
Current Understanding of Asthma Pathogenesis and Biomarkers



{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Pathological Mechanisms of Asthma
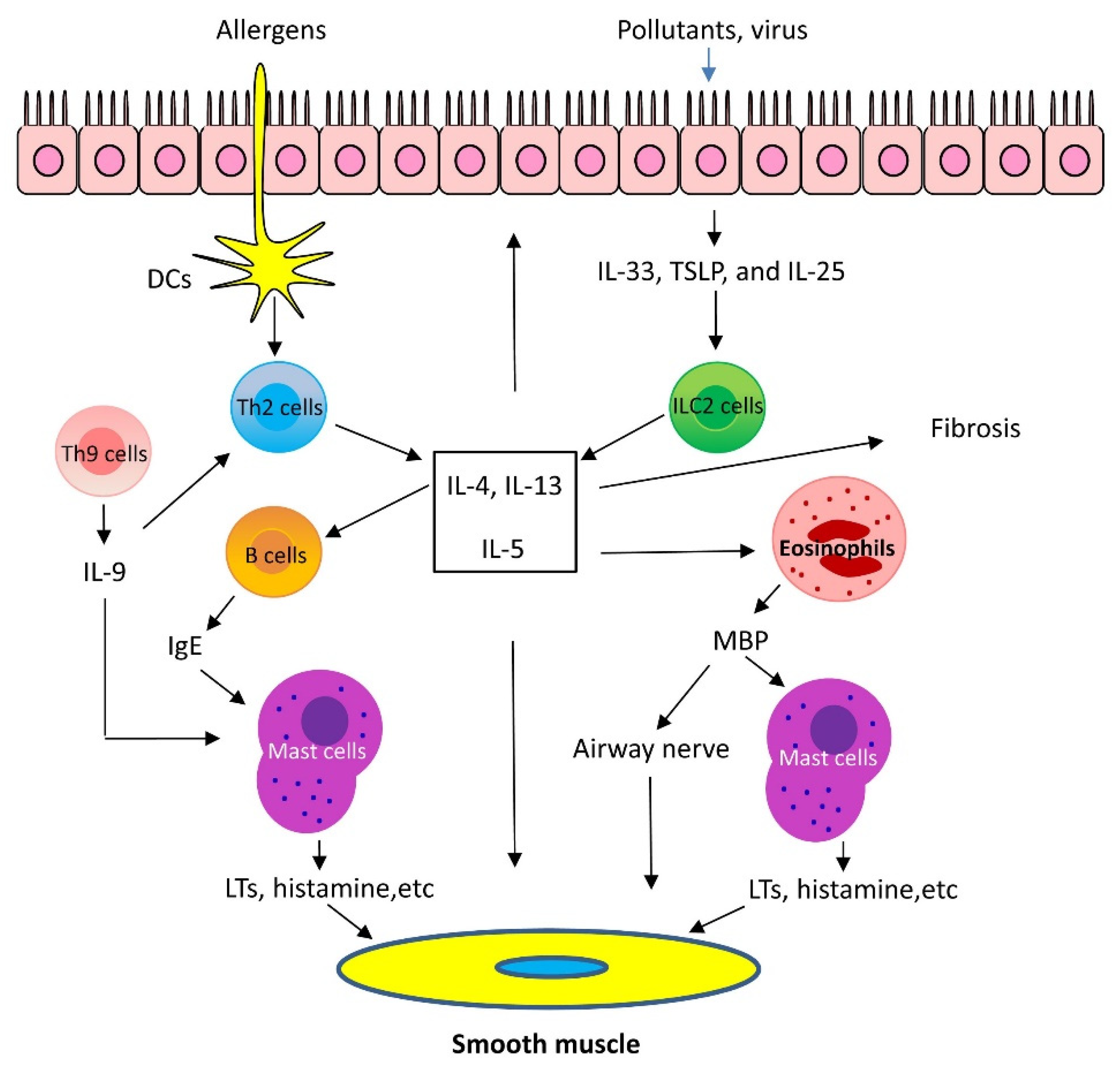
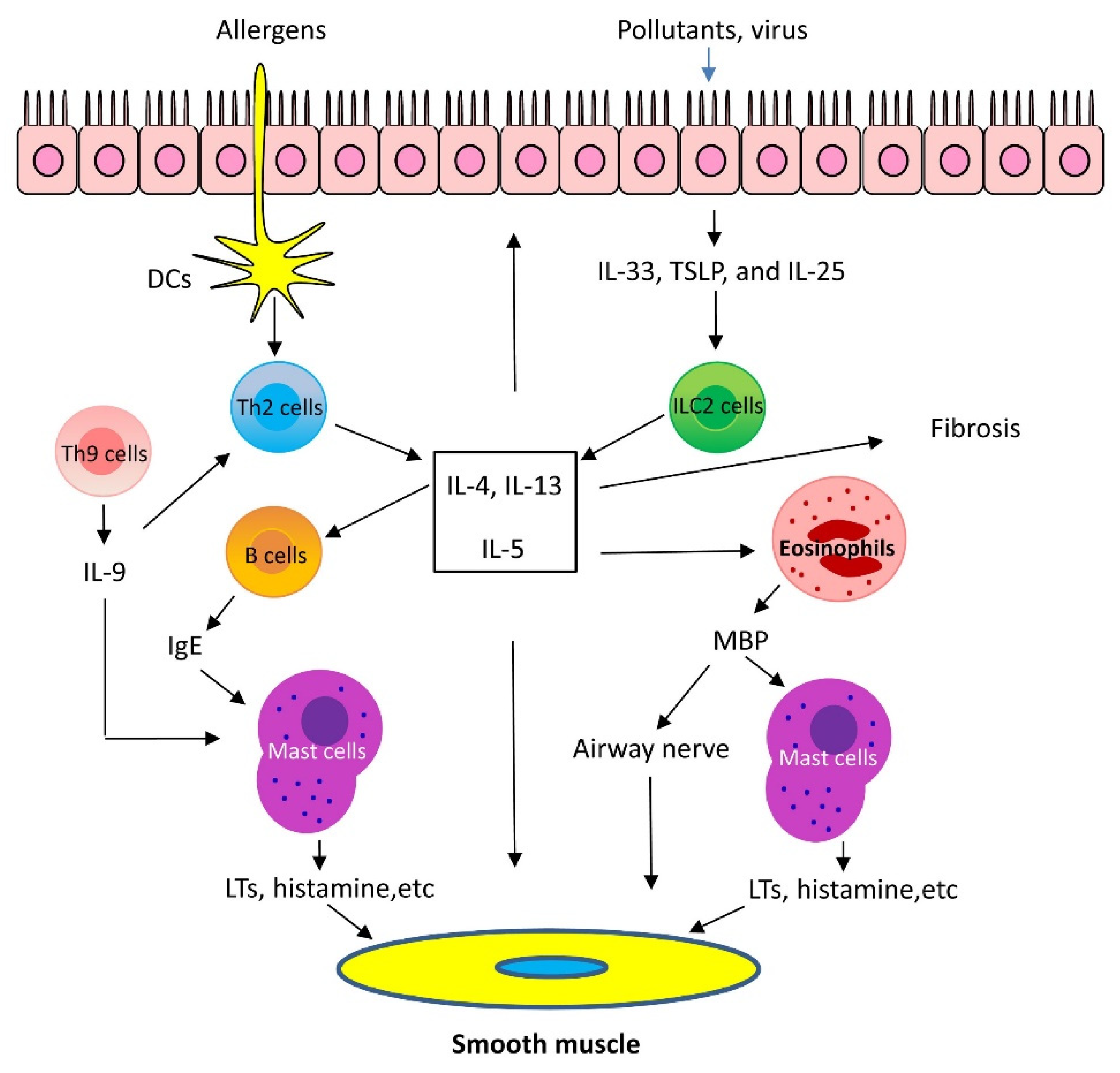
1.1. Mechanisms of Th2-High Asthma
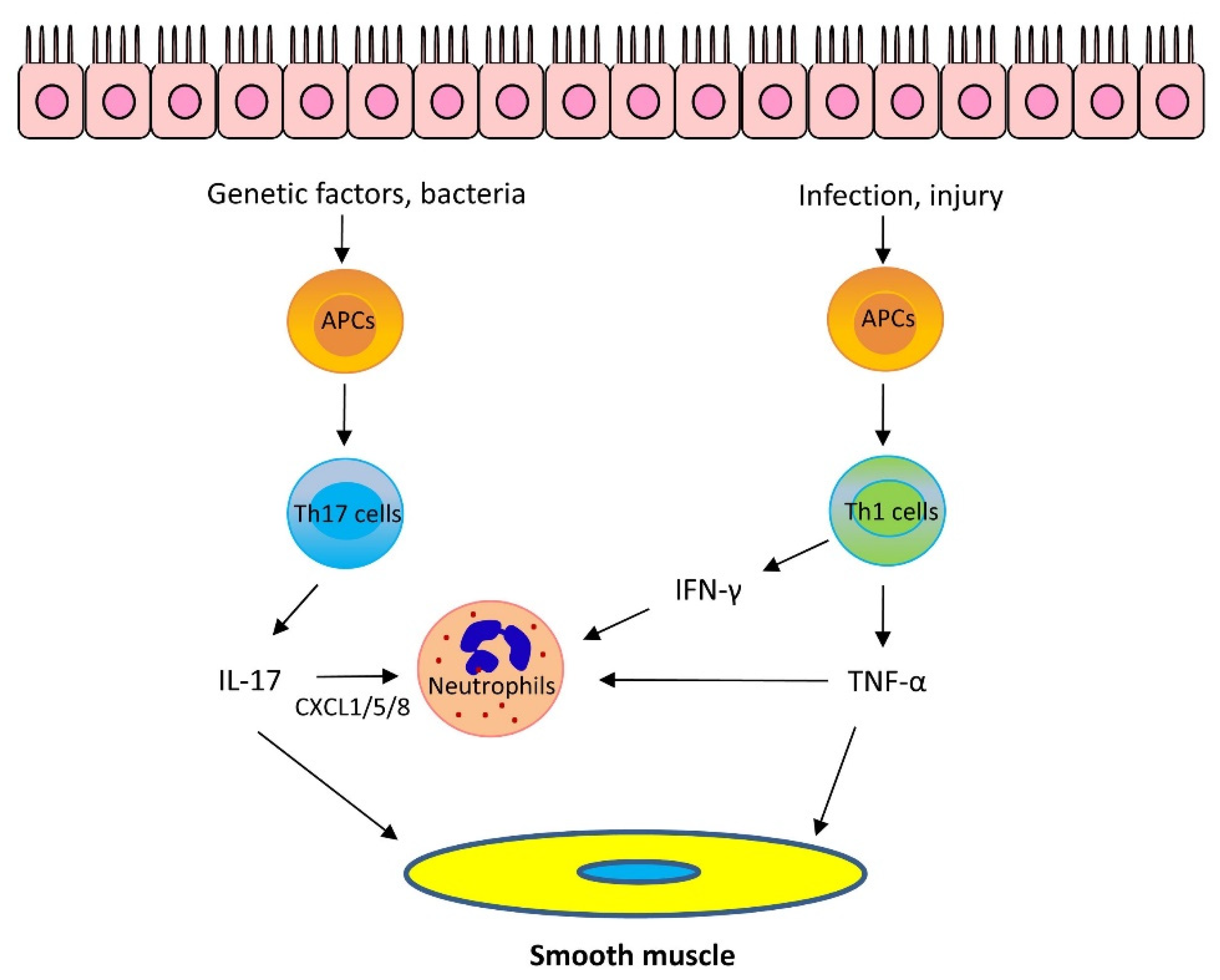
1.2. Mechanisms of Th2-Low Asthma
1.2.1. IL-17
1.2.2. Other Cytokines
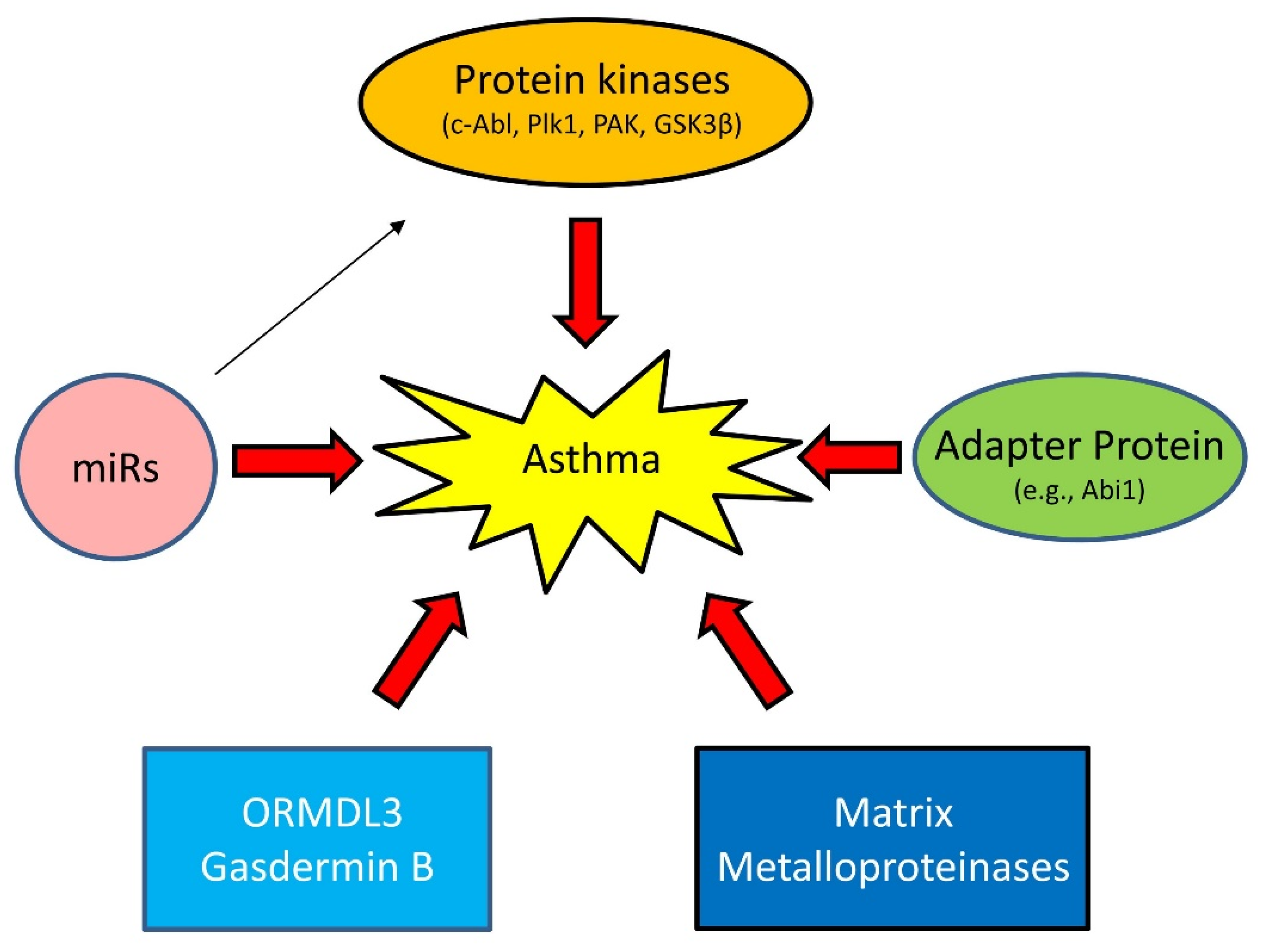
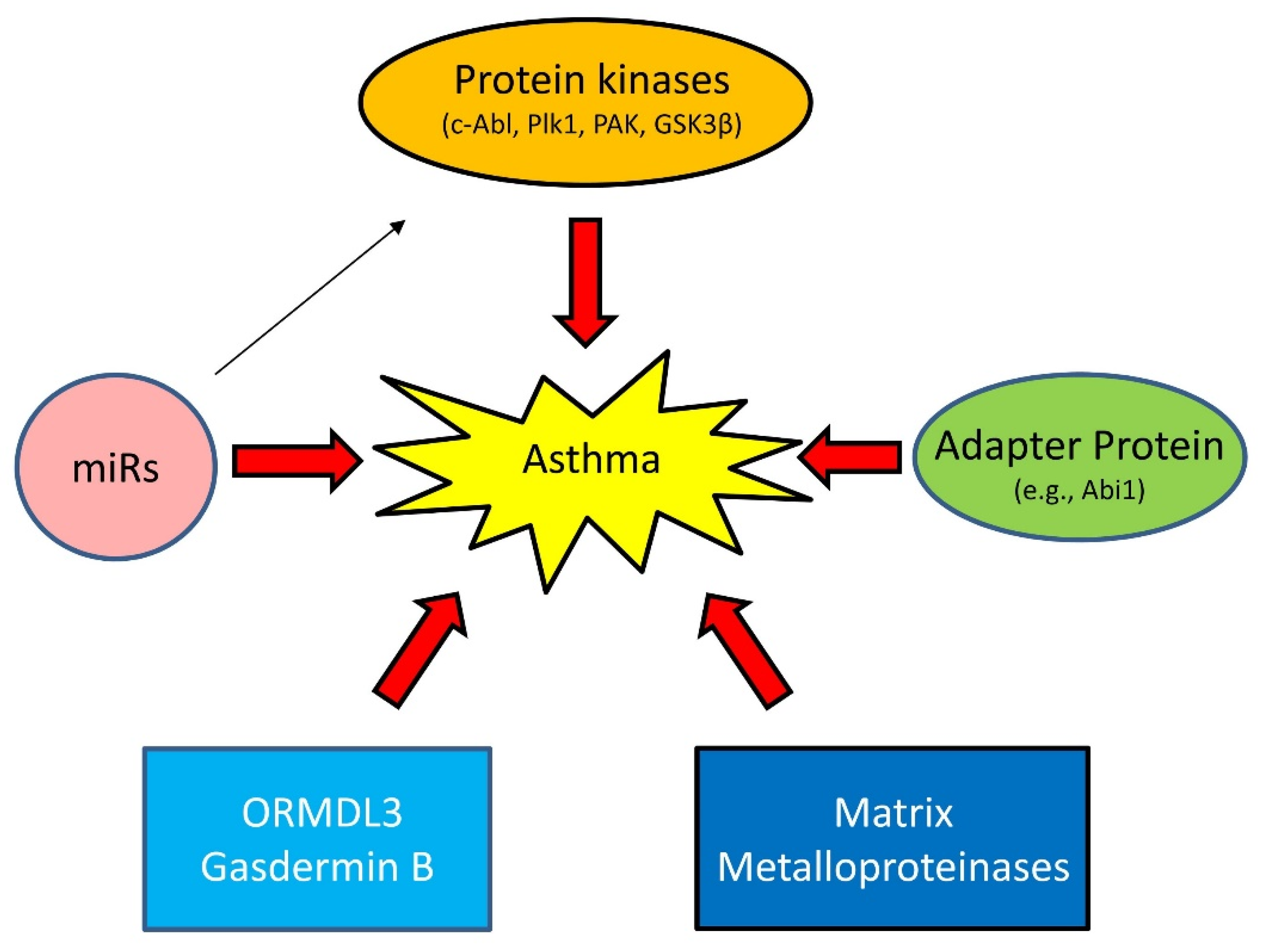
1.3. Emerging Mechanisms of Asthma
1.3.1. Proteins Kinases
1.3.2. Adapter Protein
1.3.3. MicroRNAs (miRNAs)
1.3.4. Others
2. Biomarkers of Asthma
2.1. Th2-High-Related Biomarkers
2.1.1. Sputum Eosinophils
2.1.2. Blood Total Eosinophil Count (TEC)
2.1.3. Serum IgE
2.1.4. Nitric Oxide
2.1.5. Periostin
2.1.6. Cytokines
2.2. Th2-Low-Related Biomarkers
2.2.1. Sputum Neutrophils
2.2.2. IL-17
2.2.3. Other Potential Biomarkers
2.3. Biomarkers Indicative of Airway Remodeling
2.3.1. Bronchoscopy
2.3.2. YKL-40
2.4. Genetic Risk for Asthma Development and Treatment
3. Clinical Differences in Th2-High and Th2-Low Asthma
3.1. Phenotypes of Th2-High Asthma
3.1.1. Early Onset or “Extrinsic” Allergic Asthma
3.1.2. Late-Onset Eosinophilic Asthma
3.1.3. AERD
3.2. Phenotypes of Th2-Low Asthma
3.2.1. Obesity-Associated Asthma
3.2.2. Smoking-Associated Asthma
3.2.3. Very-Late-Onset Asthma
4. Asthma-Associated Comorbidities
5. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Brusselle, G.G.; Koppelman, G.H. Biologic Therapies for Severe Asthma. N. Engl. J. Med. 2022, 386, 157–171. [Google Scholar] [CrossRef] [PubMed]
- Fahy, J.V. Type 2 inflammation in asthma—Present in most, absent in many. Nat. Rev. Immunol. 2015, 15, 57–65. [Google Scholar] [CrossRef] [PubMed]
- Lambrecht, B.N.; Hammad, H.; Fahy, J.V. The Cytokines of Asthma. Immunity 2019, 50, 975–991. [Google Scholar] [CrossRef] [PubMed]
- Gour, N.; Wills-Karp, M. IL-4 and IL-13 signaling in allergic airway disease. Cytokine 2015, 75, 68–78. [Google Scholar] [CrossRef] [PubMed]
- Pelaia, C.; Paoletti, G.; Puggioni, F.; Racca, F.; Pelaia, G.; Canonica, G.W.; Heffler, E. Interleukin-5 in the Pathophysiology of Severe Asthma. Front. Physiol. 2019, 10, 1514. [Google Scholar] [CrossRef]
- Drake, M.G.; Cook, M.; Fryer, A.D.; Jacoby, D.B.; Scott, G.D. Airway Sensory Nerve Plasticity in Asthma and Chronic Cough. Front. Physiol. 2021, 12, 720538. [Google Scholar] [CrossRef] [PubMed]
- Ingram, J.L.; Kraft, M. IL-13 in asthma and allergic disease: Asthma phenotypes and targeted therapies. J. Allergy Clin. Immunol. 2012, 130, 829–842. [Google Scholar] [CrossRef]
- Kaur, D.; Gomez, E.; Doe, C.; Berair, R.; Woodman, L.; Saunders, R.; Hollins, F.; Rose, F.R.; Amrani, Y.; May, R.; et al. IL-33 drives airway hyper-responsiveness through IL-13-mediated mast cell: Airway smooth muscle crosstalk. Allergy 2015, 70, 556–567. [Google Scholar] [CrossRef]
- Koch, S.; Sopel, N.; Finotto, S. Th9 and other IL-9-producing cells in allergic asthma. Semin. Immunopathol. 2017, 39, 55–68. [Google Scholar] [CrossRef]
- Doherty, T.A.; Broide, D.H. Insights into the biology of IL-9 in asthma. J. Allergy Clin. Immunol. 2022. [Google Scholar] [CrossRef]
- Kay, A.B. Natural killer T cells and asthma. N. Engl. J. Med. 2006, 354, 1186–1188. [Google Scholar] [CrossRef] [PubMed]
- McKnight, C.G.; Morris, S.C.; Perkins, C.; Zhu, Z.; Hildeman, D.A.; Bendelac, A.; Finkelman, F.D. NKT cells contribute to basal IL-4 production but are not required to induce experimental asthma. PLoS ONE 2017, 12, e0188221. [Google Scholar] [CrossRef]
- Pan, H.; Zhang, G.; Nie, H.; Li, S.; He, S.; Yang, J. Sulfatide-activated type II NKT cells suppress immunogenic maturation of lung dendritic cells in murine models of asthma. Am. J. Physiol. Lung Cell Mol. Physiol. 2019, 317, L578–L590. [Google Scholar] [CrossRef] [PubMed]
- Martin-Orozco, E.; Norte-Munoz, M.; Martinez-Garcia, J. Regulatory T Cells in Allergy and Asthma. Front. Pediatr. 2017, 5, 117. [Google Scholar] [CrossRef]
- Takayama, G.; Arima, K.; Kanaji, T.; Toda, S.; Tanaka, H.; Shoji, S.; McKenzie, A.N.; Nagai, H.; Hotokebuchi, T.; Izuhara, K. Periostin: A novel component of subepithelial fibrosis of bronchial asthma downstream of IL-4 and IL-13 signals. J. Allergy Clin. Immunol. 2006, 118, 98–104. [Google Scholar] [CrossRef]
- Woodruff, P.G.; Boushey, H.A.; Dolganov, G.M.; Barker, C.S.; Yang, Y.H.; Donnelly, S.; Ellwanger, A.; Sidhu, S.S.; Dao-Pick, T.P.; Pantoja, C.; et al. Genome-wide profiling identifies epithelial cell genes associated with asthma and with treatment response to corticosteroids. Proc. Natl. Acad. Sci. USA 2007, 104, 15858–15863. [Google Scholar] [CrossRef]
