

Role of the Tumor Microenvironment in Regulating Pancreatic Cancer Therapy Resistance

Abstract
:
1. Introduction
2. Therapeutic Options for PDAC Patients
3. Hallmarks of the Pancreatic Tumor Microenvironment
4. Pancreatic Stellate Cells and Therapy Resistance
5. Cancer-Associated Fibroblasts and Therapy Resistance
6. Tumor-Associated Macrophages and Therapy Resistance
7. Tumor-Associated Neutrophils and Therapy Resistance
8. Perspectives, Challenges, and Future Directions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Bengtsson, A.; Andersson, R.; Ansari, D. The actual 5-year survivors of pancreatic ductal adenocarcinoma based on real-world data. Sci. Rep. 2020, 10, 16425. [Google Scholar] [CrossRef] [PubMed]
- Mizrahi, J.D.; Surana, R.; Valle, J.W.; Shroff, R.T. Pancreatic cancer. Lancet 2020, 395, 2008–2020. [Google Scholar] [CrossRef]
- Cancer Genome Atlas Research Network. Integrated Genomic Characterization of Pancreatic Ductal Adenocarcinoma. Cancer Cell 2017, 32, 185–203.e13. [Google Scholar] [CrossRef] [PubMed]
- Ying, H.; Kimmelman, A.C.; Lyssiotis, C.A.; Hua, S.; Chu, G.C.; Fletcher-Sananikone, E.; Locasale, J.W.; Son, J.; Zhang, H.; Coloff, J.L.; et al. Oncogenic Kras maintains pancreatic tumors through regulation of anabolic glucose metabolism. Cell 2012, 149, 656–670. [Google Scholar] [CrossRef]
- Hou, P.; Wang, Y.A. Conquering oncogenic KRAS and its bypass mechanisms. Theranostics 2022, 12, 5691–5709. [Google Scholar] [CrossRef]
- Ying, H.; Dey, P.; Yao, W.; Kimmelman, A.C.; Draetta, G.F.; Maitra, A.; DePinho, R.A. Genetics and biology of pancreatic ductal adenocarcinoma. Genes Dev. 2016, 30, 355–385. [Google Scholar] [CrossRef]
- Torres, C.; Grippo, P.J. Pancreatic cancer subtypes: A roadmap for precision medicine. Ann. Med. 2018, 50, 277–287. [Google Scholar] [CrossRef]
- Wang, X.; Allen, S.; Blake, J.F.; Bowcut, V.; Briere, D.M.; Calinisan, A.; Dahlke, J.R.; Fell, J.B.; Fischer, J.P.; Gunn, R.J.; et al. Identification of MRTX1133, a Noncovalent, Potent, and Selective KRAS(G12D) Inhibitor. J. Med. Chem. 2022, 65, 3123–3133. [Google Scholar] [CrossRef]
- Truong, L.H.; Pauklin, S. Pancreatic Cancer Microenvironment and Cellular Composition: Current Understandings and Therapeutic Approaches. Cancers 2021, 13, 5028. [Google Scholar] [CrossRef]
- Opitz, F.V.; Haeberle, L.; Daum, A.; Esposito, I. Tumor Microenvironment in Pancreatic Intraepithelial Neoplasia. Cancers 2021, 13, 6188. [Google Scholar] [CrossRef]
- Hwang, R.F.; Moore, T.; Arumugam, T.; Ramachandran, V.; Amos, K.D.; Rivera, A.; Ji, B.; Evans, D.B.; Logsdon, C.D. Cancer-associated stromal fibroblasts promote pancreatic tumor progression. Cancer Res. 2008, 68, 918–926. [Google Scholar] [CrossRef] [PubMed]
- Meads, M.B.; Gatenby, R.A.; Dalton, W.S. Environment-mediated drug resistance: A major contributor to minimal residual disease. Nat. Rev. Cancer 2009, 9, 665–674. [Google Scholar] [CrossRef] [PubMed]
