
PGC-1α4 Interacts with REST to Upregulate Neuronal Genes and Augment Energy Consumption in Developing Cardiomyocytes

 , , , and
, , , and {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Gene Expression Analysis from RNA Sequencing Data
2.2. Ethical Statement
2.3. Experimental Animals
2.4. Cardiomyocyte Isolation and Culture
2.5. Real-Time Quantitative PCR
2.6. Agarose Gel Electrophoresis PCR
2.7. Western Blot
2.8. Tissue Collection
2.9. Histology
2.10. Echocardiography
2.11. Confocal Calcium Imaging
2.12. Patch-Clamp Recordings
2.13. Analysis of Energy Metabolism
2.14. Adenoviral Overexpression
2.15. RNA Library Preparation and Sequencing
2.16. Analysis of Chromatin Immunoprecipitation Data
2.17. Culture of HL-1 Cardiomyocyte Cell Line
2.18. Gene Silencing with Small Interfering RNA
2.19. Luciferase Gene Reporter Assay
2.20. Statistical Testing
3. Results
3.1. Pgc-1α Isoforms Arising from Differential Promoter Usage and mRNA Splicing Are Expressed in Cardiac Tissue
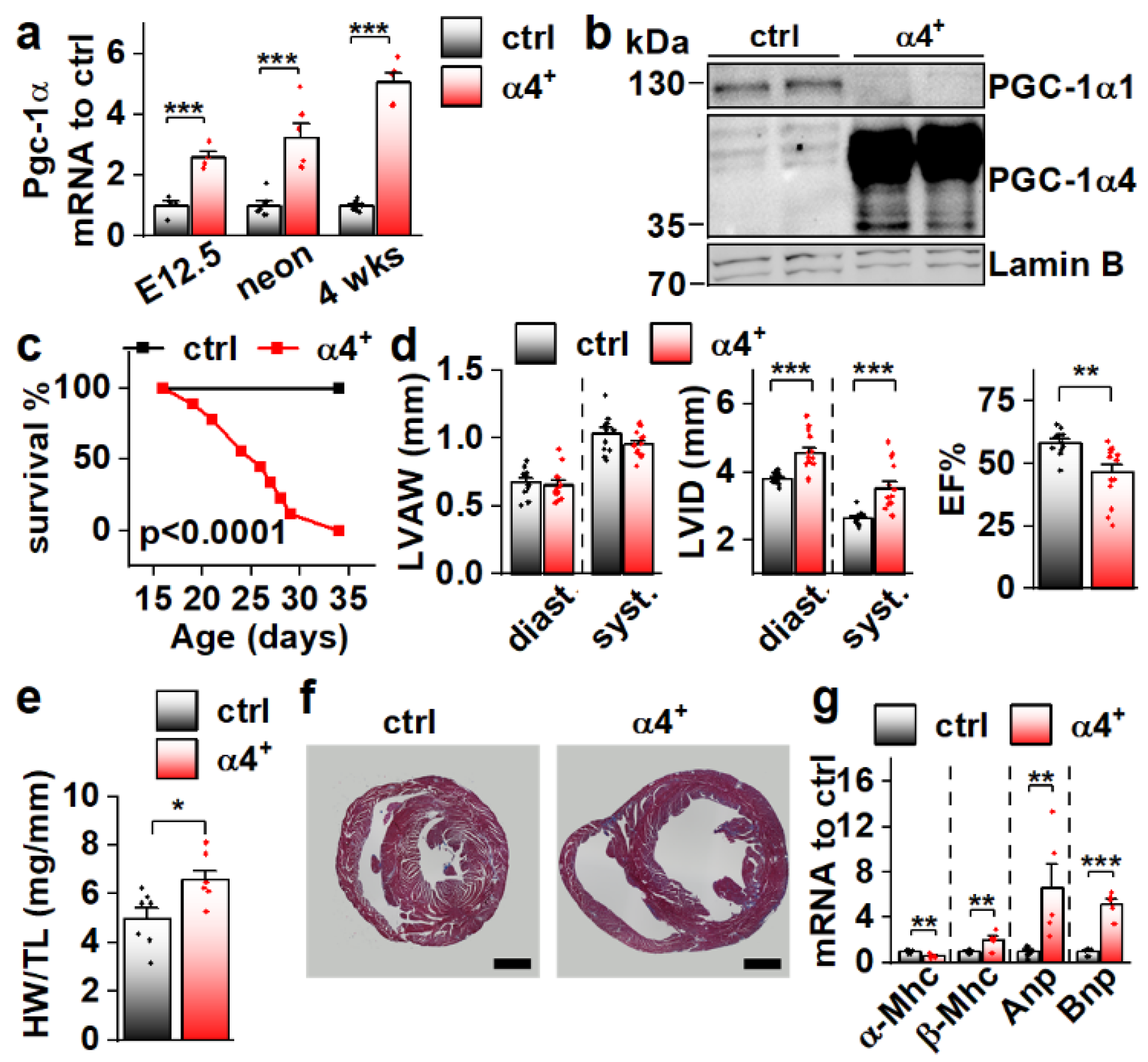
3.2. Cardiomyocyte-Specific PGC-1α4 Overexpression in Mice Leads to Early Death by Dilated Heart Failure
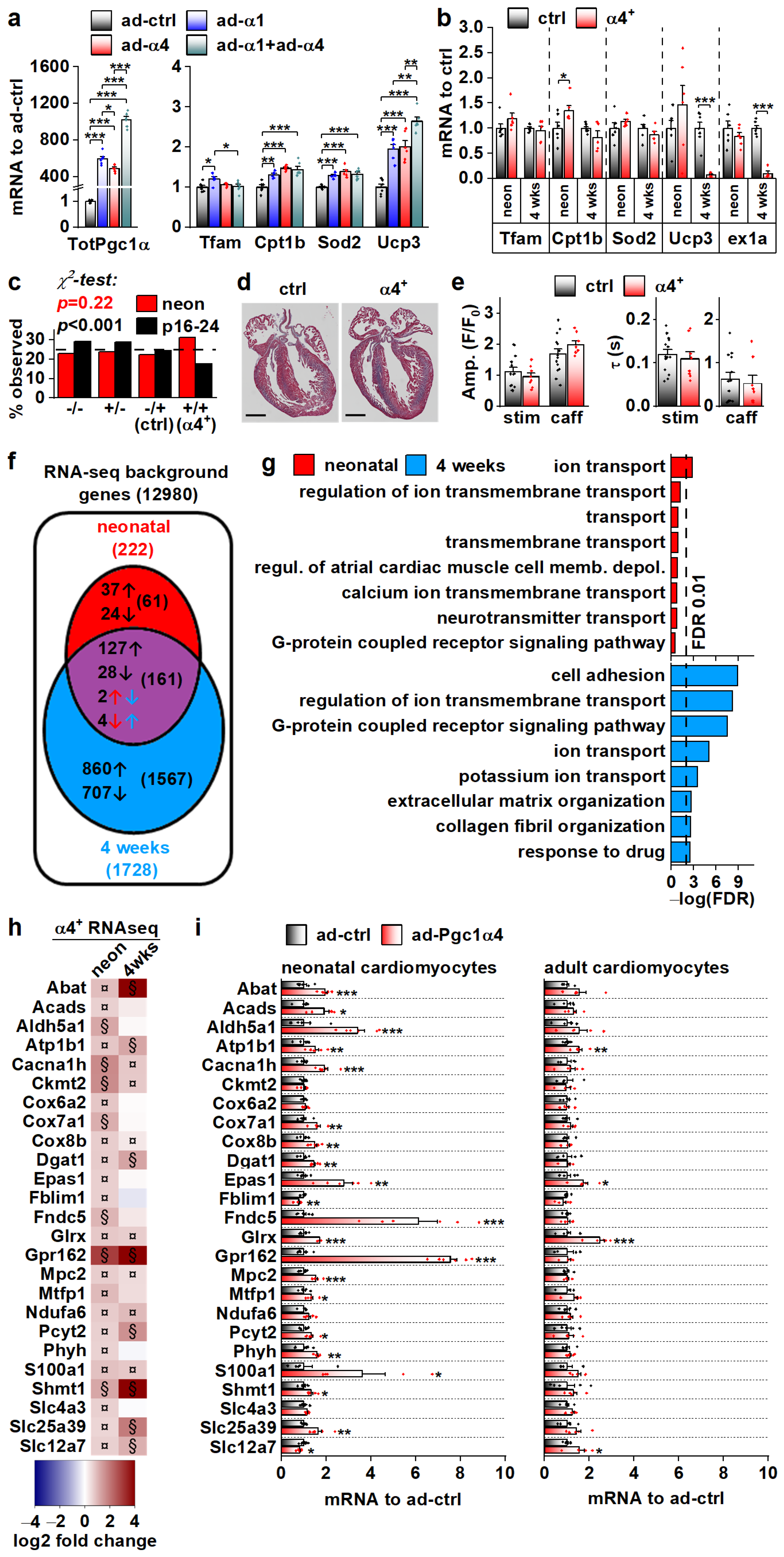
3.3. PGC-1α4 Overexpression Leads to Distinct Changes in Cardiomyocyte Phenotype without Effects on Energy Metabolic Capacity
3.4. PGC-1α4-Induced Gene Expression Response and the Resulting Phenotype Depend on Cardiomyocyte Developmental Stage
3.5. PGC-1α4 Overexpression Induces Na-K ATPase Expression and Leads to a Drastic Increase of Its Current in Neonatal Cardiomyocytes
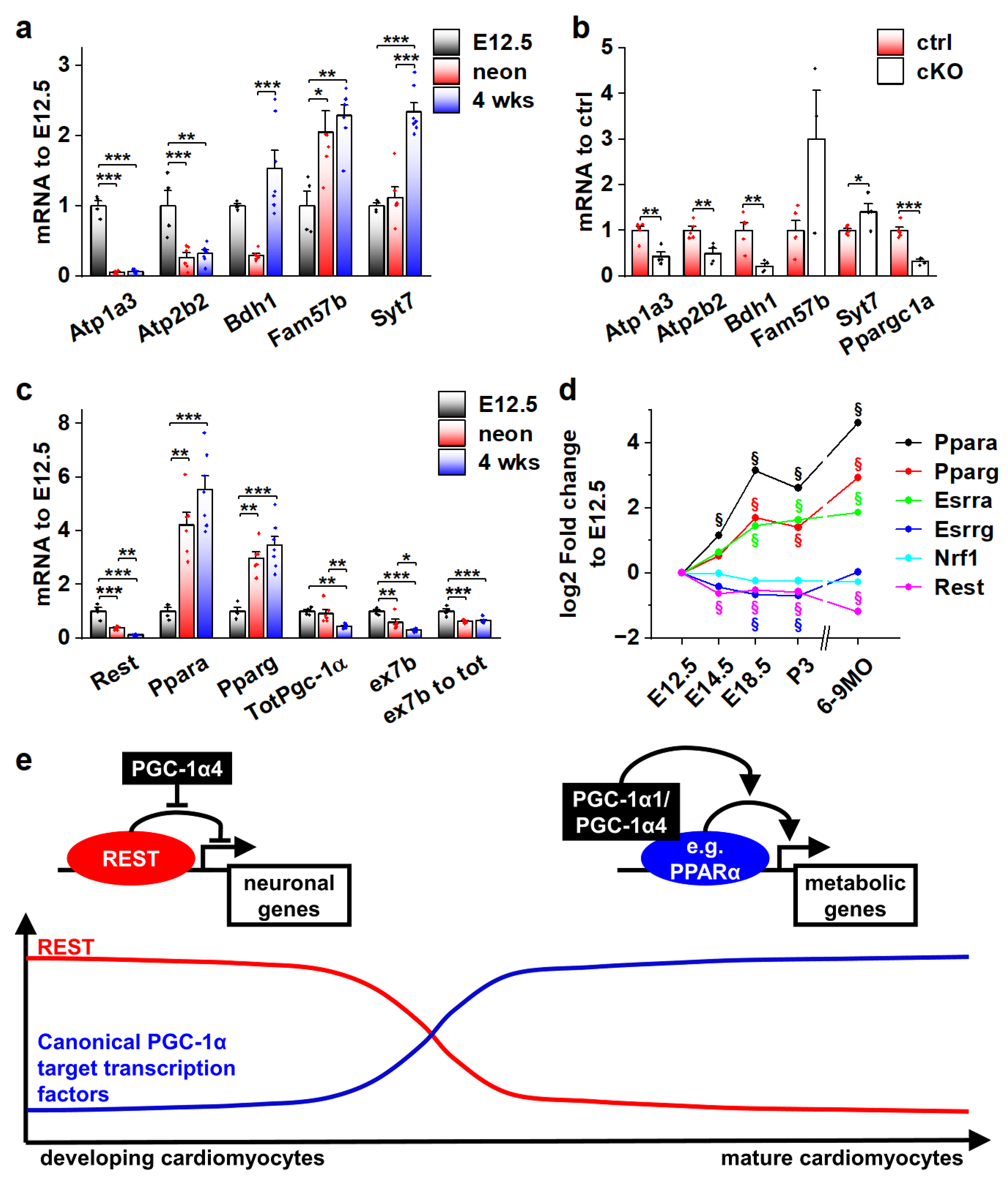
3.6. Genes Associated with the Transcriptional Repressor REST Are Induced by PGC-1α4 in Developing Cardiac Myocytes
3.7. Actions of Different PGC-1α Isoforms during Normal Cardiomyocyte Development Are Governed by the Expression of Their Target Transcription Factors
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Handschin, C.; Spiegelman, B.M. Peroxisome proliferator-activated receptor gamma coactivator 1 coactivators, energy homeostasis, and metabolism. Endocr. Rev. 2006, 27, 728–735. [Google Scholar] [CrossRef] [PubMed]