- Johansson, M.W.; Evans, M.D.; Crisafi, G.M.; Holweg, C.T.J.; Matthews, J.G.; Jarjour, N.N. Serum periostin is associated with type 2 immunity in severe asthma. J. Allergy Clin. Immunol. 2016, 137, 1904–1907. [Google Scholar] [CrossRef]
- Gordon, E.D.; Sidhu, S.S.; Wang, Z.E.; Woodruff, P.G.; Yuan, S.; Solon, M.C.; Conway, S.J.; Huang, X.; Locksley, R.M.; Fahy, J.V. A protective role for periostin and TGF-beta in IgE-mediated allergy and airway hyperresponsiveness. Clin. Exp. Allergy 2012, 42, 144–155. [Google Scholar] [CrossRef]
- Bullens, D.M.; Truyen, E.; Coteur, L.; Dilissen, E.; Hellings, P.W.; Dupont, L.J.; Ceuppens, J.L. IL-17 mRNA in sputum of asthmatic patients: Linking T cell driven inflammation and granulocytic influx? Respir. Res. 2006, 7, 135. [Google Scholar] [CrossRef]
- Finkelman, F.D.; Hogan, S.P.; Hershey, G.K.; Rothenberg, M.E.; Wills-Karp, M. Importance of cytokines in murine allergic airway disease and human asthma. J. Immunol. 2010, 184, 1663–1674. [Google Scholar] [CrossRef] [Green Version]
- Kudo, M.; Melton, A.C.; Chen, C.; Engler, M.B.; Huang, K.E.; Ren, X.; Wang, Y.; Bernstein, X.; Li, J.T.; Atabai, K.; et al. IL-17A produced by alphabeta T cells drives airway hyper-responsiveness in mice and enhances mouse and human airway smooth muscle contraction. Nat. Med. 2012, 18, 547–554. [Google Scholar] [CrossRef] [PubMed]
- Silva, M.J.; de Santana, M.B.R.; Tosta, B.R.; Espinheira, R.P.; Alcantara-Neves, N.M.; Barreto, M.L.; Figueiredo, C.A.; Costa, R.D.S. Variants in the IL17 pathway genes are associated with atopic asthma and atopy makers in a South American population. Allergy Asthma Clin. Immunol. 2019, 15, 28. [Google Scholar] [CrossRef]
- Du, J.; Han, J.C.; Zhang, Y.J.; Qi, G.B.; Li, H.B.; Zhang, Y.J.; Cai, S. Single-Nucleotide Polymorphisms of IL-17 Gene Are Associated with Asthma Susceptibility in an Asian Population. Med. Sci. Monit. 2016, 22, 780–787. [Google Scholar] [CrossRef] [PubMed]
- Hynes, G.M.; Hinks, T.S.C. The role of interleukin-17 in asthma: A protective response? ERJ Open Res. 2020, 6. [Google Scholar] [CrossRef]
- Chang, Y.; Al-Alwan, L.; Risse, P.A.; Roussel, L.; Rousseau, S.; Halayko, A.J.; Martin, J.G.; Hamid, Q.; Eidelman, D.H. TH17 cytokines induce human airway smooth muscle cell migration. J. Allergy Clin. Immunol. 2011, 127, 1046–1053. [Google Scholar] [CrossRef]
- Chang, Y.; Al-Alwan, L.; Risse, P.A.; Halayko, A.J.; Martin, J.G.; Baglole, C.J.; Eidelman, D.H.; Hamid, Q. Th17-associated cytokines promote human airway smooth muscle cell proliferation. FASEB J. 2012, 26, 5152–5160. [Google Scholar] [CrossRef]
- Niessen, N.M.; Gibson, P.G.; Baines, K.J.; Barker, D.; Yang, I.A.; Upham, J.W.; Reynolds, P.N.; Hodge, S.; James, A.L.; Jenkins, C.; et al. Sputum TNF markers are increased in neutrophilic and severe asthma and are reduced by azithromycin treatment. Allergy 2021, 76, 2090–2101. [Google Scholar] [CrossRef]
- Choi, J.P.; Kim, Y.S.; Kim, O.Y.; Kim, Y.M.; Jeon, S.G.; Roh, T.Y.; Park, J.S.; Gho, Y.S.; Kim, Y.K. TNF-alpha is a key mediator in the development of Th2 cell response to inhaled allergens induced by a viral PAMP double-stranded RNA. Allergy 2012, 67, 1138–1148. [Google Scholar] [CrossRef]
- Sieck, G.C.; Dogan, M.; Young-Soo, H.; Osorio Valencia, S.; Delmotte, P. Mechanisms underlying TNFalpha-induced enhancement of force generation in airway smooth muscle. Physiol. Rep. 2019, 7, e14220. [Google Scholar] [CrossRef]
- Guedes, A.G.; Jude, J.A.; Paulin, J.; Kita, H.; Lund, F.E.; Kannan, M.S. Role of CD38 in TNF-alpha-induced airway hyperresponsiveness. Am. J. Physiol. Lung Cell Mol. Physiol. 2008, 294, L290–L299. [Google Scholar] [PubMed]
- Deshpande, D.A.; Walseth, T.F.; Panettieri, R.A.; Kannan, M.S. CD38/cyclic ADP-ribose-mediated Ca2+ signaling contributes to airway smooth muscle hyper-responsiveness. FASEB J. 2003, 17, 452–454. [Google Scholar] [CrossRef] [PubMed]
- Wills-Karp, M. Interleukin-13 in asthma pathogenesis. Immunol. Rev. 2004, 202, 175–190. [Google Scholar] [CrossRef]
- Balenga, N.A.; Klichinsky, M.; Xie, Z.; Chan, E.C.; Zhao, M.; Jude, J.; Laviolette, M.; Panettieri, R.A., Jr.; Druey, K.M. A fungal protease allergen provokes airway hyper-responsiveness in asthma. Nat. Commun. 2015, 6, 6763. [Google Scholar] [CrossRef]
- Tang, D.D. Critical role of actin-associated proteins in smooth muscle contraction, cell proliferation, airway hyperresponsiveness and airway remodeling. Respir. Res. 2015, 16, 134. [Google Scholar] [CrossRef] [PubMed]