- Tao, J.; Yang, G.; Zhou, W.; Qiu, J.; Chen, G.; Luo, W.; Zhao, F.; You, L.; Zheng, L.; Zhang, T.; et al. Targeting hypoxic tumor microenvironment in pancreatic cancer. J. Hematol. Oncol. 2021, 14, 14. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, A.V.; Nyberg, K.D.; Scott, M.B.; Welsh, A.M.; Nguyen, A.H.; Wu, N.; Hohlbauch, S.V.; Geisse, N.A.; Gibb, E.A.; Robertson, A.G.; et al. Stiffness of pancreatic cancer cells is associated with increased invasive potential. Integr. Biol. 2016, 8, 1232–1245. [Google Scholar] [CrossRef]
- Ozdemir, B.C.; Pentcheva-Hoang, T.; Carstens, J.L.; Zheng, X.; Wu, C.C.; Simpson, T.R.; Laklai, H.; Sugimoto, H.; Kahlert, C.; Novitskiy, S.V.; et al. Depletion of carcinoma-associated fibroblasts and fibrosis induces immunosuppression and accelerates pancreas cancer with reduced survival. Cancer Cell 2014, 25, 719–734. [Google Scholar] [CrossRef]
- Rhim, A.D.; Oberstein, P.E.; Thomas, D.H.; Mirek, E.T.; Palermo, C.F.; Sastra, S.A.; Dekleva, E.N.; Saunders, T.; Becerra, C.P.; Tattersall, I.W.; et al. Stromal elements act to restrain, rather than support, pancreatic ductal adenocarcinoma. Cancer Cell 2014, 25, 735–747. [Google Scholar] [CrossRef] [PubMed]
- Liou, G.Y.; Doppler, H.; Necela, B.; Krishna, M.; Crawford, H.C.; Raimondo, M.; Storz, P. Macrophage-secreted cytokines drive pancreatic acinar-to-ductal metaplasia through NF-kappaB and MMPs. J. Cell Biol. 2013, 202, 563–577. [Google Scholar] [CrossRef]
- Zhao, F.; Obermann, S.; von Wasielewski, R.; Haile, L.; Manns, M.P.; Korangy, F.; Greten, T.F. Increase in frequency of myeloid-derived suppressor cells in mice with spontaneous pancreatic carcinoma. Immunology 2009, 128, 141–149. [Google Scholar] [CrossRef]
- Tjomsland, V.; Niklasson, L.; Sandstrom, P.; Borch, K.; Druid, H.; Bratthall, C.; Messmer, D.; Larsson, M.; Spangeus, A. The desmoplastic stroma plays an essential role in the accumulation and modulation of infiltrated immune cells in pancreatic adenocarcinoma. Clin. Dev. Immunol. 2011, 2011, 212810. [Google Scholar] [CrossRef]
- Hiraoka, N.; Onozato, K.; Kosuge, T.; Hirohashi, S. Prevalence of FOXP3+ regulatory T cells increases during the progression of pancreatic ductal adenocarcinoma and its premalignant lesions. Clin. Cancer Res. 2006, 12, 5423–5434. [Google Scholar] [CrossRef] [Green Version]
- Pylayeva-Gupta, Y.; Das, S.; Handler, J.S.; Hajdu, C.H.; Coffre, M.; Koralov, S.B.; Bar-Sagi, D. IL35-Producing B Cells Promote the Development of Pancreatic Neoplasia. Cancer Discov. 2016, 6, 247–255. [Google Scholar] [CrossRef] [PubMed]
