
A Combination Therapy of Urolithin A+EGCG Has Stronger Protective Effects than Single Drug Urolithin A in a Humanized Amyloid Beta Knockin Mice for Late-Onset Alzheimer’s Disease
 , , ,
, , ,  , and
, and
Abstract
:1. Introduction
2. Materials and Methods
Chemicals and Reagents
3. Results
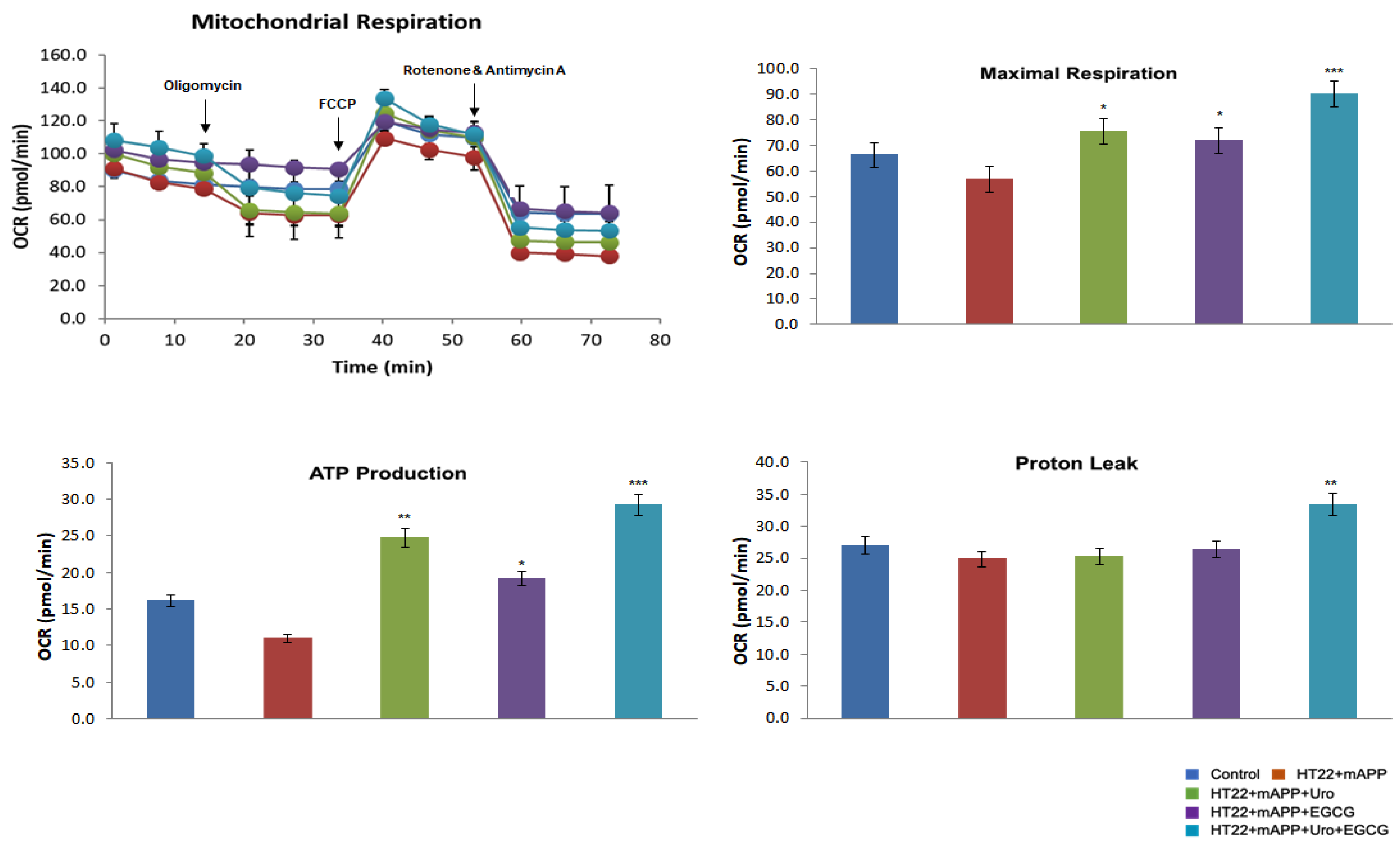
3.1. Mitochondrial Respiration in Mutant APPHTT Cells Treated with Urolithin A, EGCG, Combination of Urolithin A+EGCG
3.1.1. Phenotypic Behavior
3.1.2. mRNA Expression
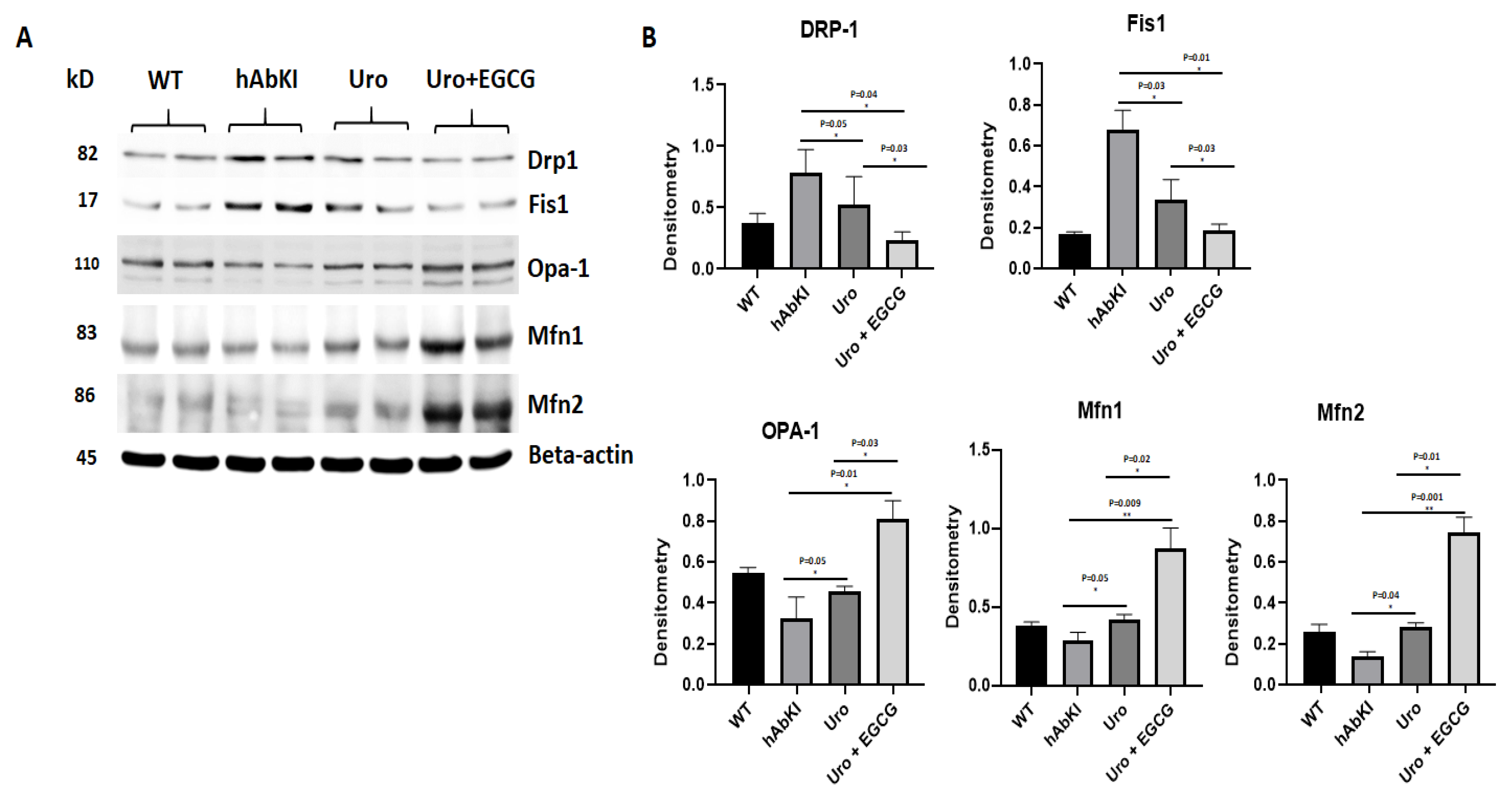
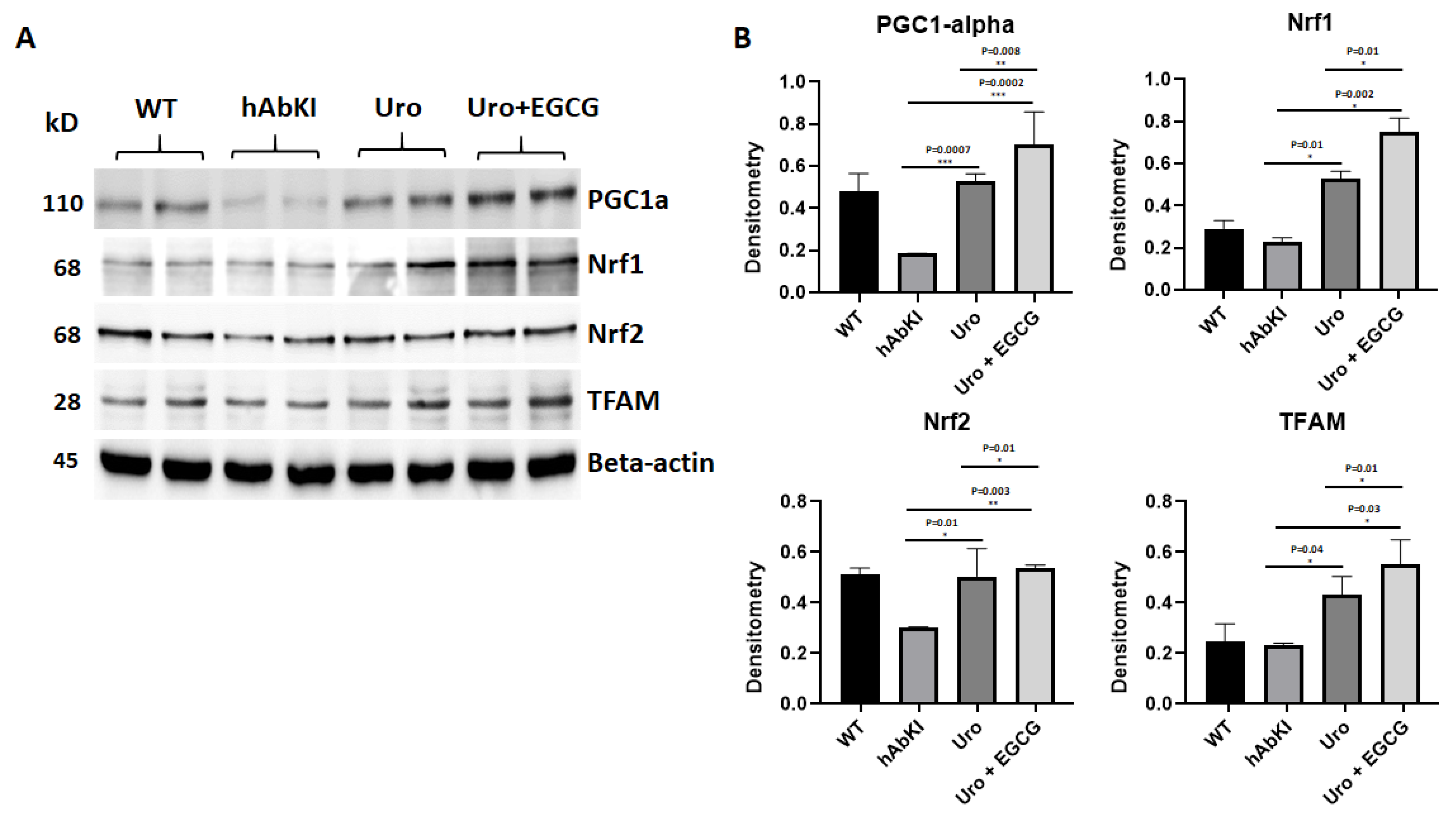
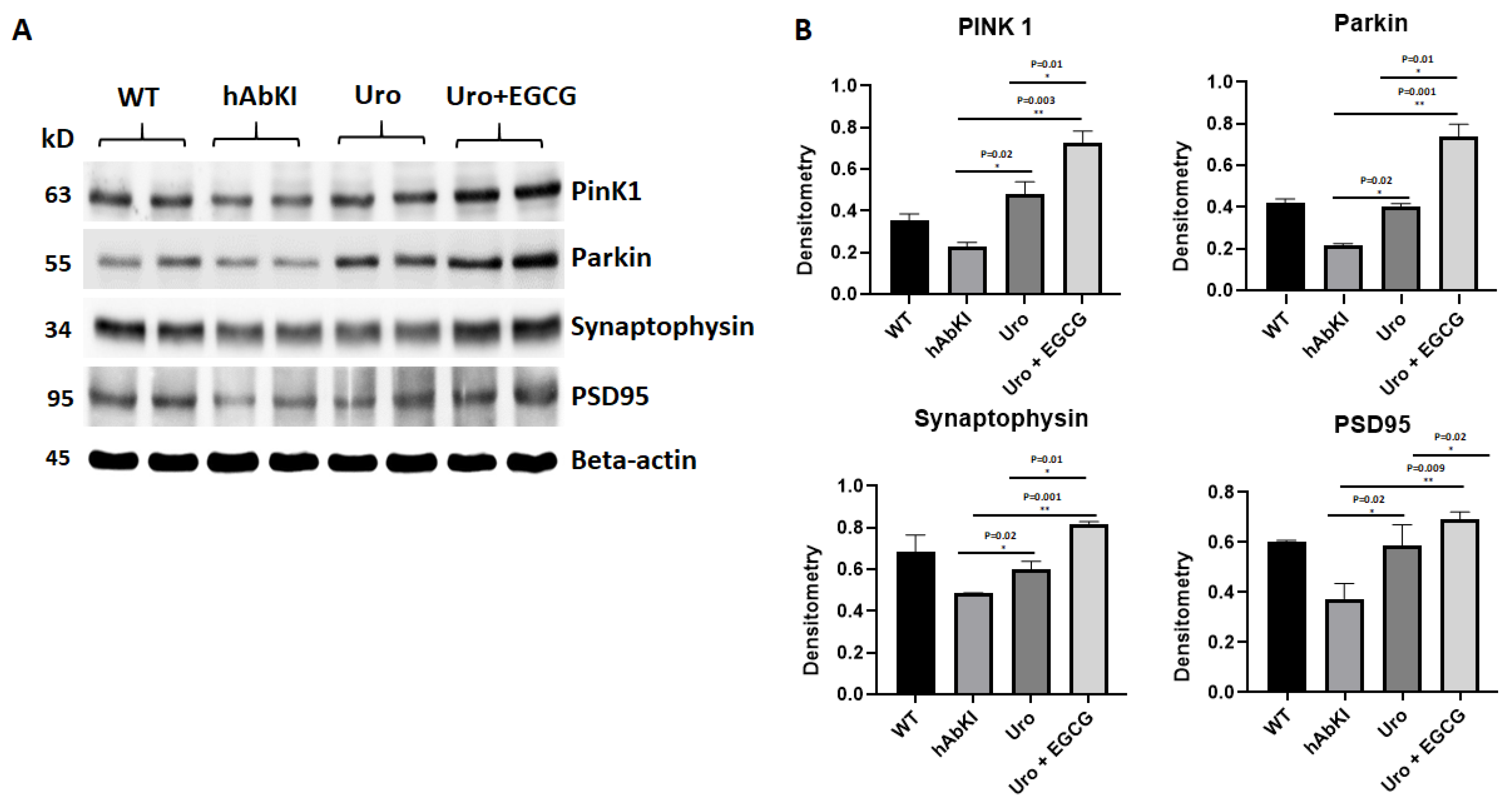
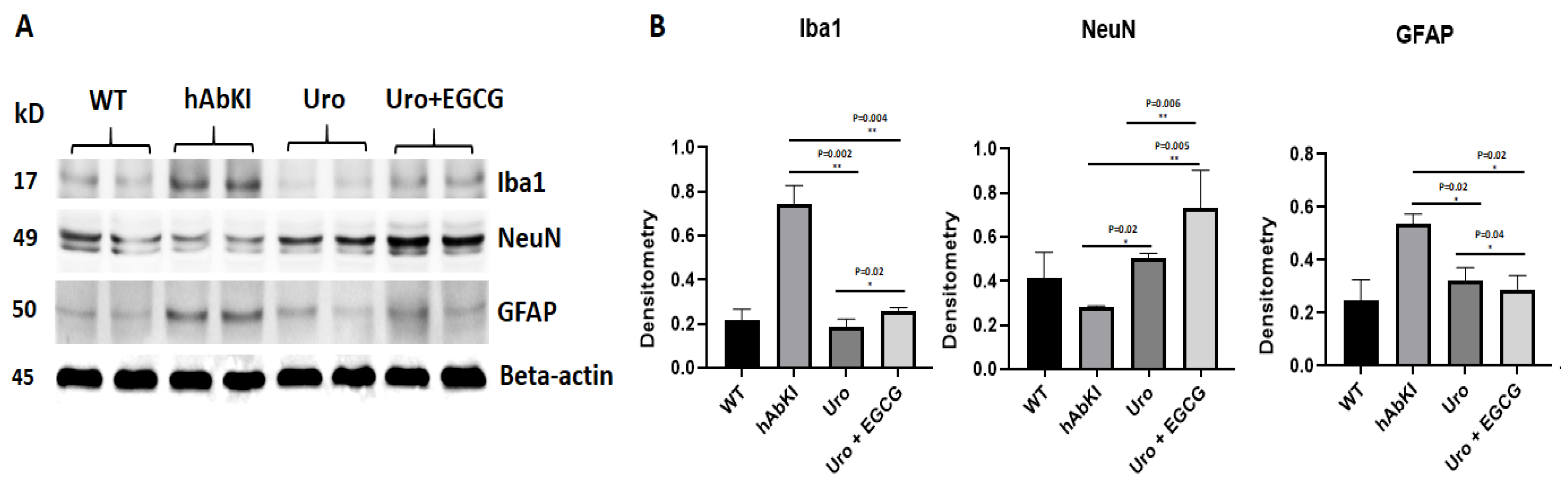
3.1.3. Immunoblotting Analysis
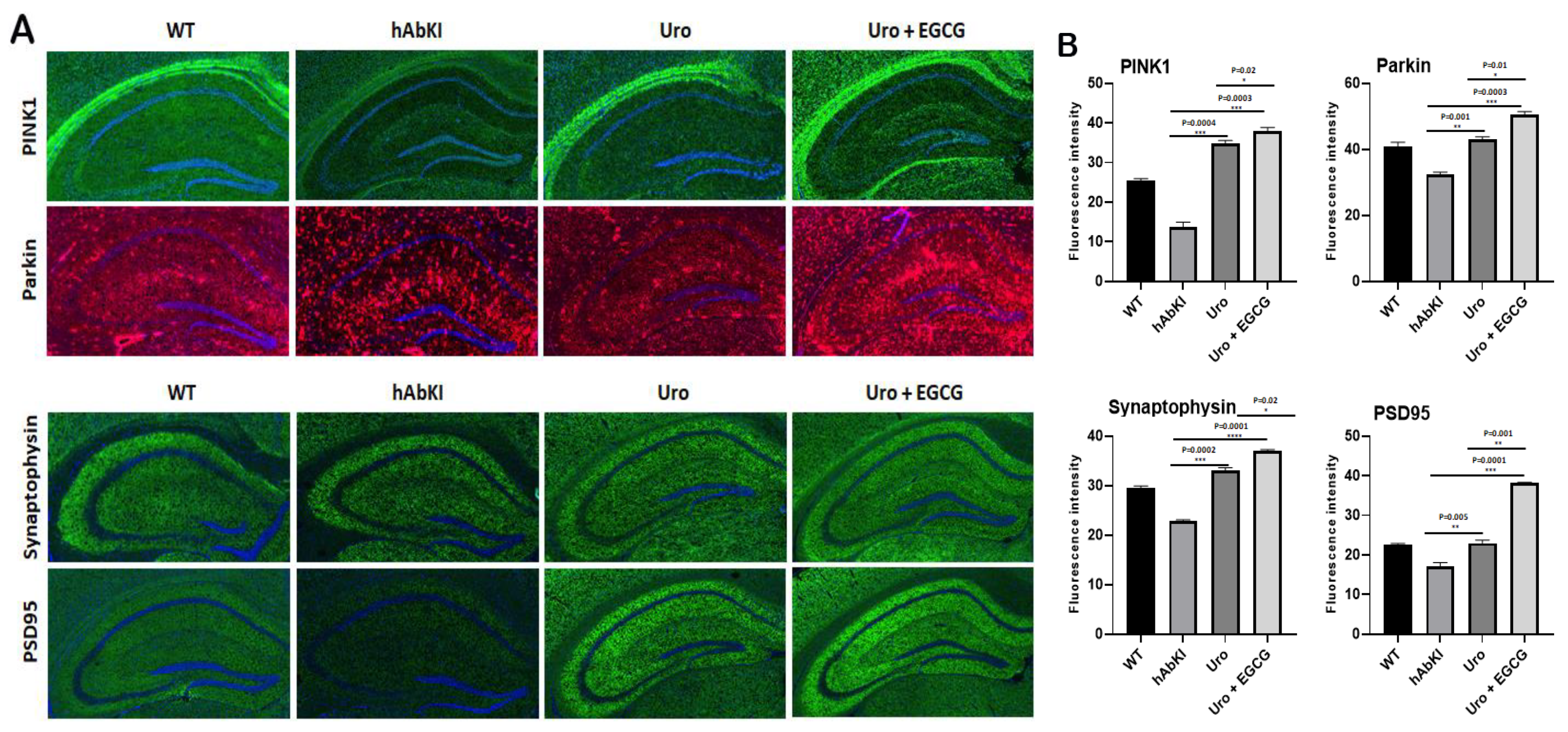
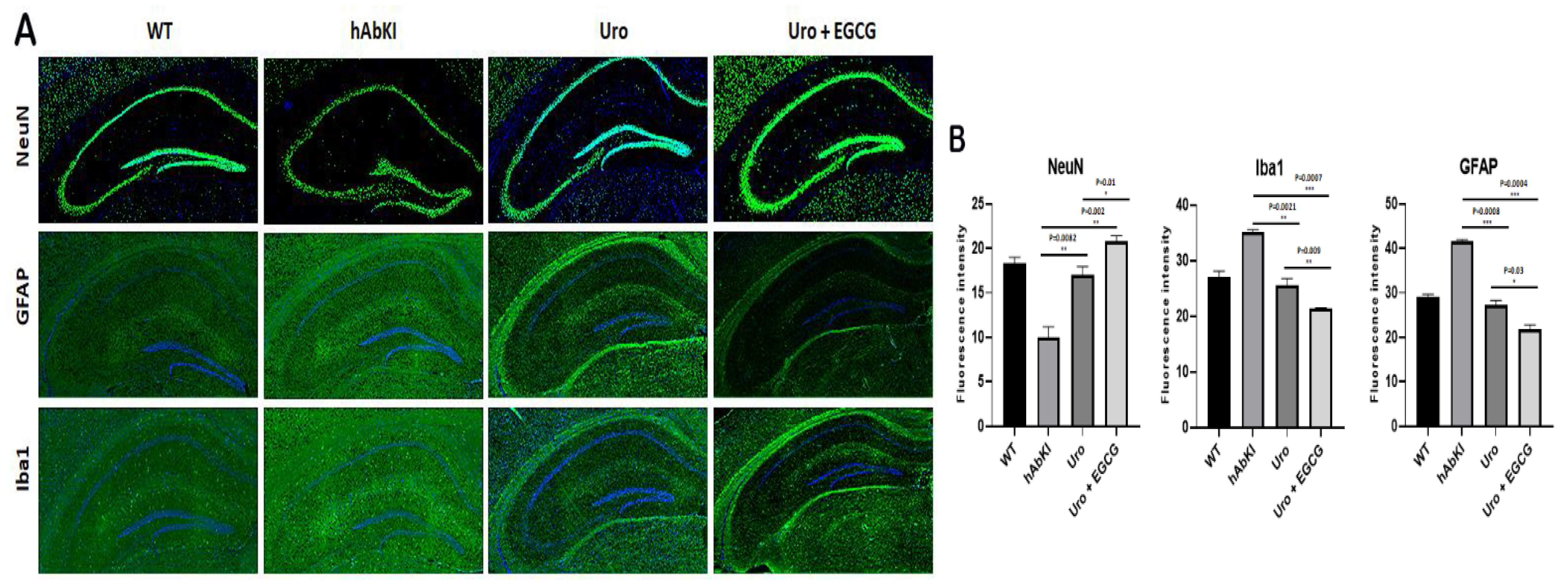
3.1.4. Immunofluorescence Analysis
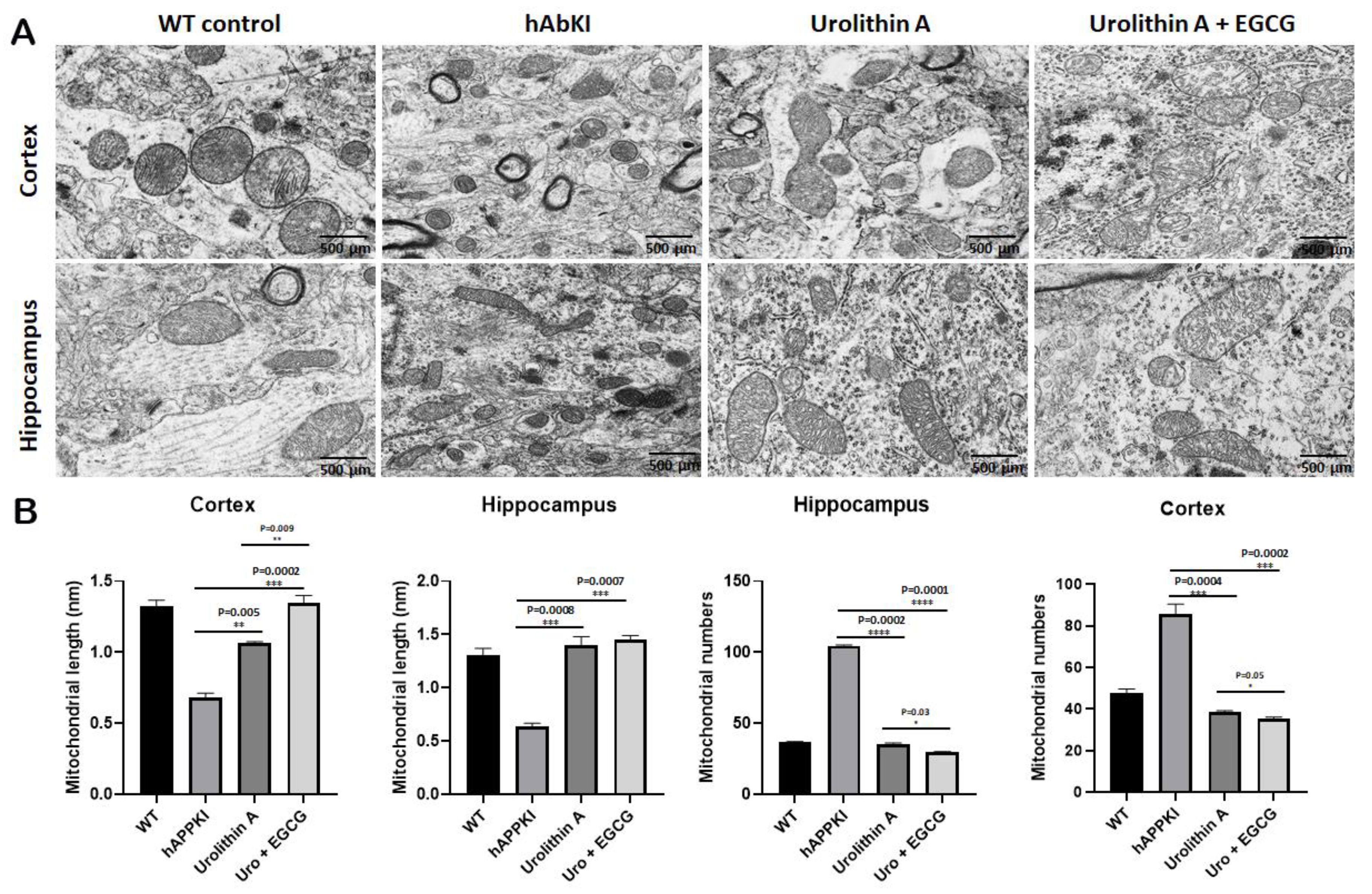
3.1.5. Transmission Electron Microscopy—Mitochondrial Number and Length
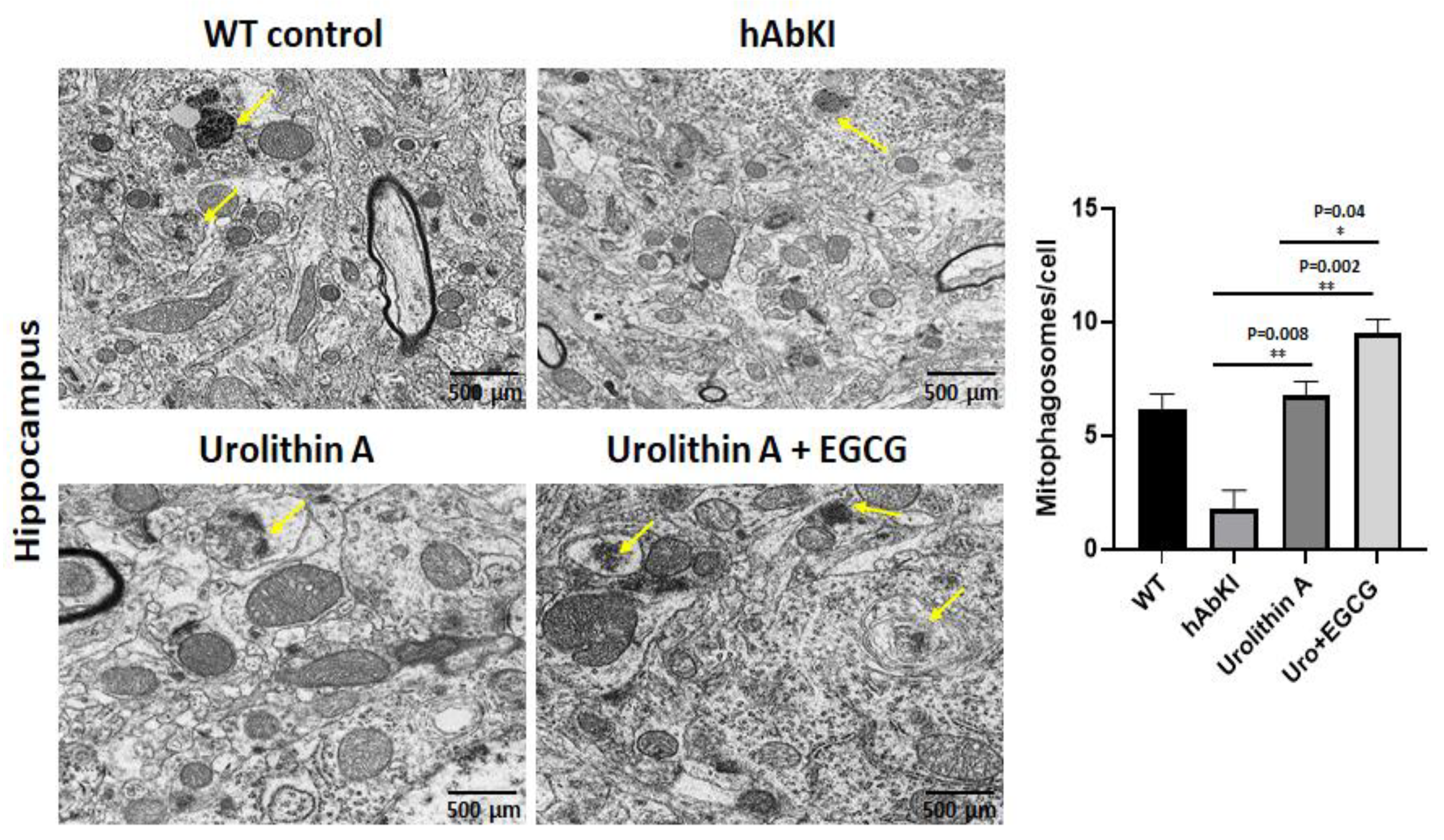
3.1.6. Transmission Electron Microscopy—Mitophagosomal Formations
3.1.7. Mitochondrial Function
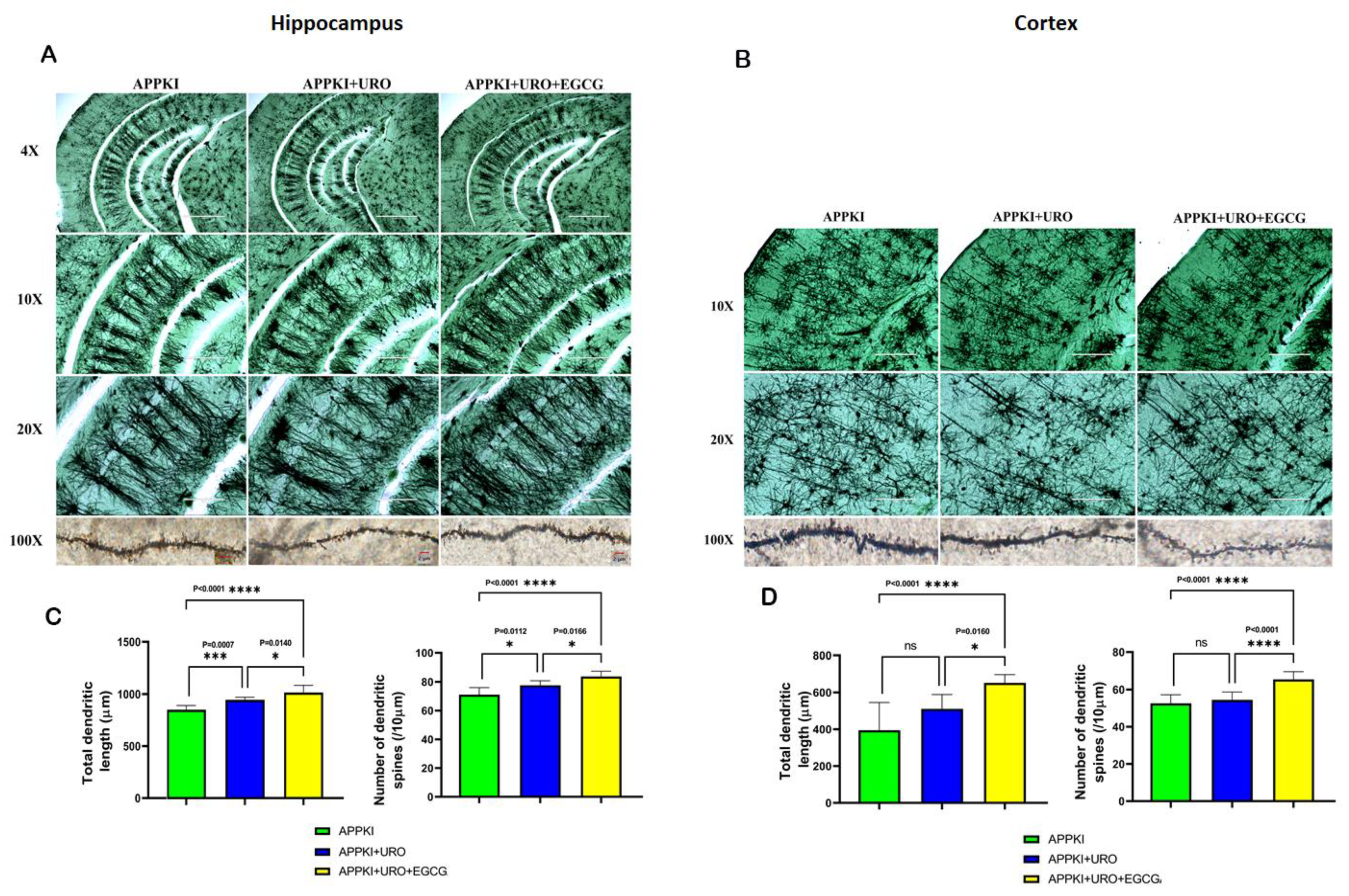
3.1.8. Golgi–Cox Staining and Dendritic Spines
3.1.9. Amyloid Beta Levels
4. Discussion
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| AAALAC | Accreditation of laboratory animal care international |
| ABAD | Amyloid beta induced alcohol dehydrogenase |
| AD | Alzheimer’s disease |
| APP | Amyloid beta precursor protein |
| Aβ | Amyloid beta |
| BBB | Blood–brain barrier |
| CHO | Chinese hamster ovary |
| CypD | Cyclophilin D |
| Drp1 | Dynamin-related protein 1 |
| EGCG | Egpigallocatechin gallate |
| Fis1 | Fission 1 |
| hAbKI | Humanized homozygous amyloid beta knockin |
| IL-1β | Interleukin 1 beta |
| IL-6 | Interleukin 6 |
| Mfn1 | Mitochondrial fusion 1 |
| Mfn2 | Mitochondrial fusion 2 |
| Nrf1 | Nuclear transcription factor 1 |
| Nrf2 | Nuclear transcription factor 2 |
| Opa1 | Optic atrophy 1 |
| p-Tau | Phosphorylated tau |
| PGC1a | Peroxisome proliferation-activated receptor gamma coactivator 1-alpha |
| PINK1 | PTEN-induced kinase 1 |
| PPARγ | Peroxisome proliferator-activated receptor gamma |
| PS1 | Presinilin 1 |
| PS2 | Presinilin 2 |
| TNFα | Tumor necrosis factor alpha |
| VDAC1 | Voltage-dependent anion channel 1 |
References
- Selkoe, D.J. Alzheimer’s disease: Genes, proteins, and therapy. Physiol. Rev. 2001, 81, 741–766. [Google Scholar] [CrossRef] [PubMed]
- John, A.; Reddy, P.H. Synaptic basis of Alzheimer’s disease: Focus on synaptic amyloid beta, P-tau and mitochondria. Ageing Res. Rev. 2021, 65, 101208. [Google Scholar] [CrossRef] [PubMed]
- Reddy, P.H.; Manczak, M.; Mao, P.; Calkins, M.J.; Reddy, A.P.; Shirendeb, U. Amyloid-beta and mitochondria in aging and Alzheimer’s disease: Implications for synaptic damage and cognitive decline. J. Alzheimer’s Dis. 2010, 20, S499–S512. [Google Scholar] [CrossRef]
- Oliver, D.M.A.; Reddy, P.H. Molecular Basis of Alzheimer’s Disease: Focus on Mitochondria. J. Alzheimer’s Dis. 2019, 72, S95–S116. [Google Scholar] [CrossRef] [PubMed]