- Lehman, J.J.; Kelly, D.P. Transcriptional activation of energy metabolic switches in the developing and hypertrophied heart. Clin. Exp. Pharmacol. Physiol. 2002, 29, 339–345. [Google Scholar] [CrossRef] [PubMed]
- Arany, Z.; He, H.; Lin, J.; Hoyer, K.; Handschin, C.; Toka, O.; Ahmad, F.; Matsui, T.; Chin, S.; Wu, P.H.; et al. Transcriptional coactivator PGC-1 alpha controls the energy state and contractile function of cardiac muscle. Cell Metab. 2005, 1, 259–271. [Google Scholar] [CrossRef]
- Karkkainen, O.; Tuomainen, T.; Mutikainen, M.; Lehtonen, M.; Ruas, J.L.; Hanhineva, K.; Tavi, P. Heart specific PGC-1alpha deletion identifies metabolome of cardiac restricted metabolic heart failure. Cardiovasc. Res. 2019, 115, 107–118. [Google Scholar] [CrossRef] [PubMed]
- Lehman, J.J.; Barger, P.M.; Kovacs, A.; Saffitz, J.E.; Medeiros, D.M.; Kelly, D.P. Peroxisome proliferator-activated receptor gamma coactivator-1 promotes cardiac mitochondrial biogenesis. J. Clin. Investig. 2000, 106, 847–856. [Google Scholar] [CrossRef] [PubMed]
- Mutikainen, M.; Tuomainen, T.; Naumenko, N.; Huusko, J.; Smirin, B.; Laidinen, S.; Kokki, K.; Hynynen, H.; Yla-Herttuala, S.; Heinaniemi, M.; et al. Peroxisome proliferator-activated receptor-gamma coactivator 1 alpha1 induces a cardiac excitation-contraction coupling phenotype without metabolic remodelling. J. Physiol. 2016, 594, 7049–7071. [Google Scholar] [CrossRef]
- Wu, Z.; Puigserver, P.; Andersson, U.; Zhang, C.; Adelmant, G.; Mootha, V.; Troy, A.; Cinti, S.; Lowell, B.; Scarpulla, R.C.; et al. Mechanisms controlling mitochondrial biogenesis and respiration through the thermogenic coactivator PGC-1. Cell 1999, 98, 115–124. [Google Scholar] [CrossRef]
- Vega, R.B.; Huss, J.M.; Kelly, D.P. The coactivator PGC-1 cooperates with peroxisome proliferator-activated receptor alpha in transcriptional control of nuclear genes encoding mitochondrial fatty acid oxidation enzymes. Mol. Cell. Biol. 2000, 20, 1868–1876. [Google Scholar] [CrossRef]
- Michael, L.F.; Wu, Z.; Cheatham, R.B.; Puigserver, P.; Adelmant, G.; Lehman, J.J.; Kelly, D.P.; Spiegelman, B.M. Restoration of insulin-sensitive glucose transporter (GLUT4) gene expression in muscle cells by the transcriptional coactivator PGC-1. Proc. Natl. Acad. Sci. USA 2001, 98, 3820–3825. [Google Scholar] [CrossRef]
