
Crosstalk between Biomolecular Condensates and Proteostasis


{kind=link}
{kind=link}
Abstract
:1. Crosstalk between Biomolecular Condensates and Proteostasis
2. An Emerging Family of Ubiquitin Condensates
2.1. The Ubiquitin-Proteasome System
2.2. Ubiquitin-Binding Shuttle Proteins Drive Formation of Nuclear Condensates
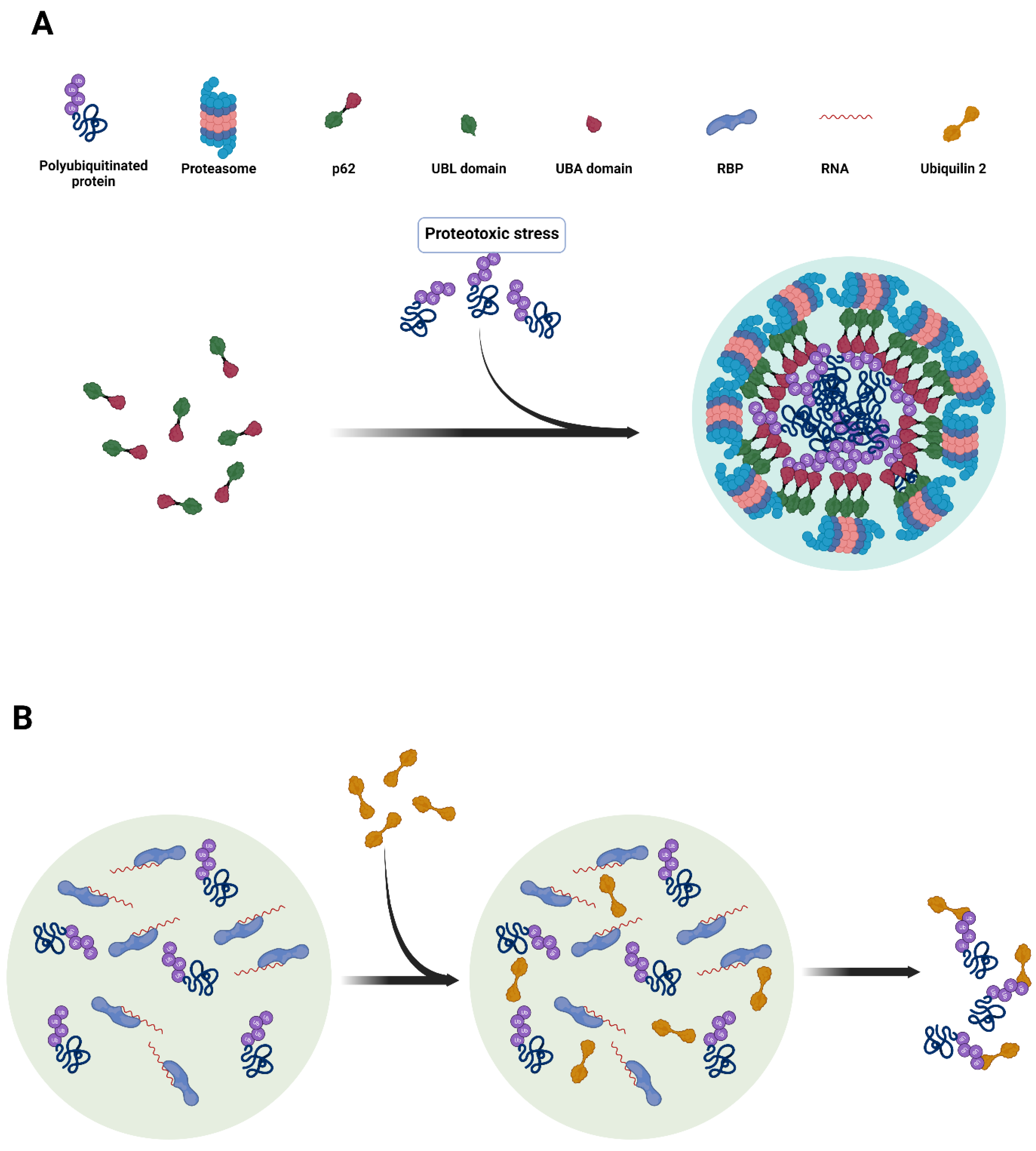
2.3. Additional Aspects of Multivalent Interactions in Ubiquitin Condensates
3. Molecular Mechanisms of Proteostasis Converge on Cytoplasmic Stress Granules
3.1. Stress Granules and the Integrated Stress Response
3.2. Proteostasis Factors Regulate SG Dynamics
3.3. Ubiquitin Signaling in SGs
3.4. Linking SUMO and StUbL to SGs
4. Stress Granules and Neuronal Inclusions in Amyotrophic Lateral Sclerosis
4.1. ALS and Cellular Proteostasis
4.2. Aberrant SG Condensation May Lead to ALS Inclusion Formation
4.3. Central Proteostasis Pathways Regulate ALS Pathogenesis
5. Summary
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Hetz, C. Adapting the proteostasis capacity to sustain brain healthspan. Cell 2021, 184, 1545–1560. [Google Scholar] [CrossRef] [PubMed]
- Hipp, M.S.; Kasturi, P.; Hartl, F.U. The proteostasis network and its decline in ageing. Nat. Rev. Mol. Cell Biol. 2019, 20, 421–435. [Google Scholar] [CrossRef] [PubMed]
- Ciryam, P.; Tartaglia, G.G.; Morimoto, R.I.; Dobson, C.M.; Vendruscolo, M. Widespread Aggregation and Neurodegenerative Diseases Are Associated with Supersaturated Proteins. Cell Rep. 2013, 5, 781–790. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ciryam, P.; Lambert-Smith, I.A.; Bean, D.M.; Freer, R.; Cid, F.; Tartaglia, G.G.; Saunders, D.N.; Wilson, M.R.; Oliver, S.G.; Morimoto, R.I.; et al. Spinal motor neuron protein supersaturation patterns are associated with inclusion body formation in ALS. Proc. Natl. Acad. Sci. USA 2017, 114, E3935–E3943. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yerbury, J.J.; Ooi, L.; Blair, I.P.; Ciryam, P.; Dobson, C.M.; Vendruscolo, M. The metastability of the proteome of spinal motor neurons underlies their selective vulnerability in ALS. Neurosci. Lett. 2019, 704, 89–94. [Google Scholar] [CrossRef] [PubMed]
- Tartaglia, G.G.; Vendruscolo, M. Correlation between mRNA expression levels and protein aggregation propensities in subcellular localisations. Mol. BioSyst. 2009, 5, 1873–1876. [Google Scholar] [CrossRef] [PubMed]
- Tartaglia, G.G.; Pechmann, S.; Dobson, C.M.; Vendruscolo, M. Life on the edge: A link between gene expression levels and aggregation rates of human proteins. Trends Biochem. Sci. 2007, 32, 204–206. [Google Scholar] [CrossRef]
- Tartaglia, G.G.; Vendruscolo, M. The Zyggregator method for predicting protein aggregation propensities. Chem. Soc. Rev. 2008, 37, 1395–1401. [Google Scholar] [CrossRef]
- Balchin, D.; Hayer-Hartl, M.; Hartl, F.U. In vivo aspects of protein folding and quality control. Science 2016, 353, aac4354. [Google Scholar] [CrossRef]
- Hartl, F.U.; Bracher, A.; Hayer-Hartl, M. Molecular chaperones in protein folding and proteostasis. Nature 2011, 475, 324–332. [Google Scholar] [CrossRef]
- Lyon, A.S.; Peeples, W.B.; Rosen, M.K. A framework for understanding the functions of biomolecular condensates across scales. Nat. Rev. Mol. Cell Biol. 2020, 22, 215–235. [Google Scholar] [CrossRef]
- Banani, S.F.; Lee, H.O.; Hyman, A.A.; Rosen, M.K. Biomolecular condensates: Organizers of cellular biochemistry. Nat. Rev. Mol. Cell Biol. 2017, 18, 285–298. [Google Scholar] [CrossRef]