- Morton, H.; Kshirsagar, S.; Orlov, E.; Bunquin, L.E.; Sawant, N.; Boleng, L.; George, M.; Basu, T.; Ramasubramanian, B.; Pradeepkiran, J.A.; et al. Defective mitophagy and synaptic degeneration in Alzheimer’s disease: Focus on aging, mitochondria and synapse. Free Radic. Biol. Med. 2021, 172, 652–667. [Google Scholar] [CrossRef]
- Wang, W.; Zhao, F.; Ma, X.; Perry, G.; Zhu, X. Mitochondria dysfunction in the pathogenesis of Alzheimer’s disease: Recent advances. Mol. Neurodegener. 2020, 15, 30. [Google Scholar] [CrossRef]
- Pradeepkiran, J.A.; Reddy, P.H. Defective mitophagy in Alzheimer’s disease. Ageing Res. Rev. 2020, 64, 101191. [Google Scholar] [CrossRef]
- Mattson, M.P. Pathways towards and away from Alzheimer’s disease. Nature 2004, 430, 631–639. [Google Scholar] [CrossRef]
- Swerdlow, R.H.; Burns, J.M.; Khan, S.M. The Alzheimer’s disease mitochondrial cascade hypothesis: Progress and perspectives. Biochim. Biophys. Acta 2014, 1842, 1219–1231. [Google Scholar] [CrossRef]
- Reddy, P.H.; Manczak, M.; Kandimalla, R. Mitochondria-targeted small molecule SS31: A potential candidate for the treatment of Alzheimer’s disease. Hum. Mol. Genet. 2017, 26, 1597. [Google Scholar] [CrossRef] [Green Version]
- Reddy, P.H.; McWeeney, S. Mapping cellular transcriptosomes in autopsied Alzheimer’s disease subjects and relevant animal models. Neurobiol. Aging. 2006, 8, 1060–1077. [Google Scholar] [CrossRef] [PubMed]
- Reddy, P.H.; Tripathi, R.; Troung, Q.; Tirumala, K.; Reddy, T.P.; Anekonda, V.; Shirendeb, U.P.; Calkins, M.J.; Reddy, A.P.; Mao, P.; et al. Abnormal mitochondrial dynamics and synaptic degeneration as early events in Alzheimer’s disease: Implications to mitochondria-targeted antioxidant therapeutics. Biochim. Biophys. Acta. 2012, 1822, 639–649. [Google Scholar] [CrossRef] [PubMed]
- Hirai, K.; Aliev, G.; Nunomura, A.; Fujioka, H.; Russell, R.L.; Atwood, C.S.; Johnson, A.B.; Kress, Y.; Vinters, H.V.; Tabaton, M.; et al. Mitochondrial Abnormalities in Alzheimer’s Disease. J. Neurosci. 2001, 21, 3017–3023. [Google Scholar] [CrossRef] [PubMed]
- Swerdlow, R.H.; Khan, S.M. The Alzheimer’s disease mitochondrial cascade hypothesis: An update. Exp. Neuro. 2009, 218, 308–315. [Google Scholar] [CrossRef]
- Silva, D.F.; Selfridge, J.E.; Lu, J.; E, L.; Roy, N.; Hutfles, L.; Burns, J.M.; Michaelis, E.K.; Yan, S.; Cardoso, S.M.; et al. Bioenergetic flux, mitochondrial mass and mitochondrial morphology dynamics in AD and MCI cybrid cell lines. Hum. Mol. Genet. 2013, 22, 3931–3946. [Google Scholar] [CrossRef]
- Wang, X.; Su, B.; Lee, H.G.; Li, X.; Perry, G.; Smith, M.A.; Zhu, X. Impaired balance of mitochondrial fission and fusion in Alzheimer’s disease. J. Neurosci. 2009, 29, 9090–9103. [Google Scholar] [CrossRef]
- Wang, X.; Su, B.; Siedlak, S.L.; Moreira, P.I.; Fujioka, H.; Wang, Y.; Casadesus, G.; Zhu, X. Amyloid-beta overproduction causes abnormal mitochondrial dynamics via differential modulation of mitochondrial fission/fusion proteins. Proc. Natl. Acad. Sci. USA 2008, 105, 19318–19323. [Google Scholar] [CrossRef]