- Tliba, O.; Deshpande, D.; Chen, H.; Van, B.C.; Kannan, M.; Panettieri, R.A., Jr.; Amrani, Y. IL-13 enhances agonist-evoked calcium signals and contractile responses in airway smooth muscle. Br. J. Pharmacol. 2003, 140, 1159–1162. [Google Scholar] [CrossRef]
- Page, C.; O’Shaughnessy, B.; Barnes, P. Pathogenesis of COPD and Asthma. Handb. Exp. Pharmacol. 2017, 237, 1–21. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Wang, R.; Gannon, O.J.; Rezey, A.C.; Jiang, S.; Gerlach, B.D.; Liao, G.; Tang, D.D. Polo-like Kinase 1 Regulates Vimentin Phosphorylation at Ser-56 and Contraction in Smooth Muscle. J. Biol. Chem. 2016, 291, 23693–23703. [Google Scholar] [CrossRef]
- Cleary, R.A.; Wang, R.; Wang, T.; Tang, D.D. Role of Abl in airway hyperresponsiveness and airway remodeling. Respir. Res. 2013, 14, 105. [Google Scholar] [CrossRef]
- Wang, R.; Wang, Y.; Liao, G.; Chen, B.; Panettieri, R.A., Jr.; Penn, R.B.; Tang, D.D. Abi1 mediates airway smooth muscle cell proliferation and airway remodeling via Jak2/STAT3 signaling. iScience 2022, 25, 103833. [Google Scholar] [CrossRef]
- Liao, G.; Wang, R.; Tang, D.D. Plk1 Regulates Caspase-9 Phosphorylation at Ser-196 and Apoptosis of Human Airway Smooth Muscle Cells. Am. J. Respir. Cell Mol. Biol. 2022, 66, 223–234. [Google Scholar] [CrossRef]
- Chen, S.; Tang, D.D. c-Abl tyrosine kinase regulates cytokinesis of human airway smooth muscle cells. Am. J. Respir. Cell Mol. Biol. 2014, 50, 1076–1083. [Google Scholar] [CrossRef] [PubMed]
- Wang, R.; Mercaitis, O.P.; Jia, L.; Panettieri, R.A.; Tang, D.D. Raf-1, Actin Dynamics and Abl in Human Airway Smooth Muscle Cells. Am. J. Respir. Cell Mol. Biol. 2013, 48, 172–178. [Google Scholar] [CrossRef] [PubMed]
- Anfinogenova, Y.; Wang, R.; Li, Q.F.; Spinelli, A.M.; Tang, D.D. Abl silencing inhibits CAS-Mediated process and constriction in resistance arteries. Circ. Res. 2007, 101, 420–428. [Google Scholar] [CrossRef]
- Jia, L.; Wang, R.; Tang, D.D. Abl regulates smooth muscle cell proliferation by modulating actin dynamics and ERK1/2 activation. Am. J. Physiol. Cell Physiol. 2012, 302, C1026–C1034. [Google Scholar] [CrossRef]
- Berlin, A.A.; Lukacs, N.W. Treatment of cockroach allergen asthma model with imatinib attenuates airway responses. Am. J. Respir. Crit. Care Med. 2005, 171, 35–39. [Google Scholar] [CrossRef]
- Liao, G.; Panettieri, R.A.; Tang, D.D. MicroRNA-203 negatively regulates c-Abl, ERK1/2 phosphorylation, and proliferation in smooth muscle cells. Physiol. Rep. 2015, 3. [Google Scholar] [CrossRef]
- Tang, D.D. The Dynamic Actin Cytoskeleton in Smooth Muscle. Adv. Pharmacol. 2018, 81, 1–38. [Google Scholar] [CrossRef] [PubMed]
- Cahill, K.N.; Katz, H.R.; Cui, J.; Lai, J.; Kazani, S.; Crosby-Thompson, A.; Garofalo, D.; Castro, M.; Jarjour, N.; DiMango, E.; et al. KIT Inhibition by Imatinib in Patients with Severe Refractory Asthma. N. Engl. J. Med. 2017, 376, 1911–1920. [Google Scholar] [CrossRef]
- de Carcer, G.; Wachowicz, P.; Martinez-Martinez, S.; Oller, J.; Mendez-Barbero, N.; Escobar, B.; Gonzalez-Loyola, A.; Takaki, T.; El Bakkali, A.; Camara, J.A.; et al. Plk1 regulates contraction of postmitotic smooth muscle cells and is required for vascular homeostasis. Nat. Med. 2017, 23, 964–974. [Google Scholar] [CrossRef]
- Liao, G.; Wang, R.; Rezey, A.C.; Gerlach, B.D.; Tang, D.D. MicroRNA miR-509 Regulates ERK1/2, the Vimentin Network, and Focal Adhesions by Targeting Plk1. Sci. Rep. 2018, 8, 12635. [Google Scholar] [CrossRef] [Green Version]
- Jiang, S.; Tang, D.D. Plk1 regulates MEK1/2 and proliferation in airway smooth muscle cells. Respir. Res. 2015, 16, 93. [Google Scholar] [CrossRef] [PubMed]
- Rezey, A.C.; Gerlach, B.D.; Wang, R.; Liao, G.; Tang, D.D. Plk1 Mediates Paxillin Phosphorylation (Ser-272), Centrosome Maturation, and Airway Smooth Muscle Layer Thickening in Allergic Asthma. Sci. Rep. 2019, 9, 7555. [Google Scholar] [CrossRef] [PubMed]
- Conduit, P.T.; Wainman, A.; Raff, J.W. Centrosome function and assembly in animal cells. Nat. Rev. Mol. Cell Biol. 2015, 16, 611–624. [Google Scholar] [CrossRef]
- Zhang, W.; Huang, Y.; Gunst, S.J. p21-Activated kinase (Pak) regulates airway smooth muscle contraction by regulating paxillin complexes that mediate actin polymerization. J. Physiol. 2016, 594, 4879–4900. [Google Scholar] [CrossRef] [PubMed]
- Wang, R.; Li, Q.F.; Anfinogenova, Y.; Tang, D.D. Dissociation of Crk-associated substrate from the vimentin network is regulated by p21-activated kinase on ACh activation of airway smooth muscle. Am. J. Physiol. Lung Cell Mol. Physiol. 2007, 292, L240–L248. [Google Scholar] [CrossRef]