- Martinez-Redondo, V.; Pettersson, A.T.; Ruas, J.L. The hitchhiker’s guide to PGC-1alpha isoform structure and biological functions. Diabetologia 2015, 58, 1969–1977. [Google Scholar] [CrossRef] [Green Version]
- Ruas, J.L.; White, J.P.; Rao, R.R.; Kleiner, S.; Brannan, K.T.; Harrison, B.C.; Greene, N.P.; Wu, J.; Estall, J.L.; Irving, B.A.; et al. A PGC-1alpha isoform induced by resistance training regulates skeletal muscle hypertrophy. Cell 2012, 151, 1319–1331. [Google Scholar] [CrossRef] [PubMed]
- Leveille, M.; Besse-Patin, A.; Jouvet, N.; Gunes, A.; Sczelecki, S.; Jeromson, S.; Khan, N.P.; Baldwin, C.; Dumouchel, A.; Correia, J.C.; et al. PGC-1alpha isoforms coordinate to balance hepatic metabolism and apoptosis in inflammatory environments. Mol. Metab. 2020, 34, 72–84. [Google Scholar] [CrossRef] [PubMed]
- Chinsomboon, J.; Ruas, J.; Gupta, R.K.; Thom, R.; Shoag, J.; Rowe, G.C.; Sawada, N.; Raghuram, S.; Arany, Z. The transcriptional coactivator PGC-1alpha mediates exercise-induced angiogenesis in skeletal muscle. Proc. Natl. Acad. Sci. USA 2009, 106, 21401–21406. [Google Scholar] [CrossRef] [PubMed]
- Liu, Z.; Tian, H.; Hua, J.; Cai, W.; Bai, Y.; Zhan, Q.; Lai, W.; Zeng, Q.; Ren, H.; Xu, D. A CRM1 Inhibitor Alleviates Cardiac Hypertrophy and Increases the Nuclear Distribution of NT-PGC-1alpha in NRVMs. Front. Pharm. 2019, 10, 465. [Google Scholar] [CrossRef] [PubMed]
- Kim, D.; Langmead, B.; Salzberg, S.L. HISAT: A fast spliced aligner with low memory requirements. Nat. Methods 2015, 12, 357–360. [Google Scholar] [CrossRef]
- Hartley, S.W.; Mullikin, J.C. QoRTs: A comprehensive toolset for quality control and data processing of RNA-Seq experiments. BMC Bioinform. 2015, 16, 224. [Google Scholar] [CrossRef]
- Love, M.I.; Huber, W.; Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome. Biol. 2014, 15, 550. [Google Scholar] [CrossRef]
- Pertea, M.; Pertea, G.M.; Antonescu, C.M.; Chang, T.C.; Mendell, J.T.; Salzberg, S.L. StringTie enables improved reconstruction of a transcriptome from RNA-seq reads. Nat. Biotechnol. 2015, 33, 290–295. [Google Scholar] [CrossRef]