- Manczak, M.; Calkins, M.J.; Reddy, P.H. Impaired mitochondrial dynamics and abnormal interaction of amyloid beta with mitochondrial protein Drp1 in neurons from patients with Alzheimer’s disease: Implications for neuronal damage. Hum. Mol. Genet. 2011, 20, 2495–2509. [Google Scholar] [CrossRef]
- Manczak, M.; Reddy, P.H. Abnormal interaction between the mitochondrial fission protein Drp1 and hyperphosphorylated tau in Alzheimer’s disease neurons: Implications for mitochondrial dysfunction and neuronal damage. Hum. Mol. Genet. 2012, 21, 2538–2547. [Google Scholar] [CrossRef]
- Manczak, M.; Reddy, P.H. Abnormal interaction of VDAC1 with amyloid beta and phosphorylated tau causes mitochondrial dysfunction in Alzheimer’s disease. Hum. Mol. Genet. 2012, 21, 5131–5146. [Google Scholar] [CrossRef]
- Lustbader, J.W.; Cirilli, M.; Lin, C.; Xu, H.W.; Takuma, K.; Wang, N.; Caspersen, C.; Chen, X.; Pollak, S.; Chaney, M.; et al. ABAD directly links Abeta to mitochondrial toxicity in Alzheimer’s disease. Science 2004, 304, 448–452. [Google Scholar] [CrossRef]
- Du, H.; Guo, L.; Fang, F.; Chen, D.; Sosunov, A.A.; McKhann, G.M.; Yan, Y.; Wang, C.; Zhang, H.; Molkentin, J.D.; et al. Cyclophilin D deficiency attenuates mitochondrial and neuronal perturbation and ameliorates learning and memory in Alzheimer’s disease. Nat. Med. 2008, 14, 1097–1105. [Google Scholar] [CrossRef] [PubMed]
- Reddy, P.H.; Yin, X.; Manczak, M.; Kumar, S.; Pradeepkiran, J.A.; Vijayan, M.; Reddy, A.P. Mutant APP and amyloid beta-induced defective autophagy, mitophagy, mitochondrial structural and functional changes and synaptic damage in hippocampal neurons from Alzheimer’s disease. Hum. Mol. Genet. 2018, 27, 2502–2516. [Google Scholar] [CrossRef] [PubMed]
- Kandimalla, R.; Manczak, M.; Yin, X.; Wang, R.; Reddy, P.H. Hippocampal phosphorylated tau induced cognitive decline, dendritic spine loss and mitochondrial abnormalities in a mouse model of Alzheimer’s disease. Hum. Mol. Genet. 2018, 27, 30–40. [Google Scholar] [CrossRef] [PubMed]
- Manczak, M.; Kandimalla, R.; Yin, X.; Reddy, P.H. Hippocampal mutant APP and amyloid beta-induced cognitive decline, dendritic spine loss, defective autophagy, mitophagy and mitochondrial abnormalities in a mouse model of Alzheimer’s disease. Hum. Mol. Genet. 2018, 27, 1332–1342. [Google Scholar] [CrossRef] [PubMed]
- Reddy, A.P.; Sawant, N.; Morton, H.; Kshirsagar, S.; Bunquin, L.E.; Yin, X.; Reddy, P.H. Selective serotonin reuptake inhibitor citalopram ameliorates cognitive decline and protects against amyloid beta-induced mitochondrial dynamics, biogenesis, autophagy, mitophagy and synaptic toxicities in a mouse model of Alzheimer’s disease. Hum. Mol. Genet. 2021, 30, 789–810. [Google Scholar] [CrossRef]
- Reddy, A.P.; Yin, X.; Sawant, N.; Reddy, P.H. Protective effects of antidepressant citalopram against abnormal APP processing and amyloid beta-induced mitochondrial dynamics, biogenesis, mitophagy and synaptic toxicities in Alzheimer’s disease. Hum. Mol. Genet. 2021, 30, 847–864. [Google Scholar] [CrossRef]
- Kshirsagar, S.; Sawant, N.; Morton, H.; Reddy, A.P.; Reddy, P.H. Protective effects of mitophagy enhancers against amyloid beta-induced mitochondrial and synaptic toxicities in Alzheimer disease. Hum. Mol. Genet. 2022, 31, 423–439. [Google Scholar] [CrossRef]
