
Phenotypic Alteration of BMDM In Vitro Using Small Interfering RNA

 ,
,  ,
, 
 , and
, and
Abstract
:1. Introduction
2. Materials and Methods
2.1. Isolation and Culture of BMDM
2.2. SiRNA Transfection of BMDM and Polarization
2.3. QPCR
2.4. Western Blot
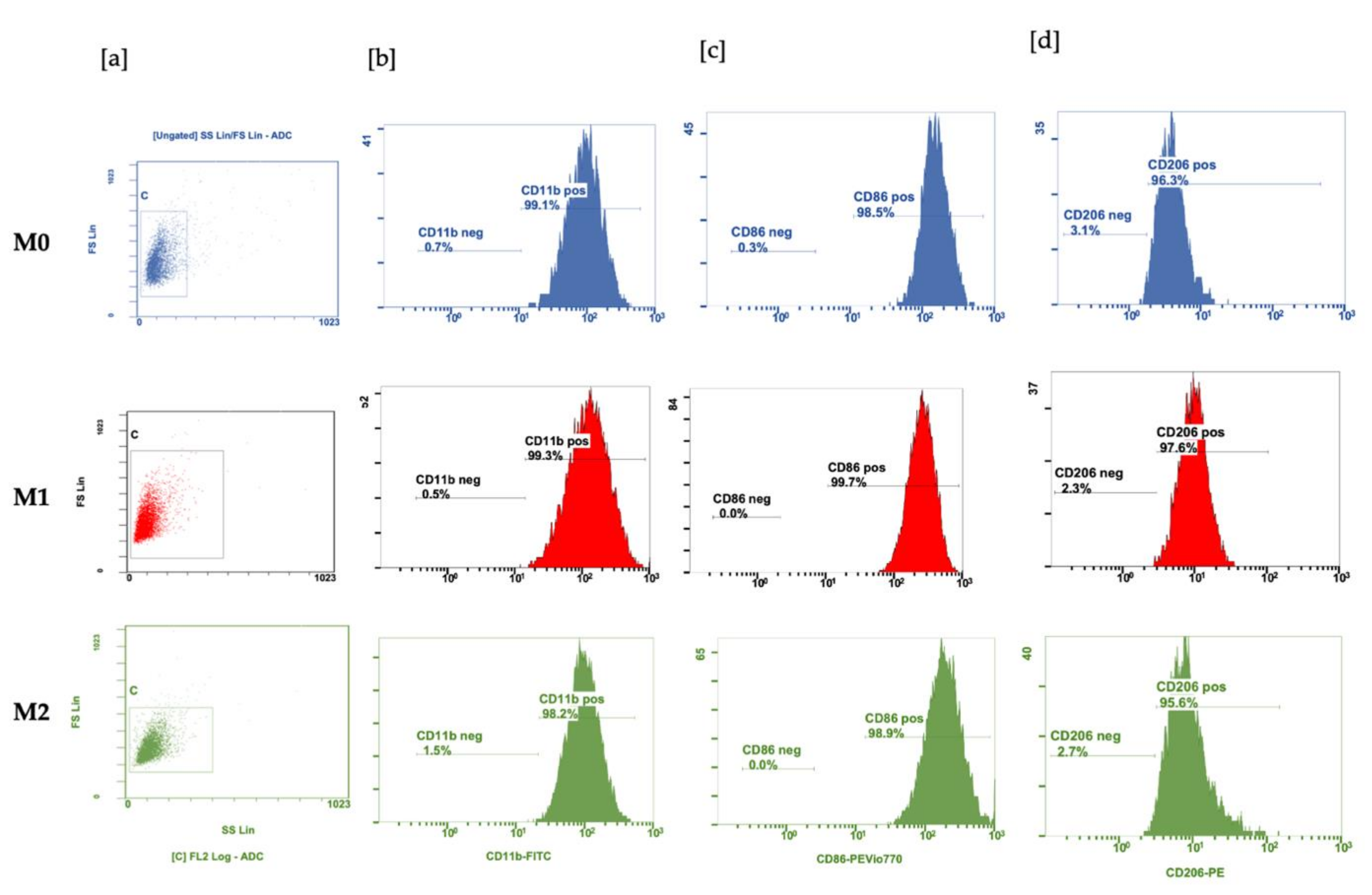
2.5. Flow Cytometry
2.6. Statistical Analysis
3. Results
3.1. Polarization of BMDM with LPS and IL-4 and IL-10
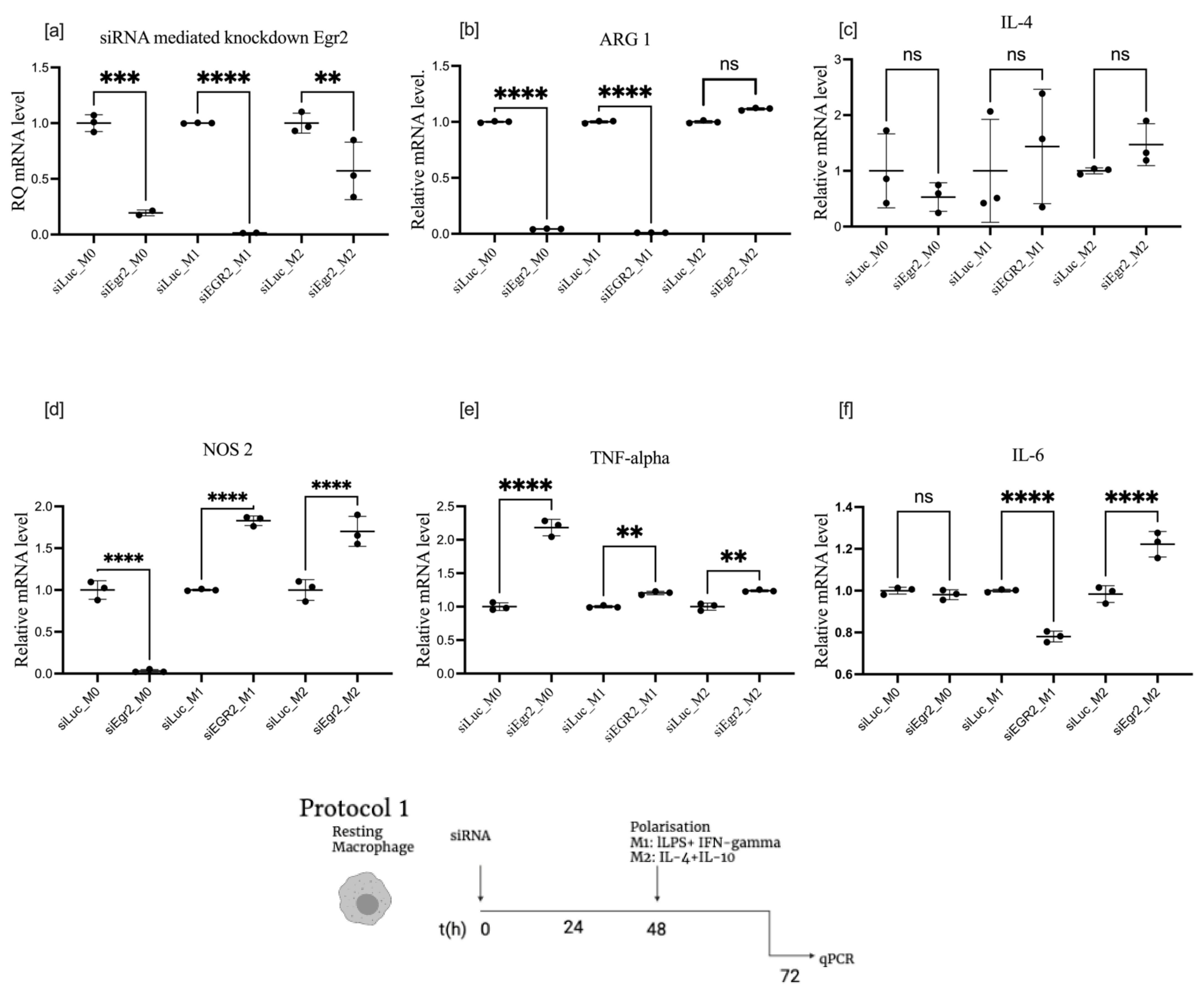
3.2. Effect of EGR2 Knockdown on BMDM Polarization
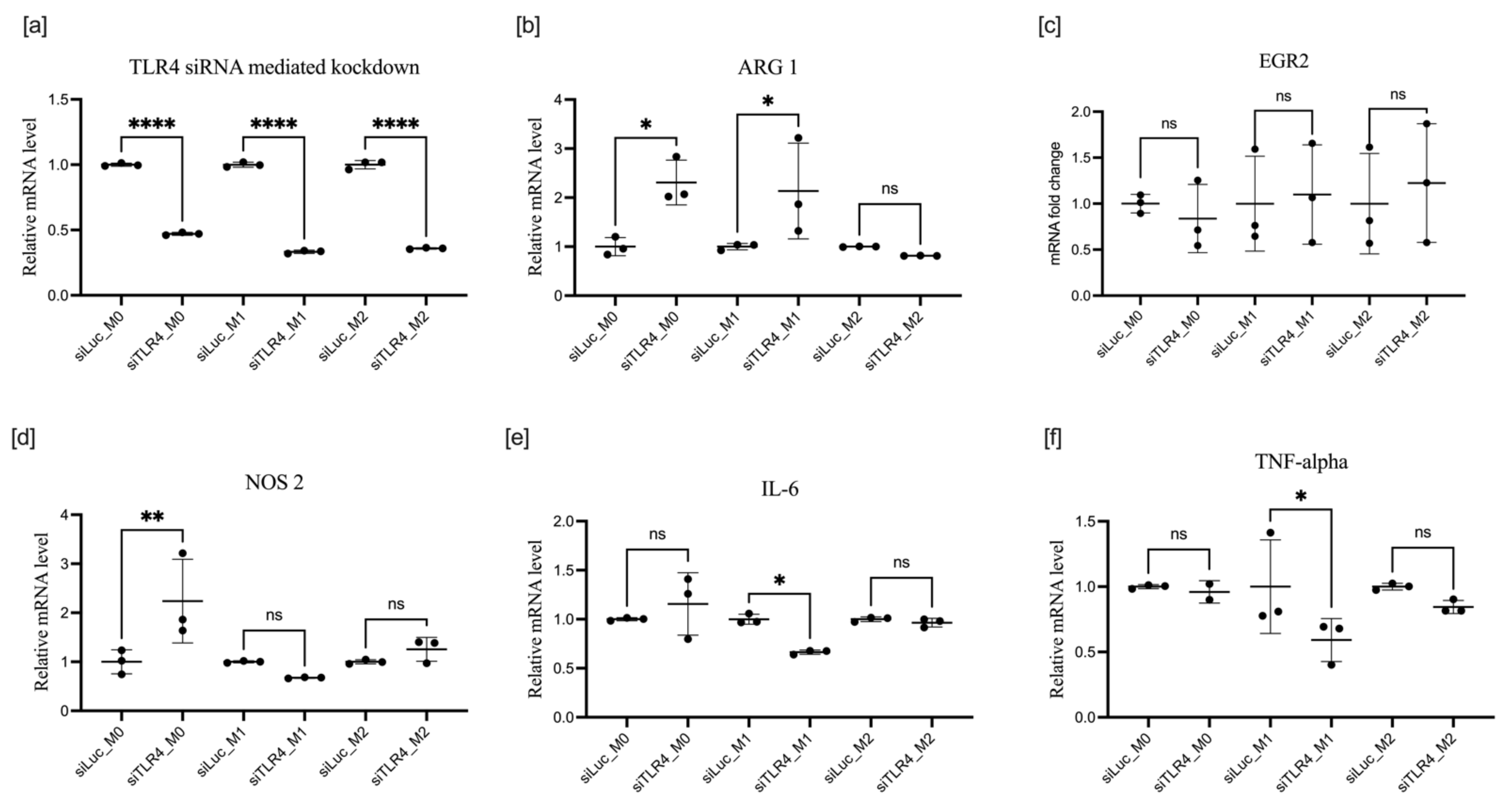
3.3. Effect of TLR4 Knockdown on BMDM Polarization
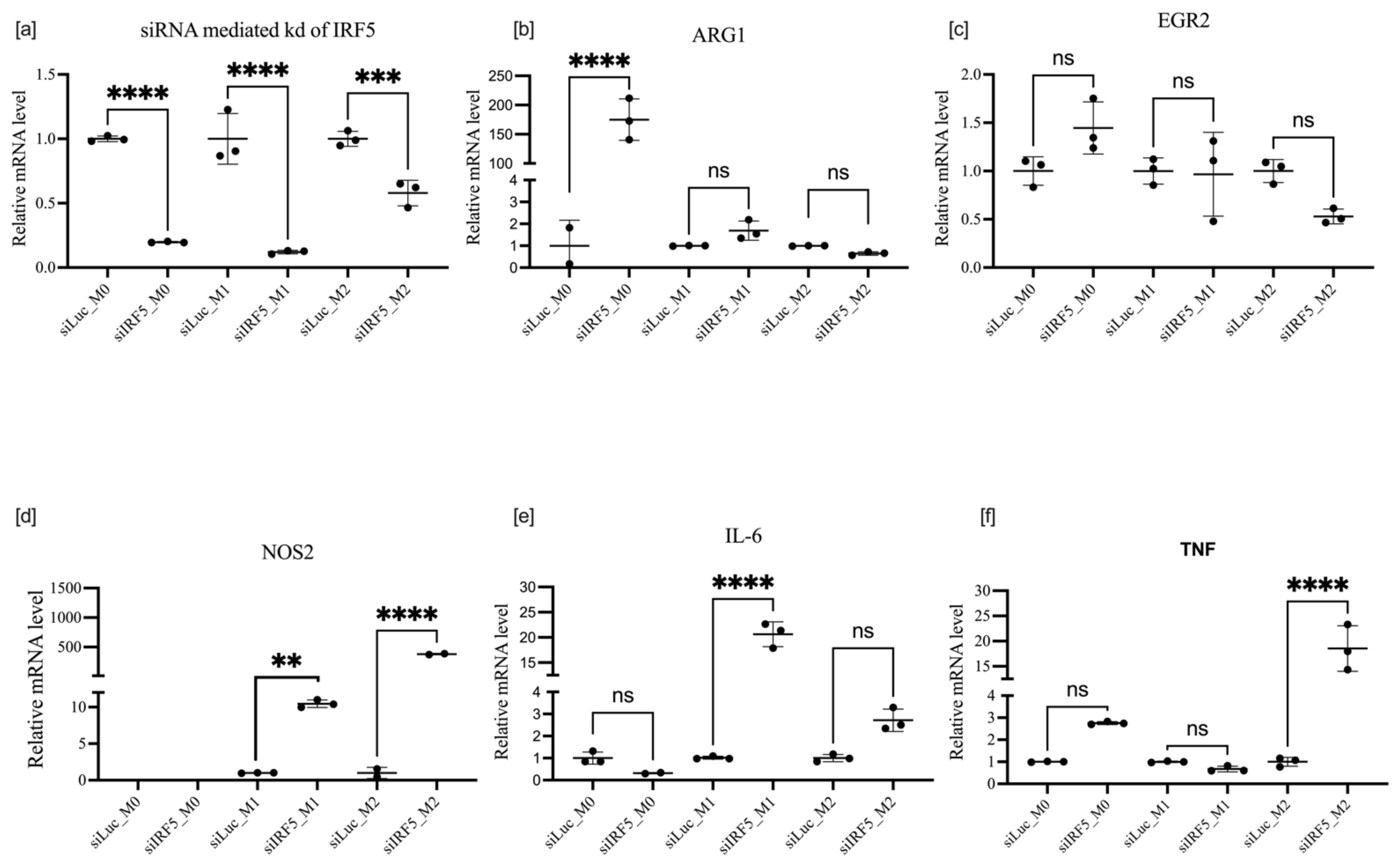
3.4. Effects of IRF5 Knockdown on BMDM Polarization
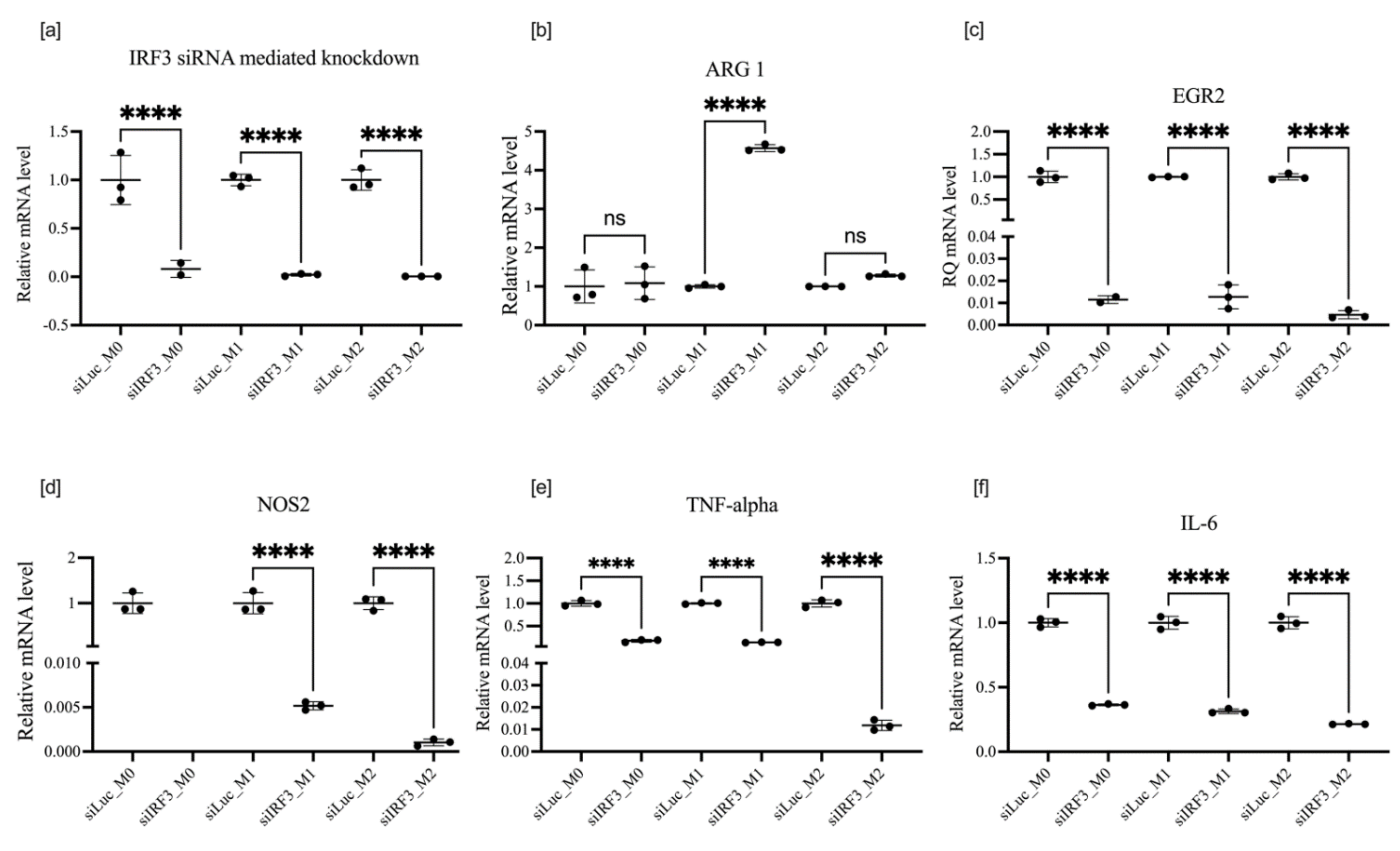
3.5. Effect of IRF3 Knockdown on BMDM Polarization
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Anderson, N.R.; Minutolo, N.G.; Gill, S.; Klichinsky, M. Macrophage-Based Approaches for Cancer Immunotherapy. Cancer Res. 2021, 81, 1201–1208. [Google Scholar] [CrossRef]
- Van der Heide, D.; Weiskirchen, R.; Bansal, R. Therapeutic Targeting of Hepatic Macrophages for the Treatment of Liver Diseases. Front. Immunol. 2019, 10, 2852. [Google Scholar] [CrossRef] [PubMed]
- Cheng, P.; Li, S.; Chen, H. Macrophages in Lung Injury, Repair, and Fibrosis. Cells 2021, 10, 436. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, T.; Arumugam, P.; Sakagami, T.; Lachmann, N.; Chalk, C.; Sallese, A.; Abe, S.; Trapnell, C.; Carey, B.; Moritz, T.; et al. Pulmonary Macrophage Transplantation Therapy. Nature 2014, 514, 450–454. [Google Scholar] [CrossRef] [PubMed]