- Huang da, W.; Sherman, B.T.; Lempicki, R.A. Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat. Protoc. 2009, 4, 44–57. [Google Scholar] [CrossRef]
- Agah, R.; Frenkel, P.A.; French, B.A.; Michael, L.H.; Overbeek, P.A.; Schneider, M.D. Gene recombination in postmitotic cells. Targeted expression of Cre recombinase provokes cardiac-restricted, site-specific rearrangement in adult ventricular muscle in vivo. J. Clin. Investig. 1997, 100, 169–179. [Google Scholar] [CrossRef] [Green Version]
- Yang, Z.; Shen, W.; Rottman, J.N.; Wikswo, J.P.; Murray, K.T. Rapid stimulation causes electrical remodeling in cultured atrial myocytes. J. Mol. Cell. Cardiol. 2005, 38, 299–308. [Google Scholar] [CrossRef] [PubMed]
- Naumenko, N.; Huusko, J.; Tuomainen, T.; Koivumaki, J.T.; Merentie, M.; Gurzeler, E.; Alitalo, K.; Kivela, R.; Yla-Herttuala, S.; Tavi, P. Vascular Endothelial Growth Factor-B Induces a Distinct Electrophysiological Phenotype in Mouse Heart. Front. Physiol. 2017, 8, 373. [Google Scholar] [CrossRef] [PubMed]
- Xu, L.; Li, X.Y.; Liu, Y.; Li, H.T.; Chen, J.; Li, X.Y.; Jiang, X.J.; Wu, G.; Tang, Y.H.; Wang, X.; et al. The mechanisms underlying ICa heterogeneity across murine left ventricle. Mol. Cell. Biochem. 2011, 352, 239–246. [Google Scholar] [CrossRef] [PubMed]
- Guo, H.C.; Guo, F.; Zhang, L.N.; Zhang, R.; Chen, Q.; Li, J.X.; Yin, J.; Wang, Y.L. Enhancement of Na/K pump activity by chronic intermittent hypobaric hypoxia protected against reperfusion injury. Am. J. Physiology. Heart Circ. Physiol. 2011, 300, H2280–H2287. [Google Scholar] [CrossRef]
- Langmead, B.; Salzberg, S.L. Fast gapped-read alignment with Bowtie 2. Nat. Methods 2012, 9, 357–359. [Google Scholar] [CrossRef]
- Heinz, S.; Benner, C.; Spann, N.; Bertolino, E.; Lin, Y.C.; Laslo, P.; Cheng, J.X.; Murre, C.; Singh, H.; Glass, C.K. Simple combinations of lineage-determining transcription factors prime cis-regulatory elements required for macrophage and B cell identities. Mol. Cell 2010, 38, 576–589. [Google Scholar] [CrossRef]
- Kondili, M.; Fust, A.; Preussner, J.; Kuenne, C.; Braun, T.; Looso, M. UROPA: A tool for Universal RObust Peak Annotation. Sci. Rep. 2017, 7, 2593. [Google Scholar] [CrossRef]