- Jang, J.E.; Hajdu, C.H.; Liot, C.; Miller, G.; Dustin, M.L.; Bar-Sagi, D. Crosstalk between Regulatory T Cells and Tumor-Associated Dendritic Cells Negates Anti-tumor Immunity in Pancreatic Cancer. Cell Rep. 2017, 20, 558–571. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Lazarus, J.; Steele, N.G.; Yan, W.; Lee, H.J.; Nwosu, Z.C.; Halbrook, C.J.; Menjivar, R.E.; Kemp, S.B.; Sirihorachai, V.R.; et al. Regulatory T-cell Depletion Alters the Tumor Microenvironment and Accelerates Pancreatic Carcinogenesis. Cancer Discov. 2020, 10, 422–439. [Google Scholar] [CrossRef] [PubMed]
- Daley, D.; Zambirinis, C.P.; Seifert, L.; Akkad, N.; Mohan, N.; Werba, G.; Barilla, R.; Torres-Hernandez, A.; Hundeyin, M.; Mani, V.R.K.; et al. gammadelta T Cells Support Pancreatic Oncogenesis by Restraining alphabeta T Cell Activation. Cell 2016, 166, 1485–1499.e15. [Google Scholar] [CrossRef] [PubMed]
- Pylayeva-Gupta, Y.; Lee, K.E.; Hajdu, C.H.; Miller, G.; Bar-Sagi, D. Oncogenic Kras-induced GM-CSF production promotes the development of pancreatic neoplasia. Cancer Cell 2012, 21, 836–847. [Google Scholar] [CrossRef]
- Bayne, L.J.; Beatty, G.L.; Jhala, N.; Clark, C.E.; Rhim, A.D.; Stanger, B.Z.; Vonderheide, R.H. Tumor-derived granulocyte-macrophage colony-stimulating factor regulates myeloid inflammation and T cell immunity in pancreatic cancer. Cancer Cell 2012, 21, 822–835. [Google Scholar] [CrossRef]
- Mathew, E.; Brannon, A.L.; Del Vecchio, A.; Garcia, P.E.; Penny, M.K.; Kane, K.T.; Vinta, A.; Buckanovich, R.J.; di Magliano, M.P. Mesenchymal Stem Cells Promote Pancreatic Tumor Growth by Inducing Alternative Polarization of Macrophages. Neoplasia 2016, 18, 142–151. [Google Scholar] [CrossRef]
- Li, J.; Yuan, S.; Norgard, R.J.; Yan, F.; Sun, Y.H.; Kim, I.K.; Merrell, A.J.; Sela, Y.; Jiang, Y.; Bhanu, N.V.; et al. Epigenetic and Transcriptional Control of the Epidermal Growth Factor Receptor Regulates the Tumor Immune Microenvironment in Pancreatic Cancer. Cancer Discov. 2021, 11, 736–753. [Google Scholar] [CrossRef]
- Ceyhan, G.O.; Bergmann, F.; Kadihasanoglu, M.; Altintas, B.; Demir, I.E.; Hinz, U.; Muller, M.W.; Giese, T.; Buchler, M.W.; Giese, N.A.; et al. Pancreatic neuropathy and neuropathic pain--a comprehensive pathomorphological study of 546 cases. Gastroenterology 2009, 136, 177–186.e1. [Google Scholar] [CrossRef]
- Liebl, F.; Demir, I.E.; Mayer, K.; Schuster, T.; D’Haese, J.G.; Becker, K.; Langer, R.; Bergmann, F.; Wang, K.; Rosenberg, R.; et al. The impact of neural invasion severity in gastrointestinal malignancies: A clinicopathological study. Ann. Surg. 2014, 260, 900–907; discussion 907–908. [Google Scholar] [CrossRef] [Green Version]
- Bapat, A.A.; Munoz, R.M.; Von Hoff, D.D.; Han, H. Blocking Nerve Growth Factor Signaling Reduces the Neural Invasion Potential of Pancreatic Cancer Cells. PLoS ONE 2016, 11, e0165586. [Google Scholar] [CrossRef] [PubMed]
- Saloman, J.L.; Albers, K.M.; Li, D.; Hartman, D.J.; Crawford, H.C.; Muha, E.A.; Rhim, A.D.; Davis, B.M. Ablation of sensory neurons in a genetic model of pancreatic ductal adenocarcinoma slows initiation and progression of cancer. Proc. Natl. Acad. Sci. USA 2016, 113, 3078–3083. [Google Scholar] [CrossRef] [PubMed]
