
SARS-CoV-2 Spike Does Not Possess Intrinsic Superantigen-like Inflammatory Activity

 ,
,  ,
,  , and
, and 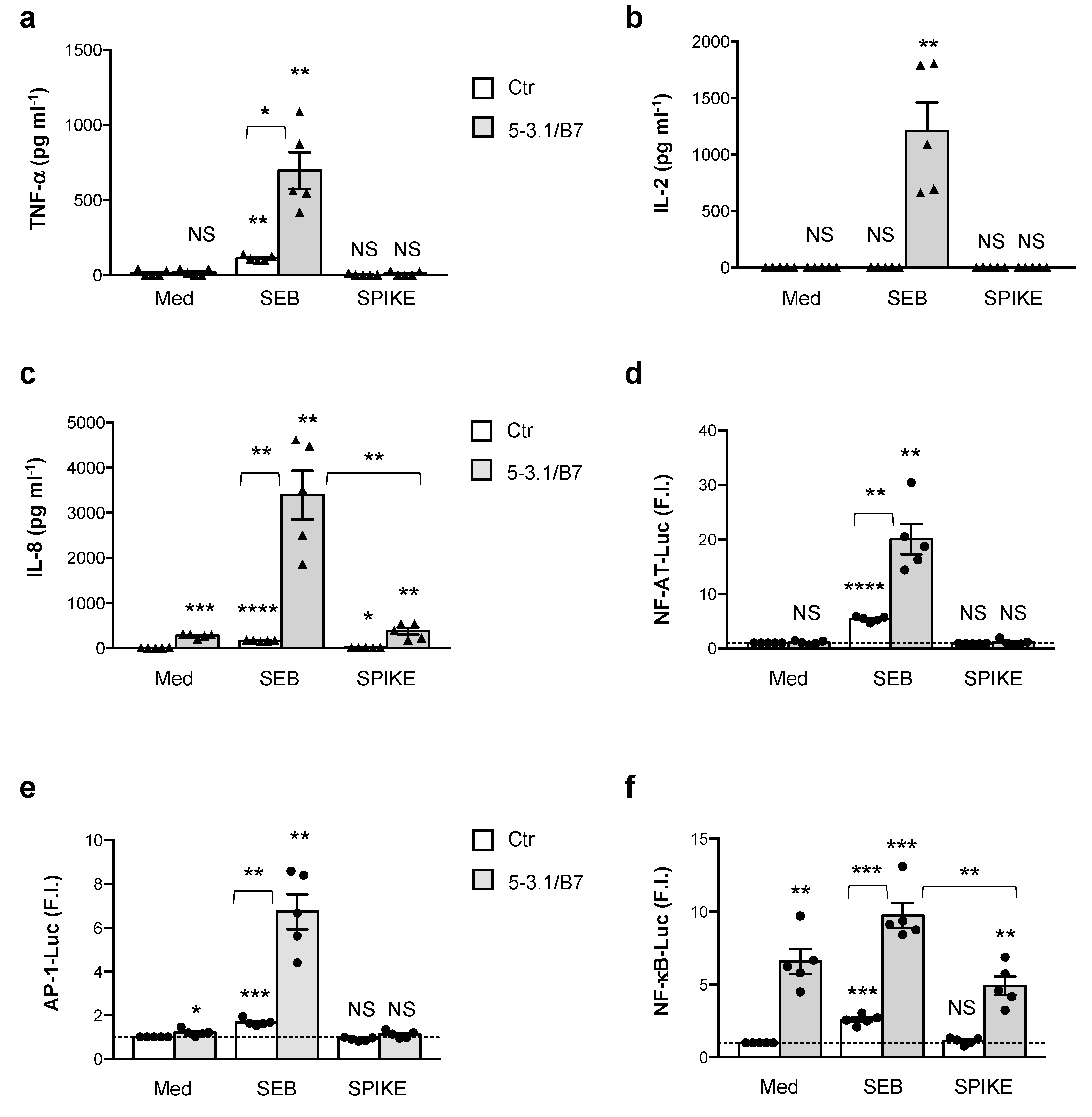{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Cells, Abs, and Reagents
2.2. Cytokine Production (ELISA)
2.3. Plasmids, Cell Transfection and Luciferase Assays
2.4. SDS-PAGE
2.5. Structural Modeling of the Putative Interactions between SARS-CoV-2 Spike and TCR as Well as between SEB and 6D3 Ab or CD28
2.6. Statistical Analysis
3. Results
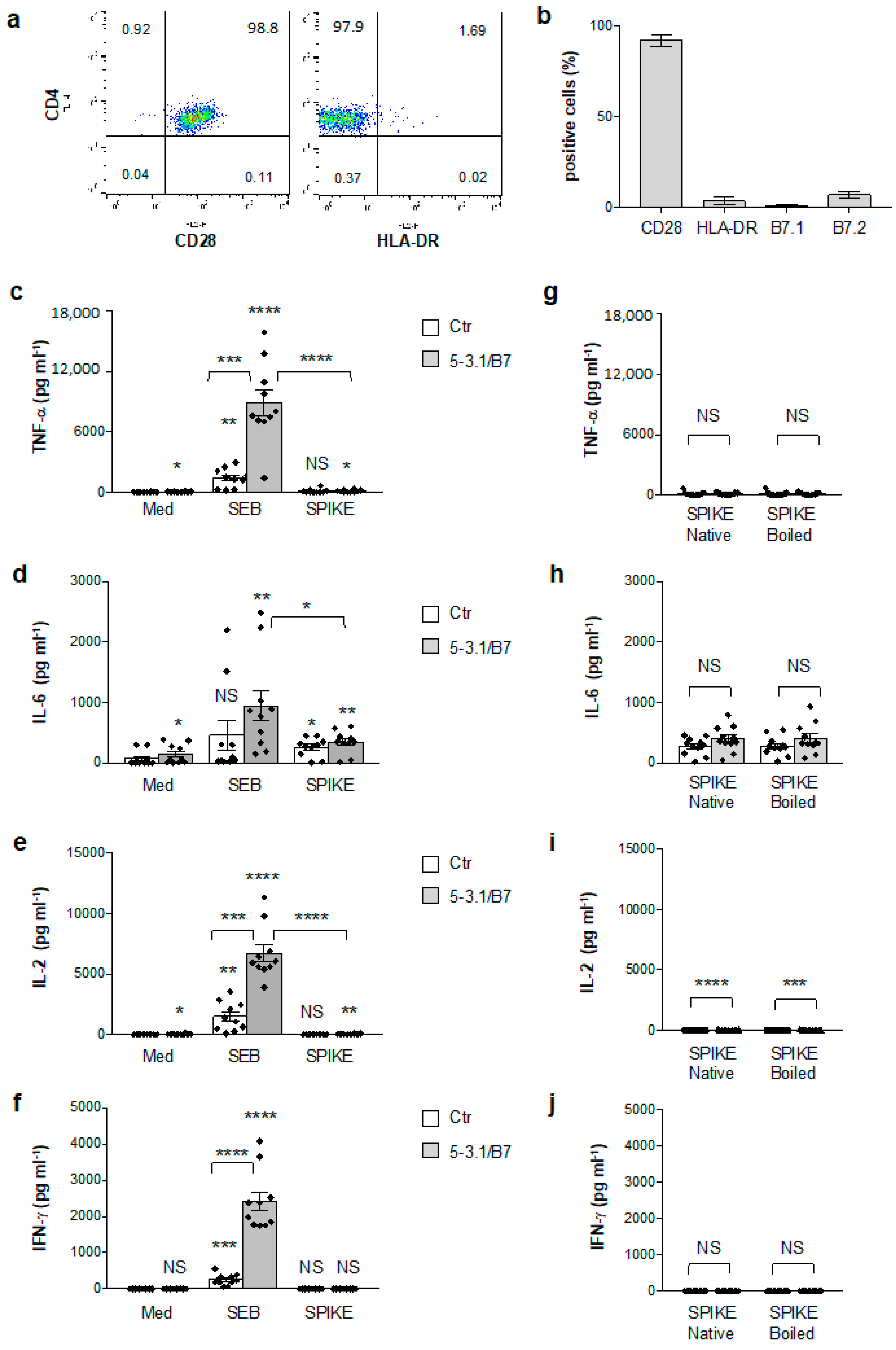
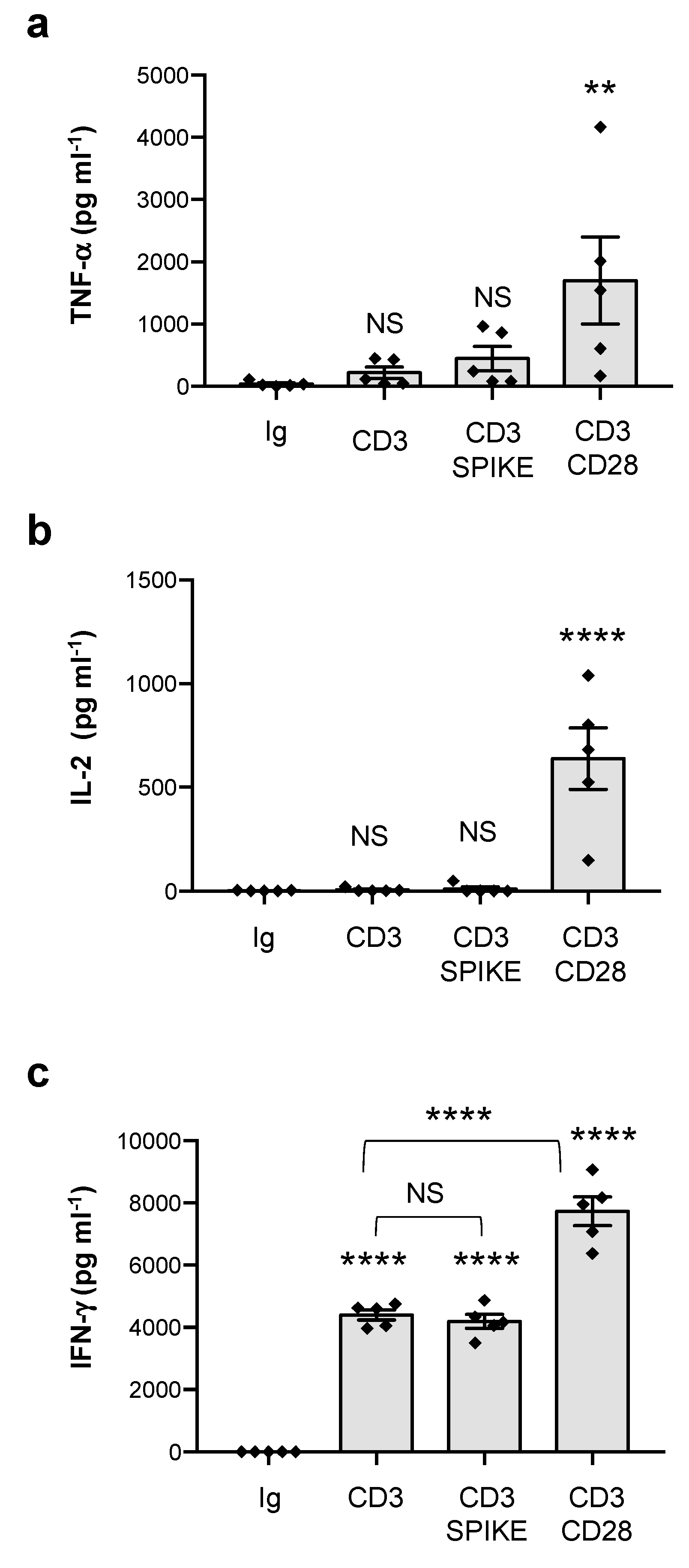
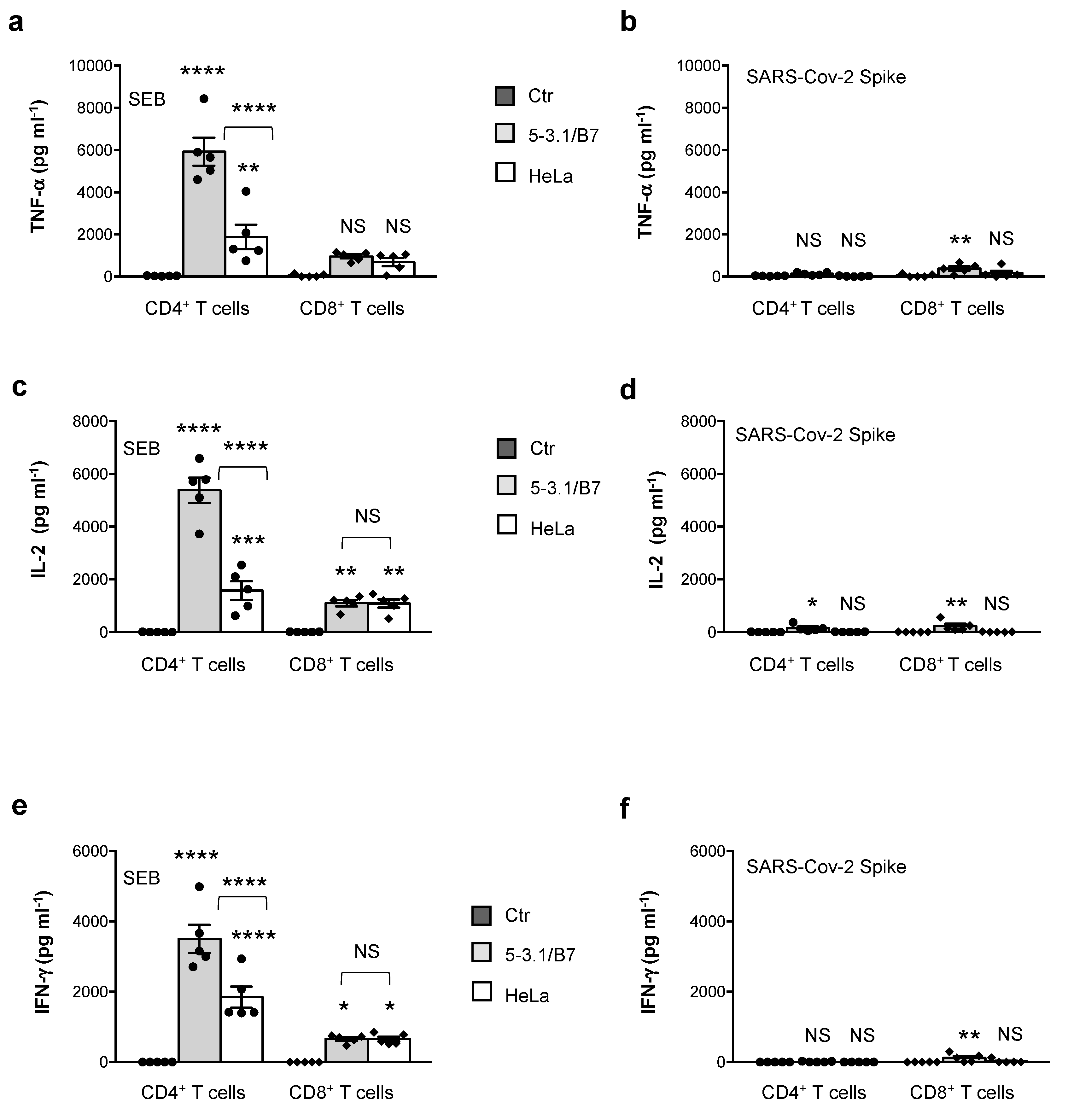
3.1. Analysis of SAg-like Inflammatory Activity of SARS-CoV-2 Spike Protein
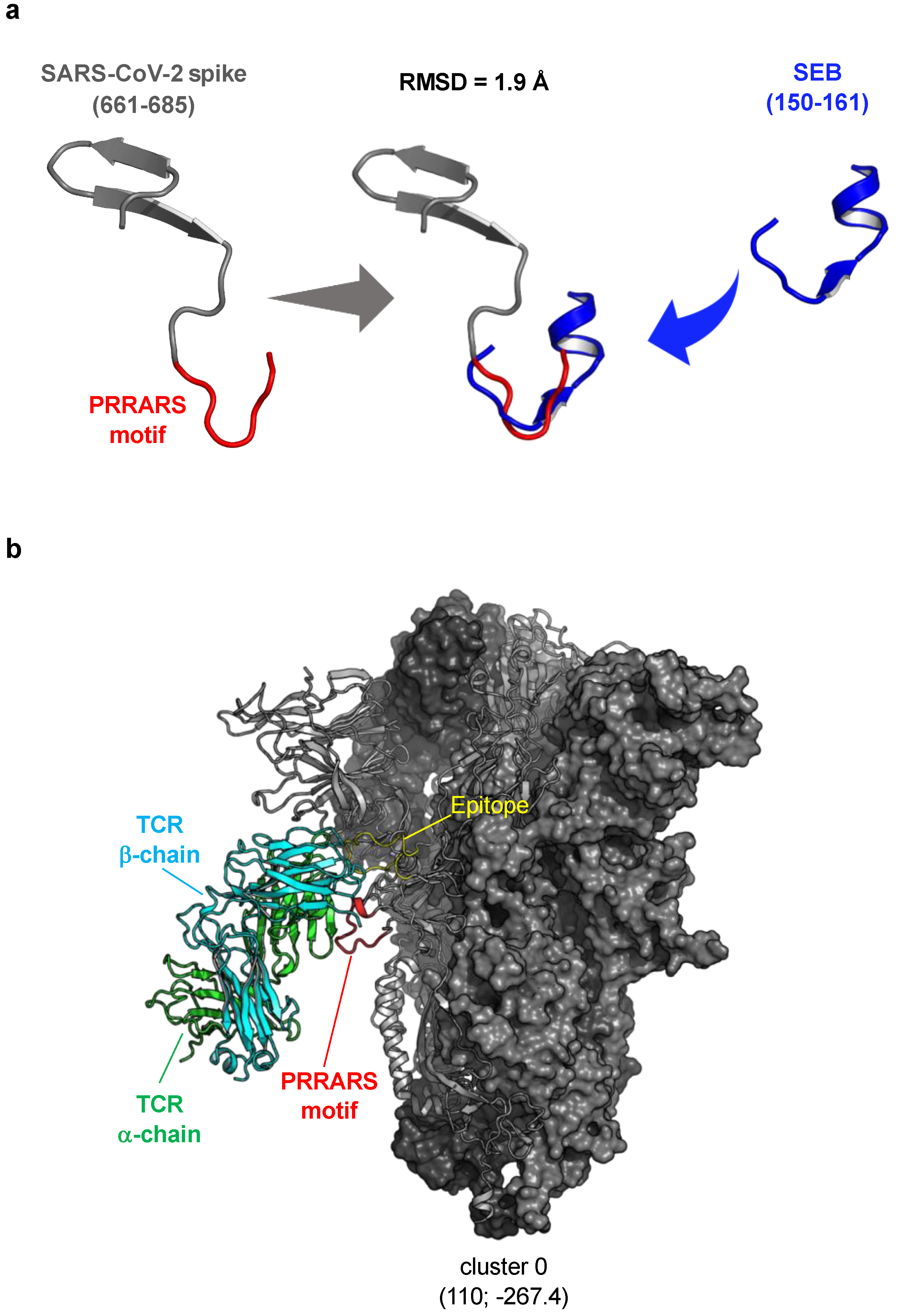
3.2. Structural Reassessment of the Putative Interaction between SARS-CoV-2 Spike and TCR
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Huang, C.; Wang, Y.; Li, X.; Ren, L.; Zhao, J.; Hu, Y.; Zhang, L.; Fan, G.; Xu, J.; Gu, X.; et al. Clinical features of patients infected with 2019 novel coronavirus in Wuhan, China. Lancet 2020, 395, 497–506. [Google Scholar] [CrossRef]
- Zhu, N.; Zhang, D.; Wang, W.; Li, X.; Yang, B.; Song, J.; Zhao, X.; Huang, B.; Shi, W.; Lu, R.; et al. A Novel Coronavirus from Patients with Pneumonia in China, 2019. N. Engl. J. Med. 2020, 382, 727–733. [Google Scholar] [CrossRef]
- Tay, M.Z.; Poh, C.M.; Renia, L.; Macary, P.A.; Ng, L.F.P. The trinity of COVID-19: Immunity, inflammation and intervention. Nat. Rev. Immunol. 2020, 20, 363–374. [Google Scholar] [CrossRef]
- Vabret, N.; Britton, G.J.; Gruber, C.; Hegde, S.; Kim, J.; Kuksin, M.; Levantovsky, R.; Malle, L.; Moreira, A.; Park, M.D.; et al. Immunology of COVID-19: Current State of the Science. Immunity 2020, 52, 910–941. [Google Scholar] [CrossRef] [PubMed]
- Gusev, E.; Sarapultsev, A.; Solomatina, L.; Chereshnev, V. SARS-CoV-2-Specific Immune Response and the Pathogenesis of COVID-19. Int. J. Mol. Sci. 2022, 23, 1716. [Google Scholar] [CrossRef]
- Morris, S.B.; Schwartz, N.G.; Patel, P.; Abbo, L.; Beauchamps, L.; Balan, S.; Lee, E.H.; Paneth-Pollak, R.; Geevarughese, A.; Lash, M.K.; et al. Case Series of Multisystem Inflammatory Syndrome in Adults Associated with SARS-CoV-2 Infection—United Kingdom and United States, March–August 2020. MMWR Morb. Mortal. Wkly. Rep. 2020, 69, 1450–1456. [Google Scholar] [CrossRef]
- Godfred-Cato, S.; Bryant, B.; Leung, J.; Oster, M.E.; Conklin, L.; Abrams, J.; Roguski, K.; Wallace, B.; Prezzato, E.; Koumans, E.H.; et al. COVID-19–Associated Multisystem Inflammatory Syndrome in Children—United States, March–July 2020. MMWR Morb. Mortal. Wkly. Rep. 2020, 69, 1074–1080. [Google Scholar] [CrossRef] [PubMed]
- Jiang, L.; Tang, K.; Levin, M.; Irfan, O.; Morris, S.K.; Wilson, K.; Klein, J.D.; Bhutta, Z.A. COVID-19 and multisystem inflammatory syndrome in children and adolescents. Lancet Infect. Dis. 2020, 20, e276–e288. [Google Scholar] [CrossRef]
- Chou, J.; Thomas, P.G.; Randolph, A.G. Immunology of SARS-CoV-2 infection in children. Nat. Immunol. 2022, 23, 177–185. [Google Scholar] [CrossRef]