- Sikkel, M.B.; Francis, D.P.; Howard, J.; Gordon, F.; Rowlands, C.; Peters, N.S.; Lyon, A.R.; Harding, S.E.; MacLeod, K.T. Hierarchical statistical techniques are necessary to draw reliable conclusions from analysis of isolated cardiomyocyte studies. Cardiovasc. Res. 2017, 113, 1743–1752. [Google Scholar] [CrossRef]
- Ma, X.; Gao, L.; Karamanlidis, G.; Gao, P.; Lee, C.F.; Garcia-Menendez, L.; Tian, R.; Tan, K. Revealing Pathway Dynamics in Heart Diseases by Analyzing Multiple Differential Networks. PLoS Comput. Biol. 2015, 11, e1004332. [Google Scholar] [CrossRef]
- Ounzain, S.; Micheletti, R.; Beckmann, T.; Schroen, B.; Alexanian, M.; Pezzuto, I.; Crippa, S.; Nemir, M.; Sarre, A.; Johnson, R.; et al. Genome-wide profiling of the cardiac transcriptome after myocardial infarction identifies novel heart-specific long non-coding RNAs. Eur. Heart J. 2015, 36, 353–368a. [Google Scholar] [CrossRef]
- Park, T.J.; Park, J.H.; Lee, G.S.; Lee, J.Y.; Shin, J.H.; Kim, M.W.; Kim, Y.S.; Kim, J.Y.; Oh, K.J.; Han, B.S.; et al. Quantitative proteomic analyses reveal that GPX4 downregulation during myocardial infarction contributes to ferroptosis in cardiomyocytes. Cell Death Dis. 2019, 10, 835. [Google Scholar] [CrossRef] [PubMed]
- Prunotto, A.; Stevenson, B.J.; Berthonneche, C.; Schupfer, F.; Beckmann, J.S.; Maurer, F.; Bergmann, S. RNAseq analysis of heart tissue from mice treated with atenolol and isoproterenol reveals a reciprocal transcriptional response. BMC Genom. 2016, 17, 717. [Google Scholar] [CrossRef] [PubMed]
- Freyermuth, F.; Rau, F.; Kokunai, Y.; Linke, T.; Sellier, C.; Nakamori, M.; Kino, Y.; Arandel, L.; Jollet, A.; Thibault, C.; et al. Splicing misregulation of SCN5A contributes to cardiac-conduction delay and heart arrhythmia in myotonic dystrophy. Nat. Commun. 2016, 7, 11067. [Google Scholar] [CrossRef] [PubMed]
- Johnson, E.K.; Matkovich, S.J.; Nerbonne, J.M. Regional Differences in mRNA and lncRNA Expression Profiles in Non-Failing Human Atria and Ventricles. Sci. Rep. 2018, 8, 13919. [Google Scholar] [CrossRef]