- Hoover, W.C.; Zhang, W.; Xue, Z.; Gao, H.; Chernoff, J.; Clapp, D.W.; Gunst, S.J.; Tepper, R.S. Inhibition of p21 activated kinase (PAK) reduces airway responsiveness in vivo and in vitro in murine and human airways. PLoS ONE 2012, 7, e42601. [Google Scholar] [CrossRef]
- Bentley, J.K.; Deng, H.; Linn, M.J.; Lei, J.; Dokshin, G.A.; Fingar, D.C.; Bitar, K.N.; Henderson, W.R., Jr.; Hershenson, M.B. Airway smooth muscle hyperplasia and hypertrophy correlate with glycogen synthase kinase-3(beta) phosphorylation in a mouse model of asthma. Am. J. Physiol. Lung Cell Mol. Physiol. 2009, 296, L176–L184. [Google Scholar] [CrossRef]
- Deng, H.; Dokshin, G.A.; Lei, J.; Goldsmith, A.M.; Bitar, K.N.; Fingar, D.C.; Hershenson, M.B.; Bentley, J.K. Inhibition of glycogen synthase kinase-3beta is sufficient for airway smooth muscle hypertrophy. J. Biol. Chem. 2008, 283, 10198–10207. [Google Scholar] [CrossRef]
- Stradal, T.; Courtney, K.D.; Rottner, K.; Hahne, P.; Small, J.V.; Pendergast, A.M. The Abl interactor proteins localize to sites of actin polymerization at the tips of lamellipodia and filopodia. Curr. Biol. 2001, 11, 891–895. [Google Scholar] [CrossRef]
- Wang, R.; Liao, G.; Wang, Y.; Tang, D.D. Distinctive roles of Abi1 in regulating actin-associated proteins during human smooth muscle cell migration. Sci. Rep. 2020, 10, 10667. [Google Scholar] [CrossRef]
- Wang, T.; Cleary, R.A.; Wang, R.; Tang, D.D. Role of the Adapter Protein Abi1 in Actin-associated Signaling and Smooth Muscle Contraction. J. Biol. Chem. 2013, 288, 20713–20722. [Google Scholar] [CrossRef] [PubMed]
- Akhabir, L.; Sandford, A.J. Genome-wide Association Studies for Discovery of Genes Involved in Asthma. Respirology 2011, 16, 396–406. [Google Scholar]
- Myers, R.A.; Scott, N.M.; Gauderman, W.J.; Qiu, W.; Mathias, R.A.; Romieu, I.; Levin, A.M.; Pino-Yanes, M.; Graves, P.E.; Villarreal, A.B.; et al. Genome-wide interaction studies reveal sex-specific asthma risk alleles. Hum. Mol. Genet. 2014, 23, 5251–5259. [Google Scholar] [CrossRef] [PubMed]
- Bartel, D.P. MicroRNAs: Target recognition and regulatory functions. Cell 2009, 136, 215–233. [Google Scholar] [CrossRef]
- Long, J.; Liao, G.; Wang, Y.; Tang, D.D. Specific protein 1, c-Abl and ERK1/2 form a regulatory loop. J. Cell Sci. 2019, 132. [Google Scholar] [CrossRef]
- Kuhn, A.R.; Schlauch, K.; Lao, R.; Halayko, A.J.; Gerthoffer, W.T.; Singer, C.A. MicroRNA expression in human airway smooth muscle cells: Role of miR-25 in regulation of airway smooth muscle phenotype. Am. J. Respir. Cell Mol. Biol. 2010, 42, 506–513. [Google Scholar] [CrossRef]
- Rodrigo-Munoz, J.M.; Gil-Martinez, M.; Lorente-Sorolla, C.; Garcia-Latorre, R.; Valverde-Monge, M.; Quirce, S.; Sastre, J.; Del Pozo, V. miR-144-3p Is a Biomarker Related to Severe Corticosteroid-Dependent Asthma. Front. Immunol. 2022, 13, 858722. [Google Scholar] [CrossRef]
- Moffatt, M.F.; Kabesch, M.; Liang, L.; Dixon, A.L.; Strachan, D.; Heath, S.; Depner, M.; von Berg, A.; Bufe, A.; Rietschel, E.; et al. Genetic variants regulating ORMDL3 expression contribute to the risk of childhood asthma. Nature 2007, 448, 470–473. [Google Scholar] [CrossRef]
- Verlaan, D.J.; Berlivet, S.; Hunninghake, G.M.; Madore, A.M.; Lariviere, M.; Moussette, S.; Grundberg, E.; Kwan, T.; Ouimet, M.; Ge, B.; et al. Allele-specific chromatin remodeling in the ZPBP2/GSDMB/ORMDL3 locus associated with the risk of asthma and autoimmune disease. Am. J. Hum. Genet. 2009, 85, 377–393. [Google Scholar] [CrossRef]
- Carreras-Sureda, A.; Cantero-Recasens, G.; Rubio-Moscardo, F.; Kiefer, K.; Peinelt, C.; Niemeyer, B.A.; Valverde, M.A.; Vicente, R. ORMDL3 modulates store-operated calcium entry and lymphocyte activation. Hum. Mol. Genet. 2013, 22, 519–530. [Google Scholar] [CrossRef]
- Ha, S.G.; Ge, X.N.; Bahaie, N.S.; Kang, B.N.; Rao, A.; Rao, S.P.; Sriramarao, P. ORMDL3 promotes eosinophil trafficking and activation via regulation of integrins and CD48. Nat. Commun. 2013, 4, 2479. [Google Scholar] [CrossRef] [PubMed]
- James, B.; Milstien, S.; Spiegel, S. ORMDL3 and allergic asthma: From physiology to pathology. J. Allergy Clin. Immunol. 2019, 144, 634–640. [Google Scholar] [CrossRef] [PubMed]
- Das, S.; Miller, M.; Beppu, A.K.; Mueller, J.; McGeough, M.D.; Vuong, C.; Karta, M.R.; Rosenthal, P.; Chouiali, F.; Doherty, T.A.; et al. GSDMB induces an asthma phenotype characterized by increased airway responsiveness and remodeling without lung inflammation. Proc. Natl. Acad. Sci. USA 2016, 113, 13132–13137. [Google Scholar] [CrossRef] [PubMed]
- Brusselle, G.G. Matrix metalloproteinase 12, asthma, and COPD. N. Engl. J. Med. 2009, 361, 2664–2665. [Google Scholar] [CrossRef]