- Manna, P.P.; Hira, S.K.; Basu, A.; Bandyopadhyay, S. Cellular Therapy by Allogeneic Macrophages against Visceral Leishmaniasis: Role of TNF-α. Cell. Immunol. 2014, 290, 152–163. [Google Scholar] [CrossRef] [PubMed]
- Moroni, F.; Dwyer, B.J.; Graham, C.; Pass, C.; Bailey, L.; Ritchie, L.; Mitchell, D.; Glover, A.; Laurie, A.; Doig, S.; et al. Safety Profile of Autologous Macrophage Therapy for Liver Cirrhosis. Nat. Med. 2019, 25, 1560–1565. [Google Scholar] [CrossRef]
- Pan, T.; Zhou, Q.; Miao, K.; Zhang, L.; Wu, G.; Yu, J.; Xu, Y.; Xiong, W.; Li, Y.; Wang, Y. Suppressing Sart1 to Modulate Macrophage Polarization by SiRNA-Loaded Liposomes: A Promising Therapeutic Strategy for Pulmonary Fibrosis. Theranostics 2021, 11, 1192–1206. [Google Scholar] [CrossRef]
- Watanabe, S.; Alexander, M.; Misharin, A.V.; Budinger, G.R.S. The Role of Macrophages in the Resolution of Inflammation. J. Clin. Investig. 2019, 129, 2619–2628. [Google Scholar] [CrossRef]
- Murray, P.J.; Wynn, T.A. Protective and Pathogenic Functions of Macrophage Subsets. Nat. Rev. Immunol. 2011, 11, 723–737. [Google Scholar] [CrossRef]
- Wynn, T.A.; Chawla, A.; Pollard, J.W. Macrophage Biology in Development, Homeostasis and Disease. Nature 2013, 496, 445–455. [Google Scholar] [CrossRef]
- Schultze, J.L.; Schmieder, A.; Goerdt, S. Macrophage Activation in Human Diseases. Semin. Immunol. 2015, 27, 249–256. [Google Scholar] [CrossRef]
- Zhang, C.; Yang, M.; Ericsson, A.C. Function of Macrophages in Disease: Current Understanding on Molecular Mechanisms. Front. Immunol. 2021, 12, 635. [Google Scholar] [CrossRef]