- Shin, Y.; Brangwynne, C.P. Liquid phase condensation in cell physiology and disease. Science 2017, 357, eaaf4382. [Google Scholar] [CrossRef] [Green Version]
- Kwon, Y.T.; Ciechanover, A. The Ubiquitin Code in the Ubiquitin-Proteasome System and Autophagy. Trends Biochem. Sci. 2017, 42, 873–886. [Google Scholar] [CrossRef]
- Dikic, I. Proteasomal and Autophagic Degradation Systems. Annu. Rev. Biochem. 2017, 86, 193–224. [Google Scholar] [CrossRef]
- Shaid, S.; Brandts, C.H.; Serve, H.; Dikic, I. Ubiquitination and selective autophagy. Cell Death Differ. 2012, 20, 21–30. [Google Scholar] [CrossRef]
- Komander, D.; Rape, M. The ubiquitin code. Annu. Rev. Biochem. 2012, 10, 203–229. [Google Scholar] [CrossRef] [Green Version]
- Zientara-Rytter, K.; Subramani, S. The Roles of Ubiquitin-Binding Protein Shuttles in the Degradative Fate of Ubiquitinated Proteins in the Ubiquitin-Proteasome System and Autophagy. Cells 2019, 8, 40. [Google Scholar] [CrossRef] [Green Version]
- Fu, A.; Cohen-Kaplan, V.; Avni, N.; Livneh, I.; Ciechanover, A. p62-containing, proteolytically active nuclear condensates, increase the efficiency of the ubiquitin–proteasome system. Proc. Natl. Acad. Sci. USA 2021, 118, e2107321118. [Google Scholar] [CrossRef]
- Dao, T.P.; Castañeda, C.A. Ubiquitin-Modulated Phase Separation of Shuttle Proteins: Does Condensate Formation Promote Protein Degradation? Bioessays 2020, 42, e2000036. [Google Scholar] [CrossRef]
- Ciuffa, R.; Lamark, T.; Tarafder, A.K.; Guesdon, A.; Rybina, S.; Hagen, W.J.; Johansen, T.; Sachse, C. The Selective Autophagy Receptor p62 Forms a Flexible Filamentous Helical Scaffold. Cell Rep. 2015, 11, 748–758. [Google Scholar] [CrossRef] [Green Version]
- Cabe, M.; Rademacher, D.J.; Karlsson, A.B.; Cherukuri, S.; Bakowska, J.C. PB1 and UBA domains of p62 are essential for aggresome-like induced structure formation. Biochem. Biophys. Res. Commun. 2018, 503, 2306–2311. [Google Scholar] [CrossRef]
- Zaffagnini, G.; Savova, A.; Danieli, A.; Romanov, J.; Tremel, S.; Ebner, M.; Peterbauer, T.; Sztacho, M.; Trapannone, R.; Tarafder, A.K.; et al. p62 filaments capture and present ubiquitinated cargos for autophagy. EMBO J. 2018, 37, e98308. [Google Scholar] [CrossRef]
- Zhang, K.; Wang, A.; Zhong, K.; Qi, S.; Wei, C.; Shu, X.; Tu, W.-Y.; Xu, W.; Xia, C.; Xiao, Y.; et al. UBQLN2-HSP70 axis reduces poly-Gly-Ala aggregates and alleviates behavioral defects in the C9ORF72 animal model. Neuron 2021, 109, 1949–1962.e6. [Google Scholar] [CrossRef]
- Hjerpe, R.; Bett, J.S.; Keuss, M.J.; Solovyova, A.; McWilliams, T.G.; Johnson, C.; Sahu, I.; Varghese, J.; Wood, N.; Wightman, M.; et al. UBQLN2 Mediates Autophagy-Independent Protein Aggregate Clearance by the Proteasome. Cell 2016, 166, 935–949. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yasuda, S.; Tsuchiya, H.; Kaiho, A.; Guo, Q.; Ikeuchi, K.; Endo, A.; Arai, N.; Ohtake, F.; Murata, S.; Inada, T.; et al. Stress- and ubiquitylation-dependent phase separation of the proteasome. Nature 2020, 578, 296–300. [Google Scholar] [CrossRef] [PubMed]
- Finley, D. Recognition and Processing of Ubiquitin-Protein Conjugates by the Proteasome. Annu. Rev. Biochem. 2009, 78, 477–513. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kageyama, S.; Gudmundsson, S.R.; Sou, Y.S.; Ichimura, Y.; Tamura, N.; Kazuno, S.; Ueno, T.; Miura, Y.; Noshiro, D.; Abe, M.; et al. p62/SQSTM1-droplet serves as a platform for autophagosome formation and anti-oxidative stress response. Nat. Commun. 2021, 12, 16. [Google Scholar] [CrossRef]
- Sun, D.; Wu, R.; Zheng, J.; Li, P.; Yu, L. Polyubiquitin chain-induced p62 phase separation drives autophagic cargo segregation. Cell Res. 2018, 28, 405–415. [Google Scholar] [CrossRef] [Green Version]