- Sinha, S.; Fu, Y.Y.; Grimont, A.; Ketcham, M.; Lafaro, K.; Saglimbeni, J.A.; Askan, G.; Bailey, J.M.; Melchor, J.P.; Zhong, Y.; et al. PanIN Neuroendocrine Cells Promote Tumorigenesis via Neuronal Cross-talk. Cancer Res. 2017, 77, 1868–1879. [Google Scholar] [CrossRef] [PubMed]
- Kim-Fuchs, C.; Le, C.P.; Pimentel, M.A.; Shackleford, D.; Ferrari, D.; Angst, E.; Hollande, F.; Sloan, E.K. Chronic stress accelerates pancreatic cancer growth and invasion: A critical role for beta-adrenergic signaling in the pancreatic microenvironment. Brain Behav. Immun. 2014, 40, 40–47. [Google Scholar] [CrossRef]
- Renz, B.W.; Takahashi, R.; Tanaka, T.; Macchini, M.; Hayakawa, Y.; Dantes, Z.; Maurer, H.C.; Chen, X.; Jiang, Z.; Westphalen, C.B.; et al. β2 Adrenergic-Neurotrophin Feedforward Loop Promotes Pancreatic Cancer. Cancer Cell 2018, 33, 75–90.e7. [Google Scholar] [CrossRef]
- Renz, B.W.; Tanaka, T.; Sunagawa, M.; Takahashi, R.; Jiang, Z.; Macchini, M.; Dantes, Z.; Valenti, G.; White, R.A.; Middelhoff, M.A.; et al. Cholinergic Signaling via Muscarinic Receptors Directly and Indirectly Suppresses Pancreatic Tumorigenesis and Cancer Stemness. Cancer Discov. 2018, 8, 1458–1473. [Google Scholar] [CrossRef]
- Banh, R.S.; Biancur, D.E.; Yamamoto, K.; Sohn, A.S.W.; Walters, B.; Kuljanin, M.; Gikandi, A.; Wang, H.; Mancias, J.D.; Schneider, R.J.; et al. Neurons Release Serine to Support mRNA Translation in Pancreatic Cancer. Cell 2020, 183, 1202–1218.e25. [Google Scholar] [CrossRef] [PubMed]
- Tape, C.J.; Ling, S.; Dimitriadi, M.; McMahon, K.M.; Worboys, J.D.; Leong, H.S.; Norrie, I.C.; Miller, C.J.; Poulogiannis, G.; Lauffenburger, D.A.; et al. Oncogenic KRAS Regulates Tumor Cell Signaling via Stromal Reciprocation. Cell 2016, 165, 910–920. [Google Scholar] [CrossRef]
- Bhattacharyya, S.; Oon, C.; Kothari, A.; Horton, W.; Link, J.; Sears, R.C.; Sherman, M.H. Acidic fibroblast growth factor underlies microenvironmental regulation of MYC in pancreatic cancer. J. Exp. Med. 2020, 217, e20191805. [Google Scholar] [CrossRef]
- Dey, P.; Li, J.; Zhang, J.; Chaurasiya, S.; Strom, A.; Wang, H.; Liao, W.T.; Cavallaro, F.; Denz, P.; Bernard, V.; et al. Oncogenic KRAS-Driven Metabolic Reprogramming in Pancreatic Cancer Cells Utilizes Cytokines from the Tumor Microenvironment. Cancer Discov. 2020, 10, 608–625. [Google Scholar] [CrossRef]
- De Monte, L.; Reni, M.; Tassi, E.; Clavenna, D.; Papa, I.; Recalde, H.; Braga, M.; Di Carlo, V.; Doglioni, C.; Protti, M.P. Intratumor T helper type 2 cell infiltrate correlates with cancer-associated fibroblast thymic stromal lymphopoietin production and reduced survival in pancreatic cancer. J. Exp. Med. 2011, 208, 469–478. [Google Scholar] [CrossRef] [PubMed]