- Poltavets, A.S.; Vishnyakova, P.A.; Elchaninov, A.V.; Sukhikh, G.T.; Fatkhudinov, T.K. Macrophage Modification Strategies for Efficient Cell Therapy. Cells 2020, 9, 1535. [Google Scholar] [CrossRef]
- Hörhold, F.; Eisel, D.; Oswald, M.; Kolte, A.; Röll, D.; Osen, W.; Eichmüller, S.B.; König, R. Reprogramming of Macrophages Employing Gene Regulatory and Metabolic Network Models. PLoS Comput. Biol. 2020, 16, e1007657. [Google Scholar] [CrossRef]
- Ying, W.; Cheruku, P.S.; Bazer, F.W.; Safe, S.H.; Zhou, B. Investigation of Macrophage Polarization Using Bone Marrow Derived Macrophages. J. Vis. Exp. 2013, 76, e50323. [Google Scholar] [CrossRef]
- Jablonski, K.A.; Amici, S.A.; Webb, L.M.; de Dios Ruiz-Rosado, J.; Popovich, P.G.; Partida-Sanchez, S.; Guerau-de-Arellano, M. Novel Markers to Delineate Murine M1 and M2 Macrophages. PLoS ONE 2015, 10, e0145342. [Google Scholar] [CrossRef]
- Daniel, B.; Czimmerer, Z.; Halasz, L.; Boto, P.; Kolostyak, Z.; Poliska, S.; Berger, W.K.; Tzerpos, P.; Nagy, G.; Horvath, A.; et al. The Transcription Factor EGR2 Is the Molecular Linchpin Connecting STAT6 Activation to the Late, Stable Epigenomic Program of Alternative Macrophage Polarization. Genes Dev. 2020, 34, 1474–1492. [Google Scholar] [CrossRef]
- Liao, J.; Hargreaves, D.C. The Alternative Macrophage Relay: STAT6 Passes the Baton to EGR2. Genes Dev. 2020, 34, 1407–1409. [Google Scholar] [CrossRef]
- Veremeyko, T.; Yung, A.W.Y.; Anthony, D.C.; Strekalova, T.; Ponomarev, E.D. Early Growth Response Gene-2 Is Essential for M1 and M2 Macrophage Activation and Plasticity by Modulation of the Transcription Factor CEBPβ. Front. Immunol. 2018, 9, 2515. [Google Scholar] [CrossRef]