- Kshirsagar, S.; Sawant, N.; Morton, H.; Reddy, A.P.; Reddy, P.H. Mitophagy enhancers against phosphorylated Tau-induced mitochondrial and synaptic toxicities in Alzheimer disease. Pharmacol. Res. 2021, 174, 105973. [Google Scholar] [CrossRef]
- Reddy, P.H.; Oliver, D.M. Amyloid Beta and Phosphorylated Tau-Induced Defective Autophagy and Mitophagy in Alzheimer’s Disease. Cells 2019, 8, 488. [Google Scholar] [CrossRef] [Green Version]
- Manczak, M.; Kandimalla, R.; Yin, X.; Reddy, P.H. Mitochondrial division inhibitor 1 reduces dynamin-related protein 1 and mitochondrial fission activity. Hum. Mol. Genet. 2019, 28, 177–199. [Google Scholar] [CrossRef] [PubMed]
- Fang, E.F.; Hou, Y.; Palikaras, K.; Adriaanse, B.A.; Kerr, J.S.; Yang, B.; Lautrup, S.; Hasan-Olive, M.M.; Caponio, D.; Dan, X.; et al. Mitophagy inhibits amyloid-β and tau pathology and reverses cognitive deficits in models of Alzheimer’s disease. Nat. Neurosci. 2019, 22, 401–412. [Google Scholar] [CrossRef] [PubMed]
- Gong, Z.; Huang, J.; Xu, B.; Ou, Z.; Zhang, L.; Lin, X.; Ye, X.; Kong, X.; Long, D.; Sun, X.; et al. Urolithin A attenuates memory impairment and neuroinflammation in APP/PS1 mice. J. Neuroinflammation 2019, 16, 62. [Google Scholar] [CrossRef] [PubMed]
- Cascella, M.; Bimonte, S.; Muzio, M.R.; Schiavone, V.; Cuomo, A. The efficacy of Epigallocatechin-3-gallate (green tea) in the treatment of Alzheimer’s disease: An overview of pre-clinical studies and translational perspectives in clinical practice. Infect. Agents Cancer 2017, 12, 36. [Google Scholar] [CrossRef]
- Wei, Y.; Liu, D.; Zheng, Y.; Hao, C.; Li, H.; Ouyang, W. Neuroprotective Effects of Kinetin Against Glutamate-Induced Oxidative Cytotoxicity in HT22 Cells: Involvement of Nrf2 and Heme Oxygenase-1. Neurotox. Res. 2018, 33, 725–737. [Google Scholar] [CrossRef]
- Ehrnhoefer, D.E.; Bieschke, J.; Boeddrich, A.; Herbst, M.; Masino, L.; Lurz, R.; Engemann, S.; Pastore, A.; Wanker, E.E. EGCG redirects amyloidogenic polypeptides into unstructured, off-pathway oligomers. Nat. Struct. Mol. Biol. 2008, 15, 558–566. [Google Scholar] [CrossRef]
- Bieschke, J.; Russ, J.; Friedrich, R.P.; Ehrnhoefer, D.E.; Wobst, H.; Neugebauer, K.; Wanker, E.E. EGCG remodels mature α-synuclein and amyloid-β fibrils and reduces cellular toxicity. Proc. Natl. Acad. Sci. USA 2010, 107, 7710–7715. [Google Scholar] [CrossRef]
- Chesser, A.S.; Ganeshan, V.; Yang, J.; Johnson, G.V. Epigallocatechin-3-gallate enhances clearance of phosphorylated tau in primary neurons. Nutr. Neurosci. 2016, 19, 21–31. [Google Scholar] [CrossRef]
- Chang, X.; Rong, C.; Chen, Y.; Yang, C.; Hu, Q.; Mo, Y.; Zhang, C.; Gu, X.; Zhang, L.; He, W.; et al. (−)-Epigallocatechin-3-gallate attenuates cognitive deterioration in Alzheimer’s disease model mice by upregulating neprilysin expression. Exp. Cell Res. 2015, 334, 136–145. [Google Scholar] [CrossRef]
- Zhang, Z.X.; Li, Y.B.; Zhao, R.P. Epigallocatechin Gallate attenuates β-Amyloid generation and oxidative stress involvement of PPARγ in N2a/APP695 cells. Neurochem. Res. 2017, 42, 468–480. [Google Scholar] [CrossRef]
