
Coding and Noncoding Genes Involved in Atrophy and Compensatory Muscle Growth in Nile Tilapia


Abstract
:1. Introduction
2. Materials and Methods
2.1. Ethics Declarations
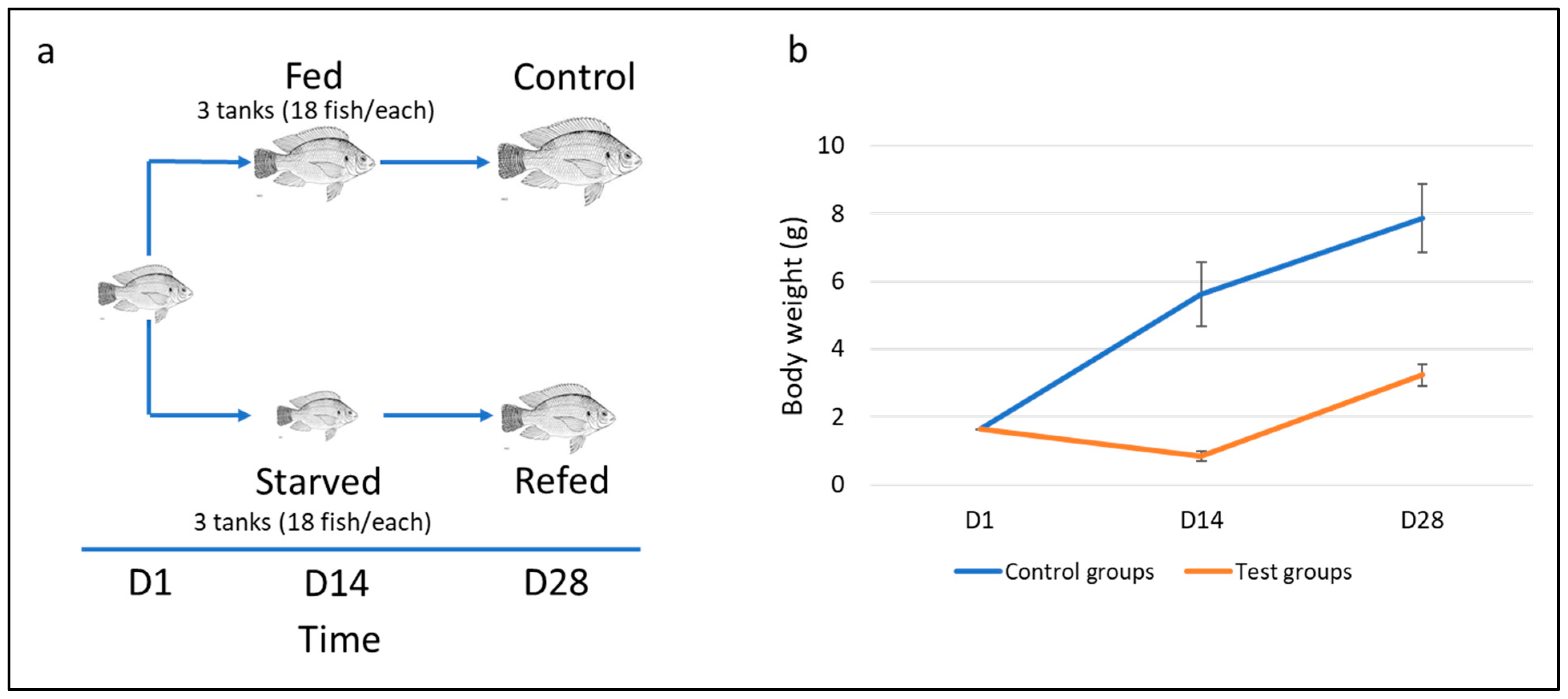
2.2. Experimental Design
2.3. RNA Extraction, Library Preparation, and Sequencing
2.4. Prediction of Novel lncRNAs
2.5. Differential Gene Expression Analyses
2.6. MicroRNA Target Prediction and Functional Enrichment Analyses
3. Results
3.1. Fish Compensatory Growth Following Fasting-Refeeding Schedule
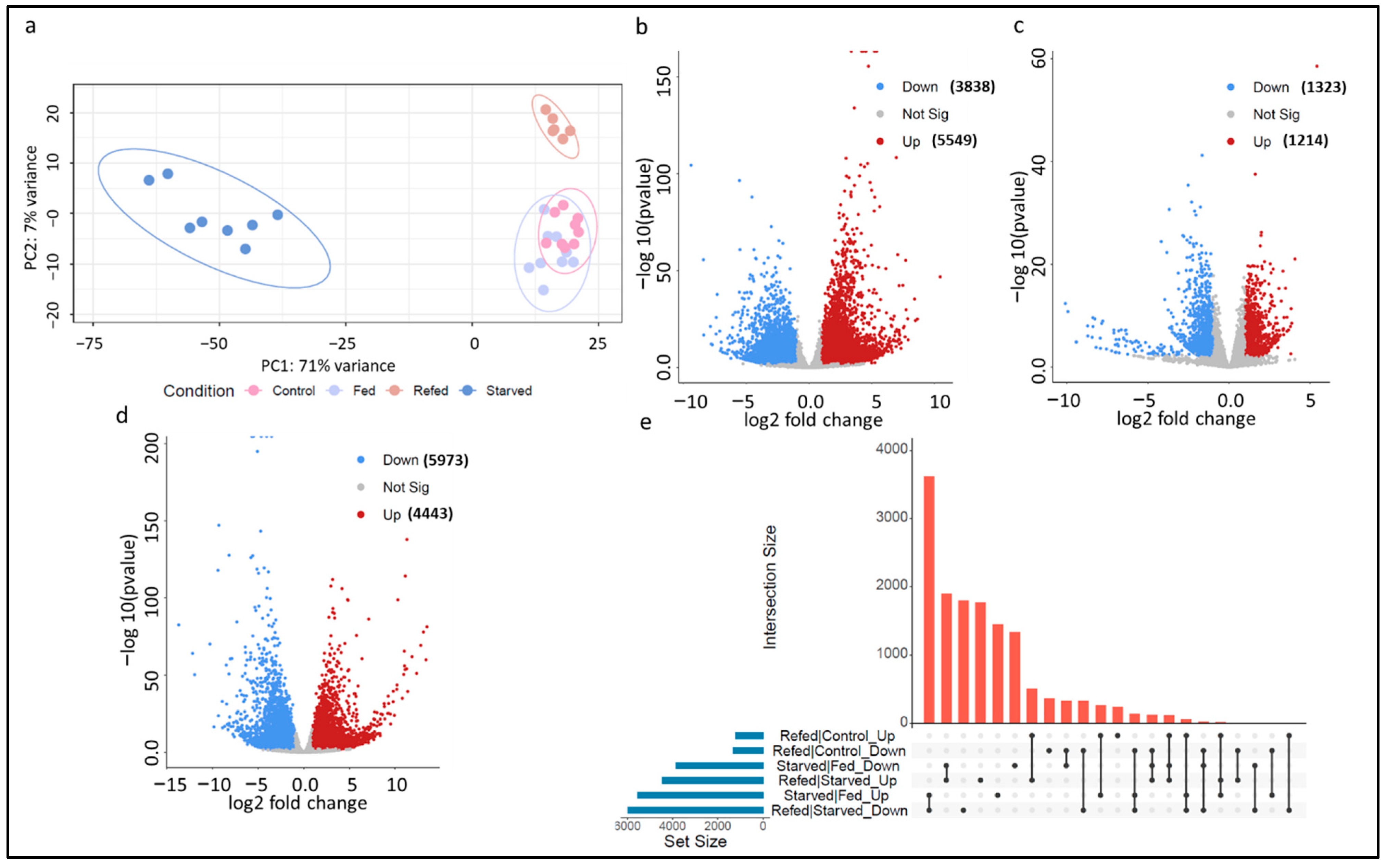
3.2. Muscle Transcriptome Sequencing and Data Processing
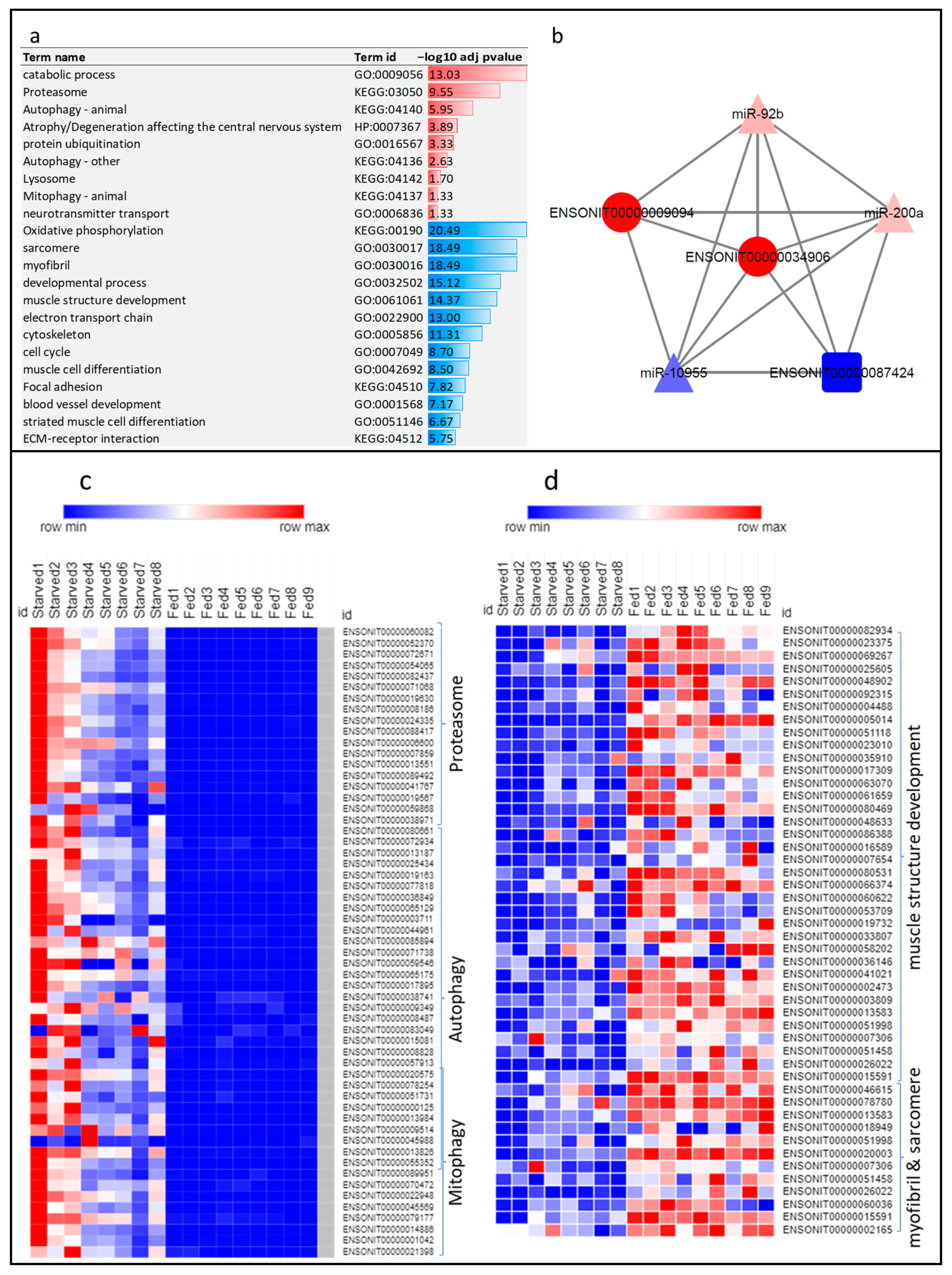
3.3. Muscle Atrophy Mediated by Ubiquitin-Proteasome and Autophagy-Lysosome Systems Accompanied by Maintenance of the Nervous System
3.4. ‘LncRNA-mRNA-microRNA’ Interactome in Atrophying Skeletal Muscle
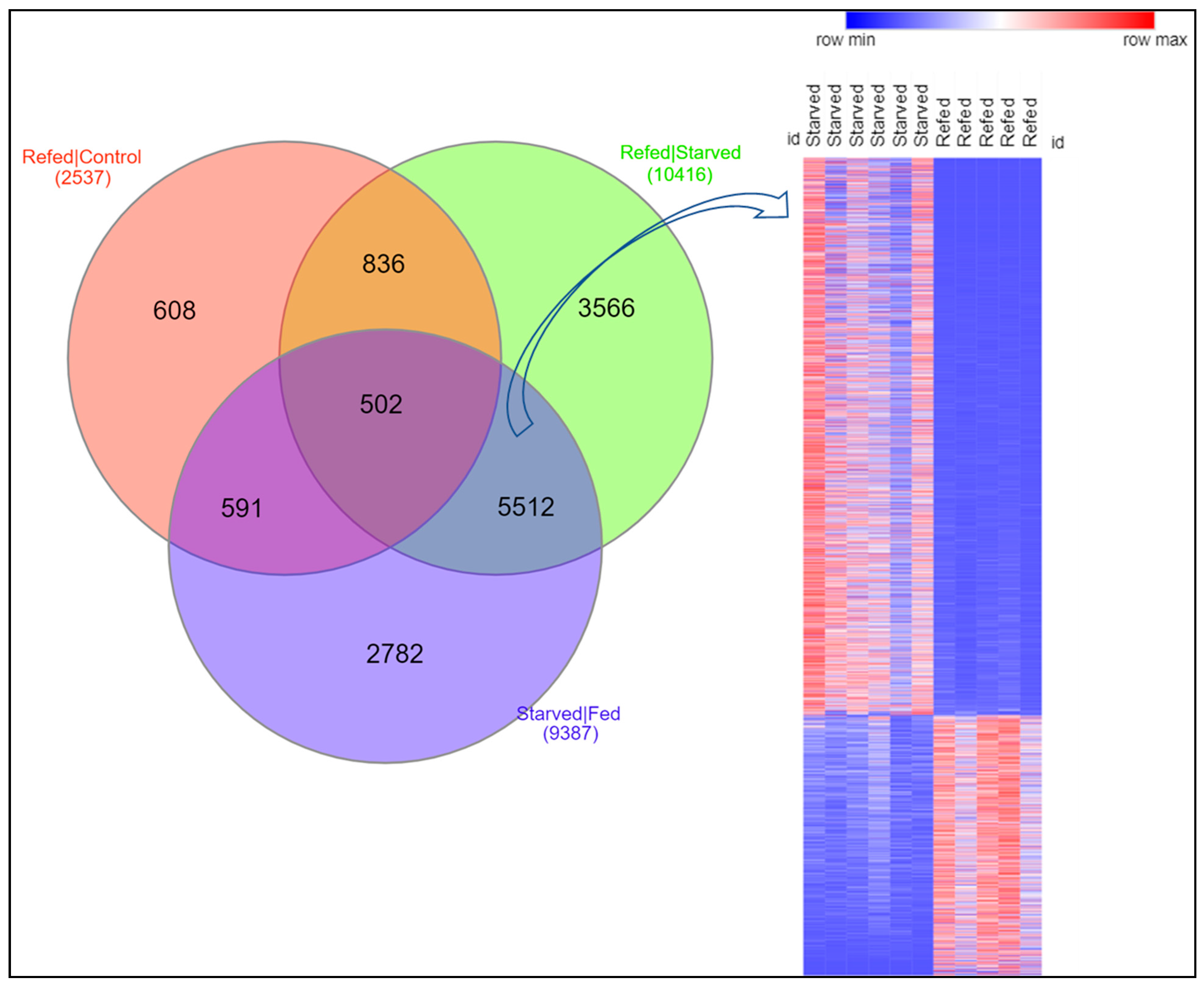
3.5. Suppression of Catabolic Processes and Induction of Muscle Hypertrophy upon Refeeding
3.6. First Cluster: Transcripts Whose Expression Levels in Refed Fish Were Restored to Normal Levels
3.7. Second Cluster: Transcripts Whose Expression Levels in Refed Fish Exceeded Normal Growth Values
3.8. Third Cluster: Transcripts Whose Expression Levels in Refed Fish Were Less than Normal Growth Values
4. Discussion and Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- da Silva-Gomes, R.N.; Gabriel Kuniyoshi, M.L.; Oliveira da Silva Duran, B.; Thomazini Zanella, B.T.; Paccielli Freire, P.; Gutierrez de Paula, T.; Fantinatti, B.E.D.A.; Salomão, R.A.S.; Carvalho, R.F.; Santos, L.D.; et al. Prolonged fasting followed by refeeding modifies proteome profile and parvalbumin expression in the fast-twitch muscle of pacu (Piaractus mesopotamicus). PLoS ONE 2019, 14, e0225864. [Google Scholar] [CrossRef] [PubMed]
- Braun, T.; Gautel, M. Transcriptional mechanisms regulating skeletal muscle differentiation, growth and homeostasis. Nat. Rev. Mol. Cell. Biol. 2011, 12, 349–361. [Google Scholar] [CrossRef] [PubMed]
- Rowlerson, A.M.; Veggetti, A. Cellular mechanisms of post-embryonic muscle growth in aquaculture species. Fish Physiol. 2001, 18, 103–140. [Google Scholar]
- Johnston, I.A.; Bower, N.I.; Macqueen, D.J. Growth and the regulation of myotomal muscle mass in teleost fish. J. Exp. Biol. 2011, 214, 1617–1628. [Google Scholar] [CrossRef] [PubMed]
- Gao, Y.; Lee, J.-Y. Compensatory Responses of Nile Tilapia Oreochromis niloticus under Different Feed-Deprivation Regimes. Fish. Aquat. Sci. 2012, 15, 305–311. [Google Scholar] [CrossRef]
- Gallardo-Collí, A.; Pérez-Fuentes, M.; Pérez-Rostro, C.I.; Hernández-Vergara, M.P. Compensatory growth of Nile tilapia Oreochromis niloticus, L. subjected to cyclic periods of feed restriction and feeding in a biofloc system. Aquac. Res. 2020, 51, 1813–1823. [Google Scholar] [CrossRef]
- Ali, M.; Nicieza, A.; Wootton, R.J. Compensatory growth in fishes: A response to growth depression. Fish Fish. 2003, 4, 147–190. [Google Scholar] [CrossRef]
- Hayward, R.S.; Noltie, D.B.; Wang, N. Use of Compensatory Growth to Double Hybrid Sunfish Growth Rates. Trans. Am. Fish. Soc. 1997, 126, 316–322. [Google Scholar] [CrossRef]
- Sevgili, H.; Hoşsu, B.; Emre, Y.; Kanyılmaz, M. Compensatory growth after various levels of dietary protein restriction in rainbow trout, Oncorhynchus mykiss. Aquaculture 2012, 344–349, 126–134. [Google Scholar] [CrossRef]