- Deng, H.-X.; Chen, W.; Hong, S.-T.; Boycott, K.M.; Gorrie, G.H.; Siddique, N.; Yang, Y.; Fecto, F.; Shi, Y.; Zhai, H.; et al. Mutations in UBQLN2 cause dominant X-linked juvenile and adult-onset ALS and ALS/dementia. Nat. 2011, 477, 211–215. [Google Scholar] [CrossRef] [Green Version]
- Riley, J.F.; Fioramonti, P.J.; Rusnock, A.K.; Hehnly, H.; Castañeda, C.A. ALS-linked mutations impair UBQLN2 stress-induced biomolecular condensate assembly in cells. J. Neurochem. 2021, 159, 145–155. [Google Scholar] [CrossRef]
- Dao, T.P.; Kolaitis, R.-M.; Kim, H.J.; O’Donovan, K.; Martyniak, B.; Colicino, E.; Hehnly, H.; Taylor, J.P.; Castañeda, C.A. Ubiquitin Modulates Liquid-Liquid Phase Separation of UBQLN2 via Disruption of Multivalent Interactions. Mol. Cell 2018, 69, 965–978.e6. [Google Scholar] [CrossRef] [Green Version]
- Dao, T.P.; Martyniak, B.; Canning, A.J.; Lei, Y.; Colicino, E.G.; Cosgrove, M.S.; Hehnly, H.; Castañeda, C.A. ALS-Linked Mutations Affect UBQLN2 Oligomerization and Phase Separation in a Position- and Amino Acid-Dependent Manner. Structure 2019, 27, 937–951.e5. [Google Scholar] [CrossRef]
- Scholefield, J.; Henriques, R.; Savulescu, A.; Fontan, E.; Boucharlat, A.; Laplantine, E.; Smahi, A.; Israël, A.; Agou, F.; Mhlanga, M.M. Super-resolution microscopy reveals a preformed NEMO lattice structure that is collapsed in incontinentia pigmenti. Nat. Commun. 2016, 7, 12629. [Google Scholar] [CrossRef] [Green Version]
- Du, M.; Ea, C.-K.; Fang, Y.; Chen, Z.J. Liquid phase separation of NEMO induced by polyubiquitin chains activates NF-κB. Mol. Cell 2022, 82, 2415–2426.e5. [Google Scholar] [CrossRef]
- Gałgański, Ł.; Urbanek-Trzeciak, M.O.; Krzyzosiak, W.J. Nuclear speckles: Molecular organization, biological function and role in disease. Nucleic Acids Res. 2017, 45, 10350–10368. [Google Scholar] [CrossRef] [Green Version]
- Liu, S.; Wang, T.; Shi, Y.; Bai, L.; Wang, S.; Guo, D.; Zhang, Y.; Qi, Y.; Chen, C.; Zhang, J.; et al. USP42 drives nuclear speckle mRNA splicing via directing dynamic phase separation to promote tumorigenesis. Cell Death Differ. 2021, 28, 2482–2498. [Google Scholar] [CrossRef]
- Nagai, Y.; Kojima, T.; Muro, Y.; Hachiya, T.; Nishizawa, Y.; Wakabayashi, T.; Hagiwara, M. Identification of a novel nuclear speckle-type protein, SPOP. FEBS Lett. 1997, 418, 23–26. [Google Scholar] [CrossRef] [Green Version]
- Marzahn, M.R.; Marada, S.; Lee, J.; Nourse, A.; Kenrick, S.; Zhao, H.; Ben-Nissan, G.; Kolaitis, R.; Peters, J.L.; Pounds, S.; et al. Higher-order oligomerization promotes localization of SPOP to liquid nuclear speckles. EMBO J. 2016, 35, 1254–1275. [Google Scholar] [CrossRef]
- Usher, E.T.; Sabri, N.; Rohac, R.; Boal, A.K.; Mittag, T.; Showalter, S.A. Intrinsically disordered substrates dictate SPOP subnuclear localization and ubiquitination activity. J. Biol. Chem. 2021, 296, 100693. [Google Scholar] [CrossRef]
- Bouchard, J.J.; Otero, J.H.; Scott, D.C.; Szulc, E.; Martin, E.W.; Sabri, N.; Granata, D.; Marzahn, M.R.; Lindorff-Larsen, K.; Salvatella, X.; et al. Cancer Mutations of the Tumor Suppressor SPOP Disrupt the Formation of Active, Phase-Separated Compartments. Mol. Cell 2018, 72, 19–36.e8. [Google Scholar] [CrossRef] [Green Version]
- Maharana, S.; Wang, J.; Papadopoulos, D.K.; Richter, D.; Pozniakovsky, A.; Poser, I.; Bickle, M.; Rizk, S.; Guillén-Boixet, J.; Franzmann, T.M.; et al. RNA buffers the phase separation behavior of prion-like RNA binding proteins. Science 2018, 360, 918–921. [Google Scholar] [CrossRef] [Green Version]