- Koopmann, T.T.; Adriaens, M.E.; Moerland, P.D.; Marsman, R.F.; Westerveld, M.L.; Lal, S.; Zhang, T.; Simmons, C.Q.; Baczko, I.; dos Remedios, C.; et al. Genome-wide identification of expression quantitative trait loci (eQTLs) in human heart. PLoS ONE 2014, 9, e97380. [Google Scholar] [CrossRef]
- Schiano, C.; Costa, V.; Aprile, M.; Grimaldi, V.; Maiello, C.; Esposito, R.; Soricelli, A.; Colantuoni, V.; Donatelli, F.; Ciccodicola, A.; et al. Heart failure: Pilot transcriptomic analysis of cardiac tissue by RNA-sequencing. Cardiol. J. 2017, 24, 539–553. [Google Scholar] [CrossRef]
- Bers, D.M.; Barry, W.H.; Despa, S. Intracellular Na+ regulation in cardiac myocytes. Cardiovasc. Res. 2003, 57, 897–912. [Google Scholar] [CrossRef]
- Verdonck, F.; Volders, P.G.; Vos, M.A.; Sipido, K.R. Intracellular Na+ and altered Na+ transport mechanisms in cardiac hypertrophy and failure. J. Mol. Cell. Cardiol. 2003, 35, 5–25. [Google Scholar] [CrossRef]
- Hilgenberg, L.G.; Pham, B.; Ortega, M.; Walid, S.; Kemmerly, T.; O’Dowd, D.K.; Smith, M.A. Agrin regulation of alpha3 sodium-potassium ATPase activity modulates cardiac myocyte contraction. J. Biol. Chem. 2009, 284, 16956–16965. [Google Scholar] [CrossRef]
- Kuwahara, K.; Saito, Y.; Takano, M.; Arai, Y.; Yasuno, S.; Nakagawa, Y.; Takahashi, N.; Adachi, Y.; Takemura, G.; Horie, M.; et al. NRSF regulates the fetal cardiac gene program and maintains normal cardiac structure and function. EMBO J. 2003, 22, 6310–6321. [Google Scholar] [CrossRef]
- Zhang, D.; Wang, Y.; Lu, P.; Wang, P.; Yuan, X.; Yan, J.; Cai, C.; Chang, C.P.; Zheng, D.; Wu, B.; et al. REST regulates the cell cycle for cardiac development and regeneration. Nat. Commun. 2017, 8, 1979. [Google Scholar] [CrossRef] [PubMed]
- Zhang, D.; Wu, B.; Wang, P.; Wang, Y.; Lu, P.; Nechiporuk, T.; Floss, T.; Greally, J.M.; Zheng, D.; Zhou, B. Non-CpG methylation by DNMT3B facilitates REST binding and gene silencing in developing mouse hearts. Nucleic. Acids. Res. 2017, 45, 3102–3115. [Google Scholar] [CrossRef] [PubMed]
- Louch, W.E.; Koivumaki, J.T.; Tavi, P. Calcium signalling in developing cardiomyocytes: Implications for model systems and disease. J. Physiol. 2015, 593, 1047–1063. [Google Scholar] [CrossRef] [PubMed]
