
FLIM for Evaluation of Difference in Metabolic Status between Native and Differentiated from iPSCs Dermal Papilla Cells

 , ,
, ,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Cell Culture and Differentiation
2.2. Isolation of Primary Dermal Cells
2.3. Obtaining of Dermal Spheroids
2.4. Immunofluorescent Staining
2.5. Isolation of RNA and Quantitative PCR Analysis
2.5.1. Isolation of RNA and Quantitative PCR Analysis of the Expression of Specialized Genes
2.5.2. Quantitative PCR Analysis of the Expression of Metabolic Genes
2.6. Analysis of Dermal Spheroid Energy Metabolism by Multiphoton Fluorescence Microscopy and FLIM
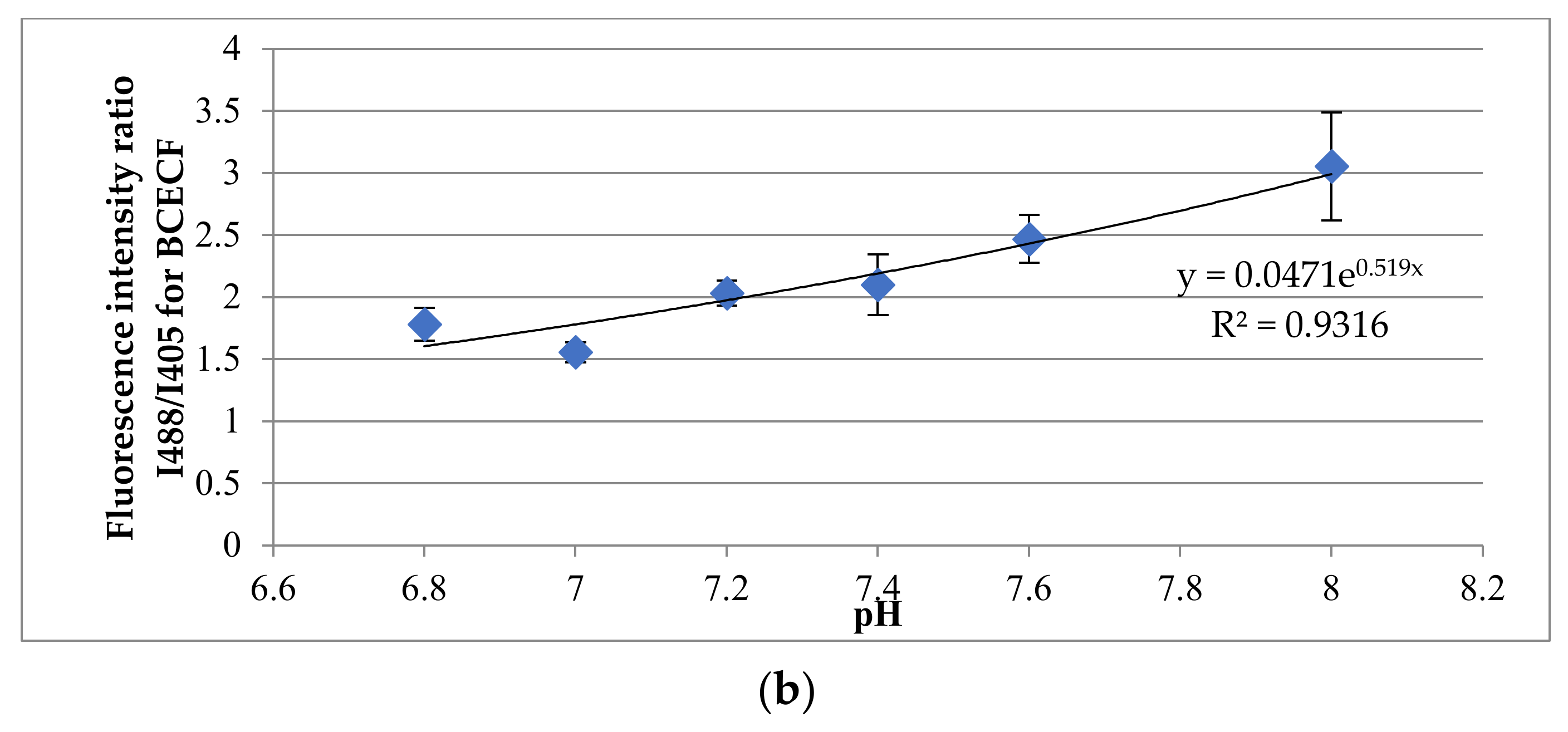
2.7. Analysis of Dermal Spheroid Intracellular pH Using Multiphoton Fluorescence Microscopy
2.8. Statistical Analysis
3. Results
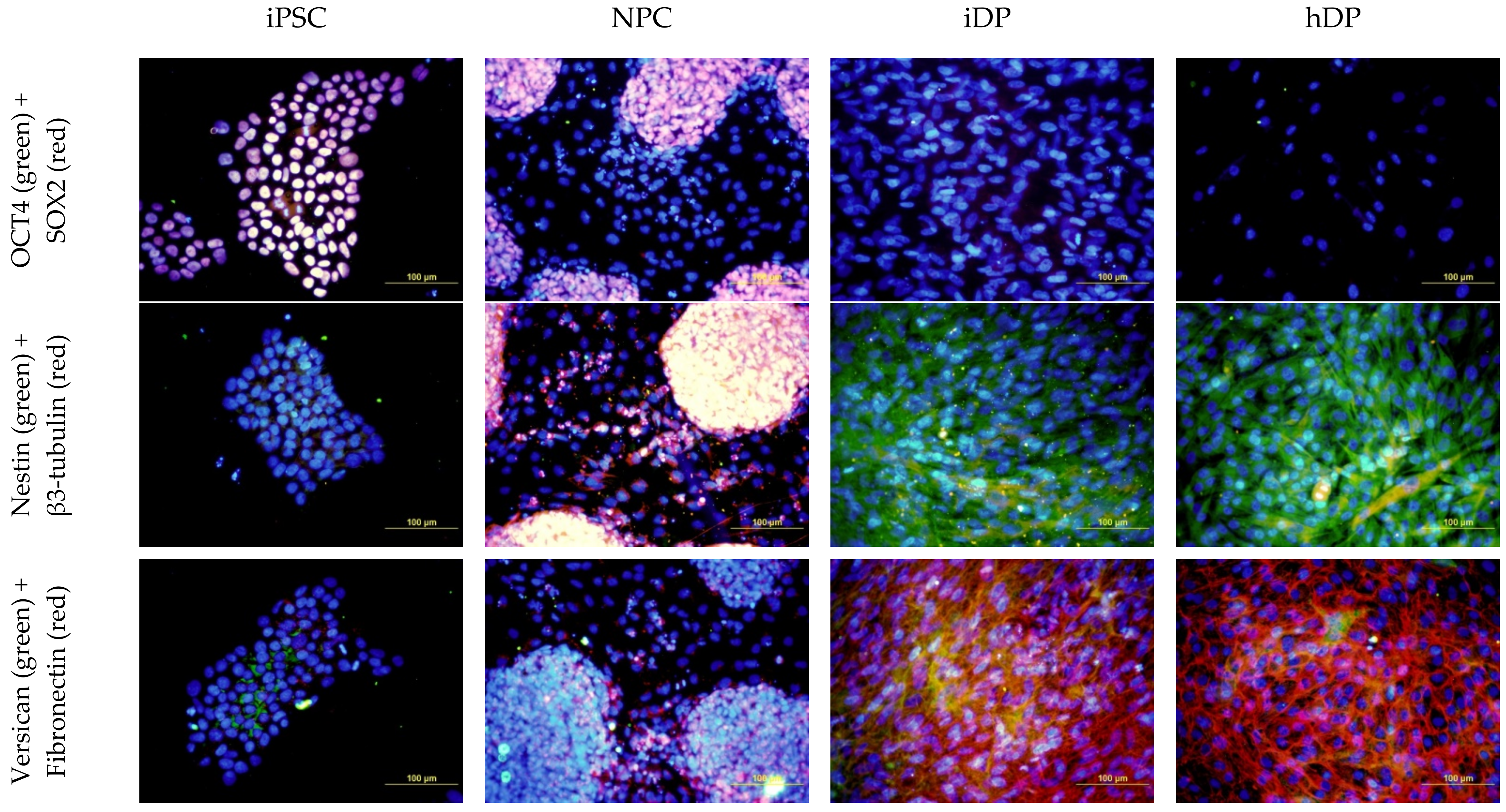
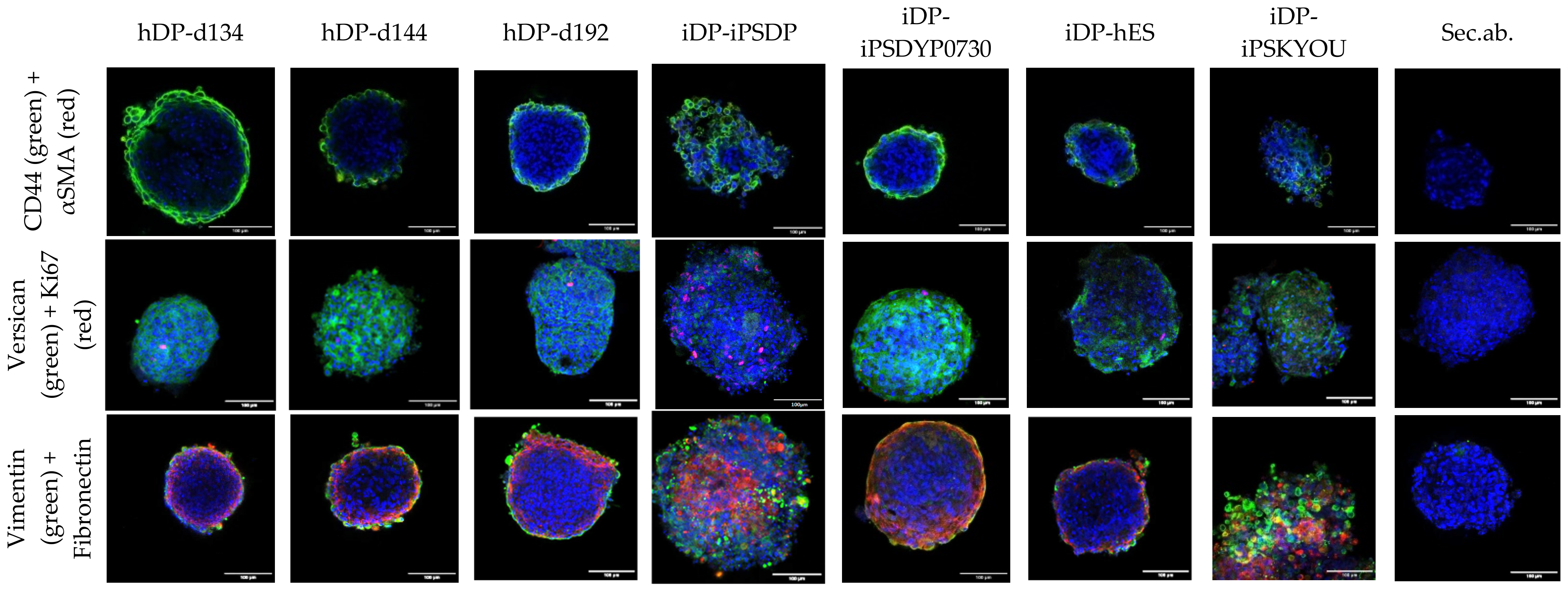
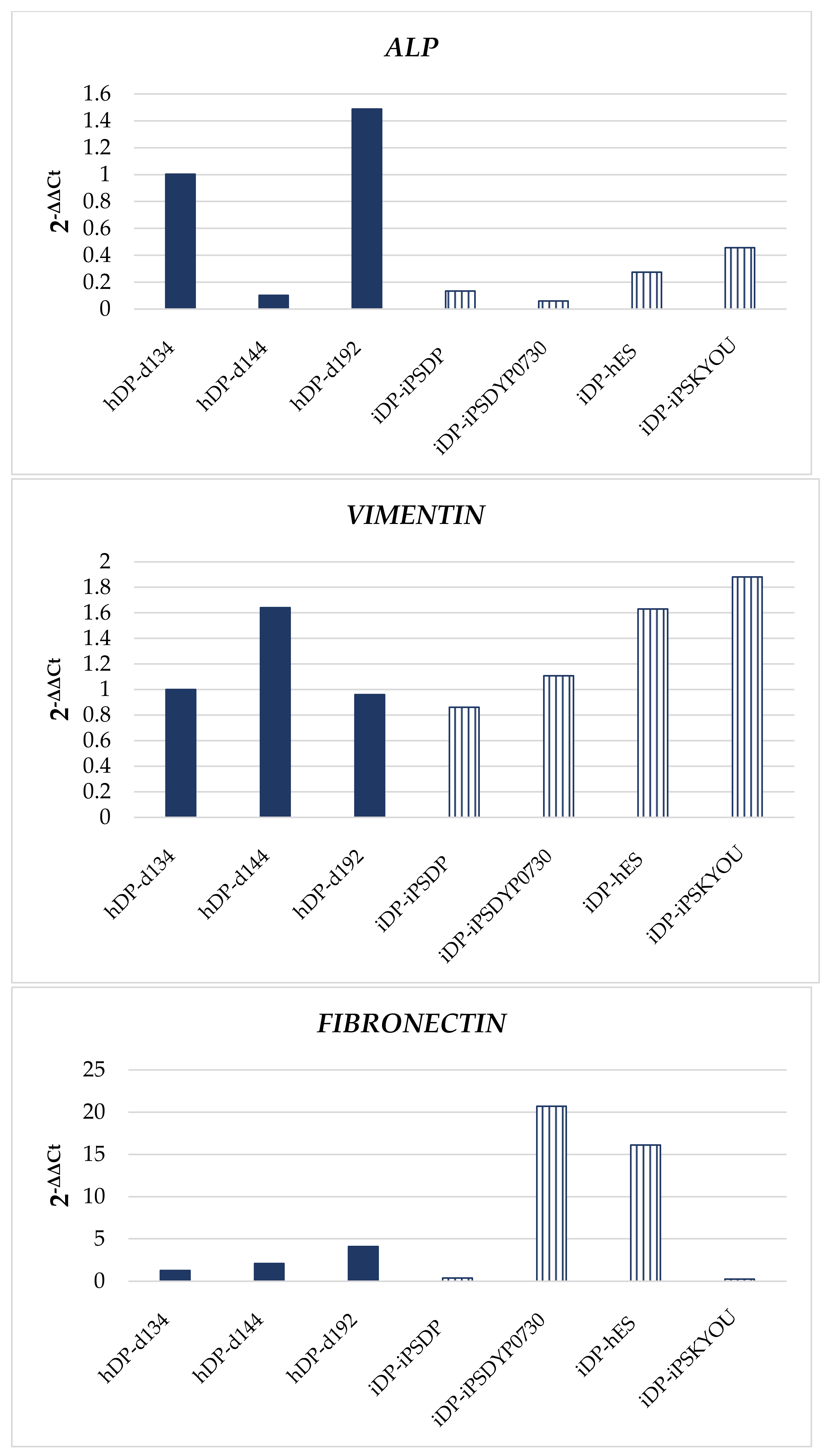
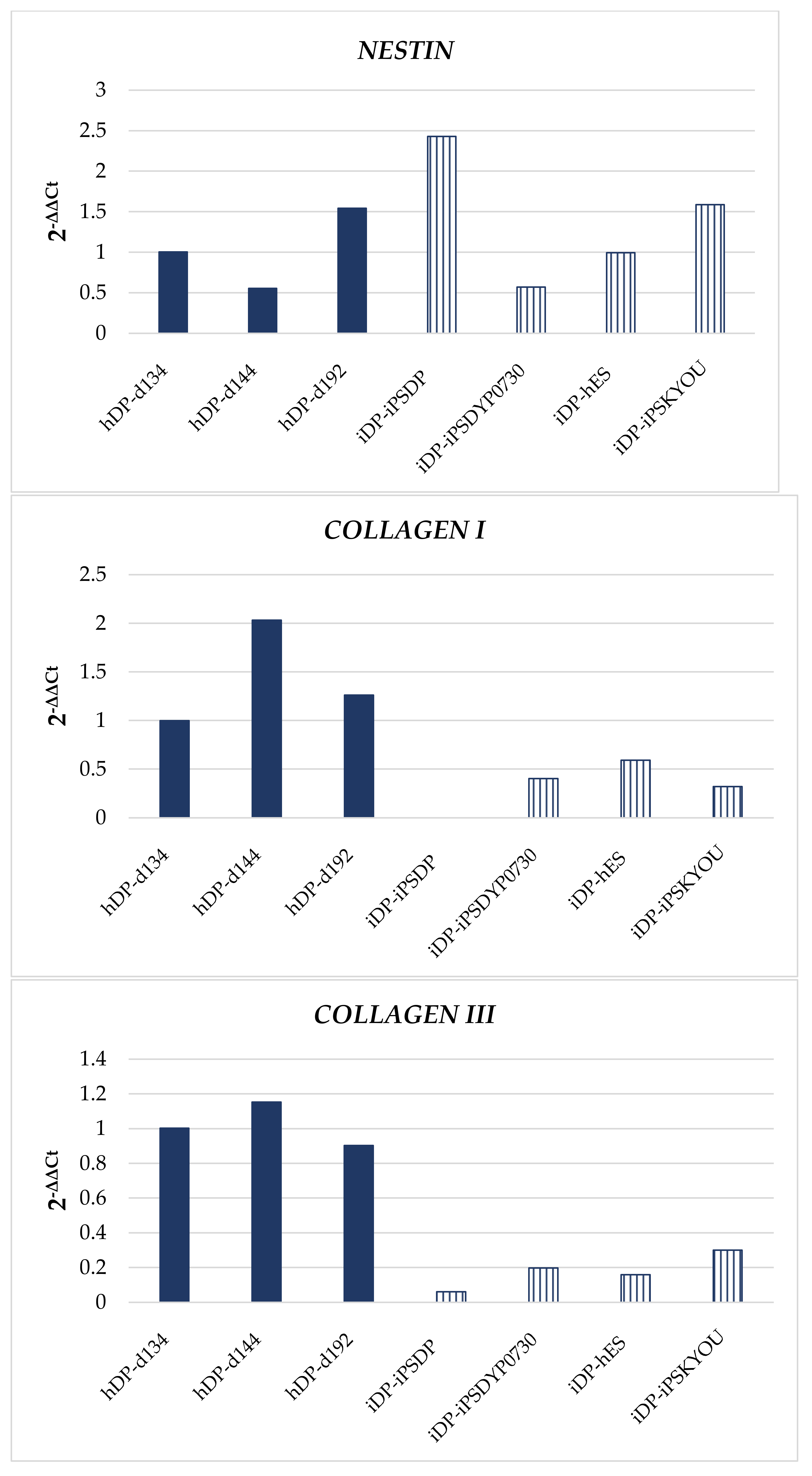
3.1. Dermal Differentiation, and Formation of hDP and iDP Spheroids
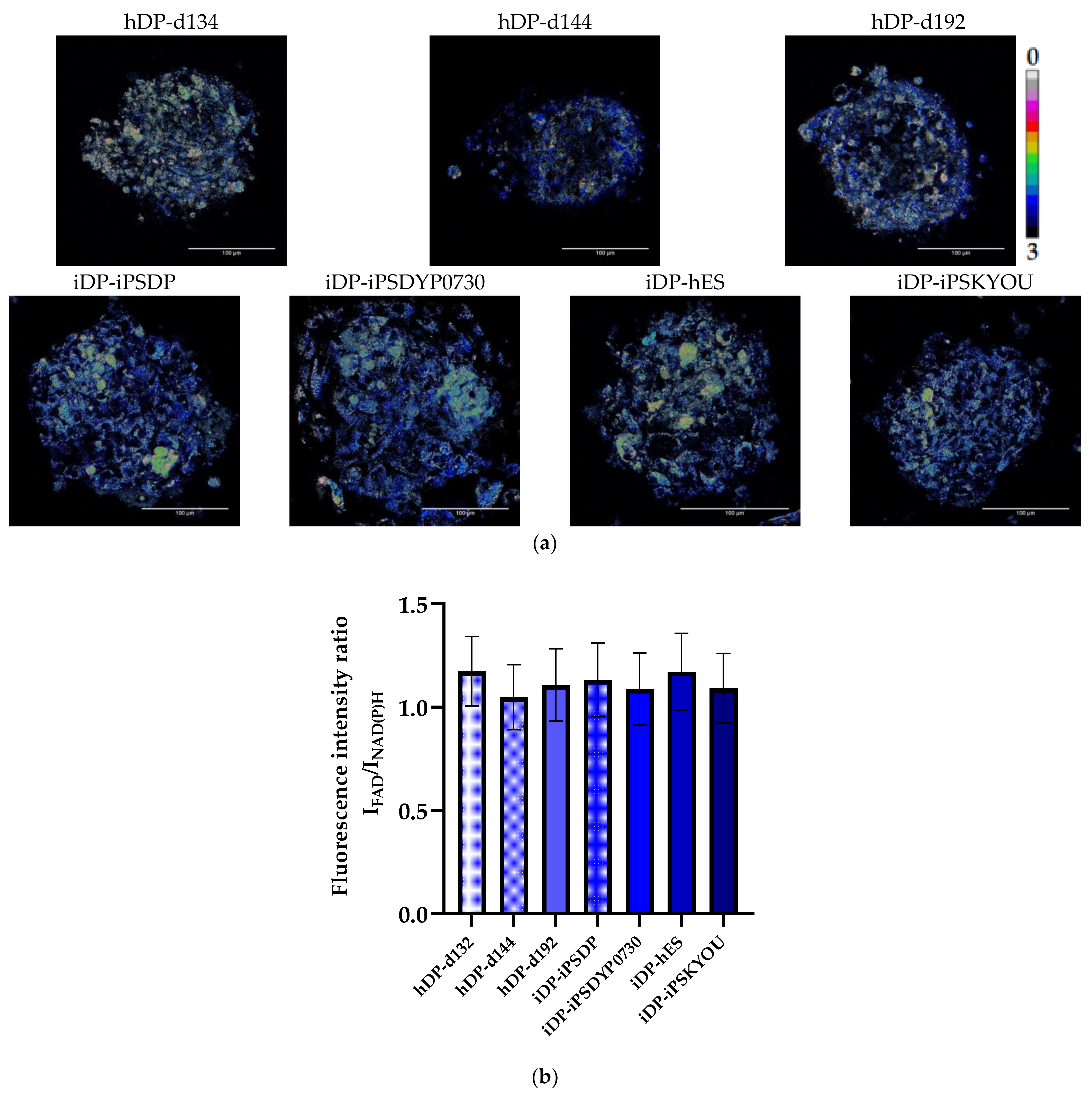
3.2. Evaluation of hDP and iDP Spheroids Energy Metabolism Using FLIM
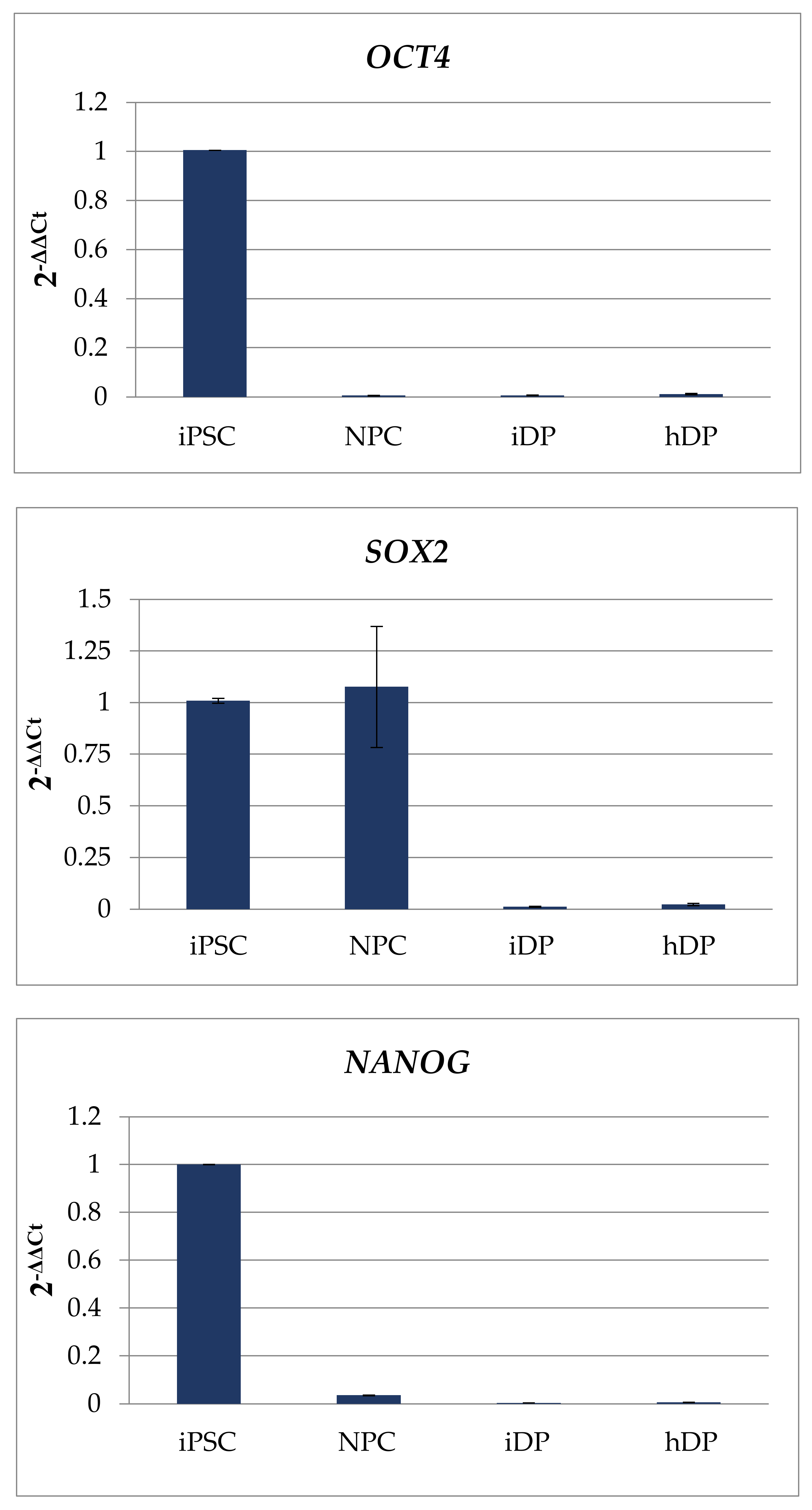
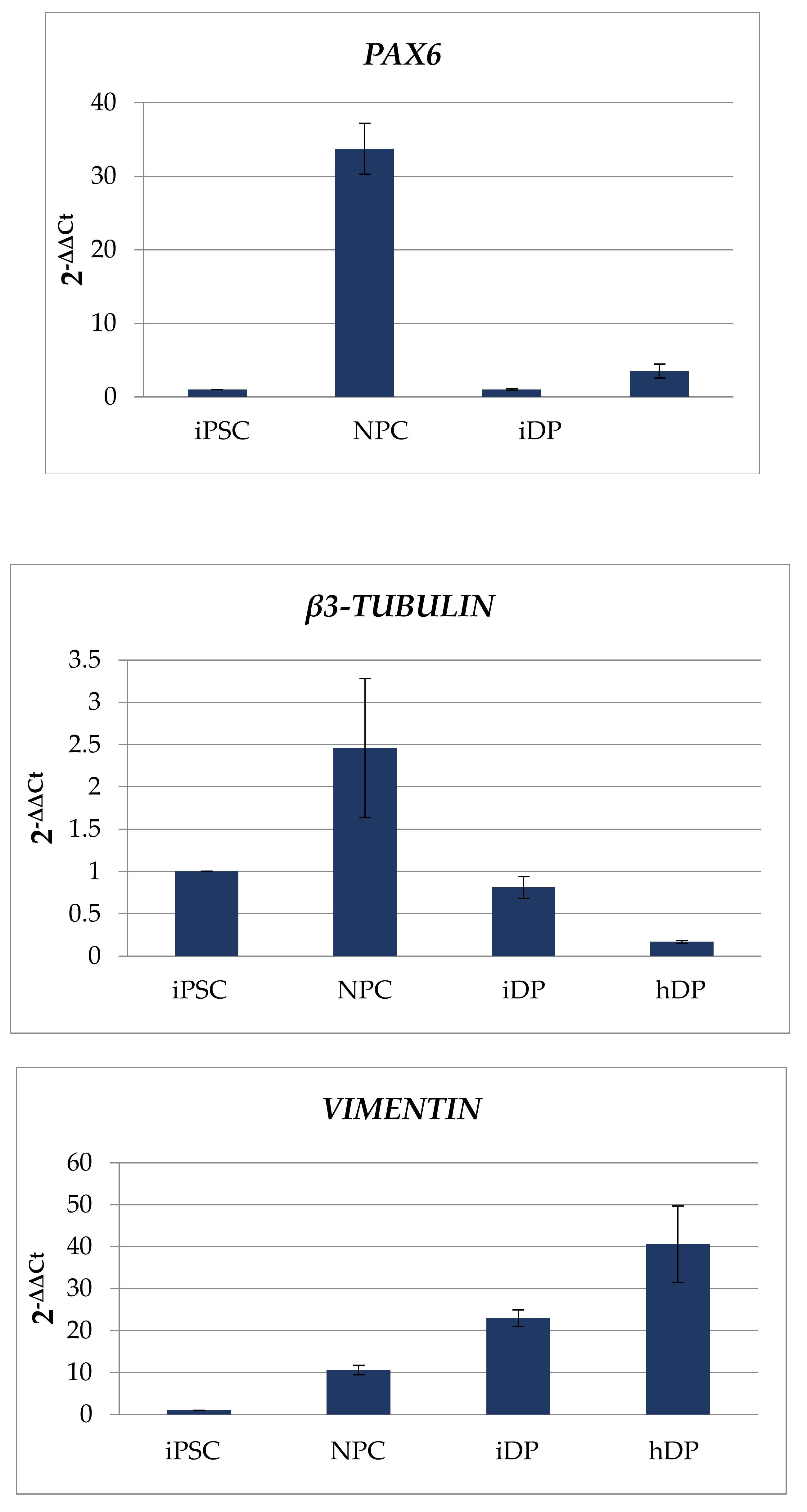
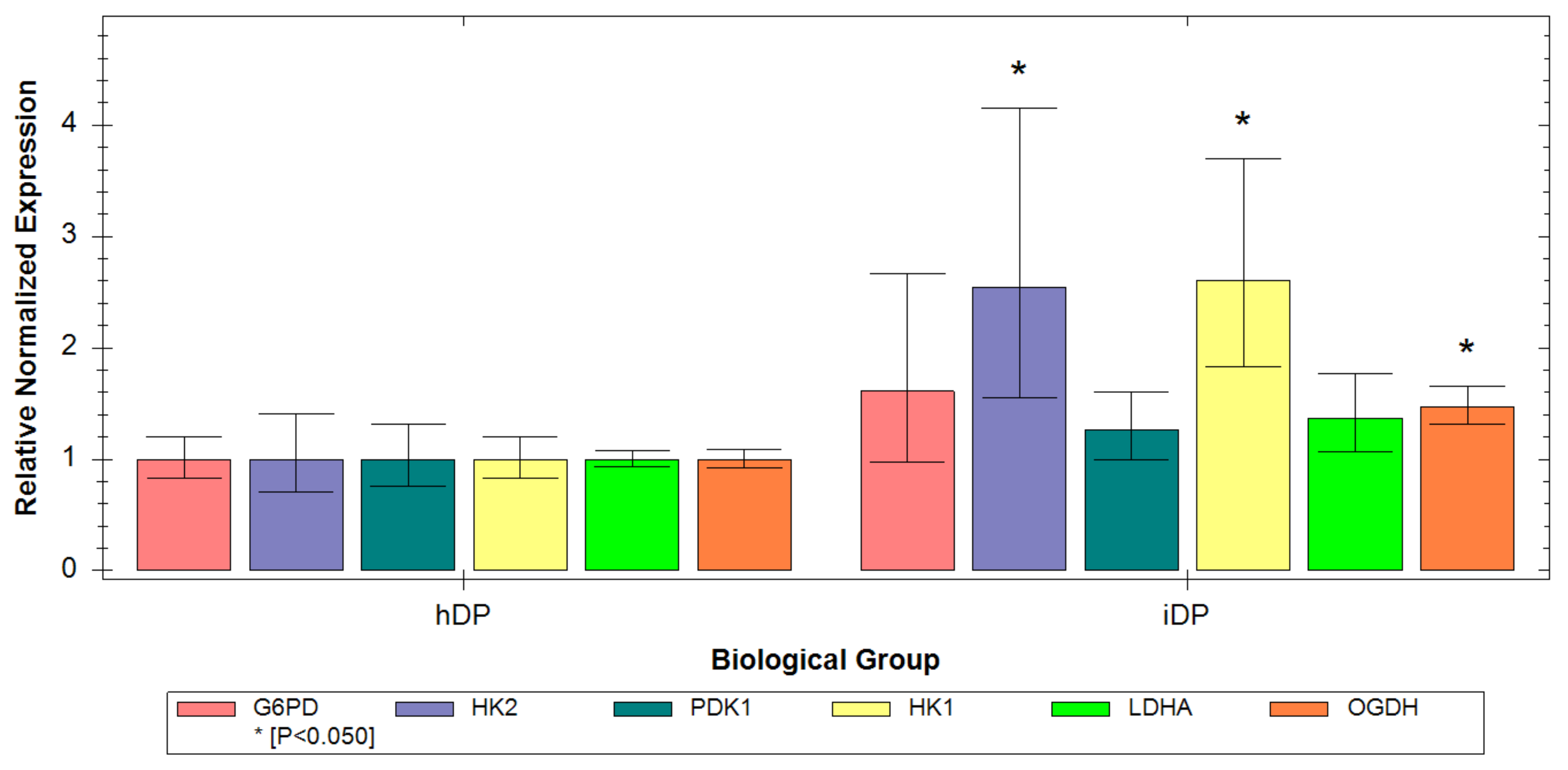
3.3. Evaluation of hDP and iDP Spheroids Energy Metabolism Using Real-Time PCR
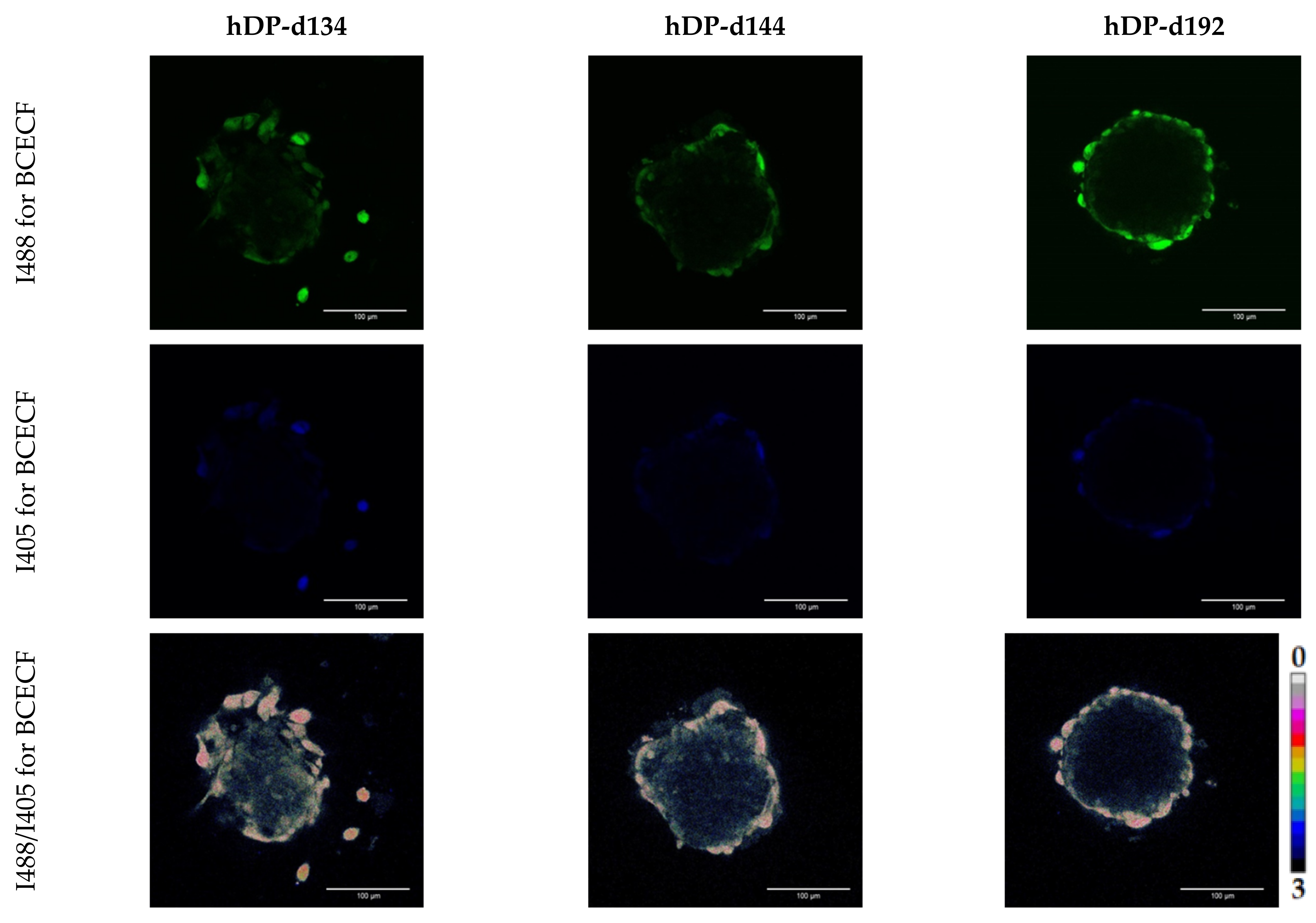
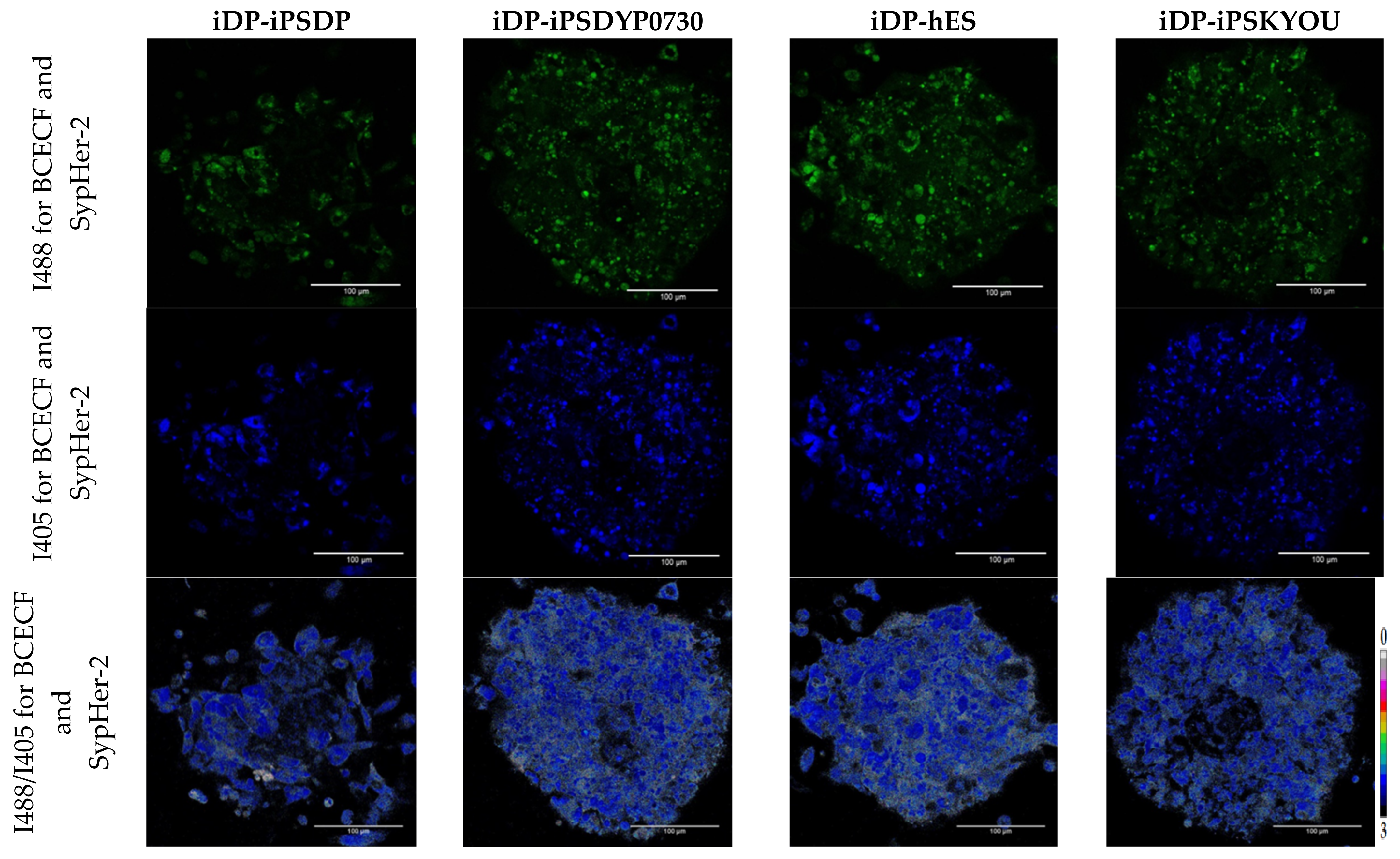
3.4. Evaluation of hDP and iDP Spheroids Intracellular pH Using Fluorescence Microscopy
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Löwa, A.; Vogt, A.; Kaessmeyer, S.; Hedtrich, S.; Roger, M. Generation of full-thickness skin equivalents using hair follicle-derived primary human keratinocytes and fibroblasts. J. Tissue Eng. Regen. Med. 2018, 12, e2134–e2146. [Google Scholar] [CrossRef] [PubMed]
- Plaza, C.; Meyrignac, C.; Botto, J.M.; Capallere, C. Characterization of a new full-thickness in vitro skin model. Tissue Eng. Part C Methods 2021, 27, 411–420. [Google Scholar] [CrossRef] [PubMed]