- Baradaran-Heravi, Y.; Van Broeckhoven, C.; van der Zee, J. Stress granule mediated protein aggregation and underlying gene defects in the FTD-ALS spectrum. Neurobiol. Dis. 2019, 134, 104639. [Google Scholar] [CrossRef]
- Alshareedah, I.; Kaur, T.; Ngo, J.; Seppala, H.; Kounatse, L.-A.D.; Wang, W.; Moosa, M.M.; Banerjee, P.R. Interplay between Short-Range Attraction and Long-Range Repulsion Controls Reentrant Liquid Condensation of Ribonucleoprotein–RNA Complexes. J. Am. Chem. Soc. 2019, 141, 14593–14602. [Google Scholar] [CrossRef]
- Varadan, R.; Assfalg, M.; Raasi, S.; Pickart, C.; Fushman, D. Structural Determinants for Selective Recognition of a Lys48-Linked Polyubiquitin Chain by a UBA Domain. Mol. Cell 2005, 18, 687–698. [Google Scholar] [CrossRef]
- Raasi, S.; Varadan, R.; Fushman, D.; Pickart, C.M. Diverse polyubiquitin interaction properties of ubiquitin-associated domains. Nat. Struct. Mol. Biol. 2005, 12, 708–714. [Google Scholar] [CrossRef]
- Lafontaine, D.L.J.; Riback, J.A.; Bascetin, R.; Brangwynne, C.P. The nucleolus as a multiphase liquid condensate. Nat. Rev. Mol. Cell Biol. 2020, 22, 165–182. [Google Scholar] [CrossRef] [PubMed]
- Marmor-Kollet, H.; Siany, A.; Kedersha, N.; Knafo, N.; Rivkin, N.; Danino, Y.M.; Moens, T.G.; Olender, T.; Sheban, D.; Cohen, N.; et al. Spatiotemporal Proteomic Analysis of Stress Granule Disassembly Using APEX Reveals Regulation by SUMOylation and Links to ALS Pathogenesis. Mol. Cell 2020, 80, 876–891.e6. [Google Scholar] [CrossRef] [PubMed]
- Jain, S.; Wheeler, J.R.; Walters, R.W.; Agrawal, A.; Barsic, A.; Parker, R. ATPase-Modulated Stress Granules Contain a Diverse Proteome and Substructure. Cell 2016, 164, 487–498. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Costa-Mattioli, M.; Walter, P. The integrated stress response: From mechanism to disease. Science 2020, 368, eaat5314. [Google Scholar] [CrossRef] [PubMed]
- Hofmann, S.; Kedersha, N.; Anderson, P.; Ivanov, P. Molecular mechanisms of stress granule assembly and disassembly. Biochim. et Biophys. Acta 2020, 1868, 118876. [Google Scholar] [CrossRef]
- Corbet, G.; Parker, R. RNP Granule Formation: Lessons from P-Bodies and Stress Granules. Cold Spring Harb. Symp. Quant. Biol. 2019, 84, 203–215. [Google Scholar] [CrossRef]
- Sanders, D.W.; Kedersha, N.; Lee, D.S.W.; Strom, A.R.; Drake, V.; Riback, J.A.; Bracha, D.; Eeftens, J.M.; Iwanicki, A.; Wang, A.; et al. Competing Protein-RNA Interaction Networks Control Multiphase Intracellular Organization. Cell 2020, 181, 306–324.e28. [Google Scholar] [CrossRef]
- Markmiller, S.; Soltanieh, S.; Server, K.L.; Mak, R.; Jin, W.; Fang, M.Y.; Luo, E.; Yeo, G.W.; Krach, F.; Yang, D.; et al. Context-dependent and Disease-specific Diversity in Stress Granules Formed from Pre-existing Protein Interactions. FASEB J. 2018, 32, 252-3. [Google Scholar] [CrossRef]
- Wheeler, J.R.; Matheny, T.; Jain, S.; Abrisch, R.; Parker, R. Distinct stages in stress granule assembly and disassembly. eLife 2016, 5, e18413. [Google Scholar] [CrossRef]
- Yang, P.; Mathieu, C.; Kolaitis, R.-M.; Zhang, P.; Messing, J.; Yurtsever, U.; Yang, Z.; Wu, J.; Li, Y.; Pan, Q.; et al. G3BP1 Is a Tunable Switch that Triggers Phase Separation to Assemble Stress Granules. Cell 2020, 181, 325–345.e28. [Google Scholar] [CrossRef]
- Kedersha, N.; Anderson, P. Chapter 4 Regulation of Translation by Stress Granules and Processing Bodies. Prog. Mol. Biol. Transl. Sci. 2009, 90, 155–185. [Google Scholar] [CrossRef]