- Turano, M.J.; Borski, R.J.; Daniels, H.V. Effects of cyclic feeding on compensatory growth of hybrid striped bass (Morone chrysops×M. saxitilis) foodfish and water quality in production ponds. Aquac. Res. 2008, 39, 1514–1523. [Google Scholar] [CrossRef]
- Yengkokpam, S.; Sahu, N.P.; Pal, A.K.; Debnath, D.; Kumar, S.; Jain, K.K. Compensatory growth, feed intake and body composition ofLabeo rohitafingerlings following feed deprivation. Aquac. Nutr. 2014, 20, 101–108. [Google Scholar] [CrossRef]
- Gonet, T. Problem of moral choice in current medicine. Arch. Hist. Filoz. Med. 1988, 51, 1–15. [Google Scholar]
- Cho, S.-H. Effects of Alternate-Week Feeding Strategies on Growth and Feed Efficiency Ratio of Juvenile Nile Tilapia Oreochromis niloticus in a Recirculating System. Fish. Aquat. Sci. 2005, 8, 128–131. [Google Scholar] [CrossRef]
- Wang, Y.; Li, C.; Qin, J.G.; Han, H. Cyclical feed deprivation and refeeding fails to enhance compensatory growth in Nile tilapia, Oreochromis niloticus L. Aquac. Res. 2009, 40, 204–210. [Google Scholar] [CrossRef]
- Elbialy, Z.I.; Gamal, S.; Al-Hawary, I.I.; Shukry, M.; Salah, A.S.; Aboshosha, A.A.; Assar, D.H. Exploring the impacts of different fasting and refeeding regimes on Nile tilapia (Oreochromis niloticus L.): Growth performance, histopathological study, and expression levels of some muscle growth-related genes. Fish Physiol. Biochem. 2022. [Google Scholar] [CrossRef]
- Won, E.T.; Borski, R.J. Endocrine regulation of compensatory growth in fish. Front. Endocrinol. 2013, 4, 74. [Google Scholar] [CrossRef]
- Liu, W.; Lu, X.; Jiang, M.; Wu, F.; Tian, J.; Yang, C.; Yu, L.; Wen, H. Effects of dietary manipulation on compensatory growth of juvenile genetically improved farmed tilapia (Oreochromis niloticus). Fish Physiol. Biochem. 2019, 45, 21–32. [Google Scholar] [CrossRef]
- Hvas, M.; Nilsson, J.; Vågseth, T.; Nola, V.; Fjelldal, P.G.; Hansen, T.J.; Oppedal, F.; Stien, L.H.; Folkedal, O. Full compensatory growth before harvest and no impact on fish welfare in Atlantic salmon after an 8-week fasting period. Aquaculture 2022, 546, 737415. [Google Scholar] [CrossRef]
- Reigh, R.C.; Williams, M.B.; Jacob, B.J. Influence of repetitive periods of fasting and satiation feeding on growth and production characteristics of channel catfish, Ictalurus punctatus. Aquaculture 2006, 254, 506–516. [Google Scholar] [CrossRef]
- Xie, S.; Zhu, X.; Cui, Y.; Wootton, R.J.; Lei, W.; Yang, Y. Compensatory growth in the gibel carp following feed deprivation: Temporal patterns in growth, nutrient deposition, feed intake and body composition. J. Fish Biol. 2001, 58, 999–1009. [Google Scholar] [CrossRef]
- Tian, X.; Fang, J.; Dong, S. Effects of starvation and recovery on the growth, metabolism and energy budget of juvenile tongue sole (Cynoglossus semilaevis). Aquaculture 2010, 310, 122–129. [Google Scholar] [CrossRef]
- Yang, Y.; Zhou, H.; Hou, L.; Xing, K.; Shu, H. Transcriptional profiling of skeletal muscle reveals starvation response and compensatory growth in Spinibarbus hollandi. BMC Genom. 2019, 20, 938. [Google Scholar] [CrossRef] [PubMed]
- Russell, N.R.; Wootton, R.J. Appetite and growth compensation in the European minnow, Phoxinus phoxinus (Cyprinidae), following short periods of food restriction. Environ. Biol. Fishes 1992, 34, 277–285. [Google Scholar] [CrossRef]
- Tian, X.; Qin, J.G. A single phase of food deprivation provoked compensatory growth in barramundi Lates calcarifer. Aquaculture 2003, 224, 169–179. [Google Scholar] [CrossRef]