- Verdoni, L.; Mazza, A.; Gervasoni, A.; Martelli, L.; Ruggeri, M.; Ciuffreda, M.; Bonanomi, E.; D’Antiga, L. An outbreak of severe Kawasaki-like disease at the Italian epicentre of the SARS-CoV-2 epidemic: An observational cohort study. Lancet 2020, 395, 1771–1778. [Google Scholar] [CrossRef]
- Toubiana, J.; Poirault, C.; Corsia, A.; Bajolle, F.; Fourgeaud, J.; Angoulvant, F.; Debray, A.; Basmaci, R.; Salvador, E.; Biscardi, S.; et al. Kawasaki-like multisystem inflammatory syndrome in children during the COVID-19 pandemic in Paris, France: Prospective observational study. BMJ 2020, 369, m2094–m2101. [Google Scholar] [CrossRef]
- Whittaker, E.; Bamford, A.; Kenny, J.; Kaforou, M.; Jones, C.E.; Shah, P.; Ramnarayan, P.; Fraisse, A.; Miller, O.; Davies, P.; et al. Clinical Characteristics of 58 Children with a Pediatric Inflammatory Multisystem Syndrome Temporally Associated With SARS-CoV-2. JAMA 2020, 324, 259–269. [Google Scholar] [CrossRef]
- Riphagen, S.; Gomez, X.; Gonzalez-Martinez, C.; Wilkinson, N.; Theocharis, P. Hyperinflammatory shock in children during COVID-19 pandemic. Lancet 2020, 395, 1607–1608. [Google Scholar] [CrossRef]
- Belay, E.D.; Cato, S.G.; Rao, A.K.; Abrams, J.; Wilson, W.W.; Lim, S.; Newton-Cheh, C.; Melgar, M.; DeCuir, J.; Webb, B.; et al. Multisystem Inflammatory Syndrome in Adults after SARS-CoV-2 Infection and COVID-19 Vaccination. Clin. Infect. Dis. 2021, ciab936–ciab948. [Google Scholar] [CrossRef]
- Cheung, E.W.; Zachariah, P.; Gorelik, M.; Boneparth, A.; Kernie, S.G.; Orange, J.S.; Milner, J.D. Multisystem Inflammatory Syndrome Related to COVID-19 in Previously Healthy Children and Adolescents in New York City. JAMA 2020, 324, 294–296. [Google Scholar] [CrossRef]
- Belhadjer, Z.; Meot, M.; Bajolle, F.; Khraiche, D.; Legendre, A.; Abakka, S.; Auriau, J.; Grimaud, M.; Oualha, M.; Beghetti, M.; et al. Acute Heart Failure in Multisystem Inflammatory Syndrome in Children in the Context of Global SARS-CoV-2 Pandemic. Circulation 2020, 142, 429–436. [Google Scholar] [CrossRef]
- McMurray, J.C.; May, J.W.; Cunningham, M.W.; Jones, O.Y. Multisystem Inflammatory Syndrome in Children (MIS-C), a Post-viral Myocarditis and Systemic Vasculitis—A Critical Review of Its Pathogenesis and Treatment. Front. Pediatr. 2020, 8, 626182–626198. [Google Scholar] [CrossRef] [PubMed]
- Consiglio, C.R.; Cotugno, N.; Sardh, F.; Pou, C.; Amodio, D.; Rodriguez, L.; Tan, Z.; Zicari, S.; Ruggiero, A.; Pascucci, G.R.; et al. The Immunology of Multisystem Inflammatory Syndrome in Children with COVID-19. Cell 2020, 183, 968–981.e967. [Google Scholar] [CrossRef] [PubMed]
- Cheng, M.H.; Zhang, S.; Porritt, R.A.; Rivas, M.N.; Paschold, L.; Willscher, E.; Binder, M.; Arditi, M.; Bahar, I. Superantigenic character of an insert unique to SARS-CoV-2 spike supported by skewed TCR repertoire in patients with hyperinflammation. Proc. Natl. Acad. Sci. USA 2020, 117, 25254–25262. [Google Scholar] [CrossRef]
- Porritt, R.A.; Binek, A.; Paschold, L.; Rivas, M.N.; McArdle, A.; Yonker, L.M.; Alter, G.; Chandnani, H.K.; Lopez, M.; Fasano, A.; et al. The autoimmune signature of hyperinflammatory multisystem inflammatory syndrome in children. J. Clin. Investig. 2021, 131, e151520–e151537. [Google Scholar] [CrossRef]