- Gwon, Y.; Maxwell, B.A.; Kolaitis, R.-M.; Zhang, P.; Kim, H.J.; Taylor, J.P. Ubiquitination of G3BP1 mediates stress granule disassembly in a context-specific manner. Science 2021, 372, eabf6548. [Google Scholar] [CrossRef]
- Maxwell, B.A.; Gwon, Y.; Mishra, A.; Peng, J.; Nakamura, H.; Zhang, K.; Kim, H.J.; Taylor, J.P. Ubiquitination is essential for recovery of cellular activities after heat shock. Science 2021, 372, eabc3593. [Google Scholar] [CrossRef]
- Tolay, N.; Buchberger, A. Comparative profiling of stress granule clearance reveals differential contributions of the ubiquitin system. Life Sci. Alliance 2021, 4, e202000927. [Google Scholar] [CrossRef]
- Mediani, L.; Guillén-Boixet, J.; Vinet, J.; Franzmann, T.M.; Bigi, I.; Mateju, D.; Carrà, A.D.; Morelli, F.F.; Tiago, T.; Poser, I.; et al. Defective ribosomal products challenge nuclear function by impairing nuclear condensate dynamics and immobilizing ubiquitin. EMBO J. 2019, 38, e101341. [Google Scholar] [CrossRef]
- Ganassi, M.; Mateju, D.; Bigi, I.; Mediani, L.; Poser, I.; Lee, H.O.; Seguin, S.J.; Morelli, F.F.; Vinet, J.; Leo, G.; et al. A Surveillance Function of the HSPB8-BAG3-HSP70 Chaperone Complex Ensures Stress Granule Integrity and Dynamism. Mol. Cell 2016, 63, 796–810. [Google Scholar] [CrossRef] [Green Version]
- Mateju, D.; Franzmann, T.M.; Patel, A.; Kopach, A.; E Boczek, E.; Maharana, S.; O Lee, H.; Carra, S.; A Hyman, A.; Alberti, S. An aberrant phase transition of stress granules triggered by misfolded protein and prevented by chaperone function. EMBO J. 2017, 36, 1669–1687. [Google Scholar] [CrossRef]
- Samanta, N.; Ribeiro, S.S.; Becker, M.; Laborie, E.; Pollak, R.; Timr, S.; Sterpone, F.; Ebbinghaus, S. Sequestration of Proteins in Stress Granules Relies on the In-Cell but Not the In Vitro Folding Stability. J. Am. Chem. Soc. 2021, 143, 19909–19918. [Google Scholar] [CrossRef]
- Cabrera, M.; Boronat, S.; Marte, L.; Vega, M.; Pérez, P.; Ayté, J.; Hidalgo, E. Chaperone-Facilitated Aggregation of Thermo-Sensitive Proteins Shields Them from Degradation during Heat Stress. Cell Rep. 2020, 30, 2430–2443.e4. [Google Scholar] [CrossRef] [Green Version]
- Seguin, S.J.; Morelli, F.F.; Vinet, J.; Amore, D.; De Biasi, S.; Poletti, A.; Rubinsztein, D.C.; Carra, S. Inhibition of autophagy, lysosome and VCP function impairs stress granule assembly. Cell Death Differ. 2014, 21, 1838–1851. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rozales, K.; Younis, A.; Saida, N.; Meller, A.; Goldman, H.; Kellerman, L.; Heinrich, R.; Berlin, S.; Shalgi, R. Differential roles for DNAJ isoforms in HTT-polyQ and FUS aggregation modulation revealed by chaperone screens. Nat. Commun. 2022, 13, 516. [Google Scholar] [CrossRef]
- Gu, J.; Liu, Z.; Zhang, S.; Li, Y.; Xia, W.; Wang, C.; Xiang, H.; Liu, Z.; Tan, L.; Fang, Y.; et al. Hsp40 proteins phase separate to chaperone the assembly and maintenance of membraneless organelles. Proc. Natl. Acad. Sci. USA 2020, 117, 31123–31133. [Google Scholar] [CrossRef] [PubMed]
- Yoo, H.; Bard, J.A.; Pilipenko, E.V.; Drummond, D.A. Chaperones directly and efficiently disperse stress-triggered biomolecular condensates. Mol. Cell 2022, 82, 741–755.e11. [Google Scholar] [CrossRef] [PubMed]
- Walters, R.W.; Muhlrad, D.; Garcia, J.; Parker, R. Differential effects of Ydj1 and Sis1 on Hsp70-mediated clearance of stress granules in Saccharomyces cerevisiae. RNA 2015, 21, 1660–1671. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- VanPelt, J.; Page, R.C. Unraveling the CHIP:Hsp70 complex as an information processor for protein quality control. Biochim. et Biophys. Acta (BBA)-Proteins Proteom. 2017, 1865, 133–141. [Google Scholar] [CrossRef] [Green Version]