- Rescan, P.Y.; Montfort, J.; Rallière, C.; Le Cam, A.; Esquerré, D.; Hugot, K. Dynamic gene expression in fish muscle during recovery growth induced by a fasting-refeeding schedule. BMC Genom. 2007, 8, 438. [Google Scholar] [CrossRef] [PubMed]
- Mendez, K.N.; Zuloaga, R.; Valenzuela, C.A.; Bastias-Molina, M.; Meneses, C.; Vizoso, P.; Valdés, J.A.; Molina, A. RNA-seq analysis of compensatory growth in the skeletal muscle of fine flounder (Paralichthys adspersus). Aquaculture 2018, 490, 270–280. [Google Scholar] [CrossRef]
- He, L.; Pei, Y.; Jiang, Y.; Li, Y.; Liao, L.; Zhu, Z.; Wang, Y. Global gene expression patterns of grass carp following compensatory growth. BMC Genom. 2015, 16, 184. [Google Scholar] [CrossRef] [PubMed]
- Paneru, B.; Ali, A.; Al-Tobasei, R.; Kenney, B.; Salem, M. Crosstalk among lncRNAs, microRNAs and mRNAs in the muscle ‘degradome’ of rainbow trout. Sci. Rep. 2018, 8, 8416. [Google Scholar] [CrossRef]
- Chen, J.F.; Mandel, E.M.; Thomson, J.M.; Wu, Q.; Callis, T.E.; Hammond, S.M.; Conlon, F.L.; Wang, D.-Z. The role of microRNA-1 and microRNA-133 in skeletal muscle proliferation and differentiation. Nat. Genet. 2006, 38, 228–233. [Google Scholar] [CrossRef]
- McCarthy, J.J.; Esser, K.A. MicroRNA-1 and microRNA-133a expression are decreased during skeletal muscle hypertrophy. J. Appl. Physiol. (1985) 2007, 102, 306–313. [Google Scholar] [CrossRef]
- Liu, C.; Chen, M.; Wang, M.; Pi, W.; Li, N.; Meng, Q. MiR-18a regulates myoblasts proliferation by targeting Fgf1. PLoS ONE 2018, 13, e0201551. [Google Scholar] [CrossRef]
- Hudson, M.B.; Woodworth-Hobbs, M.E.; Zheng, B.; Rahnert, J.A.; Blount, M.A.; Gooch, J.L.; Searles, C.D.; Price, S.R. miR-23a is decreased during muscle atrophy by a mechanism that includes calcineurin signaling and exosome-mediated export. Am. J. Physiol. Cell. Physiol. 2014, 306, C551–C558. [Google Scholar] [CrossRef]
- Hou, L.; Xu, J.; Jiao, Y.; Li, H.; Pan, Z.; Duan, J.; Gu, T.; Hu, C.; Wang, C. MiR-27b Promotes Muscle Development by Inhibiting MDFI Expression. Cell. Physiol. Biochem. 2018, 46, 2271–2283. [Google Scholar] [CrossRef]
- Li, J.; Chan, M.C.; Yu, Y.; Bei, Y.; Chen, P.; Zhou, Q.; Cheng, L.; Chen, L.; Ziegler, O.; Rowe, G.; et al. miR-29b contributes to multiple types of muscle atrophy. Nat. Commun. 2017, 8, 15201. [Google Scholar] [CrossRef]
- Taetzsch, T.; Shapiro, D.; Eldosougi, R.; Myers, T.; Settlage, R.E.; Valdez, G. The microRNA miR-133b functions to slow Duchenne muscular dystrophy pathogenesis. J. Physiol. 2021, 599, 171–192. [Google Scholar] [CrossRef]
- Antoniou, A.; Mastroyiannopoulos, N.P.; Uney, J.B.; Phylactou, L.A. miR-186 inhibits muscle cell differentiation through myogenin regulation. J. Biol. Chem. 2014, 289, 3923–3935. [Google Scholar] [CrossRef]
- Kim, H.K.; Lee, Y.S.; Sivaprasad, U.; Malhotra, A.; Dutta, A. Muscle-specific microRNA miR-206 promotes muscle differentiation. J. Cell. Biol. 2006, 174, 677–687. [Google Scholar] [CrossRef]
- Chen, J.F.; Tao, Y.; Li, J.; Deng, Z.; Yan, Z.; Xiao, X.; Wang, D.-Z. microRNA-1 and microRNA-206 regulate skeletal muscle satellite cell proliferation and differentiation by repressing Pax7. J. Cell. Biol. 2010, 190, 867–879. [Google Scholar] [CrossRef]
- Yan, B.; Zhu, C.D.; Guo, J.T.; Zhao, L.H.; Zhao, J.L. miR-206 regulates the growth of the teleost tilapia (Oreochromis niloticus) through the modulation of IGF-1 gene expression. J. Exp. Biol. 2013, 216, 1265–1269. [Google Scholar] [CrossRef]
- Yan, B.; Guo, J.T.; Zhu, C.D.; Zhao, L.H.; Zhao, J.L. miR-203b: A novel regulator of MyoD expression in tilapia skeletal muscle. J. Exp. Biol. 2013, 216, 447–451. [Google Scholar] [CrossRef]