- Kshirsagar, S.; Alvir, R.V.; Hindle, A.; Kumar, S.; Vijayan, M.; Pradeepkiran, J.A.; Reddy, A.P.; Ramasubramanian, B.; Reddy, P.H. Early Cellular, Molecular, Morphological and Behavioral Changes in the Humanized Amyloid-Beta-Knock-In Mouse Model of Late-Onset Alzheimer’s Disease. Cells 2022, 11, 733. [Google Scholar] [CrossRef] [PubMed]
- Baglietto-Vargas, D.; Forner, S.; Cai, L.; Martini, A.C.; Trujillo-Estrada, L.; Swarup, V.; Nguyen, M.M.T.; do Huynh, K.; Javonillo, D.I.; Tran, K.M.; et al. Generation of a humanized Aβ expressing mouse demonstrating aspects of Alzheimer’s disease-like pathology. Nat. Commun. 2021, 12, 2421. [Google Scholar] [CrossRef] [PubMed]
- Kandimalla, R.; Manczak, M.; Fry, D.; Suneetha, Y.; Sesaki, H.; Reddy, P.H. Reduced dynamin-related protein 1 protects against phosphorylated Tau-induced mitochondrial dysfunction and synaptic damage in Alzheimer’s disease. Hum. Mol. Genet. 2016, 25, 4881–4897. [Google Scholar] [CrossRef] [PubMed]
- Kandimalla, R.; Manczak, M.; Pradeepkiran, J.A.; Morton, H.; Reddy, P.H. A partial reduction of Drp1 improves cognitive behavior and enhances mitophagy, autophagy and dendritic spines in a transgenic Tau mouse model of Alzheimer disease. Hum. Mol. Genet. 2022, 31, 1788–1805. [Google Scholar] [CrossRef] [PubMed]
- Vijayan, M.; Bose, C.; Reddy, P.H. Protective effects of a small molecule inhibitor, DDQ against amyloid beta in Alzheimer’s disease. Mitochondrion 2021, 59, 17–29. [Google Scholar] [CrossRef] [PubMed]
- Kumar, S.; Morton, H.; Sawant, N.; Orlov, E.; Bunquin, L.E.; Pradeepkiran, J.A.; Alvir, R.; Reddy, P.H. MicroRNA-455-3p improves synaptic, cognitive functions and extends lifespan: Relevance to Alzheimer’s disease. Redox Biol. 2021, 48, 102182. [Google Scholar] [CrossRef] [PubMed]
- Vijayan, M.; George, M.; Bunquin, L.E.; Bose, C.; Reddy, P.H. Protective effects of a small-molecule inhibitor DDQ against tau-induced toxicities in a transgenic tau mouse model of Alzheimer’s disease. Hum. Mol. Genet. 2022, 31, 1022–1034. [Google Scholar] [CrossRef]
- Manczak, M.; Kandimalla, R.; Fry, D.; Sesaki, H.; Reddy, P.H. Protective effects of reduced dynamin-related protein 1 against amyloid beta-induced mitochondrial dysfunction and synaptic damage in Alzheimer’s disease. Hum. Mol. Genet. 2016, 23, 5148–5166. [Google Scholar] [CrossRef]
- Mega, M.S.; Cummings, J.L.; Fiorello, T.; Gornbein, J. The spectrum of behavioral changes in Alzheimer’s disease. Neurology 1996, 46, 130–305. [Google Scholar] [CrossRef]
- Abdulrahman, A.O.; Alzubaidi, M.Y.; Nadeem, M.S.; Khan, J.A.; Rather, I.A.; Khan, M.I. The Utilization of Urolithin A—A Natural Polyphenol Metabolite of Ellagitannins as a Modulator of the Gut Microbiota for Its Potential Use in Obesity Therapy. Proceedings 2021, 79, 12. [Google Scholar]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene | DNA Sequence (5′-3′) | PCR Product Size |
|---|---|---|
| Mitochondrial Structural Genes | ||
| Drp1 | Forward Primer ATGCCAGCAAGTCCACAGAA | 86 |
| Reverse Primer TGTTCTCGGGCAGACAGTTT | ||
| Fis1 | Forward Primer CAAAGAGGAACAGCGGGACT | 95 |