- Meyer, H.; Weihl, C.C. The VCP/p97 system at a glance: Connecting cellular function to disease pathogenesis. J. Cell Sci. 2014, 127, 3877–3883. [Google Scholar] [CrossRef] [Green Version]
- Buchan, J.R.; Kolaitis, R.M.; Taylor, J.P.; Parker, R. Eukaryotic stress granules are cleared by autophagy and Cdc48/VCP function. Cell 2013, 153, 1461–1474. [Google Scholar] [CrossRef] [Green Version]
- Turakhiya, A.; Meyer, S.R.; Marincola, G.; Böhm, S.; Vanselow, J.T.; Schlosser, A.; Hofmann, K.; Buchberger, A. ZFAND1 Recruits p97 and the 26S Proteasome to Promote the Clearance of Arsenite-Induced Stress Granules. Mol Cell. 2018, 70, 906–919.e7. [Google Scholar] [CrossRef] [Green Version]
- Xie, X.; Matsumoto, S.; Endo, A.; Fukushima, T.; Kawahara, H.; Saeki, Y.; Komada, M. Deubiquitinases USP5 and USP13 are recruited to and regulate heat-induced stress granules by deubiquitinating activities. J. Cell Sci. 2018, 131, jcs.210856. [Google Scholar] [CrossRef] [Green Version]
- Chitiprolu, M.; Jagow, C.; Tremblay, V.; Bondy-Chorney, E.; Paris, G.; Savard, A.; Palidwor, G.; Barry, F.A.; Zinman, L.; Keith, J.; et al. A complex of C9ORF72 and p62 uses arginine methylation to eliminate stress granules by autophagy. Nat. Commun. 2018, 9, 1–18. [Google Scholar] [CrossRef] [Green Version]
- Markmiller, S.; Fulzele, A.; Higgins, R.; Leonard, M.; Yeo, G.W.; Bennett, E.J. Active Protein Neddylation or Ubiquitylation Is Dispensable for Stress Granule Dynamics. Cell Rep. 2019, 27, 1356–1363.e3. [Google Scholar] [CrossRef] [Green Version]
- Alexander, E.J.; Niaki, A.G.; Zhang, T.; Sarkar, J.; Liu, Y.; Nirujogi, R.S.; Pandey, A.; Myong, S.; Wang, J. Ubiquilin 2 modulates ALS/FTD-linked FUS–RNA complex dynamics and stress granule formation. Proc. Natl. Acad. Sci. USA 2018, 115, E11485–E11494. [Google Scholar] [CrossRef] [Green Version]
- Mee Hayes, E.; Sirvio, L.; Ye, Y. A Potential Mechanism for Targeting Aggregates With Proteasomes and Disaggregases in Liquid Droplets. Front. Aging Neurosci. 2022, 14, 854380. [Google Scholar] [CrossRef]
- Weidtkamp-Peters, S.; Lenser, T.; Negorev, D.; Gerstner, N.; Hofmann, T.G.; Schwanitz, G.; Hoischen, C.; Maul, G.; Dittrich, P.; Hemmerich, P. Dynamics of component exchange at PML nuclear bodies. J. Cell Sci. 2008, 121, 2731–2743. [Google Scholar] [CrossRef] [Green Version]
- Sung, K.S.; Lee, Y.-A.; Kim, E.T.; Lee, S.-R.; Ahn, J.-H.; Choi, C.Y. Role of the SUMO-interacting motif in HIPK2 targeting to the PML nuclear bodies and regulation of p53. Exp. Cell Res. 2011, 317, 1060–1070. [Google Scholar] [CrossRef]
- Keiten-Schmitz, J.; Röder, L.; Hornstein, E.; Müller-McNicoll, M.; Müller, S. SUMO: Glue or Solvent for Phase-Separated Ribonucleoprotein Complexes and Molecular Condensates? Front. Mol. Biosci. 2021, 8, 673038. [Google Scholar] [CrossRef]
- Keiten-Schmitz, J.; Wagner, K.; Piller, T.; Kaulich, M.; Alberti, S.; Müller, S. The Nuclear SUMO-Targeted Ubiquitin Quality Control Network Regulates the Dynamics of Cytoplasmic Stress Granules. Mol. Cell 2020, 79, 54–67.e7. [Google Scholar] [CrossRef]
- Lallemand-Breitenbach, V.; Jeanne, M.; Benhenda, S.; Nasr, R.; Lei, M.; Peres, L.; Zhou, J.; Zhu, J.; Raught, B.; de Thé, H. Arsenic degrades PML or PML-RARalpha through a SUMO-triggered RNF4/ubiquitin-mediated pathway. Nat. Cell Biol. 2008, 10, 547–555. [Google Scholar] [CrossRef]
- Lallemand-Breitenbach, V.; de Thé, H. PML nuclear bodies: From architecture to function. Curr. Opin. Cell. Biol. 2018, 52, 154–161. [Google Scholar] [CrossRef]