- Leirós, G.J.; Kusinsky, A.G.; Drago, H.; Bossi, S.; Sturla, F.; Castellanos, M.L.; Stella, I.Y.; Balañá, M.E. Dermal papilla cells improve the wound healing process and generate hair bud-like structures in grafted skin substitutes using hair follicle stem cells. Stem Cells Transl. Med. 2014, 3, 1209–1219. [Google Scholar] [CrossRef] [PubMed]
- Chermnykh, E.S.; Vorotelyak, E.A.; Gnedeva, K.Y.; Moldaver, M.V.; Yegorov, Y.E.; Vasiliev, A.V.; Terskikh, V.V. Dermal papilla cells induce keratinocyte tubulogenesis in culture. Histochem. Cell Biol. 2010, 133, 567–576. [Google Scholar] [CrossRef] [PubMed]
- Yang, C.C.; Cotsarelis, G. Review of hair follicle dermal cells. J. Dermatol. Sci. 2010, 57, 2–11. [Google Scholar] [CrossRef] [PubMed]
- Driskell, R.R.; Giangreco, A.; Jensen, K.B.; Mulder, K.W.; Watt, F.M. Sox2-positive dermal papilla cells specify hair follicle type in mammalian epidermis. Development 2009, 136, 2815–2823. [Google Scholar] [CrossRef]
- Ohyama, M.; Okano, H. Promise of human induced pluripotent stem cells in skin regeneration and investigation. J. Investig. Dermatol. 2014, 134, 605–609. [Google Scholar] [CrossRef]
- Higgins, C.A.; Chen, J.C.; Cerise, J.E.; Jahoda, C.A.; Christiano, A.M. Microenvironmental reprogramming by three-dimensional culture enables dermal papilla cells to induce de novo human hair-follicle growth. Proc. Natl. Acad. Sci. USA 2013, 110, 19679–19688. [Google Scholar] [CrossRef]
- Thangapazham, R.L.; Klover, P.; Li, S.; Wang, J.A.; Sperling, L.; Darling, T.N. A model system to analyse the ability of human keratinocytes to form hair follicles. Exp. Dermatol. 2014, 23, 443–446. [Google Scholar] [CrossRef]
- Ohyama, M. Use of human intra-tissue stem/progenitor cells and induced pluripotent stem cells for hair follicle regeneration. Inflamm. Regen. 2019, 39, 4. [Google Scholar] [CrossRef] [Green Version]