- Fitzgerald, K.A.; Kagan, J.C. Toll-like Receptors and the Control of Immunity. Cell 2020, 180, 1044–1066. [Google Scholar] [CrossRef]
- Leifer, C.A.; Medvedev, A.E. Molecular Mechanisms of Regulation of Toll-like Receptor Signaling. J. Leukoc. Biol. 2016, 100, 927–941. [Google Scholar] [CrossRef]
- El Kasmi, K.C.; Qualls, J.E.; Pesce, J.T.; Smith, A.M.; Thompson, R.W.; Henao-Tamayo, M.; Basaraba, R.J.; König, T.; Schleicher, U.; Koo, M.S.; et al. Toll-like Receptor–Induced Arginase 1 in Macrophages Thwarts Effective Immunity against Intracellular Pathogens. Nat. Immunol. 2008, 9, 1399–1406. [Google Scholar] [CrossRef]
- Jiang, N.; Xie, F.; Chen, L.; Chen, F.; Sui, L. The Effect of TLR4 on the Growth and Local Inflammatory Microenvironment of HPV-Related Cervical Cancer in Vivo. Infect. Agents Cancer 2020, 15, 12. [Google Scholar] [CrossRef]
- Krausgruber, T.; Blazek, K.; Smallie, T.; Alzabin, S.; Lockstone, H.; Sahgal, N.; Hussell, T.; Feldmann, M.; Udalova, I.A. IRF5 Promotes Inflammatory Macrophage Polarization and TH1-TH17 Responses. Nat. Immunol. 2011, 12, 231–238. [Google Scholar] [CrossRef]
- Weiss, M.; Blazek, K.; Byrne, A.J.; Perocheau, D.P.; Udalova, I.A. IRF5 Is a Specific Marker of Inflammatory Macrophages in Vivo. Mediat. Inflamm. 2013, 2013, 245804. [Google Scholar] [CrossRef]
- Saliba, D.G.; Heger, A.; Eames, H.L.; Oikonomopoulos, S.; Teixeira, A.; Blazek, K.; Androulidaki, A.; Wong, D.; Goh, F.G.; Weiss, M.; et al. IRF5:RelA Interaction Targets Inflammatory Genes in Macrophages. Cell Rep. 2014, 8, 1308–1317. [Google Scholar] [CrossRef]