- Takahashi, Y.; Kobayashi, T.; D’Alessandro-Gabazza, C.N.; Toda, M.; Fujiwara, K.; Okano, T.; Fujimoto, H.; Asayama, K.; Takeshita, A.; Yasuma, T.; et al. Protective Role of Matrix Metalloproteinase-2 in Allergic Bronchial Asthma. Front. Immunol. 2019, 10, 1795. [Google Scholar] [CrossRef]
- Hunninghake, G.M.; Cho, M.H.; Tesfaigzi, Y.; Soto-Quiros, M.E.; Avila, L.; Lasky-Su, J.; Stidley, C.; Melen, E.; Soderhall, C.; Hallberg, J.; et al. MMP12, lung function, and COPD in high-risk populations. N. Engl. J. Med. 2009, 361, 2599–2608. [Google Scholar] [CrossRef]
- Naveed, S.U.; Clements, D.; Jackson, D.J.; Philp, C.; Billington, C.K.; Soomro, I.; Reynolds, C.; Harrison, T.W.; Johnston, S.L.; Shaw, D.E.; et al. Matrix Metalloproteinase-1 Activation Contributes to Airway Smooth Muscle Growth and Asthma Severity. Am. J. Respir. Crit. Care Med. 2017, 195, 1000–1009. [Google Scholar] [CrossRef]
- Ogulur, I.; Pat, Y.; Ardicli, O.; Barletta, E.; Cevhertas, L.; Fernandez-Santamaria, R.; Huang, M.; Bel Imam, M.; Koch, J.; Ma, S.; et al. Advances and highlights in biomarkers of allergic diseases. Allergy 2021, 76, 3659–3686. [Google Scholar] [CrossRef]
- Hur, G.Y.; Ye, Y.M.; Yang, E.; Park, H.S. Serum potential biomarkers according to sputum inflammatory cell profiles in adult asthmatics. Korean J. Intern. Med. 2020, 35, 988–997. [Google Scholar] [CrossRef]
- Pizzichini, M.M.; Popov, T.A.; Efthimiadis, A.; Hussack, P.; Evans, S.; Pizzichini, E.; Dolovich, J.; Hargreave, F.E. Spontaneous and induced sputum to measure indices of airway inflammation in asthma. Am. J. Respir. Crit. Care Med. 1996, 154, 866–869. [Google Scholar] [CrossRef]
- Green, R.H.; Brightling, C.E.; McKenna, S.; Hargadon, B.; Parker, D.; Bradding, P.; Wardlaw, A.J.; Pavord, I.D. Asthma exacerbations and sputum eosinophil counts: A randomised controlled trial. Lancet 2002, 360, 1715–1721. [Google Scholar] [CrossRef] [PubMed]
- Chiappori, A.; De Ferrari, L.; Folli, C.; Mauri, P.; Riccio, A.M.; Canonica, G.W. Biomarkers and severe asthma: A critical appraisal. Clin. Mol. Allergy 2015, 13, 20. [Google Scholar] [CrossRef] [PubMed]
- Tran, T.N.; Khatry, D.B.; Ke, X.; Ward, C.K.; Gossage, D. High blood eosinophil count is associated with more frequent asthma attacks in asthma patients. Ann. Allergy Asthma Immunol. 2014, 113, 19–24. [Google Scholar] [CrossRef]
- Horn, B.R.; Robin, E.D.; Theodore, J.; Van Kessel, A. Total eosinophil counts in the management of bronchial asthma. N. Engl. J. Med. 1975, 292, 1152–1155. [Google Scholar] [CrossRef] [PubMed]
- Ortega, H.G.; Liu, M.C.; Pavord, I.D.; Brusselle, G.G.; FitzGerald, J.M.; Chetta, A.; Humbert, M.; Katz, L.E.; Keene, O.N.; Yancey, S.W.; et al. Mepolizumab treatment in patients with severe eosinophilic asthma. N. Engl. J. Med. 2014, 371, 1198–1207. [Google Scholar] [CrossRef] [PubMed]
- Pavord, I.D.; Korn, S.; Howarth, P.; Bleecker, E.R.; Buhl, R.; Keene, O.N.; Ortega, H.; Chanez, P. Mepolizumab for severe eosinophilic asthma (DREAM): A multicentre, double-blind, placebo-controlled trial. Lancet 2012, 380, 651–659. [Google Scholar] [CrossRef]
- Nair, P.; Wenzel, S.; Rabe, K.F.; Bourdin, A.; Lugogo, N.L.; Kuna, P.; Barker, P.; Sproule, S.; Ponnarambil, S.; Goldman, M. Oral Glucocorticoid-Sparing Effect of Benralizumab in Severe Asthma. N. Engl. J. Med. 2017, 376, 2448–2458. [Google Scholar] [CrossRef]
- Biomarkers and surrogate endpoints: Preferred definitions and conceptual framework. Clin. Pharm. 2001, 69, 89–95. [CrossRef]
- Lehtimäki, L.; Shrimanker, R.; Moran, A.; Hynes, G.; Thulborn, S.; Borg, C.; Connolly, C.; Gittins, A.; Downs, T.; Russell, R. P13 Exhaled nitric oxide and blood eosinophil count in predicting sputum inflammatory type in a heterogeneous airways disease population. Thorax 2019, 74, A95. [Google Scholar]
- Couillard, S.; Shrimanker, R.; Chaudhuri, R.; Mansur, A.H.; McGarvey, L.P.; Heaney, L.G.; Fowler, S.J.; Bradding, P.; Pavord, I.D.; Hinks, T.S.C. Fractional Exhaled Nitric Oxide Nonsuppression Identifies Corticosteroid-Resistant Type 2 Signaling in Severe Asthma. Am. J. Respir. Crit. Care Med. 2021, 204, 731–734. [Google Scholar] [CrossRef]
- Burrows, B.; Martinez, F.D.; Halonen, M.; Barbee, R.A.; Cline, M.G. Association of asthma with serum IgE levels and skin-test reactivity to allergens. N. Engl. J. Med. 1989, 320, 271–277. [Google Scholar] [CrossRef] [PubMed]
- Sherrill, D.L.; Lebowitz, M.D.; Halonen, M.; Barbee, R.A.; Burrows, B. Longitudinal evaluation of the association between pulmonary function and total serum IgE. Am. J. Respir. Crit. Care Med. 1995, 152, 98–102. [Google Scholar] [CrossRef] [PubMed]