| Reverse Primer ACAGCCCTCGCACATACTTT | ||
| Mfn1 | Forward Primer GCAGACAGCACATGGAGAGA | 83 |
| Reverse Primer GATCCGATTCCGAGCTTCCG | ||
| Mfn2 | Forward Primer TGCACCGCCATATAGAGGAAG | 78 |
| Reverse Primer TCTGCAGTGAACTGGCAATG | ||
| Opa1 | Forward Primer ACCTTGCCAGTTTAGCTCCC | 82 |
| Reverse Primer TTGGGACCTGCAGTGAAGAA | ||
| Mitochondrial Biogenesis Genes | ||
| PGC1α | Forward Primer GCAGTCGCAACATGCTCAAG | 83 |
| Reverse Primer GGGAACCCTTGGGGTCATTT | ||
| Nrf1 | Forward Primer AGAAACGGAAACGGCTCAT | 96 |
| Reverse Primer CATCCAACGTGGCTCTGAGT | ||
| Nrf2 | Forward Primer ATGGAGCAAGTTTGGCAGGA | 96 |
| Reverse Primer GCTGGGAACAGAGGTAGTAT | ||
| TFAM | Forward Primer TCCACAGAACAGCTACCCAA | 84 |
| Reverse Primer CCACAGGGCTGCAATTTTCC | ||
| Synaptic Genes | ||
| Synaptophysin | Forward Primer CTGCGTTAAAGGGGGCACTA | 81 |
| Reverse Primer ACAGCCACGGTGACAAAGAA | ||
| PSD95 | Forward Primer CTTCATCCTTGCTGGGGGTC | 90 |
| Reverse Primer TTGCGGAGGTCAACACCATT | ||
| Synapsin 1 | Forward Primer TGAGGACATCAGTGTCGGGTAA | 64 |
| Reverse Primer GGCAATCTGCTCAAGCATAGC | ||
| Synapsin 2 | Forward Primer TCCCACTCATTGAGCAGACATACT | |
| Reverse Primer GGGAACGTAGGAAGCGTAAGC | ||
| Synaptobrevin 1 | Forward Primer TGCTGCCAAGCTAAAAAGGAA | 68 |
| Reverse Primer CAGATAGCTCCCAGCATGATCA | ||
| Synaptobrevin 2 | Forward Primer GGGACCAGAAGTTGTCGGAG | 89 |
| Reverse Primer CTTGAGCTTGGCTGCACTTG | ||
| Neurogranin | Forward Primer CTCCAAGCCAGACGACGATA | 83 |
| Reverse Primer AACTCGCCTGGATTTTGGCT | ||
| Mitophagy genes | ||
| PINK1 | Forward Primer CCATCGGGATCTCAAGTCCG | 70 |
| Reverse Primer GATCACTAGCCAGGGACAGC | ||
| Parkin | Forward Primer AGAGGTCCAGTTAAACCCACC | 90 |
| Reverse Primer GAGGGTTGCTTGTTTGCAGG | ||
| ATG5 | Forward Primer TCCATCCAAGGATGCGGTTG | 95 |
| Reverse Primer TCTGCATTTCGTTGATCACTTGAC | ||
| BCL2 | Forward Primer TCCTTCCAGCCTGAGAGCAA | 73 |
| Reverse Primer GCCTGAGAGGAGACGTCCTG | ||
| LC3B | Forward Primer TCCACTCCCATCTCCGAAGT | 94 |
| Reverse Primer TTGCTGTCCCGAATGTCTCC | ||
| Marker Primary Antibody—Species and Dilution | Purchased from Company, City and State | Secondary Antibody, Dilution | Purchased from Company, City and State |
|---|---|---|---|
| Drp1 Rabbit polyclonal (#NB110-55288) 1:100 | Novus Biological, Littleton, CO | Donkey anti-rabbit HRP 1:200 | Invitrogen |
| Fis1 Rabbit polyclonal (#10956-1-AP) 1:100 | Protein Tech Group, Inc., Chicago, IL | Donkey anti-rabbit HRP 1:200 | GE Healthcare Amersham, Piscataway, NJ |
| Mfn1 Rabbit polyclonal (#ab191853) 1:100 | Abcam, Cambridge, MA | Donkey anti-rabbit HRP 1:200 | GE Healthcare Amersham, Piscataway, NJ |
| Mfn2 Rabbit polyclonal (#Ab205236) 1:100 | Abcam, Cambridge, MA | Donkey anti-rabbit HRP 1:200 | GE Healthcare Amersham, Piscataway, NJ |
| OPA1 Rabbit polyclonal (#NBP2-59770) 1:100 | Novus Biological, Littleton, CO | Donkey anti-rabbit HRP 1:200 | GE Healthcare Amersham, Piscataway, NJ |
| SYN Rabbit monoclonal (#Ab32127) 1:400 | Abcam, Cambridge, MA | Donkey anti-rabbit HRP 1:200 | GE Healthcare Amersham, Piscataway, NJ |
| PGC1a Rabbit polyclonal (#NBP1-04676) 1:100 | Novus Biological, Littleton, CO | Donkey anti-rabbit HRP 1:200 | GE Healthcare Amersham, Piscataway, NJ |