- Almuttaqi, H.; Udalova, I.A. Advances and Challenges in Targeting IRF5, a Key Regulator of Inflammation. FEBS J. 2019, 286, 1624–1637. [Google Scholar] [CrossRef]
- Paun, A.; Bankoti, R.; Joshi, T.; Pitha, P.M.; Stäger, S. Critical Role of IRF-5 in the Development of T Helper 1 Responses to Leishmania Donovani Infection. PLoS Pathog. 2011, 7, e1001246. [Google Scholar] [CrossRef]
- Paun, A.; Reinert, J.T.; Jiang, Z.; Medin, C.; Balkhi, M.Y.; Fitzgerald, K.A.; Pitha, P.M. Functional Characterization of Murine Interferon Regulatory Factor 5 (IRF-5) and Its Role in the Innate Antiviral Response. J. Biol. Chem. 2008, 283, 14295–14308. [Google Scholar] [CrossRef]
- Hedl, M.; Yan, J.; Witt, H.; Abraham, C. IRF5 Is Required for Bacterial Clearance in Human M1-Polarized Macrophages, and IRF5 Immune-Mediated Disease Risk Variants Modulate This Outcome. J. Immunol. 2019, 202, 920–930. [Google Scholar] [CrossRef]
- Viola, A.; Munari, F.; Sánchez-Rodríguez, R.; Scolaro, T.; Castegna, A. The Metabolic Signature of Macrophage Responses. Front. Immunol. 2019, 10, 1462. [Google Scholar] [CrossRef] [PubMed]
- Guiteras, J.; Ripoll, É.; Bolaños, N.; de Ramon, L.; Fontova, P.; Loberas, N.; Cruzado, J.M.; Aràn, J.M.; Aviñó, A.; Eritja, R.; et al. The Gene Silencing of IRF5 and BLYSS Effectively Modulates the Outcome of Experimental Lupus Nephritis. Mol. Ther.-Nucleic Acids 2021, 24, 807–821. [Google Scholar] [CrossRef] [PubMed]
- Alzaid, F.; Lagadec, F.; Albuquerque, M.; Ballaire, R.; Orliaguet, L.; Hainault, I.; Blugeon, C.; Lemoine, S.; Lehuen, A.; Saliba, D.G.; et al. IRF5 Governs Liver Macrophage Activation That Promotes Hepatic Fibrosis in Mice and Humans. JCI Insight 2016, 1, e88689. [Google Scholar] [CrossRef] [PubMed]