- Moreews, M.; Le Gouge, K.; Khaldi-Plassart, S.; Pescarmona, R.; Mathieu, A.L.; Malcus, C.; Djebali, S.; Bellomo, A.; Dauwalder, O.; Perret, M.; et al. Polyclonal expansion of TCR Vbeta 21.3+ CD4+ and CD8+ T cells is a hallmark of multisystem inflammatory syndrome in children. Sci. Immunol. 2021, 6, eabh1516–eabh1527. [Google Scholar] [CrossRef] [PubMed]
- Porritt, R.A.; Paschold, L.; Rivas, M.N.; Cheng, M.H.; Yonker, L.M.; Chandnani, H.; Lopez, M.; Simnica, D.; Schultheiß, C.; Santiskulvong, C.; et al. HLA class I-associated expansion of TRBV11-2 T cells in Multisystem Inflammatory Syndrome in Children. J. Clin. Investig. 2021, 131, e146614–e146627. [Google Scholar] [CrossRef]
- Carter, M.J.; Fish, M.; Jennings, A.; Doores, K.J.; Wellman, P.; Seow, J.; Acors, S.; Graham, C.; Timms, E.; Kenny, J.; et al. Peripheral immunophenotypes in children with multisystem inflammatory syndrome associated with SARS-CoV-2 infection. Nat. Med. 2020, 26, 1701–1707. [Google Scholar] [CrossRef]
- Buonsenso, D.; Riitano, F.; Valentini, P. Pediatric Inflammatory Multisystem Syndrome Temporally Related With SARS-CoV-2: Immunological Similarities with Acute Rheumatic Fever and Toxic Shock Syndrome. Front. Pediatr. 2020, 8, 574–579. [Google Scholar] [CrossRef] [PubMed]
- Cheng, M.H.; Porritt, R.A.; Rivas, M.N.; Krieger, J.M.; Ozdemir, A.B.; Garcia, G., Jr.; Arumugaswami, V.; Fries, B.C.; Arditi, M.; Bahar, I. A monoclonal antibody against staphylococcal enterotoxin B superantigen inhibits SARS-CoV-2 entry in vitro. Structure 2021, 29, 951–962.e953. [Google Scholar] [CrossRef]
- Rivas, M.N.; Porritt, R.A.; Cheng, M.H.; Bahar, I.; Arditi, M. COVID-19—Associated multisystem inflammatory syndrome in children (MIS-C): A novel disease that mimics toxic shock syndrome—the superantigen hypothesis. J. Allergy Clin. Immunol. 2021, 147, 57–59. [Google Scholar] [CrossRef]
- Kouo, T.; Chaisawangwong, W. SARS-CoV-2 as a superantigen in multisystem inflammatory syndrome in children (MIS-C). J. Clin. Investig. 2021, 131, e149327–e149330. [Google Scholar] [CrossRef] [PubMed]
- Jardetzky, T.S.; Brown, J.H.; Gorga, J.C.; Stern, L.J.; Urban, R.G.; Chi, Y.I.; Stauffacher, C.; Strominger, J.L.; Wiley, D.C. Three-dimensional structure of a human class II histocompatibility molecule complexed with superantigen. Nature 1994, 368, 711–718. [Google Scholar] [CrossRef] [PubMed]
- Seth, A.; Stern, L.J.; Ottenhoff, T.H.M.; Engel, I.; Owen, M.J.; Lamb, J.R.; Klausner, R.D.; Wiley, N.C. Binary and ternary complexes between T-cell receptor, class II MHC and superantigen in vitro. Nature 1994, 369, 324–327. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Llera, A.; Tsuchiya, D.; Leder, L.; Ysern, X.; Schlievert, P.M.; Karjalainen, K.; Mariuzza, R.A. Three-Dimensional Structure of the Complex between a T Cell Receptor β Chain and the Superantigen Staphylococcal Enterotoxin B. Immunity 1998, 9, 807–816. [Google Scholar] [CrossRef]