| NRF1 Rabbit polyclonal (#NBP1-77822) 1:100 | Novus Biological, Littleton, CO | Donkey anti-rabbit HRP 1:200 | GE Healthcare Amersham, Piscataway, NJ |
| NRF2 Rabbit polyclonal (#NBP1-32822) 1:100 | Novus Biological, Littleton, CO | Donkey anti-rabbit HRP 1:200 | GE Healthcare Amersham, Piscataway, NJ |
| TFAM Rabbit polyclonal (#NBP2-59770) 1:100 | Novus Biological, Littleton, CO | Donkey anti-rabbit HRP 1:200 | GE Healthcare Amersham, Piscataway, NJ |
| PINK1 Rabbit polyclonal (#BC100-494) 1:100 | Novus Biological, Littleton, CO | Donkey anti-rabbit HRP 1:200 | GE Healthcare Amersham, Piscataway, NJ |
| Parkin Mouse polyclonal (#NBP2-29838) 1:100 | Novus Biological, Littleton, CO | Sheep anti-mouse HRP 1:200 | GE Healthcare Amersham, Piscataway, NJ |
| Iba1/AIF-12 Rabbit monoclonal (#17178S) 1:100 | Cell Signaling Technology, Inc., MA | Donkey Anti-rabbit HRP 1:200 | GE Healthcare Amersham, Piscataway, NJ |
| Anti-NeuN Rabbit monoclonal (#Ab177487) 1:100 | Abcam, Cambridge, MA | Donkey Anti-rabbit HRP 1:200 | GE Healthcare Amersham, Piscataway, NJ |
| Marker Primary Antibody—Species and Dilution | Purchased from Company, City and State | Secondary Antibody, Dilution | Purchased from Company, City and State |
|---|---|---|---|
| Drp1 Rabbit polyclonal (#NB110-55288) 1:500 | Novus Biological, Littleton, CO | Donkey anti-rabbit HRP 1:10,000 | GE Healthcare Amersham, Piscataway, NJ |
| Fis1 Rabbit polyclonal (#10956-1-AP) 1:500 | Protein Tech Group, Inc., Chicago, IL | Donkey anti-rabbit HRP 1:10,000 | GE Healthcare Amersham, Piscataway, NJ |
| Mfn1 Rabbit polyclonal (#Ab221661) 1:400 | Abcam, Cambridge, MA | Donkey anti-rabbit HRP 1:10,000 | GE Healthcare Amersham, Piscataway, NJ |
| Mfn2 Rabbit polyclonal (#Ab205236) 1:400 | Abcam, Cambridge, MA | Donkey anti-rabbit HRP 1:10,000 | GE Healthcare Amersham, Piscataway, NJ |
| OPA1 Rabbit polyclonal (#NBP2-59770) 1:500 | Novus Biological, Littleton, CO | Donkey anti-rabbit HRP 1:10,000 | GE Healthcare Amersham, Piscataway, NJ |
| SYN Rabbit monoclonal (#Ab32127) 1:400 | Abcam, Cambridge, MA | Donkey anti-rabbit HRP 1:10,000 | GE Healthcare Amersham, Piscataway, NJ |
| PGC1a Rabbit polyclonal (#NBP1-04676) 1:500 | Novus Biological, Littleton, CO | Donkey anti-rabbit HRP 1:10,000 | GE Healthcare Amersham, Piscataway, NJ |
| NRF1 Rabbit polyclonal (#NBP1-77822) 1:300 | Novus Biological, Littleton, CO | Donkey anti-rabbit HRP 1:10,000 | GE Healthcare Amersham, Piscataway, NJ |
| NRF2 Rabbit polyclonal (#NBP1-32822) 1:300 | Novus Biological, Littleton, CO | Donkey anti-rabbit HRP 1:10,000 | GE Healthcare Amersham, Piscataway, NJ |
| TFAM Rabbit polyclonal (#NBP2-59770) 1:300 | Novus Biological, Littleton, CO | Donkey anti-rabbit HRP 1:10,000 | GE Healthcare Amersham, Piscataway, NJ |
| PINK1 Rabbit polyclonal (#BC100-494) 1:500 | Novus Biological, Littleton, CO | Donkey anti-rabbit HRP 1:10,000 | GE Healthcare Amersham, Piscataway, NJ |
| Parkin Mouse polyclonal (#NBP2-29838) 1:500 | Novus Biological, Littleton, CO | Sheep anti-mouse HRP 1:10,000 | GE Healthcare Amersham, Piscataway, NJ |