- Sun, K.; Qu, J.; Chen, J.; Dang, S.; He, S.; Zhang, J.; Xie, R.; Wang, Y.; Zhang, J. [Interferon Regulatory Factor 5(IRF5) Regulates the Differentiation of Bone Marrow-Derived Macrophages in Mice]. Xi Bao Yu Fen Zi Mian Yi Xue Za Zhi 2017, 33, 168–173. [Google Scholar]
- Günthner, R.; Anders, H.J. Interferon-Regulatory Factors Determine Macrophage Phenotype Polarization. Mediat. Inflamm. 2013, 2013, 731023. [Google Scholar] [CrossRef]
- Petro, T.M. IFN Regulatory Factor 3 in Health and Disease. J. Immunol. 2020, 205, 1981–1989. [Google Scholar] [CrossRef]
- Petrasek, J.; Dolganiuc, A.; Csak, T.; Nath, B.; Hritz, I.; Kodys, K.; Catalano, D.; Kurt-Jones, E.; Mandrekar, P.; Szabo, G. Interferon Regulatory Factor 3 and Type I Interferons Are Protective in Alcoholic Liver Injury in Mice by Way of Crosstalk of Parenchymal and Myeloid Cells. Hepatology 2011, 53, 649–660. [Google Scholar] [CrossRef]
- Iracheta-Vellve, A.; Petrasek, J.; Gyongyosi, B.; Satishchandran, A.; Lowe, P.; Kodys, K.; Catalano, D.; Calenda, C.D.; Kurt-Jones, E.A.; Fitzgerald, K.A.; et al. Endoplasmic Reticulum Stress-Induced Hepatocellular Death Pathways Mediate Liver Injury and Fibrosis via Stimulator of Interferon Genes. J. Biol. Chem. 2016, 291, 26794–26805. [Google Scholar] [CrossRef]
- Sanz-Garcia, C.; McMullen, M.R.; Chattopadhyay, S.; Roychowdhury, S.; Sen, G.; Nagy, L.E. Non-transcriptional Activity of Interferon Regulatory Factor 3 Protects Mice From High-Fat Diet-Induced Liver Injury. Hepatol. Commun. 2019, 3, 1626–1641. [Google Scholar] [CrossRef]