- Marrack, P.; Blackman, M.; Kushnir, E.; Kappler, J. The toxicity of staphylococcal enterotoxin B in mice is mediated by T cells. J. Exp. Med. 1990, 171, 455–464. [Google Scholar] [CrossRef] [PubMed]
- Krakauer, T. Staphylococcal Superantigens: Pyrogenic Toxins Induce Toxic Shock. Toxins 2019, 11, 178. [Google Scholar] [CrossRef]
- Szabo, P.A.; Goswami, A.; Mazzuca, D.M.; Kim, K.; O’Gorman, D.B.; Hess, D.A.; Welch, I.D.; Young, H.A.; Singh, B.; McCormick, J.K.; et al. Rapid and Rigorous IL-17A Production by a Distinct Subpopulation of Effector Memory T Lymphocytes Constitutes a Novel Mechanism of Toxic Shock Syndrome Immunopathology. J. Immunol. 2017, 198, 2805–2818. [Google Scholar] [CrossRef]
- Arad, G.; Levy, R.; Nasie, I.; Hillman, D.; Rotfogel, Z.; Barash, U.; Supper, E.; Shpilka, T.; Minis, A.; Kaempfer, R. Binding of Superantigen Toxins into the CD28 Homodimer Interface Is Essential for Induction of Cytokine Genes That Mediate Lethal Shock. PLoS Biol. 2011, 9, e1001149–e1001162. [Google Scholar] [CrossRef]
- Kaempfer, R.; Arad, G.; Levy, R.; Hillman, D.; Nasie, I.; Rotfogel, Z. CD28: Direct and Critical Receptor for Superantigen Toxins. Toxins 2013, 5, 1531–1542. [Google Scholar] [CrossRef]
- Kaempfer, R.; Popugailo, A.; Levy, R.; Arad, G.; Hillman, D.; Rotfogel, Z. Bacterial superantigen toxins induce a lethal cytokine storm by enhancing B7-2/CD28 costimulatory receptor engagement, a critical immune checkpoint. Recept. Clin. Investig. 2017, 4, e1500–e1509. [Google Scholar]
- Levy, R.; Rotfogel, Z.; Hillman, D.; Popugailo, A.; Arad, G.; Supper, E.; Osman, F.; Kaempfer, R. Superantigens hyperinduce inflammatory cytokines by enhancing the B7-2/CD28 costimulatory receptor interaction. Proc. Natl. Acad. Sci. USA 2016, 113, E6437–E6446. [Google Scholar] [CrossRef] [PubMed]
- Kunkl, M.; Amormino, C.; Caristi, S.; Tedeschi, V.; Fiorillo, M.T.; Levy, R.; Popugailo, A.; Kaempfer, R.; Tuosto, L. Binding of Staphylococcal Enterotoxin B (SEB) to B7 Receptors Triggers TCR- and CD28-Mediated Inflammatory Signals in the Absence of MHC Class II Molecules. Front. Immunol. 2021, 12, 723689–723705. [Google Scholar] [CrossRef]
- Acuto, O.; Michel, F.M. CD28-mediated co-stimulation: A quantitative support for TCR signalling. Nat. Rev. Immunol. 2003, 3, 939–951. [Google Scholar] [CrossRef]
- Porciello, N.; Tuosto, L. CD28 costimulatory signals in T lymphocyte activation: Emerging functions beyond a qualitative and quantitative support to TCR signalling. Cytokine Growth Factor Rev. 2016, 28, 11–19. [Google Scholar] [CrossRef] [PubMed]
- Porciello, N.; Grazioli, P.; Campese, A.F.; Kunkl, M.; Caristi, S.; Mastrogiovanni, M.; Muscolini, M.; Spadaro, F.; Favre, C.; Nunes, J.A.; et al. A non-conserved amino acid variant regulates differential signalling between human and mouse CD28. Nat. Commun. 2018, 9, 1080. [Google Scholar] [CrossRef]
- Kunkl, M.; Sambucci, M.; Ruggieri, S.; Amormino, C.; Tortorella, C.; Gasperini, C.; Battistini, L.; Tuosto, L. CD28 Autonomous Signaling Up-Regulates C-Myc Expression and Promotes Glycolysis Enabling Inflammatory T Cell Responses in Multiple Sclerosis. Cells 2019, 8, 575. [Google Scholar] [CrossRef]