- Neumann, M.; Sampathu, D.M.; Kwong, L.K.; Truax, A.C.; Micsenyi, M.C.; Chou, T.T.; Bruce, J.; Schuck, T.; Grossman, M.; Clark, C.M.; et al. Ubiquitinated TDP-43 in Frontotemporal Lobar Degeneration and Amyotrophic Lateral Sclerosis. Science 2006, 314, 130–133. [Google Scholar] [CrossRef] [Green Version]
- Li, F.; Xu, D.; Wang, Y.; Zhou, Z.; Liu, J.; Hu, S.; Gong, Y.; Yuan, J.; Pan, L. Structural insights into the ubiquitin recognition by OPTN (optineurin) and its regulation by TBK1-mediated phosphorylation. Autophagy 2018, 14, 66–79. [Google Scholar] [CrossRef] [Green Version]
- Keller, B.A.; Volkening, K.; Droppelmann, C.A.; Ang, L.C.; Rademakers, R.; Strong, M.J. Co-aggregation of RNA binding proteins in ALS spinal motor neurons: Evidence of a common pathogenic mechanism. Acta Neuropathol. 2012, 124, 733–747. [Google Scholar] [CrossRef]
- Watanabe, M.; Dykes-Hoberg, M.; Cizewski Culotta, V.; Price, D.L.; Wong, P.C.; Rothstein, J.D. Histological Evidence of Protein Aggregation in Mutant SOD1 Transgenic Mice and in Amyotrophic Lateral Sclerosis Neural Tissues. Neurobiol. Dis. 2001, 8, 933–941. [Google Scholar] [CrossRef] [Green Version]
- Weisberg, S.J.; Lyakhovetsky, R.; Werdiger, A.-C.; Gitler, A.D.; Soen, Y.; Kaganovich, D. Compartmentalization of superoxide dismutase 1 (SOD1G93A) aggregates determines their toxicity. Proc. Natl. Acad. Sci. USA 2012, 109, 15811–15816. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Basso, M.; Samengo, G.; Nardo, G.; Massignan, T.; D’Alessandro, G.; Tartari, S.; Cantoni, L.; Marino, M.; Cheroni, C.; De Biasi, S.; et al. Characterization of Detergent-Insoluble Proteins in ALS Indicates a Causal Link between Nitrative Stress and Aggregation in Pathogenesis. PLoS ONE 2009, 4, e8130. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guo, Q.; Lehmer, C.; Martínez-Sánchez, A.; Rudack, T.; Beck, F.; Hartmann, H.; Pérez-Berlanga, M.; Frottin, F.; Hipp, M.S.; Hartl, F.U.; et al. In Situ Structure of Neuronal C9orf72 Poly-GA Aggregates Reveals Proteasome Recruitment. Cell 2018, 172, 696–705.e12. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gupta, R.; Lan, M.; Mojsilovic-Petrovic, J.; Choi, W.H.; Safren, N.; Barmada, S.; Lee, M.J.; Kalb, R. The Proline/Arginine Dipeptide from Hexanucleotide Repeat Expanded C9ORF72 Inhibits the Proteasome. Eneuro 2017, 4. [Google Scholar] [CrossRef] [Green Version]
- Farrawell, N.E.; Lambert-Smith, I.; Mitchell, K.; McKenna, J.; McAlary, L.; Ciryam, P.; Vine, K.L.; Saunders, D.N.; Yerbury, J.J. SOD1A4V aggregation alters ubiquitin homeostasis in a cell model of ALS. J. Cell Sci. 2018, 131, jcs209122. [Google Scholar] [CrossRef] [Green Version]
- Farrawell, N.E.; McAlary, L.; Lum, J.S.; Chisholm, C.G.; Warraich, S.T.; Blair, I.P.; Vine, K.L.; Saunders, D.N.; Yerbury, J.J. Ubiquitin Homeostasis Is Disrupted in TDP-43 and FUS Cell Models of ALS. iScience 2020, 23, 101700. [Google Scholar] [CrossRef]
- Gami-Patel, P.; Dijken, I.; Meeter, L.H.; Melhem, S.; Morrema, T.H.J.; Scheper, W.; Swieten, J.C.; Rozemuller, A.J.M.; Dijkstra, A.A.; Hoozemans, J.J.M. Unfolded protein response activation in C9orf72 frontotemporal dementia is associated with dipeptide pathology and granulovacuolar degeneration in granule cells. Brain Pathol. 2020, 31, 163–173. [Google Scholar] [CrossRef]
- Farhan, S.M.K.; Howrigan, D.P.; Abbott, L.E.; Klim, J.R.; Topp, S.D.; Byrnes, A.E.; Churchhouse, C.; Phatnani, H.; Smith, B.N.; Rampersaud, E.; et al. Exome sequencing in amyotrophic lateral sclerosis implicates a novel gene, DNAJC7, encoding a heat-shock protein. Nat. Neurosci. 2019, 22, 1966–1974. [Google Scholar] [CrossRef]