- Sun, B.K.; Siprashvili, Z.; Khavari, P.A. Advances in skin grafting and treatment of cutaneous wounds. Science 2014, 346, 941–945. [Google Scholar] [CrossRef]
- Itoh, M.; Umegaki-Arao, N.; Guo, Z.; Liu, L.; Higgins, C.A.; Christiano, A.M. Generation of 3D skin equivalents fully reconstituted from human induced pluripotent stem cells (iPSCs). PLoS ONE 2013, 8, e77673. [Google Scholar]
- Zhao, A.; Yang, Y.; Pan, X.; Pan, Y.; Cai, S. Generation of keratinocyte stem-like cells from human fibroblasts via a direct reprogramming approach. Biotechnol. Prog. 2020, 36, e2961. [Google Scholar] [CrossRef]
- Gnedeva, K.; Vorotelyak, E.; Cimadamore, F.; Cattarossi, G.; Giusto, E.; Terskikh, V.V.; Terskikh, A.V. Derivation of hair-inducing cell from human pluripotent stem cells. PLoS ONE 2015, 10, e0116892. [Google Scholar] [CrossRef]
- Fukuyama, M.; Tsukashima, A.; Kimishima, M.; Yamazaki, Y.; Okano, H.; Ohyama, M. Human iPS cell-derived cell aggregates exhibited dermal papilla cell properties in in vitro three-dimensional assemblage mimicking hair follicle structures. Front. Cell Dev. Biol. 2021, 9, 590333. [Google Scholar] [CrossRef]
- Veraitch, O.; Mabuchi, Y.; Matsuzaki, Y.; Sasaki, T.; Okuno, H.; Tsukashima, A. Induction of hair follicle dermal papilla cell properties in human induced pluripotent stem cell-derived multipotent LNGFR( + )THY-1( + ) mesenchymal cells. Sci. Rep. 2017, 7, 42777. [Google Scholar] [CrossRef]
- Quinn, K.P.; Sridharan, G.V.; Hayden, R.S.; Kaplan, D.L.; Lee, K.; Georgakoudi, I. Quantitative metabolic imaging using en-dogenous fluorescence to detect stem cell differentiation. Sci. Rep. 2013, 3, 3432. [Google Scholar] [CrossRef]
- Meleshina, A.V.; Dudenkova, V.V.; Shirmanova, M.V.; Shcheslavskiy, V.I.; Becker, W.; Bystrova, A.S.; Cherkasova, E.I.; Zagaynova, E.V. Probing metabolic states of differentiating stem cells using two-photon FLIM. Sci. Rep. 2016, 6, 21853. [Google Scholar] [CrossRef]