- Zhao, Z.; Yu, X.; Jia, J.; Yang, G.; Sun, C.; Li, W. miR-181b-5p May Regulate Muscle Growth in Tilapia by Targeting Myostatin b. Front. Endocrinol. 2019, 10, 812. [Google Scholar] [CrossRef] [PubMed]
- Huang, C.W.; Li, Y.H.; Hu, S.Y.; Chi, J.R.; Lin, G.H.; Lin, C.C.; Gong, H.Y.; Chen, J.Y.; Chen, R.H.; Chang, S.J.; et al. Differential expression patterns of growth-related microRNAs in the skeletal muscle of Nile tilapia (Oreochromis niloticus). J. Anim. Sci. 2012, 90, 4266–4279. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; An, X.; Bao, L.; Li, Y.; Pan, Y.; He, J.; Liu, L.; Zhu, X.; Zhang, J.; Cheng, J.; et al. MiR-125a-3p-KLF15-BCAA Regulates the Skeletal Muscle Branched-Chain Amino Acid Metabolism in Nile Tilapia (Oreochromis niloticus) During Starvation. Front. Genet. 2020, 11, 852. [Google Scholar] [CrossRef] [PubMed]
- Zhu, X.; Chu, W.Y.; Wu, P.; Yi, T.; Chen, T.; Zhang, J.S. MicroRNA signature in response to nutrient restriction and re-feeding in fast skeletal muscle of grass carp (Ctenopharyngodon idella). Dongwuxue Yanjiu 2014, 35, 404–410. [Google Scholar]
- Ali, A.; Al-Tobasei, R.; Kenney, B.; Leeds, T.D.; Salem, M. Integrated analysis of lncRNA and mRNA expression in rainbow trout families showing variation in muscle growth and fillet quality traits. Sci. Rep. 2018, 8, 12111. [Google Scholar] [CrossRef]
- Cesana, M.; Cacchiarelli, D.; Legnini, I.; Santini, T.; Sthandier, O.; Chinappi, M.; Tramontano, A.; Bozzoni, I. A long noncoding RNA controls muscle differentiation by functioning as a competing endogenous RNA. Cell 2011, 147, 358–369. [Google Scholar] [CrossRef]
- Zhu, M.; Liu, J.; Xiao, J.; Yang, L.; Cai, M.; Shen, H.; Chen, X.; Ma, Y.; Hu, S.; Wang, Z.; et al. Lnc-mg is a long non-coding RNA that promotes myogenesis. Nat. Commun. 2017, 8, 14718. [Google Scholar] [CrossRef]
- Sun, L.; Si, M.; Liu, X.; Choi, J.M.; Wang, Y.; Thomas, S.S.; Peng, H.; Hu, Z. Long-noncoding RNA Atrolnc-1 promotes muscle wasting in mice with chronic kidney disease. J. Cachexia Sarcopenia Muscle 2018, 9, 962–974. [Google Scholar] [CrossRef]
- Sert, N.P.d.; Hurst, V.; Ahluwalia, A.; Alam, S.; Avey, M.T.; Baker, M.; Browne, W.J.; Clark, A.; Cuthill, I.C.; Dirnagl, U.; et al. The ARRIVE guidelines 2.0: Updated guidelines for reporting animal research. PLoS Biol. 2020, 18, e3000410. [Google Scholar]
- Shaalan, W.M.; El-Hameid, N.A.A.; El-Serafy, S.S.; Salem, M. Expressions and characterization of MuRFs, Atrogin-1, F-box25 genes in tilapia, Oreochromis niloticus, in response to starvation. Fish Physiol. Biochem. 2019, 45, 1321–1330. [Google Scholar] [CrossRef]
- Al-Tobasei, R.; Paneru, B.; Salem, M. Genome-Wide Discovery of Long Non-Coding RNAs in Rainbow Trout. PLoS ONE 2016, 11, e0148940. [Google Scholar] [CrossRef]
- Raudvere, U.; Kolberg, L.; Kuzmin, I.; Arak, T.; Adler, P.; Peterson, H.; Vito, J. g:Profiler: A web server for functional enrichment analysis and conversions of gene lists (2019 update). Nucleic Acids Res. 2019, 47, W191–W198. [Google Scholar] [CrossRef]
- Gaetani, L.; Blennow, K.; Calabresi, P.; di Filippo, M.; Parnetti, L.; Zetterberg, H. Neurofilament light chain as a biomarker in neurological disorders. J. Neurol. Neurosurg. Psychiatry 2019, 90, 870–881. [Google Scholar] [CrossRef]
- Jeanne, M.; Demory, H.; Moutal, A.; Vuillaume, M.L.; Blesson, S.; Thepault, R.A.; Marouillat, S.; Halewa, J.; Maas, S.M.; Motazacker, M.M.; et al. Missense variants in DPYSL5 cause a neurodevelopmental disorder with corpus callosum agenesis and cerebellar abnormalities. Am. J. Hum. Genet. 2021, 108, 951–961. [Google Scholar] [CrossRef]