- Kunkl, M.; Porciello, N.; Mastrogiovanni, M.; Capuano, C.; Lucantoni, F.; Moretti, C.; Persson, J.L.; Galandrini, R.; Buzzetti, R.; Tuosto, L. ISA-2011B, a Phosphatidylinositol 4-Phosphate 5-Kinase α Inhibitor, Impairs CD28-Dependent Costimulatory and Pro-inflammatory Signals in Human T Lymphocytes. Front. Immunol. 2017, 8, 502–512. [Google Scholar] [CrossRef]
- Kunkl, M.; Mastrogiovanni, M.; Porciello, N.; Caristi, S.; Monteleone, E.; Arcieri, S.; Tuosto, L. CD28 Individual Signaling Up-regulates Human IL-17A Expression by Promoting the Recruitment of RelA/NF-κB and STAT3 Transcription Factors on the Proximal Promoter. Front. Immunol. 2019, 10, 864–881. [Google Scholar] [CrossRef] [PubMed]
- Kunkl, M.; Amormino, C.; Frascolla, S.; Sambucci, M.; De Bardi, M.; Caristi, S.; Arcieri, S.; Battistini, L.; Tuosto, L. CD28 Autonomous Signaling Orchestrates IL-22 Expression and IL-22-Regulated Epithelial Barrier Functions in Human T Lymphocytes. Front. Immunol. 2020, 11, 590964–590977. [Google Scholar] [CrossRef] [PubMed]
- Souza, P.F.N.; Mesquita, F.P.; Amaral, J.L.; Landim, P.G.C.; Lima, K.R.P.; Costa, M.B.; Farias, I.R.; Belém, M.O.; Pinto, Y.O.; Moreira, H.H.; et al. The spike glycoprotein of SARS-CoV-2: A review of how mutations of spike glycoproteins have driven the emergence of variants with high transmissibility and immune escape. Int. J. Biol. Macromol. 2022, 208, 105–125. [Google Scholar] [CrossRef] [PubMed]
- Michel, F.; Mangino, G.; Attal-Bonnefoy, G.; Tuosto, L.; Alcover, A.; Roumier, A.; Olive, D.; Acuto, O. CD28 Utilizes Vav-1 to Enhance TCR-Proximal Signaling and NF-AT Activation. J. Immunol. 2000, 165, 3820–3829. [Google Scholar] [CrossRef]
- Hewitt, C.R.; Lamb, J.R.; Hayball, J.; Hill, M.; Owen, M.J.; O’Hehir, R.E. Major histocompatibility complex independent clonal T cell anergy by direct interaction of Staphylococcus aureus enterotoxin B with the T cell antigen receptor. J. Exp. Med. 1992, 175, 1493–1499. [Google Scholar] [CrossRef]
- Karr, R.W.; Gregersen, P.K.; Obata, F.; Goldberg, D.; Maccari, J.; Alber, C.; Silver, J. Analysis of DR beta and DQ beta chain cDNA clones from a DR7 haplotype. J. Immunol. 1986, 137, 2886–2890. [Google Scholar]
- Tuosto, L.; Piazza, C.; Moretti, S.; Modesti, A.; Greenlaw, R.; Lechler, R.; Lombardi, G.; Piccolella, E. Ligation of either CD2 or CD28 rescues CD4+ T cells from HIV-gp120-induced apoptosis. Eur. J. Immunol. 1995, 25, 2917–2922. [Google Scholar] [CrossRef]
- Muscolini, M.; Camperio, C.; Porciello, N.; Caristi, S.; Capuano, C.; Viola, A.; Galandrini, R.; Tuosto, L. Phosphatidylinositol 4–Phosphate 5–Kinase α and Vav1 Mutual Cooperation in CD28-Mediated Actin Remodeling and Signaling Functions. J. Immunol. 2015, 194, 1323–1333. [Google Scholar] [CrossRef]
- Magnacca, A.; Persiconi, I.; Nurzia, E.; Caristi, S.; Meloni, F.; Barnaba, V.; Paladini, F.; Raimondo, D.; Fiorillo, M.T.; Sorrentino, R. Characterization of a Proteasome and TAP-independent Presentation of Intracellular Epitopes by HLA-B27 Molecules. J. Biol. Chem. 2012, 287, 30358–30367. [Google Scholar] [CrossRef]