- Busse, W.; Corren, J.; Lanier, B.Q.; McAlary, M.; Fowler-Taylor, A.; Cioppa, G.D.; van As, A.; Gupta, N. Omalizumab, anti-IgE recombinant humanized monoclonal antibody, for the treatment of severe allergic asthma. J. Allergy Clin. Immunol. 2001, 108, 184–190. [Google Scholar] [CrossRef]
- Normansell, R.; Walker, S.; Milan, S.J.; Walters, E.H.; Nair, P. Omalizumab for asthma in adults and children. Cochrane Database Syst. Rev. 2014, Cd003559. [Google Scholar] [CrossRef]
- Alving, K.; Weitzberg, E.; Lundberg, J.M. Increased amount of nitric oxide in exhaled air of asthmatics. Eur. Respir. J. 1993, 6, 1368–1370. [Google Scholar]
- Chibana, K.; Trudeau, J.B.; Mustovich, A.T.; Hu, H.; Zhao, J.; Balzar, S.; Chu, H.W.; Wenzel, S.E. IL-13 induced increases in nitrite levels are primarily driven by increases in inducible nitric oxide synthase as compared with effects on arginases in human primary bronchial epithelial cells. Clin. Exp. Allergy 2008, 38, 936–946. [Google Scholar] [CrossRef] [PubMed]
- Suresh, V.; Mih, J.D.; George, S.C. Measurement of IL-13-induced iNOS-derived gas phase nitric oxide in human bronchial epithelial cells. Am. J. Respir. Cell Mol. Biol. 2007, 37, 97–104. [Google Scholar] [CrossRef]
- Pasha, M.A.; Smith, T.C.; Feustel, P.J.; Jourd’heuil, D. Effects of low-dose fluticasone propionate/salmeterol combination therapy on exhaled nitric oxide and nitrite/nitrate in breath condensates from patients with mild persistent asthma. J. Asthma 2013, 50, 64–70. [Google Scholar] [CrossRef]
- Dweik, R.A.; Boggs, P.B.; Erzurum, S.C.; Irvin, C.G.; Leigh, M.W.; Lundberg, J.O.; Olin, A.C.; Plummer, A.L.; Taylor, D.R. An official ATS clinical practice guideline: Interpretation of exhaled nitric oxide levels (FENO) for clinical applications. Am. J. Respir. Crit. Care Med. 2011, 184, 602–615. [Google Scholar] [CrossRef]
- Bjermer, L.; Alving, K.; Diamant, Z.; Magnussen, H.; Pavord, I.; Piacentini, G.; Price, D.; Roche, N.; Sastre, J.; Thomas, M.; et al. Current evidence and future research needs for FeNO measurement in respiratory diseases. Respir. Med. 2014, 108, 830–841. [Google Scholar] [CrossRef]
- Zuiker, R.G.; Boot, J.D.; Calderon, C.; Piantone, A.; Petty, K.; de Kam, M.; Diamant, Z. Sputum induction with hypertonic saline reduces fractional exhaled nitric oxide in chronic smokers and non-smokers. Respir. Med. 2010, 104, 917–920. [Google Scholar] [CrossRef] [PubMed]
- Boot, J.D.; de Kam, M.L.; Mascelli, M.A.; Miller, B.; van Wijk, R.G.; de Groot, H.; Cohen, A.F.; Diamant, Z. Nasal nitric oxide: Longitudinal reproducibility and the effects of a nasal allergen challenge in patients with allergic rhinitis. Allergy 2007, 62, 378–384. [Google Scholar] [CrossRef] [PubMed]
- Kostikas, K.; Minas, M.; Papaioannou, A.I.; Papiris, S.; Dweik, R.A. Exhaled nitric oxide in asthma in adults: The end is the beginning? Curr. Med. Chem. 2011, 18, 1423–1431. [Google Scholar] [CrossRef]
- Menzies-Gow, A.; Mansur, A.H.; Brightling, C.E. Clinical utility of fractional exhaled nitric oxide in severe asthma management. Eur. Respir. J. 2020, 55. [Google Scholar] [CrossRef]
- Petsky, H.L.; Cates, C.J.; Kew, K.M.; Chang, A.B. Tailoring asthma treatment on eosinophilic markers (exhaled nitric oxide or sputum eosinophils): A systematic review and meta-analysis. Thorax 2018, 73, 1110–1119. [Google Scholar] [CrossRef] [PubMed]
- Cevhertas, L.; Ogulur, I.; Maurer, D.J.; Burla, D.; Ding, M.; Jansen, K.; Koch, J.; Liu, C.; Ma, S.; Mitamura, Y.; et al. Advances and recent developments in asthma in 2020. Allergy 2020, 75, 3124–3146. [Google Scholar] [CrossRef]
- Jia, G.; Erickson, R.W.; Choy, D.F.; Mosesova, S.; Wu, L.C.; Solberg, O.D.; Shikotra, A.; Carter, R.; Audusseau, S.; Hamid, Q.; et al. Periostin is a systemic biomarker of eosinophilic airway inflammation in asthmatic patients. J. Allergy Clin. Immunol. 2012, 130, 647–654.e10. [Google Scholar] [CrossRef]
- Corren, J.; Lemanske, R.F.; Hanania, N.A.; Korenblat, P.E.; Parsey, M.V.; Arron, J.R.; Harris, J.M.; Scheerens, H.; Wu, L.C.; Su, Z.; et al. Lebrikizumab treatment in adults with asthma. N. Engl. J. Med. 2011, 365, 1088–1098. [Google Scholar] [CrossRef]
- Hanania, N.A.; Noonan, M.; Corren, J.; Korenblat, P.; Zheng, Y.; Fischer, S.K.; Cheu, M.; Putnam, W.S.; Murray, E.; Scheerens, H.; et al. Lebrikizumab in moderate-to-severe asthma: Pooled data from two randomised placebo-controlled studies. Thorax 2015, 70, 748–756. [Google Scholar] [CrossRef]