- Dubinski, A.; Vande Velde, C. Altered stress granule disassembly: Links to neurodegenerative disease? Trends Neurosci. 2021, 44, 765–766. [Google Scholar] [CrossRef]
- Khalfallah, Y.; Kuta, R.; Grasmuck, C.; Prat, A.; Durham, H.D.; Velde, C.V. TDP-43 regulation of stress granule dynamics in neurodegenerative disease-relevant cell types. Sci. Rep. 2018, 8, 7551. [Google Scholar] [CrossRef] [Green Version]
- Li, Y.R.; King, O.D.; Shorter, J.; Gitler, A.D. Stress granules as crucibles of ALS pathogenesis. J. Cell Biol. 2013, 201, 361–372. [Google Scholar] [CrossRef]
- Wolozin, B.; Ivanov, P. Stress granules and neurodegeneration. Nat. Rev. Neurosci. 2019, 20, 649–666. [Google Scholar] [CrossRef] [PubMed]
- Deng, H.-X.; Zhai, H.; Bigio, E.H.; Yan, J.; Fecto, F.; Ajroud, K.; Mishra, M.; Ajroud-Driss, S.; Heller, S.; Sufit, R.; et al. FUS-immunoreactive inclusions are a common feature in sporadic and non-SOD1 familial amyotrophic lateral sclerosis. Ann. Neurol. 2010, 67, 739–748. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu-Yesucevitz, L.; Bilgutay, A.; Zhang, Y.J.; Vanderweyde, T.; Citro, A.; Mehta, T.; Zaarur, N.; McKee, A.; Bowser, R.; Sherman, M.; et al. Tar DNA binding protein-43 (TDP-43) associates with stress granules: Analysis of cultured cells and pathological brain tissue. PLoS ONE 2010, 5, e13250. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fujita, K.; Ito, H.; Nakano, S.; Kinoshita, Y.; Wate, R.; Kusaka, H. Immunohistochemical identification of messenger RNA-related proteins in basophilic inclusions of adult-onset atypical motor neuron disease. Acta Neuropathol. 2008, 116, 439–445. [Google Scholar] [CrossRef]
- Lee, K.-H.; Zhang, P.; Kim, H.J.; Mitrea, D.M.; Sarkar, M.; Freibaum, B.D.; Cika, J.; Coughlin, M.; Messing, J.; Molliex, A.; et al. C9orf72 Dipeptide Repeats Impair the Assembly, Dynamics, and Function of Membrane-Less Organelles. Cell 2016, 167, 774–788.e17. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Y.J.; Gendron, T.F.; Ebbert, M.T.W.; O’Raw, A.D.; Yue, M.; Jansen-West, K.; Zhang, X.; Prudencio, M.; Chew, J.; Cook, C.N.; et al. Poly(GR) impairs protein translation and stress granule dynamics in C9orf72-associated frontotemporal dementia and amyotrophic lateral sclerosis. Nat. Med. 2018, 24, 1136–1142. [Google Scholar] [CrossRef]
- Prudencio, M.; Belzil, V.V.; Batra, R.; Ross, C.A.; Gendron, T.F.; Pregent, L.J.; Murray, M.E.; Overstreet, K.K.; Piazza-Johnston, A.E.; Desaro, P.; et al. Distinct brain transcriptome profiles in C9orf72-associated and sporadic ALS. Nat. Neurosci. 2015, 18, 1175–1182. [Google Scholar] [CrossRef]
- Wang, R.; Xu, X.; Hao, Z.; Zhang, S.; Wu, D.; Sun, H.; Mu, C.; Ren, H.; Wang, G. Poly-PR in C9ORF72-Related Amyotrophic Lateral Sclerosis/Frontotemporal Dementia Causes Neurotoxicity by Clathrin-Dependent Endocytosis. Neurosci. Bull. 2019, 35, 889–900. [Google Scholar] [CrossRef]
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Amzallag, E.; Hornstein, E. Crosstalk between Biomolecular Condensates and Proteostasis. Cells 2022, 11, 2415. https://doi.org/10.3390/cells11152415
Amzallag E, Hornstein E. Crosstalk between Biomolecular Condensates and Proteostasis. Cells. 2022; 11(15):2415. https://doi.org/10.3390/cells11152415
Chicago/Turabian StyleAmzallag, Emmanuel, and Eran Hornstein. 2022. "Crosstalk between Biomolecular Condensates and Proteostasis" Cells 11, no. 15: 2415. https://doi.org/10.3390/cells11152415