.; et al. Cholinergic chemosensory cells in the trachea regulate breathing. Proc. Natl. Acad. Sci. USA 2011, 108, 9478–9483. [Google Scholar] [CrossRef] [Green Version]
- Baral, P.; Umans, B.D.; Li, L.; Wallrapp, A.; Bist, M.; Kirschbaum, T.; Wei, Y.; Zhou, Y.; Kuchroo, V.K.; Burkett, P.R.; et al. Nociceptor sensory neurons suppress neutrophil and γδ T cell responses in bacterial lung infections and lethal pneumonia. Nat. Med. 2018, 24, 417–426. [Google Scholar] [CrossRef]
- Chou, M.Z.; Mtui, T.; Gao, Y.D.; Kohler, M.; Middleton, R.E. Resiniferatoxin Binds to the Capsaicin Receptor (TRPV1) near the Extracellular Side of the S4 Transmembrane Domain. Biochemistry 2004, 43, 2501–2511. [Google Scholar] [CrossRef]
- Katada, T. The inhibitory G protein Gi identified as pertussis toxin-catalyzed ADP-ribosylation. Biol. Pharm. Bull. 2012, 35, 2103–2111. [Google Scholar] [CrossRef] [Green Version]
- Pleli, T.; Mondorf, A.; Ferreiros, N.; Thomas, D.; Dvorak, K.; Biondi, R.M.; Heringdorf, D.M.Z.; Zeuzem, S.; Geisslinger, G.; Zimmermann, H.; et al. Activation of Adenylyl Cyclase Causes Stimulation of Adenosine Receptors. Cell. Physiol. Biochem. 2018, 45, 2516–2528. [Google Scholar] [CrossRef] [PubMed]
- Clapp, T.R.; Trubey, K.R.; Vandenbeuch, A.; Stone, L.M.; Margolskee, R.F.; Chaudhari, N.; Kinnamon, S.C. Tonic activity of Galpha-gustducin regulates taste cell responsivity. FEBS Lett. 2008, 582, 3783–3787. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Krasteva, G.; Canning, B.J.; Papadakis, T.; Kummer, W. Cholinergic brush cells in the trachea mediate respiratory responses to quorum sensing molecules. Life Sci. 2012, 91, 992–996. [Google Scholar] [CrossRef] [PubMed]
- Kinnamon, S.C. Taste receptor signalling—from tongues to lungs. Acta Physiol. 2012, 204, 158–168. [Google Scholar] [CrossRef]
- Chandrashekar, J.; Mueller, K.L.; Hoon, M.A.; Adler, E.; Feng, L.; Guo, W.; Zuker, C.S.; Ryba, N.J.P. T2Rs function as bitter taste receptors. Cell 2000, 100, 703–711. [Google Scholar] [CrossRef] [Green Version]
- Lossow, K.; Hübner, S.; Roudnitzky, N.; Slack, J.P.; Pollastro, F.; Behrens, M.; Meyerhof, W. Comprehensive Analysis of Mouse Bitter Taste Receptors Reveals Different Molecular Receptive Ranges for Orthologous Receptors in Mice and Humans. J. Biol. Chem. 2016, 291, 15358–15377. [Google Scholar] [CrossRef] [Green Version]
- Meyerhof, W.; Batram, C.; Kuhn, C.; Brockhoff, A.; Chudoba, E.; Bufe, B.; Appendino, G.; Behrens, M. The molecular receptive ranges of human TAS2R bitter taste receptors. Chem. Senses 2009, 35, 157–170. [Google Scholar] [CrossRef]
- Prawitt, D.; Monteilh-Zoller, M.K.; Brixel, L.; Spangenberg, C.; Zabel, B.; Fleig, A.; Penner, R. TRPM5 is a transient Ca2+-activated cation channel responding to rapid changes in [Ca2+]i. Proc. Natl. Acad. Sci. USA 2003, 100, 15166–15171. [Google Scholar] [CrossRef] [Green Version]
- Nadjsombati, M.S.; McGinty, J.W.; Lyons-Cohen, M.R.; Jaffe, J.B.; DiPeso, L.; Schneider, C.; Miller, C.N.; Pollack, J.L.; Nagana Gowda, G.A.; Fontana, M.F.; et al. Detection of Succinate by Intestinal Tuft Cells Triggers a Type 2 Innate Immune Circuit. Immunity 2018, 49, 33–41. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shah, A.S.; Ben-Shahar, Y.; Moninger, T.O.; Kline, J.N.; Welsh, M.J. Motile Cilia of Human Airway Epithelia Are Chemosensory. Science 2009, 325, 1131–1134. [Google Scholar] [CrossRef] [Green Version]
- Büsst, C.J. Blood pressure regulation via the epithelial sodium channel: From gene to kidney and beyond. Clin. Exp. Pharmacol. Physiol. 2013, 40, 495–503. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hobbs, C.A.; Da Tan, C.; Tarran, R. Does epithelial sodium channel hyperactivity contribute to cystic fibrosis lung disease? J. Physiol. 2013, 591, 4377–4387. [Google Scholar] [CrossRef] [PubMed]