- Zhu, P.P.; Soderblom, C.; Tao-Cheng, J.H.; Stadler, J.; Blackstone, C. SPG3A protein atlastin-1 is enriched in growth cones and promotes axon elongation during neuronal development. Hum. Mol. Genet. 2006, 15, 1343–1353. [Google Scholar] [CrossRef]
- Wang, X.H. MicroRNA in myogenesis and muscle atrophy. Curr. Opin. Clin. Nutr. Metab. Care 2013, 16, 258–266. [Google Scholar] [CrossRef]
- Georgantas, R.W.; Streicher, K.; Greenberg, S.A.; Greenlees, L.M.; Zhu, W.; Brohawn, P.Z.; Higgs, B.; Czapiga, M.; Morehouse, C.A.; Amato, A.; et al. Inhibition of myogenic microRNAs 1, 133, and 206 by inflammatory cytokines links inflammation and muscle degeneration in adult inflammatory myopathies. Arthritis Rheumatol. 2014, 66, 1022–1033. [Google Scholar] [CrossRef]
- Yamaguchi, J.; Suzuki, C.; Nanao, T.; Kakuta, S.; Ozawa, K.; Tanida, I.; Saitoh, T.; Sunabori, T.; Komatsu, M.; Tanaka, K.; et al. Atg9a deficiency causes axon-specific lesions including neuronal circuit dysgenesis. Autophagy 2018, 14, 764–777. [Google Scholar] [CrossRef]
- Li, Y.; Zhang, Y.; Wang, L.; Wang, P.; Xue, Y.; Li, X.; Qiao, X.; Zhang, X.; Xu, T.; Liu, G.-H.; et al. Autophagy impairment mediated by S-nitrosation of ATG4B leads to neurotoxicity in response to hyperglycemia. Autophagy 2017, 13, 1145–1160. [Google Scholar] [CrossRef]
- Eshel, O.; Shirak, A.; Dor, L.; Band, M.; Zak, T.; Markovich-Gordon, M.; Chalifa-Caspi, V.; Feldmesser, E.; Weller, I.J.; Seroussi, E.; et al. Identification of male-specific amh duplication, sexually differentially expressed genes and microRNAs at early embryonic development of Nile tilapia (Oreochromis niloticus). BMC Genom. 2014, 15, 774. [Google Scholar] [CrossRef]
- Liedtke, W.; Leman, E.E.; Fyffe, R.E.; Raine, C.S.; Schubart, U.K. Stathmin-deficient mice develop an age-dependent axonopathy of the central and peripheral nervous systems. Am. J. Pathol. 2002, 160, 469–480. [Google Scholar] [CrossRef]
- Cai, H.; Reim, K.; Varoqueaux, F.; Tapechum, S.; Hill, K.; Sorensen, J.B.; Brose, N.; Chow, R.H. Complexin II plays a positive role in Ca2+-triggered exocytosis by facilitating vesicle priming. Proc. Natl. Acad. Sci. USA 2008, 105, 19538–19543. [Google Scholar] [CrossRef] [PubMed]
- Shaker, M.R.; Kahtan, A.; Prasad, R.; Lee, J.H.; Pietrogrande, G.; Leeson, H.C.; Sun, W.; Wolvetang, E.J.; Slonchak, A. Neural Epidermal Growth Factor-Like Like Protein 2 Is Expressed in Human Oligodendroglial Cell Types. Front. Cell. Dev. Biol. 2022, 10, 803061. [Google Scholar] [CrossRef] [PubMed]
- Gomes, M.D.; Lecker, S.H.; Jagoe, R.T.; Navon, A.; Goldberg, A.L. Atrogin-1, a muscle-specific F-box protein highly expressed during muscle atrophy. Proc. Natl. Acad. Sci. USA 2001, 98, 14440–14445. [Google Scholar] [CrossRef]
- Rescan, P.Y.; le Cam, A.; Ralliere, C.; Montfort, J. Global gene expression in muscle from fasted/refed trout reveals up-regulation of genes promoting myofibre hypertrophy but not myofibre production. BMC Genom. 2017, 18, 447. [Google Scholar] [CrossRef]
- Ge, Y.; Chen, J. MicroRNAs in skeletal myogenesis. Cell Cycle 2011, 10, 441–448. [Google Scholar] [CrossRef]
- Bai, L.; Liang, R.; Yang, Y.; Hou, X.; Wang, Z.; Zhu, S.; Wang, C.; Tang, Z.; Li, K. MicroRNA-21 Regulates PI3K/Akt/mTOR Signaling by Targeting TGFbetaI during Skeletal Muscle Development in Pigs. PLoS ONE 2015, 10, e0119396. [Google Scholar] [CrossRef]