- Meleshina, A.V.; Dudenkova, V.V.; Bystrova, A.S.; Kuznetsova, D.S.; Shirmanova, M.V.; Zagaynova, E.V. Two-photon FLIM of NAD(P)H and FAD in mesenchymal stem cells undergoing either osteogenic or chondrogenic differentiation. Stem Cell Res. Ther. 2017, 8, 15. [Google Scholar] [CrossRef]
- Kashirina, A.; Gavrina, A.; Kryukov, E.; Elagin, V.; Kolesova, Y.; Artyuhov, A.; Momotyuk, E.; Abdyyev, V.; Meshcheryakova, N.; Zagaynova, E.; et al. Energy Metabolism and Intracellular pH Alteration in Neural Spheroids Carrying Down Syndrome. Biomedicines 2021, 9, 1741. [Google Scholar] [CrossRef]
- Muchkaeva, I.A.; Dashinimaev, E.B.; Artyuhov, A.S.; Myagkova, E.P.; Vorotelyak, E.A.; Yegorov, Y.Y.; Vishnyakova, K.S.; Kravchenko, I.E.; Chumakov, P.M.; Terskikh, V.V.; et al. Generation of iPS Cells from Human Hair Follice Dermal Papilla Cells. Acta Nat. 2014, 6, 45–53. [Google Scholar] [CrossRef]
- Rodimova, S.; Kuznetsova, D.; Bobrov, N.; Elagin, V.; Shcheslavskiy, V.; Zagainov, V.; Zagaynova, E. Mapping metabolism of liver tissue using two-photon FLIM. Biomed. Opt. Express 2020, 11, 4458–4470. [Google Scholar] [CrossRef]
- Topouzi, H.; Logan, N.J.; Williams, G.; Higgins, C.A. Methods for the isolation and 3D culture of dermal papilla cells from human hair follicles. Exp. Dermatol. 2017, 26, 491–496. [Google Scholar] [CrossRef]
- Gupta, A.C.; Chawla, S.; Hegde, A.; Singh, D.; Bandyopadhyay, B.; Lakshmanan, C.C.; Kalsi, G.; Ghosh, S. Establishment of an in vitro organoid model of dermal papilla of human hair follicle. J. Cell Physiol. 2018, 233, 9015–9030. [Google Scholar] [CrossRef]
- Higgins, C.A.; Richardson, G.D.; Ferdinando, D.; Westgate, G.E.; Jahoda, C.A. Modelling the hair follicle dermal papilla using spheroid cell cultures. Exp. Dermatol. 2010, 19, 546–548. [Google Scholar] [CrossRef]
- Choi, M.; Choi, Y.M.; Choi, S.Y.; An, I.S.; Bae, S.; An, S.; Jung, J.H. Glucose metabolism regulates expression of hair-inductive genes of dermal papilla spheres via histone acetylation. Sci. Rep. 2020, 10, 4887. [Google Scholar] [CrossRef]