- Lopaschuk, G.D.; Jaswal, J.S. Energy metabolic phenotype of the cardiomyocyte during development, differentiation, and postnatal maturation. J. Cardiovasc. Pharmacol. 2010, 56, 130–140. [Google Scholar] [CrossRef]
- Kolwicz, S.C., Jr. An “Exercise” in Cardiac Metabolism. Front. Cardiovasc. Med. 2018, 5, 66. [Google Scholar] [CrossRef]
- Doenst, T.; Nguyen, T.D.; Abel, E.D. Cardiac metabolism in heart failure: Implications beyond ATP production. Circ. Res. 2013, 113, 709–724. [Google Scholar] [CrossRef]
- Chong, J.A.; Tapia-Ramirez, J.; Kim, S.; Toledo-Aral, J.J.; Zheng, Y.; Boutros, M.C.; Altshuller, Y.M.; Frohman, M.A.; Kraner, S.D.; Mandel, G. REST: A mammalian silencer protein that restricts sodium channel gene expression to neurons. Cell 1995, 80, 949–957. [Google Scholar] [CrossRef]
- Chen, Z.F.; Paquette, A.J.; Anderson, D.J. NRSF/REST is required in vivo for repression of multiple neuronal target genes during embryogenesis. Nat. Genet. 1998, 20, 136–142. [Google Scholar] [CrossRef]
- Stadler, M.B.; Murr, R.; Burger, L.; Ivanek, R.; Lienert, F.; Scholer, A.; van Nimwegen, E.; Wirbelauer, C.; Oakeley, E.J.; Gaidatzis, D.; et al. DNA-binding factors shape the mouse methylome at distal regulatory regions. Nature 2011, 480, 490–495. [Google Scholar] [CrossRef]
- Vanzan, L.; Soldati, H.; Ythier, V.; Anand, S.; Braun, S.M.G.; Francis, N.; Murr, R. High throughput screening identifies SOX2 as a super pioneer factor that inhibits DNA methylation maintenance at its binding sites. Nat. Commun. 2021, 12, 3337. [Google Scholar] [CrossRef]
- Stafford, N.; Wilson, C.; Oceandy, D.; Neyses, L.; Cartwright, E.J. The Plasma Membrane Calcium ATPases and Their Role as Major New Players in Human Disease. Physiol. Rev. 2017, 97, 1089–1125. [Google Scholar] [CrossRef] [PubMed]
- De Carvalho, A.; Bassaneze, V.; Forni, M.F.; Keusseyan, A.A.; Kowaltowski, A.J.; Krieger, J.E. Early Postnatal Cardiomyocyte Proliferation Requires High Oxidative Energy Metabolism. Sci. Rep. 2017, 7, 15434. [Google Scholar] [CrossRef] [PubMed]