- Rescan, P.Y.; Montfort, J.; Fautrel, A.; Ralliere, C.; Lebret, V. Gene expression profiling of the hyperplastic growth zones of the late trout embryo myotome using laser capture microdissection and microarray analysis. BMC Genom. 2013, 14, 173. [Google Scholar] [CrossRef]
- Verbrugge, S.A.J.; Schonfelder, M.; Becker, L.; Nezhad, F.Y.; de Angelis, M.H.; Wackerhage, H. Genes Whose Gain or Loss-Of-Function Increases Skeletal Muscle Mass in Mice: A Systematic Literature Review. Front. Physiol. 2018, 9, 553. [Google Scholar] [CrossRef]
- Raffaello, A.; Milan, G.; Masiero, E.; Carnio, S.; Lee, D.; Lanfranchi, G.; Goldberg, A.L.; Sandri, M. JunB transcription factor maintains skeletal muscle mass and promotes hypertrophy. J. Cell. Biol. 2010, 191, 101–113. [Google Scholar] [CrossRef]
- Goll, D.E.; Thompson, V.F.; Taylor, R.G.; Ouali, A. The calpain system and skeletal muscle growth. Can. J. Anim. Sci. 1998, 78, 503–512. [Google Scholar] [CrossRef]
- Bruscoli, S.; Donato, V.; Velardi, E.; di Sante, M.; Migliorati, G.; Donato, R.; Riccardi, C. Glucocorticoid-induced leucine zipper (GILZ) and long GILZ inhibit myogenic differentiation and mediate anti-myogenic effects of glucocorticoids. J. Biol. Chem. 2010, 285, 10385–10396. [Google Scholar] [CrossRef]
- Marceca, G.P.; Nigita, G.; Calore, F.; Croce, C.M. MicroRNAs in Skeletal Muscle and Hints on Their Potential Role in Muscle Wasting During Cancer Cachexia. Front. Oncol. 2020, 10, 607196. [Google Scholar] [CrossRef]
- Falcone, G. A new role of miR-29c as a potent inducer of skeletal muscle hypertrophy. Acta Physiol. 2019, 226, e13320. [Google Scholar] [CrossRef]
- Shi, J.Y.; Chen, C.; Xu, X.; Lu, Q. miR-29a promotes pathological cardiac hypertrophy by targeting the PTEN/AKT/mTOR signalling pathway and suppressing autophagy. Acta Physiol. 2019, 227, e13323. [Google Scholar] [CrossRef]
- Leger, B.; Cartoni, R.; Praz, M.; Lamon, S.; Deriaz, O.; Crettenand, A.; Gobelet, C.; Rohmer, P.; Konzelmann, M.; Luthi, F.; et al. Akt signalling through GSK-3beta, mTOR and Foxo1 is involved in human skeletal muscle hypertrophy and atrophy. J. Physiol. 2006, 576, 923–933. [Google Scholar] [CrossRef]
- Cohen, A.W.; Park, D.S.; Woodman, S.E.; Williams, T.M.; Chandra, M.; Shirani, J.; de Souza, A.P.; Kitsis, R.N.; Russell, R.G.; Weiss, L.M.; et al. Caveolin-1 null mice develop cardiac hypertrophy with hyperactivation of p42/44 MAP kinase in cardiac fibroblasts. Am. J. Physiol. Cell. Physiol. 2003, 284, C457–C474. [Google Scholar] [CrossRef]
- Matsushima, S.; Kuroda, J.; Zhai, P.; Liu, T.; Ikeda, S.; Nagarajan, N.; Oka, S.-I.; Yokota, T.; Kinugawa, S.; Hsu, C.-P.; et al. Tyrosine kinase FYN negatively regulates NOX4 in cardiac remodeling. J. Clin. Investig. 2016, 126, 3403–3416. [Google Scholar] [CrossRef]
- Eguchi, A.; Eguchi, S.; Tilley, D.G. Unexpected cardiac hypertrophy by epidermal growth factor receptor silencing. Hypertension 2013, 61, e46. [Google Scholar] [CrossRef]
- Holness, M.J.; MacLennan, P.A.; Palmer, T.N.; Sugden, M.C. The disposition of carbohydrate between glycogenesis, lipogenesis and oxidation in liver during the starved-to-fed transition. Biochem. J. 1988, 252, 325–330. [Google Scholar] [CrossRef]
- Kuwajima, M.; Newgard, C.B.; Foster, D.W.; McGarry, J.D. Time course and significance of changes in hepatic fructose-2,6-bisphosphate levels during refeeding of fasted rats. J. Clin. Investig. 1984, 74, 1108–1111. [Google Scholar] [CrossRef] [PubMed]