- Skala, M.C.; Riching, K.M.; Gendron-Fitzpatrick, A.; Eickhoff, J.; Eliceiri, K.W.; White, J.G.; Ramanujam, N. In vivo multiphoton microscopy of NADH and FAD redox states, fluorescence lifetimes, and cellular morphology in precancerous epithelia. Proc. Natl. Acad. Sci. USA 2007, 104, 19494–19499. [Google Scholar] [CrossRef] [PubMed]
- Stringari, C.; Wang, H.; Geyfman, M.; Crosignani, V.; Kumar, V.; Takahashi, J.S.; Andersen, B.; Gratton, E. In vivo single-cell detection of metabolic oscillations in stem cells. Cell Rep. 2015, 10, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Okkelman, I.A.; Puschhof, J.; Papkovsky, D.B.; Dmitriev, R.I. Visualization of stem cell niche by fluorescence lifetime imaging microscopy. Methods Mol. Biol. 2020, 2171, 65–97. [Google Scholar] [PubMed]
- Berezin, M.Y.; Achilefu, S. Fluorescence lifetime measurements and biological imaging. Chem. Rev. 2010, 110, 2641–2684. [Google Scholar] [CrossRef] [PubMed]
- Datta, R.; Heaster, T.M.; Sharick, J.T.; Gillette, A.A.; Skala, M.C. Fluorescence lifetime imaging microscopy: Fundamentals and advances in instrumentation, analysis, and applications. J. Biomed. Opt. 2020, 25, 1–43. [Google Scholar] [CrossRef]
- Meleshina, A.V.; Rogovaya, O.S.; Dudenkova, V.V.; Sirotkina, M.A.; Lukina, M.M.; Bystrova, A.S.; Krut, V.G.; Kuznetsova, D.S.; Kalabusheva, E.P.; Vasiliev, A.V.; et al. Multimodal label-free imaging of living dermal equivalents including dermal papilla cells. Stem Cell Res. Ther. 2018, 9, 84. [Google Scholar] [CrossRef]
- Wieland, J.G.; Naskar, N.; Rück, A.; Walther, P. Fluorescence lifetime imaging and electron microscopy: A correlative approach. Histochem. Cell Biol. 2022; Online ahead of print. [Google Scholar]
- Liu, Z.; Pouli, D.; Alonzo, C.A.; Varone, A.; Karaliota, S.; Quinn, K.P.; Georgakoudi, I. Mapping metabolic changes by noninvasive, multiparametric, high-resolution imaging using endogenous contrast. Sci. Adv. 2018, 4, eaap9302. [Google Scholar] [CrossRef]