- Yanai, H.; Chiba, S.; Hangai, S.; Kometani, K.; Inoue, A.; Kimura, Y.; Abe, T.; Kiyonari, H.; Nishio, J.; Taguchi-Atarashi, N.; et al. Revisiting the Role of IRF3 in Inflammation and Immunity by Conditional and Specifically Targeted Gene Ablation in Mice. Proc. Natl. Acad. Sci. USA 2018, 115, 5253–5258. [Google Scholar] [CrossRef]
- Farlik, M.; Reutterer, B.; Schindler, C.; Greten, F.; Vogl, C.; Müller, M.; Decker, T. Nonconventional Initiation Complex Assembly by STAT and NF-ΚB Transcription Factors Regulates Nitric Oxide Synthase Expression. Immunity 2010, 33, 25–34. [Google Scholar] [CrossRef]
- Moore, T.C.; Petro, T.M. IRF3 and ERK MAP-Kinases Control Nitric Oxide Production from Macrophages in Response to Poly-I:C. FEBS Lett. 2013, 587, 3014–3020. [Google Scholar] [CrossRef]
- Freed, S.M.; Baldi, D.S.; Snow, J.A.; Athen, S.R.; Guinn, Z.P.; Pinkerton, T.S.; Petro, T.M.; Moore, T.C. MEK/ERK MAP Kinase Limits Poly I:C-Induced Antiviral Gene Expression in RAW264.7 Macrophages by Reducing Interferon-Beta Expression. FEBS Lett. 2021, 595, 2665–2674. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Protocol of Treatment | Target Knocked Down | Overall Impact on M1 Markers | Overall Impact on M2 Markers | Phenotype Enhanced |
|---|---|---|---|---|
| Transfection followed by polarization | EGR2 | NOS2  | ARG1  | M1 |
TNF-α  | IL-4  | |||
IL-6  | ||||
| Polarization followed by transfection | IRF3 | NOS2  | ARG1  | M2 |
TNF-α  | EGR2  | |||
IL-6  | ||||
| Transfection followed by polarization | IRF5 | NOS2  | ARG1  | M1 |
TNF-α  | EGR2  | |||
IL-6  | ||||