- Breiteneder, H.; Peng, Y.Q.; Agache, I.; Diamant, Z.; Eiwegger, T.; Fokkens, W.J.; Traidl-Hoffmann, C.; Nadeau, K.; O’Hehir, R.E.; O’Mahony, L.; et al. Biomarkers for diagnosis and prediction of therapy responses in allergic diseases and asthma. Allergy 2020, 75, 3039–3068. [Google Scholar] [CrossRef]
- Wan, X.C.; Woodruff, P.G. Biomarkers in Severe Asthma. Immunol. Allergy Clin. N. Am. 2016, 36, 547–557. [Google Scholar] [CrossRef] [PubMed]
- McGrath, K.W.; Icitovic, N.; Boushey, H.A.; Lazarus, S.C.; Sutherland, E.R.; Chinchilli, V.M.; Fahy, J.V. A large subgroup of mild-to-moderate asthma is persistently noneosinophilic. Am. J. Respir. Crit. Care Med. 2012, 185, 612–619. [Google Scholar] [CrossRef] [PubMed]
- Berry, M.; Morgan, A.; Shaw, D.E.; Parker, D.; Green, R.; Brightling, C.; Bradding, P.; Wardlaw, A.J.; Pavord, I.D. Pathological features and inhaled corticosteroid response of eosinophilic and non-eosinophilic asthma. Thorax 2007, 62, 1043–1049. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barbato, A.; Turato, G.; Baraldo, S.; Bazzan, E.; Calabrese, F.; Panizzolo, C.; Zanin, M.E.; Zuin, R.; Maestrelli, P.; Fabbri, L.M.; et al. Epithelial damage and angiogenesis in the airways of children with asthma. Am. J. Respir. Crit. Care Med. 2006, 174, 975–981. [Google Scholar] [CrossRef] [PubMed]
- Riccio, A.M.; Mauri, P.; De Ferrari, L.; Rossi, R.; Di Silvestre, D.; Benazzi, L.; Chiappori, A.; Dal Negro, R.W.; Micheletto, C.; Canonica, G.W. Galectin-3: An early predictive biomarker of modulation of airway remodeling in patients with severe asthma treated with omalizumab for 36 months. Clin. Transl. Allergy 2017, 7, 6. [Google Scholar] [CrossRef] [PubMed]
- Bossley, C.J.; Fleming, L.; Gupta, A.; Regamey, N.; Frith, J.; Oates, T.; Tsartsali, L.; Lloyd, C.M.; Bush, A.; Saglani, S. Pediatric severe asthma is characterized by eosinophilia and remodeling without T(H)2 cytokines. J. Allergy Clin. Immunol 2012, 129, 974–982.e13. [Google Scholar] [CrossRef]
- Payne, D.N.; Rogers, A.V.; Adelroth, E.; Bandi, V.; Guntupalli, K.K.; Bush, A.; Jeffery, P.K. Early thickening of the reticular basement membrane in children with difficult asthma. Am. J. Respir. Crit Care Med. 2003, 167, 78–82. [Google Scholar] [CrossRef] [PubMed]
- Mauri, P.; Riccio, A.M.; Rossi, R.; Di Silvestre, D.; Benazzi, L.; De Ferrari, L.; Dal Negro, R.W.; Holgate, S.T.; Canonica, G.W. Proteomics of bronchial biopsies: Galectin-3 as a predictive biomarker of airway remodelling modulation in omalizumab-treated severe asthma patients. Immunol. Lett. 2014, 162, 2–10. [Google Scholar] [CrossRef]
- Loftheim, H.; Midtvedt, K.; Hartmann, A.; Reisæter, A.V.; Falck, P.; Holdaas, H.; Jenssen, T.; Reubsaet, L.; Asberg, A. Urinary proteomic shotgun approach for identification of potential acute rejection biomarkers in renal transplant recipients. Transpl. Res. 2012, 1, 9. [Google Scholar] [CrossRef]
- Konradsen, J.R.; James, A.; Nordlund, B.; Reinius, L.E.; Soderhall, C.; Melen, E.; Wheelock, A.M.; Lodrup Carlsen, K.C.; Lidegran, M.; Verhoek, M.; et al. The chitinase-like protein YKL-40: A possible biomarker of inflammation and airway remodeling in severe pediatric asthma. J. Allergy Clin. Immunol. 2013, 132, 328–335.e5. [Google Scholar] [CrossRef]
- Farzan, N.; Vijverberg, S.J.; Hernandez-Pacheco, N.; Bel, E.H.D.; Berce, V.; Bonnelykke, K.; Bisgaard, H.; Burchard, E.G.; Canino, G.; Celedon, J.C.; et al. 17q21 variant increases the risk of exacerbations in asthmatic children despite inhaled corticosteroids use. Allergy 2018, 73, 2083–2088. [Google Scholar] [CrossRef] [PubMed]
- Hur, G.Y.; Pham, A.; Miller, M.; Weng, N.; Hu, J.; Kurten, R.C.; Broide, D.H. ORMDL3 but not neighboring 17q21 gene LRRC3C is expressed in human lungs and lung cells of asthmatics. Allergy 2020, 75, 2061–2065. [Google Scholar] [CrossRef]
- Dijk, F.N.; Vijverberg, S.J.; Hernandez-Pacheco, N.; Repnik, K.; Karimi, L.; Mitratza, M.; Farzan, N.; Nawijn, M.C.; Burchard, E.G.; Engelkes, M.; et al. IL1RL1 gene variations are associated with asthma exacerbations in children and adolescents using inhaled corticosteroids. Allergy 2020, 75, 984–989. [Google Scholar] [CrossRef] [Green Version]
- Ye, L.; Pan, J.; Pasha, M.A.; Shen, X.; D’Souza, S.S.; Fung, I.T.H.; Wang, Y.; Guo, B.; Tang, D.D.; Yang, Q. Mucosal-associated invariant T cells restrict allergic airway inflammation. J. Allergy Clin. Immunol. 2019. [Google Scholar] [CrossRef]
- Ye, L.; Pan, J.; Liang, M.; Pasha, M.A.; Shen, X.; D’Souza, S.S.; Fung, I.T.H.; Wang, Y.; Patel, G.; Tang, D.D.; et al. A critical role for c-Myc in group 2 innate lymphoid cell activation. Allergy 2020, 75, 841–852. [Google Scholar] [CrossRef] [PubMed]