| ATG5 Rabbit Polyclonal (#NBP2-54702) 1:1000 | Novus Biological, Littleton, CO | Donkey Anti-rabbit HRP 1:10,000 | GE Healthcare Amersham, Piscataway, NJ |
| LC3B Rabbit Polyclonal (#NB100-2220) 1:1000 | Novus Biological, Littleton, CO | Donkey Anti-rabbit HRP 1:10,000 | GE Healthcare Amersham, Piscataway, NJ |
| Beclin1 Rabbit Polyclonal (#NB500-249) 1:1000 | Novus Biological, Littleton, CO | Donkey Anti-rabbit HRP 1:10,000 | GE Healthcare Amersham, Piscataway, NJ |
| Bcl-2 Rabbit Polyclonal (#NB100-56098) 1:1000 | Novus Biological, Littleton, CO | Donkey Anti-rabbit HRP 1:10,000 | GE Healthcare Amersham, Piscataway, NJ |
| Iba1/AIF-12 Rabbit monoclonal (#17178S) 1:1000 | Cell Signaling Technology, Inc., MA | Donkey Anti-rabbit HRP 1:10,000 | GE Healthcare Amersham, Piscataway, NJ |
| Anti-NeuN Rabbit monoclonal (#Ab177487) 1:1000 | Abcam, Cambridge, MA | Donkey Anti-rabbit HRP 1:10,000 | GE Healthcare Amersham, Piscataway, NJ |
| Genes | mRNA Fold Change in Urolithin A-Treated hAbKI Mice | mRNA Fold Change in Urolithin A+EGCG-Treated hAbKI Mice | |
|---|---|---|---|
| Mitochondrial genes | Drp1 | −1.96 * | −2.12 * |
| Fis1 | −2.32 ** | −2.6 ** | |
| Mfn1 | 2.03 * | 8.9 ** | |
| Mfn2 | 3.4 ** | 5.23 ** | |
| OPA1 | 2.91 * | 3.76 * | |
| Biogenesis genes | PGC1 alpha | 3.03 ** | 4.1 ** |
| Nrf1 | 1.46 * | 2.76 *** | |
| Nrf2 | 2.39 ** | 3.25 *** | |
| TFAM | 1.8 * | 2.2 ** | |
| Synaptic genes | Synaptophysin | 3.45 * | 3.9 * |
| PSD95 | 2.9 * | 3.5 ** | |
| Snap25 | 3.7 ** | 6.34 ** | |
| Synapsin 1 Synapsin 2 | 2.25 10.2 *** | 17.03 *** 14.6 *** | |
| Synaptobrevin 1 Synaptobrevin 2 | 2.7 * 6.6 ** | 4.67 ** 7.87 *** | |
| Neurogranin | 3.81 ** | 8.5 *** | |
| Mitophagy genes | PINK1 | 2.63 ** | 3.24 ** |
| Parkin | 2.3 * | 2.33 * | |
| Autophagy genes | ATG5 | 2.07 * | 2.23 * |
| LC3B | 2.08 ** | 4.34 *** | |
| BCL2 | 2.25 * | 4.8 ** |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kshirsagar, S.; Alvir, R.V.; Pradeepkiran, J.A.; Hindle, A.; Vijayan, M.; Ramasubramaniam, B.; Kumar, S.; Reddy, A.P.; Reddy, P.H. A Combination Therapy of Urolithin A+EGCG Has Stronger Protective Effects than Single Drug Urolithin A in a Humanized Amyloid Beta Knockin Mice for Late-Onset Alzheimer’s Disease. Cells 2022, 11, 2660. https://doi.org/10.3390/cells11172660
Kshirsagar S, Alvir RV, Pradeepkiran JA, Hindle A, Vijayan M, Ramasubramaniam B, Kumar S, Reddy AP, Reddy PH. A Combination Therapy of Urolithin A+EGCG Has Stronger Protective Effects than Single Drug Urolithin A in a Humanized Amyloid Beta Knockin Mice for Late-Onset Alzheimer’s Disease. Cells. 2022; 11(17):2660. https://doi.org/10.3390/cells11172660
Chicago/Turabian StyleKshirsagar, Sudhir, Rainier Vladlen Alvir, Jangampalli Adi Pradeepkiran, Ashly Hindle, Murali Vijayan, Bhagavathi Ramasubramaniam, Subodh Kumar, Arubala P. Reddy, and P. Hemachandra Reddy. 2022. "A Combination Therapy of Urolithin A+EGCG Has Stronger Protective Effects than Single Drug Urolithin A in a Humanized Amyloid Beta Knockin Mice for Late-Onset Alzheimer’s Disease" Cells 11, no. 17: 2660. https://doi.org/10.3390/cells11172660