- Ung, T.P.L.; Lim, S.; Solinas, X.; Mahou, P.; Chessel, A.; Marionnet, C.; Bornschlögl, T.; Beaurepaire, E.; Bernerd, F.; Pena, A.M.; et al. Simultaneous NAD(P)H and FAD fluorescence lifetime microscopy of long UVA-induced metabolic stress in reconstructed human skin. Sci. Rep. 2021, 11, 22171. [Google Scholar] [CrossRef]
- Dietl, K.; Renner, K.; Dettmer, K.; Timischl, B.; Eberhart, K.; Dorn, C.; Hellerbrand, C.; Kastenberger, M.; Kunz-Schughart, L.A.; Oefner, P.J.; et al. Lactic acid and acidification inhibit TNF secretion and glycolysis of human monocytes. J. Immunol. 2010, 184, 1200–1209. [Google Scholar] [CrossRef]
- Webb, B.A.; Chimenti, M.; Jacobson, M.P.; Barber, D.L. Dysregulated pH: A perfect storm for cancer progression. Nat Rev Cancer. 2011, 11, 671–677. [Google Scholar] [CrossRef]
- Hanson, G.T.; McAnaney, T.B.; Park, E.S.; Rendell, M.E.; Yarbrough, D.K.; Chu, S.; Xi, L.; Boxer, S.G.; Montrose, M.H.; Remington, S.J. Green fluorescent protein variants as ratiometric dual emission pH sensors. 1. Structural characterization and preliminary application. Biochemistry 2002, 41, 15477–15488. [Google Scholar] [CrossRef]
- Van Erp, P.E.; Jansen, M.J.; de Jongh, G.J.; Boezeman, J.B.; Schalkwijk, J. Ratiometric measurement of intracellular pH in cultured human keratinocytes using carboxy-SNARF-1 and flow cytometry. Cytometry 1991, 12, 127–132. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Primer Target | Primer Sequence (5′→3′) |
|---|---|
| OCT4 | Forward Primer-ACCCACACTGCAGCAGATCA |
| Reverse Primer-CACACTCGGACCACATCCTTCT | |
| SOX2 | Forward Primer-TGCGAGCGCTGCACAT |
| Reverse Primer-GCAGCGTGTACTTATCCTTCTTCA | |
| NANOG | Forward Primer-GTCTCGTATTTGCTGCATCGT |
| Reverse Primer-AACACTCGGTGAAATCAGGGT | |
| TUBB3 | Forward Primer-FW CAACAGCGACGGAGGTCTC |
| Reverse Primer-AAGACAGAGACAGGAGCAGC | |
| PAX6 | Forward Primer-AGTGCCCGTCCATCTTTGC |
| Reverse Primer-CGCTTGGTATGTTATCGTTGGT | |
| FIBRONECTIN | Forward Primer-CCAGTTTTGTGACATTCCCTT |
| Reverse Primer-GCATTTGCTTATTTCCTTGTG | |