- Candotti, M.; Perez, A.; Ferrer-Costa, C.; Rueda, M.; Meyer, T.; Gelpí, J.L.; Orozco, M. Exploring Early Stages of the Chemical Unfolding of Proteins at the Proteome Scale. PLoS Comput. Biol. 2013, 9, e1003393–e1003404. [Google Scholar] [CrossRef]
- Traenckner, E.B.; Pahl, H.L.; Henkel, T.; Schmidt, K.N.; Wilk, S.; Baeuerle, P.A. Phosphorylation of human I kappa B-alpha on serines 32 and 36 controls I kappa B-alpha proteolysis and NF-kappa B activation in response to diverse stimuli. EMBO J. 1995, 14, 2876–2883. [Google Scholar] [CrossRef]
- Emmel, E.A.; Verweij, C.L.; Durand, D.B.; Higgins, K.M.; Lacy, E.; Crabtree, G.R. Cyclosporin A Specifically Inhibits Function of Nuclear Proteins Involved in T Cell Activation. Science 1989, 246, 1617–1620. [Google Scholar] [CrossRef]
- Rincon, M.; Flavell, R.A. AP-1 transcriptional activity requires both T-cell receptor-mediated and co-stimulatory signals in primary T lymphocytes. EMBO J. 1994, 13, 4370–4381. [Google Scholar] [CrossRef]
- Jumper, J.; Evans, R.; Pritzel, A.; Green, T.; Figurnov, M.; Ronneberger, O.; Tunyasuvunakool, K.; Bates, R.; Žídek, A.; Potapenko, A.; et al. Highly accurate protein structure prediction with AlphaFold. Nature 2021, 596, 583–589. [Google Scholar] [CrossRef] [PubMed]
- Cai, Y.; Zhang, J.; Xiao, T.; Lavine, C.L.; Rawson, S.; Peng, H.; Zhu, H.; Anand, K.; Tong, P.; Gautam, A.; et al. Structural basis for enhanced infectivity and immune evasion of SARS-CoV-2 variants. Science 2021, 373, 642–648. [Google Scholar] [CrossRef]
- Brenke, R.; Hall, D.R.; Chuang, G.-Y.; Comeau, S.R.; Bohnuud, T.; Beglov, D.; Schueler-Furman, O.; Vajda, S.; Kozakov, D. Application of asymmetric statistical potentials to antibody–protein docking. Bioinformatics 2012, 28, 2608–2614. [Google Scholar] [CrossRef] [PubMed]
- Wrapp, D.; Wang, N.; Corbett, K.S.; Goldsmith, J.A.; Hsieh, C.L.; Abiona, O.; Graham, B.S.; McLellan, J.S. Cryo-EM structure of the 2019-nCoV spike in the prefusion conformation. Science 2020, 367, 1260–1263. [Google Scholar] [CrossRef] [PubMed]
- Saline, M.; Rodstrom, K.E.; Fischer, G.; Orekhov, V.Y.; Karlsson, B.G.; Lindkvist-Petersson, K. The structure of superantigen complexed with TCR and MHC reveals novel insights into superantigenic T cell activation. Nat. Commun. 2010, 1, 119–128. [Google Scholar] [CrossRef] [PubMed]
- Popugailo, A.; Rotfogel, Z.; Supper, E.; Hillman, D.; Kaempfer, R. Staphylococcal and Streptococcal Superantigens Trigger B7/CD28 Costimulatory Receptor Engagement to Hyperinduce Inflammatory Cytokines. Front. Immunol. 2019, 10, 942–951. [Google Scholar] [CrossRef] [PubMed]
- Hamdy, A.; Leonardi, A. Superantigens and SARS-CoV-2. Pathogens 2022, 11, 390. [Google Scholar] [CrossRef]
- Shajahan, A.; Supekar, N.T.; Gleinich, A.S.; Azadi, P. Deducing the N- and O-glycosylation profile of the spike protein of novel coronavirus SARS-CoV-2. Glycobiology 2020, 30, 981–988. [Google Scholar] [CrossRef]
- Watanabe, Y.; Allen, J.D.; Wrapp, D.; McLellan, J.S.; Crispin, M. Site-specific analysis of the SARS-CoV-2 glycan shield. Science 2020, 369, 330–333. [Google Scholar] [CrossRef]