- Joo, N.S.; Krouse, M.E.; Choi, J.Y.; Cho, H.J.; Wine, J.J. Inhibition of airway surface fluid absorption by cholinergic stimulation. Sci. Rep. 2016, 6, 20735. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gupta, A.; Anderson, H.; Buo, A.M.; Moorer, M.C.; Ren, M.; Stains, J.P. Communication of cAMP by connexin43 gap junctions regulates osteoblast signaling and gene expression. Cell. Signal. 2016, 28, 1048–1057. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, Y.J.; Lee, J.S.; Kim, H.; Jang, J.H.; Choung, Y.H. Gap junction-mediated intercellular communication of cAMP prevents CDDP-induced ototoxicity via cAMP/PKA/CREB pathway. Int. J. Mol. Sci. 2021, 22, 6327. [Google Scholar] [CrossRef] [PubMed]
- Xu, J.; Yang, G.; Li, T.; Liu, L. Myoendothelial gap junctions mediate regulation of angiopoietin-2-induced vascular hyporeactivity after hypoxia through connexin 43-gated cAMP transfer. Am. J. Physiol. Lung Cell Physiol. 2017, 313, C262–C273. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, L.M.; Rinke, R.; Korbmacher, C. Stimulation of the epithelial sodium channel (ENaC) by cAMP involves putative ERK phosphorylation sites in the C termini of the channel’s β- and γ-subunit. J. Biol. Chem. 2006, 281, 9859–9868. [Google Scholar] [CrossRef] [Green Version]
- Butterworth, M.B.; Edinger, R.S.; Johnson, J.P.; Frizzell, R.A. Acute ENaC stimulation by cAMP in a kidney cell line is mediated by exocytic insertion from a recycling channel pool. J. Gen. Physiol. 2005, 125, 81–101. [Google Scholar] [CrossRef] [Green Version]
- Liang, J.; Chen, F.; Gu, F.; Liu, X.; Li, F.; Du, D. Expression and functional activity of bitter taste receptors in primary renal tubular epithelial cells and M-1 cells. Mol. Cell. Biochem. 2017, 428, 193–202. [Google Scholar] [CrossRef]
- Maouche, K.; Medjber, K.; Zahm, J.-M.; Delavoie, F.; Terryn, C.; Coraux, C.; Pons, S.; Cloez-Tayarani, I.; Maskos, U.; Birembaut, P.; et al. Contribution of 7 nicotinic receptor to airway epithelium dysfunction under nicotine exposure. Proc. Natl. Acad. Sci. USA 2013, 110, 4099–4104. [Google Scholar] [CrossRef] [Green Version]
- Grahammer, F.; Warth, R.; Barhanin, J.; Bleich, M.; Hug, M.J. The Small Conductance K+ Channel, KCNQ1. Expression, function, and subunit composition in murine trachea. J. Biol. Chem. 2001, 276, 42268–42275. [Google Scholar] [CrossRef] [PubMed] [Green Version]
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hollenhorst, M.I.; Kumar, P.; Zimmer, M.; Salah, A.; Maxeiner, S.; Elhawy, M.I.; Evers, S.B.; Flockerzi, V.; Gudermann, T.; Chubanov, V.; et al. Taste Receptor Activation in Tracheal Brush Cells by Denatonium Modulates ENaC Channels via Ca2+, cAMP and ACh. Cells 2022, 11, 2411. https://doi.org/10.3390/cells11152411
Hollenhorst MI, Kumar P, Zimmer M, Salah A, Maxeiner S, Elhawy MI, Evers SB, Flockerzi V, Gudermann T, Chubanov V, et al. Taste Receptor Activation in Tracheal Brush Cells by Denatonium Modulates ENaC Channels via Ca2+, cAMP and ACh. Cells. 2022; 11(15):2411. https://doi.org/10.3390/cells11152411
Chicago/Turabian StyleHollenhorst, Monika I., Praveen Kumar, Maxim Zimmer, Alaa Salah, Stephan Maxeiner, Mohamed Ibrahem Elhawy, Saskia B. Evers, Veit Flockerzi, Thomas Gudermann, Vladimir Chubanov, and et al. 2022. "Taste Receptor Activation in Tracheal Brush Cells by Denatonium Modulates ENaC Channels via Ca2+, cAMP and ACh" Cells 11, no. 15: 2411. https://doi.org/10.3390/cells11152411
APA StyleHollenhorst, M. I., Kumar, P., Zimmer, M., Salah, A., Maxeiner, S., Elhawy, M. I., Evers, S. B., Flockerzi, V., Gudermann, T., Chubanov, V., Boehm, U., & Krasteva-Christ, G. (2022). Taste Receptor Activation in Tracheal Brush Cells by Denatonium Modulates ENaC Channels via Ca2+, cAMP and ACh. Cells, 11(15), 2411. https://doi.org/10.3390/cells11152411