| VERSICAN | Forward Primer-TGCCACCCAGTTACAACACC |
| Reverse Primer-TGCCACCCAGTTACAACACC | |
| VIMENTIN | Forward Primer-GATGTTTCCAAGCCTGACCT |
| Reverse Primer-TACCATTCTTCTGCCTCCTG | |
| NESTIN | Forward Primer-GAGAAACAGGGCCTACAGAGC |
| Reverse Primer-GGCTGAGGGACATCTTGAGG | |
| ALKALINE PHOSPHATASE | Forward Primer-ATGAGGCGGTGGAGATGGAC |
| Reverse Primer-AATGTGAAGACGTGGGAATGGT | |
| Collagen I | Forward Primer-AGAAAGGGGTCTCCATGGTG |
| Reverse Primer-AGGACCTCGGCTTCCAATAG | |
| Collagen III | Forward Primer-CCAGGAGCTAACGGTCTCAG |
| Reverse Primer-TGATCCAGGGTTTCCATCTC | |
| GAPDH | Forward Primer-CCATGTTCGTCATGGGTGTG |
| Reverse Primer-GGTGCTAAGCAGTTGGTGGTG | |
| PSMB4 | Forward Primer-CATTCCGTCCACTCCCGATT |
| Reverse Primer-CGAACTTAACGCCGAGGACT | |
| REEP5 | Forward Primer-ACTGCATGACTGACCTTCTGG |
| Reverse Primer-AGTCCGATGACACCAAGAGC | |
| C1ORF43 | Forward Primer-ACGCCTTTCAAGGGTGTACG |
| Reverse Primer-CAAAGACCCCTGTCCCATAGC | |
| ACTB | Forward Primer-TGCGTTGTTACAGGAAGTCCC |
| Reverse Primer-GCTATCACCTCCCCTGTGTG |
| Primer Target | Primer Sequence (5’→3’) |
|---|---|
| HK1 | Forward Primer-CTGCTGGTGAAAATCCGTAGTGG |
| Reverse Primer-GTCCAAGAAGTCAGAGATGCAGG | |
| HK2 | Forward Primer-GAGTTTGACCTGGATGTGGTTGC |
| Reverse Primer-CCTCCATGTAGCAGGCATTGCT | |
| PDK1 | Forward Primer-CATGTCACGCTGGGTAATGAGG |
| Reverse Primer-CTCAACACGAGGTCTTGGTGCA | |
| LDHA | Forward Primer-GGATCTCCAACATGGCAGCCTT |
| Reverse Primer-AGACGGCTTTCTCCCTCTTGCT | |
| G6PD | Forward Primer-CTGTTCCGTGAGGACCAGATCT |
| Reverse Primer-TGAAGGTGAGGATAACGCAGGC | |
| OGDH | Forward Primer-GAGGCTGTCATGTACGTGTGCA |
| Reverse Primer-TACATGAGCGGCTGCGTGAACA | |
| ABL1 | Forward Primer-CCAGGTGTATGAGCTGCTAGAG |
| Reverse Primer-GTCAGAGGGATTCCACTGCCAA | |
| EIF2B | Forward Primer-CTACTCCAGAGTGGTCCTGAGA |
| Reverse Primer-GTTGAGGTGGCAGAGGGCTTTG |
| Type of Spheroids | Redox Ratio | τm (ps) | τ1 (ps) | τ2 (ps) | α2, % | |
|---|---|---|---|---|---|---|
| hDP | hDP-d134 | 1.17 ± 0.17 | 840.58 ± 48.83 | 324.85 ± 31.34 | 2169.36 ± 78.82 | 27.77± 1.31 |