- Murrison, L.B.; Brandt, E.B.; Myers, J.B.; Hershey, G.K.K. Environmental exposures and mechanisms in allergy and asthma development. J. Clin. Investig. 2019, 129, 1504–1515. [Google Scholar] [CrossRef]
- Lin, C.H.; Li, Y.R.; Kor, C.T.; Lin, S.H.; Ji, B.C.; Lin, M.T.; Chai, W.H. The Mediating Effect of Cytokines on the Association between Fungal Sensitization and Poor Clinical Outcome in Asthma. Biomedicines 2022, 10, 1452. [Google Scholar] [CrossRef]
- Kuruvilla, M.E.; Lee, F.E.; Lee, G.B. Understanding Asthma Phenotypes, Endotypes, and Mechanisms of Disease. Clin. Rev. Allergy Immunol. 2019, 56, 219–233. [Google Scholar] [CrossRef]
- Miranda, C.; Busacker, A.; Balzar, S.; Trudeau, J.; Wenzel, S.E. Distinguishing severe asthma phenotypes: Role of age at onset and eosinophilic inflammation. J. Allergy Clin. Immunol. 2004, 113, 101–108. [Google Scholar] [CrossRef]
- Wenzel, S.E.; Schwartz, L.B.; Langmack, E.L.; Halliday, J.L.; Trudeau, J.B.; Gibbs, R.L.; Chu, H.W. Evidence that severe asthma can be divided pathologically into two inflammatory subtypes with distinct physiologic and clinical characteristics. Am. J. Respir. Crit. Care Med. 1999, 160, 1001–1008. [Google Scholar] [CrossRef]
- Peters, M.C.; Kerr, S.; Dunican, E.M.; Woodruff, P.G.; Fajt, M.L.; Levy, B.D.; Israel, E.; Phillips, B.R.; Mauger, D.T.; Comhair, S.A.; et al. Refractory airway type 2 inflammation in a large subgroup of asthmatic patients treated with inhaled corticosteroids. J. Allergy Clin. Immunol. 2019, 143, 104–113.e14. [Google Scholar] [CrossRef] [PubMed]
- Hastie, A.T.; Moore, W.C.; Meyers, D.A.; Vestal, P.L.; Li, H.; Peters, S.P.; Bleecker, E.R.; National Heart, L.; Blood Institute Severe Asthma Research Program. Analyses of asthma severity phenotypes and inflammatory proteins in subjects stratified by sputum granulocytes. J. Allergy Clin. Immunol. 2010, 125, 1028–1036.e13. [Google Scholar] [CrossRef] [PubMed]
- Rastogi, D.; Fraser, S.; Oh, J.; Huber, A.M.; Schulman, Y.; Bhagtani, R.H.; Khan, Z.S.; Tesfa, L.; Hall, C.B.; Macian, F. Inflammation, metabolic dysregulation, and pulmonary function among obese urban adolescents with asthma. Am. J. Respir. Crit. Care Med. 2015, 191, 149–160. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Peters, U.; Dixon, A.E.; Forno, E. Obesity and asthma. J. Allergy Clin. Immunol. 2018, 141, 1169–1179. [Google Scholar] [CrossRef]
- Takahashi, K.; Pavlidis, S.; Ng Kee Kwong, F.; Hoda, U.; Rossios, C.; Sun, K.; Loza, M.; Baribaud, F.; Chanez, P.; Fowler, S.J.; et al. Sputum proteomics and airway cell transcripts of current and ex-smokers with severe asthma in U-BIOPRED: An exploratory analysis. Eur. Respir. J. 2018, 51. [Google Scholar] [CrossRef]
- Pite, H.; Pereira, A.M.; Morais-Almeida, M.; Nunes, C.; Bousquet, J.; Fonseca, J.A. Prevalence of asthma and its association with rhinitis in the elderly. Respir. Med. 2014, 108, 1117–1126. [Google Scholar] [CrossRef]
- Gibson, P.G.; McDonald, V.M.; Marks, G.B. Asthma in older adults. Lancet 2010, 376, 803–813. [Google Scholar] [CrossRef]
- Nyenhuis, S.M.; Schwantes, E.A.; Evans, M.D.; Mathur, S.K. Airway neutrophil inflammatory phenotype in older subjects with asthma. J. Allergy Clin. Immunol. 2010, 125, 1163–1165. [Google Scholar] [CrossRef]
- Dunn, R.M.; Busse, P.J.; Wechsler, M.E. Asthma in the elderly and late-onset adult asthma. Allergy 2018, 73, 284–294. [Google Scholar] [CrossRef]
- Boulet, L.P.; Boulay, M.E. Asthma-related comorbidities. Expert. Rev. Respir. Med. 2011, 5, 377–393. [Google Scholar] [CrossRef]
- Kaplan, A.; Szefler, S.J.; Halpin, D.M.G. Impact of comorbid conditions on asthmatic adults and children. NPJ Prim. Care Respir. Med. 2020, 30, 36. [Google Scholar] [CrossRef] [PubMed]
- Langdon, C.; Mullol, J. Nasal polyps in patients with asthma: Prevalence, impact, and management challenges. J. Asthma Allergy 2016, 9, 45–53. [Google Scholar] [CrossRef] [PubMed] [Green Version]

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Habib, N.; Pasha, M.A.; Tang, D.D. Current Understanding of Asthma Pathogenesis and Biomarkers. Cells 2022, 11, 2764. https://doi.org/10.3390/cells11172764
Habib N, Pasha MA, Tang DD. Current Understanding of Asthma Pathogenesis and Biomarkers. Cells. 2022; 11(17):2764. https://doi.org/10.3390/cells11172764
Chicago/Turabian StyleHabib, Nazia, Muhammad Asghar Pasha, and Dale D. Tang. 2022. "Current Understanding of Asthma Pathogenesis and Biomarkers" Cells 11, no. 17: 2764. https://doi.org/10.3390/cells11172764
APA StyleHabib, N., Pasha, M. A., & Tang, D. D. (2022). Current Understanding of Asthma Pathogenesis and Biomarkers. Cells, 11(17), 2764. https://doi.org/10.3390/cells11172764