- Maffei, M.; Montemiglio, L.C.; Vitagliano, G.; Fedele, L.; Sellathurai, S.; Bucci, F.; Compagnone, M.; Chiarini, V.; Exertier, C.; Muzi, A.; et al. The Nuts and Bolts of SARS-CoV-2 Spike Receptor-Binding Domain Heterologous Expression. Biomolecules 2021, 11, 1812. [Google Scholar] [CrossRef]
- Choi, Y.W.; Kotzin, B.; Herron, L.; Callahan, J.; Marrack, P.; Kappler, J. Interaction of Staphylococcus aureus toxin “superantigens” with human T cells. Proc. Natl. Acad. Sci. USA 1989, 86, 8941–8945. [Google Scholar] [CrossRef]
- Tuosto, L.; Acuto, O. CD28 affects the earliest signaling events generated by TCR engagement. Eur. J. Immunol. 1998, 28, 2131–2142. [Google Scholar] [CrossRef]
- Ramaswamy, A.; Brodsky, N.N.; Sumida, T.S.; Comi, M.; Asashima, H.; Hoehn, K.B.; Li, N.; Liu, Y.; Shah, A.; Ravindra, N.G.; et al. Immune dysregulation and autoreactivity correlate with disease severity in SARS-CoV-2-associated multisystem inflammatory syndrome in children. Immunity 2021, 54, 1083–1095.e1087. [Google Scholar] [CrossRef]
- Thomas, D.; Dauwalder, O.; Brun, V.; Badiou, C.; Ferry, T.; Etienne, J.; Vandenesch, F.; Lina, G. Staphylococcus aureus Superantigens Elicit Redundant and Extensive Human Vβ Patterns. Infect. Immun. 2009, 77, 2043–2050. [Google Scholar] [CrossRef]
- Ochsenreither, S.; Fusi, A.; Busse, A.; Nagorsen, D.; Schrama, D.; Becker, J.; Thiel, E.; Keilholz, U. Relative quantification of TCR Vbeta-chain families by real time PCR for identification of clonal T-cell populations. J. Transl. Med. 2008, 6, 34–42. [Google Scholar] [CrossRef]
- Dutta, K.; Varshney, A.K.; Franklin, M.C.; Goger, M.; Wang, X.; Fries, B.C. Mechanisms Mediating Enhanced Neutralization Efficacy of Staphylococcal Enterotoxin B by Combinations of Monoclonal Antibodies. J. Biol. Chem. 2015, 290, 6715–6730. [Google Scholar] [CrossRef] [PubMed]
- Evans, E.J.; Esnouf, R.M.; Manso-Sancho, R.; Gilbert, R.J.; James, J.R.; Yu, C.; Fennelly, J.A.; Vowles, C.; Hanke, T.; Walse, B.; et al. Crystal structure of a soluble CD28-Fab complex. Nat. Immunol. 2005, 6, 271–279. [Google Scholar] [CrossRef]
- Herrmann, T.; Accolla, R.S.; Macdonald, H.R. Different staphylococcal enterotoxins bind preferentially to distinct major histocompatibility complex class ii isotypes. Eur. J. Immunol. 1989, 19, 2171–2174. [Google Scholar] [CrossRef] [PubMed]
- Kaempfer, R. Bacterial Superantigen Toxins, CD28, and Drug Development. Toxins 2018, 10, 459. [Google Scholar] [CrossRef] [PubMed]
- Arad, G.; Levy, R.; Hillman, D.; Kaempfer, R. Superantigen antagonist protects against lethal shock and defines a new domain for T-cell activation. Nat. Med. 2000, 6, 414–421. [Google Scholar] [CrossRef]
- Rodstrom, K.E.; Elbing, K.; Lindkvist-Petersson, K. Structure of the Superantigen Staphylococcal Enterotoxin B in Complex with TCR and Peptide–MHC Demonstrates Absence of TCR–Peptide Contacts. J. Immunol. 2014, 193, 1998–2004. [Google Scholar] [CrossRef]
- Ghosh, P.; Katkar, G.D.; Shimizu, C.; Kim, J.; Khandelwal, S.; Tremoulet, A.H.; Kanegaye, J.T.; Abe, N.; Austin-Page, L.; Bryl, A.; et al. An Artificial Intelligence-guided signature reveals the shared host immune response in MIS-C and Kawasaki disease. Nat. Commun. 2022, 13, 2687–2705. [Google Scholar] [CrossRef] [PubMed]