| hDP-d144 | 1.05 ± 0.16 | 807.24 ± 41.02 | 318.72 ± 23.77 | 2147.32 ± 90.50 | 27.09 ± 1.22 | |
| hDP-d192 | 1.11 ± 0.17 | 795.98 ± 31.42 | 322.29 ± 27.29 | 2124.53 ± 75.41 | 26.46 ± 1.03 | |
| iDP | iDP-iPSDP | 1.13 ± 0.18 | 628.96 ± 84.35 | 257.28 ± 42.79 | 2062.52 ± 185.35 | 21.43 ± 1.70 |
| iDP-iPSDYP0730 | 1.09 ± 0.17 | 664.74 ± 76.88 | 259.73 ± 41.14 | 2016.94 ± 116.72 | 23.02 ± 1.41 | |
| iDP-hES | 1.17 ± 0.19 | 714.27 ± 62.76 | 292.66 ± 29.67 | 2068.76 ± 67.66 | 23.63 ± 1.28 | |
| iDP-iPSKYOU | 1.09 ± 0.17 | 670.92 ± 54.01 | 279.09 ± 28.34 | 2028.40 ± 79.29 | 22.42 ± 1.00 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kashirina, A.; Gavrina, A.; Mozherov, A.; Kozlov, D.; Kuznetsova, D.; Vorotelyak, E.; Zagaynova, E.; Kalabusheva, E.; Kashina, A. FLIM for Evaluation of Difference in Metabolic Status between Native and Differentiated from iPSCs Dermal Papilla Cells. Cells 2022, 11, 2730. https://doi.org/10.3390/cells11172730
Kashirina A, Gavrina A, Mozherov A, Kozlov D, Kuznetsova D, Vorotelyak E, Zagaynova E, Kalabusheva E, Kashina A. FLIM for Evaluation of Difference in Metabolic Status between Native and Differentiated from iPSCs Dermal Papilla Cells. Cells. 2022; 11(17):2730. https://doi.org/10.3390/cells11172730
Chicago/Turabian StyleKashirina, Alena, Alena Gavrina, Artem Mozherov, Dmitriy Kozlov, Daria Kuznetsova, Ekaterina Vorotelyak, Elena Zagaynova, Ekaterina Kalabusheva, and Aleksandra Kashina. 2022. "FLIM for Evaluation of Difference in Metabolic Status between Native and Differentiated from iPSCs Dermal Papilla Cells" Cells 11, no. 17: 2730. https://doi.org/10.3390/cells11172730
APA StyleKashirina, A., Gavrina, A., Mozherov, A., Kozlov, D., Kuznetsova, D., Vorotelyak, E., Zagaynova, E., Kalabusheva, E., & Kashina, A. (2022). FLIM for Evaluation of Difference in Metabolic Status between Native and Differentiated from iPSCs Dermal Papilla Cells. Cells, 11(17), 2730. https://doi.org/10.3390/cells11172730