- Neubauer, S.; Horn, M.; Cramer, M.; Harre, K.; Newell, J.B.; Peters, W.; Pabst, T.; Ertl, G.; Hahn, D.; Ingwall, J.S.; et al. Myocardial phosphocreatine-to-ATP ratio is a predictor of mortality in patients with dilated cardiomyopathy. Circ. 1997, 96, 2190–2196. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Huypens, P.; Adamson, A.W.; Chang, J.S.; Henagan, T.M.; Boudreau, A.; Lenard, N.R.; Burk, D.; Klein, J.; Perwitz, N.; et al. Alternative mRNA splicing produces a novel biologically active short isoform of PGC-1alpha. J. Biol. Chem. 2009, 284, 32813–32826. [Google Scholar] [CrossRef]
- Chang, J.S.; Fernand, V.; Zhang, Y.; Shin, J.; Jun, H.J.; Joshi, Y.; Gettys, T.W. NT-PGC-1alpha protein is sufficient to link beta3-adrenergic receptor activation to transcriptional and physiological components of adaptive thermogenesis. J. Biol. Chem. 2012, 287, 9100–9111. [Google Scholar] [CrossRef]
- Kuwahara, K. Role of NRSF/REST in the regulation of cardiac gene expression and function. Circ. J. 2013, 77, 2682–2686. [Google Scholar] [CrossRef] [Green Version]




Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tuomainen, T.; Naumenko, N.; Mutikainen, M.; Shakirzyanova, A.; Sczelecki, S.; Estall, J.L.; Ruas, J.L.; Tavi, P. PGC-1α4 Interacts with REST to Upregulate Neuronal Genes and Augment Energy Consumption in Developing Cardiomyocytes. Cells 2022, 11, 2944. https://doi.org/10.3390/cells11192944
Tuomainen T, Naumenko N, Mutikainen M, Shakirzyanova A, Sczelecki S, Estall JL, Ruas JL, Tavi P. PGC-1α4 Interacts with REST to Upregulate Neuronal Genes and Augment Energy Consumption in Developing Cardiomyocytes. Cells. 2022; 11(19):2944. https://doi.org/10.3390/cells11192944
Chicago/Turabian StyleTuomainen, Tomi, Nikolay Naumenko, Maija Mutikainen, Anastasia Shakirzyanova, Sarah Sczelecki, Jennifer L. Estall, Jorge L. Ruas, and Pasi Tavi. 2022. "PGC-1α4 Interacts with REST to Upregulate Neuronal Genes and Augment Energy Consumption in Developing Cardiomyocytes" Cells 11, no. 19: 2944. https://doi.org/10.3390/cells11192944
APA StyleTuomainen, T., Naumenko, N., Mutikainen, M., Shakirzyanova, A., Sczelecki, S., Estall, J. L., Ruas, J. L., & Tavi, P. (2022). PGC-1α4 Interacts with REST to Upregulate Neuronal Genes and Augment Energy Consumption in Developing Cardiomyocytes. Cells, 11(19), 2944. https://doi.org/10.3390/cells11192944