- Vella, L.A.; Giles, J.R.; Baxter, A.E.; Oldridge, D.A.; Diorio, C.; Kuri-Cervantes, L.; Alanio, C.; Pampena, M.B.; Wu, J.E.; Chen, Z.; et al. Deep immune profiling of MIS-C demonstrates marked but transient immune activation compared to adult and pediatric COVID-19. Sci. Immunol. 2021, 6, e7570–e7585. [Google Scholar] [CrossRef]
- Yonker, L.M.; Gilboa, T.; Ogata, A.F.; Senussi, Y.; Lazarovits, R.; Boribong, B.P.; Bartsch, Y.C.; Loiselle, M.; Rivas, M.N.; Porritt, R.A.; et al. Multisystem inflammatory syndrome in children is driven by zonulin-dependent loss of gut mucosal barrier. J. Clin. Investig. 2021, 131, e149633–e149645. [Google Scholar] [CrossRef] [PubMed]
- Sahoo, D.; Katkar, G.D.; Khandelwal, S.; Behroozikhah, M.; Claire, A.; Castillo, V.; Tindle, C.; Fuller, M.; Taheri, S.; Rogers, T.F.; et al. AI-guided discovery of the invariant host response to viral pandemics. eBioMedicine 2021, 68, 103390–103410. [Google Scholar] [CrossRef]
- Gruber, C.N.; Patel, R.S.; Trachtman, R.; Lepow, L.; Amanat, F.; Krammer, F.; Wilson, K.M.; Onel, K.; Geanon, D.; Tuballes, K.; et al. Mapping Systemic Inflammation and Antibody Responses in Multisystem Inflammatory Syndrome in Children (MIS-C). Cell 2020, 183, 982–995.e914. [Google Scholar] [CrossRef]
- Bartsch, Y.C.; Wang, C.; Zohar, T.; Fischinger, S.; Atyeo, C.; Burke, J.S.; Kang, J.; Edlow, A.G.; Fasano, A.; Baden, L.R.; et al. Humoral signatures of protective and pathological SARS-CoV-2 infection in children. Nat. Med. 2021, 27, 454–462. [Google Scholar] [CrossRef]
- Junqueira, C.; Crespo, A.; Ranjbar, S.; de Lacerda, L.B.; Lewandrowski, M.; Ingber, J.; Parry, B.; Ravid, S.; Clark, S.; Schrimpf, M.R.; et al. FcγR-mediated SARS-CoV-2 infection of monocytes activates inflammation. Nature 2022, 606, 576–584. [Google Scholar] [CrossRef] [PubMed]





Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Amormino, C.; Tedeschi, V.; Paldino, G.; Arcieri, S.; Fiorillo, M.T.; Paiardini, A.; Tuosto, L.; Kunkl, M. SARS-CoV-2 Spike Does Not Possess Intrinsic Superantigen-like Inflammatory Activity. Cells 2022, 11, 2526. https://doi.org/10.3390/cells11162526
Amormino C, Tedeschi V, Paldino G, Arcieri S, Fiorillo MT, Paiardini A, Tuosto L, Kunkl M. SARS-CoV-2 Spike Does Not Possess Intrinsic Superantigen-like Inflammatory Activity. Cells. 2022; 11(16):2526. https://doi.org/10.3390/cells11162526
Chicago/Turabian StyleAmormino, Carola, Valentina Tedeschi, Giorgia Paldino, Stefano Arcieri, Maria Teresa Fiorillo, Alessandro Paiardini, Loretta Tuosto, and Martina Kunkl. 2022. "SARS-CoV-2 Spike Does Not Possess Intrinsic Superantigen-like Inflammatory Activity" Cells 11, no. 16: 2526. https://doi.org/10.3390/cells11162526
APA StyleAmormino, C., Tedeschi, V., Paldino, G., Arcieri, S., Fiorillo, M. T., Paiardini, A., Tuosto, L., & Kunkl, M. (2022). SARS-CoV-2 Spike Does Not Possess Intrinsic Superantigen-like Inflammatory Activity. Cells, 11(16), 2526. https://doi.org/10.3390/cells11162526