| Polarization followed by transfection | NOS2  | ARG1  | M2 | |
TNF-α  | EGR2  | |||
IL-6  | ||||
| Transfection followed by polarization | TLR4 | NOS2  | ARG1  | M2 |
TNF-α  | EGR2  | |||
IL-6  | ||||
| Polarization followed by transfection | NOS2  |  ARG1 ARG1 | M2 | |
TNF-α  | EGR2  | |||
IL-6  |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Halimani, N.; Nesterchuk, M.; Andreichenko, I.N.; Tsitrina, A.A.; Elchaninov, A.; Lokhonina, A.; Fatkhudinov, T.; Dashenkova, N.O.; Brezgina, V.; Zatsepin, T.S.; et al. Phenotypic Alteration of BMDM In Vitro Using Small Interfering RNA. Cells 2022, 11, 2498. https://doi.org/10.3390/cells11162498
Halimani N, Nesterchuk M, Andreichenko IN, Tsitrina AA, Elchaninov A, Lokhonina A, Fatkhudinov T, Dashenkova NO, Brezgina V, Zatsepin TS, et al. Phenotypic Alteration of BMDM In Vitro Using Small Interfering RNA. Cells. 2022; 11(16):2498. https://doi.org/10.3390/cells11162498
Chicago/Turabian StyleHalimani, Noreen, Mikhail Nesterchuk, Irina N. Andreichenko, Alexandra A. Tsitrina, Andrey Elchaninov, Anastasia Lokhonina, Timur Fatkhudinov, Nataliya O. Dashenkova, Vera Brezgina, Timofei S. Zatsepin, and et al. 2022. "Phenotypic Alteration of BMDM In Vitro Using Small Interfering RNA" Cells 11, no. 16: 2498. https://doi.org/10.3390/cells11162498
APA StyleHalimani, N., Nesterchuk, M., Andreichenko, I. N., Tsitrina, A. A., Elchaninov, A., Lokhonina, A., Fatkhudinov, T., Dashenkova, N. O., Brezgina, V., Zatsepin, T. S., Mikaelyan, A. S., & Kotelevtsev, Y. V. (2022). Phenotypic Alteration of BMDM In Vitro Using Small Interfering RNA. Cells, 11(16), 2498. https://doi.org/10.3390/cells11162498