- Scott, G.K.; Yau, C.; Becker, B.C.; Khateeb, S.; Mahoney, S.; Jensen, M.B.; Hann, B.; Cowen, B.J.; Pegan, S.D.; Benz, C.C. Targeting Mitochondrial Proline Dehydrogenase with a Suicide Inhibitor to Exploit Synthetic Lethal Interactions with p53 Upregulation and Glutaminase Inhibition. Mol. Cancer Ther. 2019, 18, 1374–1385. [Google Scholar] [CrossRef] [PubMed]
- Stamatikos, A.D.; Paton, C.M. Role of stearoyl-CoA desaturase-1 in skeletal muscle function and metabolism. Am. J. Physiol. Endocrinol. Metab. 2013, 305, E767–E775. [Google Scholar] [CrossRef] [PubMed]
- Johnston, I.A. Muscle development and growth: Potential implications for flesh quality in fish. Aquaculture 1999, 177, 99–115. [Google Scholar] [CrossRef]
- Montfort, J.; le Cam, A.; Gabillard, J.C.; Rescan, P.Y. Gene expression profiling of trout regenerating muscle reveals common transcriptional signatures with hyperplastic growth zones of the post-embryonic myotome. BMC Genom. 2016, 17, 810. [Google Scholar] [CrossRef] [PubMed]
- Krauss, R.S.; Cole, F.; Gaio, U.; Takaesu, G.; Zhang, W.; Kang, J.S. Close encounters: Regulation of vertebrate skeletal myogenesis by cell-cell contact. J. Cell. Sci. 2005, 118, 2355–2362. [Google Scholar] [CrossRef]
- Srinivas, B.P.; Woo, J.; Leong, W.Y.; Roy, S. A conserved molecular pathway mediates myoblast fusion in insects and vertebrates. Nat. Genet. 2007, 39, 781–786. [Google Scholar] [CrossRef]
- Powell, G.T.; Wright, G.J. Jamb and jamc are essential for vertebrate myocyte fusion. PLoS Biol. 2011, 9, e1001216. [Google Scholar] [CrossRef]
- Johansen, K.A.; Overturf, K. Alterations in expression of genes associated with muscle metabolism and growth during nutritional restriction and refeeding in rainbow trout. Comp. Biochem. Physiol. B Biochem. Mol. Biol. 2006, 144, 119–127. [Google Scholar] [CrossRef]
- Mallappa, C.; Hu, Y.J.; Shamulailatpam, P.; Tae, S.; Sif, S.; Imbalzano, A.N. The expression of myogenic microRNAs indirectly requires protein arginine methyltransferase (Prmt)5 but directly requires Prmt4. Nucleic Acids Res. 2011, 39, 1243–1255. [Google Scholar] [CrossRef]
- Nakada, S.; Ogasawara, R.; Kawada, S.; Maekawa, T.; Ishii, N. Correlation between Ribosome Biogenesis and the Magnitude of Hypertrophy in Overloaded Skeletal Muscle. PLoS ONE 2016, 11, e0147284. [Google Scholar] [CrossRef]
- Chaillou, T.; Kirby, T.J.; McCarthy, J.J. Ribosome biogenesis: Emerging evidence for a central role in the regulation of skeletal muscle mass. J. Cell. Physiol. 2014, 229, 1584–1594. [Google Scholar] [CrossRef]
- Senf, S.M.; Howard, T.M.; Ahn, B.; Ferreira, L.F.; Judge, A.R. Loss of the inducible Hsp70 delays the inflammatory response to skeletal muscle injury and severely impairs muscle regeneration. PLoS ONE 2013, 8, e62687. [Google Scholar] [CrossRef]
- Hitachi, K.; Tsuchida, K. Role of microRNAs in skeletal muscle hypertrophy. Front. Physiol. 2013, 4, 408. [Google Scholar] [CrossRef]
- Wehbe, N.; Nasser, S.A.; Pintus, G.; Badran, A.; Eid, A.H.; Baydoun, E. MicroRNAs in Cardiac Hypertrophy. Int. J. Mol. Sci. 2019, 20, 4714. [Google Scholar] [CrossRef]
- Kim, J.O.; Song, D.W.; Kwon, E.J.; Hong, S.E.; Song, H.K.; Min, C.K.; Kim, D.H. miR-185 plays an anti-hypertrophic role in the heart via multiple targets in the calcium-signaling pathways. PLoS ONE 2015, 10, e0122509. [Google Scholar] [CrossRef]
- Guess, M.G.; Barthel, K.K.; Harrison, B.C.; Leinwand, L.A. miR-30 family microRNAs regulate myogenic differentiation and provide negative feedback on the microRNA pathway. PLoS ONE 2015, 10, e0118229. [Google Scholar] [CrossRef]
- Kwon, I.; Jang, Y.; Cho, J.Y.; Jang, Y.C.; Lee, Y. Long-term resistance exercise-induced muscular hypertrophy is associated with autophagy modulation in rats. J. Physiol. Sci. 2018, 68, 269–280. [Google Scholar] [CrossRef]
- McPherron, A.C.; Lee, S.J. Double muscling in cattle due to mutations in the myostatin gene. Proc. Natl. Acad. Sci. USA 1997, 94, 12457–12461. [Google Scholar] [CrossRef]
- Nowak, J.; Archange, C.; Tardivel-Lacombe, J.; Pontarotti, P.; Pebusque, M.J.; Vaccaro, M.I.; Vetasco, G.; Dagorn, J.-C.; Iovanna, J.L. The TP53INP2 protein is required for autophagy in mammalian cells. Mol. Biol. Cell. 2009, 20, 870–881. [Google Scholar] [CrossRef]
- Yamamoto, H.; Williams, E.G.; Mouchiroud, L.; Canto, C.; Fan, W.; Downes, M.; Héligon, C.; Barish, G.D.; Desvergne, B.; Evans, R.; et al. NCoR1 is a conserved physiological modulator of muscle mass and oxidative function. Cell 2011, 147, 827–839. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Transcript Id | Annotation | Starved/Fed | Refed/Control | Correlation with Body Weight | |||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| RNA-Seq | qPCR | RNA-Seq | qPCR | ||||||||
| log2 fc | padj | log2 fc | p-Value | log2 fc | padj | log2 fc | p-Value | R | p-Value | ||
| ENSONIT00000034906 | F-box only protein 32 | 6.68 | 7.94 × 10−24 | 9.67 | 7.05 × 10−9 | −2.36 | 5.2 × 10−3 | −3.18 | 1.7 × 10−2 | −0.69 | 9.83 × 10−5 |
| ENSONIT00000080661 | autophagy related 2A | 7.51 | 1.18 × 10−19 | 3.29 | 1.93 × 10−7 | N/A | N/A | −1.87 | 8.5 × 10−3 | −0.66 | 2.2 × 10−4 |
| ENSONIT00000062024 | nuclear receptor corepressor 1 | 1.80 | 3.26 × 10−2 | 1.56 | 1.71 × 10−4 | −3.53 | 4.23 × 10−7 | −1.47 | 7.1 × 10−3 | −0.40 | 4.2 × 10−2 |
| ENSONIT00000072045 | neurofilament, light polypeptide b | 7.20 | 8.38 × 10−14 | 4.55 | 4.48 × 10−3 | N/A | N/A | N/A | N/A | −0.41 | 3.8 × 10−2 |
| ENSONIT00000022026 | tumor protein p53 inducible nuclear protein 2 | −3.34 | 2.26 × 10−38 | −3.64 | 1.09 × 10−5 | −2.67 | 2.98 × 10−12 | −3.14 | 4.7 × 10−5 | 0.82 | 2.36 × 10−7 |
| Transcript Id | Fold Change | p Value | Padj | Annotation |
|---|---|---|---|---|
| ENSONIT00000060950 | 3.12 | 2.00 × 10−5 | 2.88 × 10−4 | Transcription Factor Jun-B (AP-1) |
| ENSONIT00000039894 | 2.31 | 6.07 × 10−3 | 3.59 × 10−2 | Calpastatin |
| ENSONIT00000016158 | −2.03 | 6.12 × 10−6 | 1.03 × 10−4 | Nuclear receptor corepressor 1 |
| ENSONIT00000055439 | −3.07 | 2.30 × 10−3 | 1.64 × 10−2 | Diacylglycerol O-acyltransferase 1 |
| ENSONIT00000076842 | −3.53 | 6.51 × 10−8 | 1.92 × 10−6 | Bradykinin receptor B2 |
| ENSONIT00000045589 | −3.61 | 1.08 × 10−3 | 8.78 × 10−3 | Inhibin beta B chain |
| ENSONIT00000048569 | −3.76 | 5.74 × 10−6 | 9.70 × 10−5 | Myostatin |
| ENSONIT00000022026 | −6.36 | 2.2 × 10−14 | 2.98 × 10−12 | Tumor protein p53 inducible nuclear protein 2 |
| ENSONIT00000062024 | −11.55 | 1.19 × 10−8 | 4.23 × 10−7 | Nuclear receptor corepressor 1 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ali, A.; Shaalan, W.M.; Al-Tobasei, R.; Salem, M. Coding and Noncoding Genes Involved in Atrophy and Compensatory Muscle Growth in Nile Tilapia. Cells 2022, 11, 2504. https://doi.org/10.3390/cells11162504
Ali A, Shaalan WM, Al-Tobasei R, Salem M. Coding and Noncoding Genes Involved in Atrophy and Compensatory Muscle Growth in Nile Tilapia. Cells. 2022; 11(16):2504. https://doi.org/10.3390/cells11162504
Chicago/Turabian StyleAli, Ali, Walaa M. Shaalan, Rafet Al-Tobasei, and Mohamed Salem. 2022. "Coding and Noncoding Genes Involved in Atrophy and Compensatory Muscle Growth in Nile Tilapia" Cells 11, no. 16: 2504. https://doi.org/10.3390/cells11162504