- De Monte, L.; Wormann, S.; Brunetto, E.; Heltai, S.; Magliacane, G.; Reni, M.; Paganoni, A.M.; Recalde, H.; Mondino, A.; Falconi, M.; et al. Basophil Recruitment into Tumor-Draining Lymph Nodes Correlates with Th2 Inflammation and Reduced Survival in Pancreatic Cancer Patients. Cancer Res. 2016, 76, 1792–1803. [Google Scholar] [CrossRef] [PubMed]
- Li, F.; Simon, M.C. Cancer Cells Don’t Live Alone: Metabolic Communication within Tumor Microenvironments. Dev. Cell 2020, 54, 183–195. [Google Scholar] [CrossRef] [PubMed]
- Son, J.; Lyssiotis, C.A.; Ying, H.; Wang, X.; Hua, S.; Ligorio, M.; Perera, R.M.; Ferrone, C.R.; Mullarky, E.; Shyh-Chang, N.; et al. Glutamine supports pancreatic cancer growth through a KRAS-regulated metabolic pathway. Nature 2013, 496, 101–105. [Google Scholar] [CrossRef]
- Hui, S.; Ghergurovich, J.M.; Morscher, R.J.; Jang, C.; Teng, X.; Lu, W.; Esparza, L.A.; Reya, T.; Le, Z.; Yanxiang Guo, J.; et al. Glucose feeds the TCA cycle via circulating lactate. Nature 2017, 551, 115–118. [Google Scholar] [CrossRef]
- Kamphorst, J.J.; Nofal, M.; Commisso, C.; Hackett, S.R.; Lu, W.; Grabocka, E.; Vander Heiden, M.G.; Miller, G.; Drebin, J.A.; Bar-Sagi, D.; et al. Human pancreatic cancer tumors are nutrient poor and tumor cells actively scavenge extracellular protein. Cancer Res. 2015, 75, 544–553. [Google Scholar] [CrossRef]
- Commisso, C.; Davidson, S.M.; Soydaner-Azeloglu, R.G.; Parker, S.J.; Kamphorst, J.J.; Hackett, S.; Grabocka, E.; Nofal, M.; Drebin, J.A.; Thompson, C.B.; et al. Macropinocytosis of protein is an amino acid supply route in Ras-transformed cells. Nature 2013, 497, 633–637. [Google Scholar] [CrossRef]
- Pavlides, S.; Whitaker-Menezes, D.; Castello-Cros, R.; Flomenberg, N.; Witkiewicz, A.K.; Frank, P.G.; Casimiro, M.C.; Wang, C.; Fortina, P.; Addya, S.; et al. The reverse Warburg effect: Aerobic glycolysis in cancer associated fibroblasts and the tumor stroma. Cell Cycle 2009, 8, 3984–4001. [Google Scholar] [CrossRef]
- Sousa, C.M.; Biancur, D.E.; Wang, X.; Halbrook, C.J.; Sherman, M.H.; Zhang, L.; Kremer, D.; Hwang, R.F.; Witkiewicz, A.K.; Ying, H.; et al. Pancreatic stellate cells support tumour metabolism through autophagic alanine secretion. Nature 2016, 536, 479–483. [Google Scholar] [CrossRef]
- Li, Y.; Zhang, J.; Xu, J.; Liu, S. The Metabolism Symbiosis Between Pancreatic Cancer and Tumor Microenvironment. Front. Oncol. 2021, 11, 759376. [Google Scholar] [CrossRef]
- Geiger, R.; Rieckmann, J.C.; Wolf, T.; Basso, C.; Feng, Y.; Fuhrer, T.; Kogadeeva, M.; Picotti, P.; Meissner, F.; Mann, M.; et al. L-Arginine Modulates T Cell Metabolism and Enhances Survival and Anti-tumor Activity. Cell 2016, 167, 829–842.e13. [Google Scholar] [CrossRef] [PubMed]
- Shah, V.M.; Sheppard, B.C.; Sears, R.C.; Alani, A.W. Hypoxia: Friend or Foe for drug delivery in Pancreatic Cancer. Cancer Lett. 2020, 492, 63–70. [Google Scholar] [CrossRef] [PubMed]
- Daniel, S.K.; Sullivan, K.M.; Labadie, K.P.; Pillarisetty, V.G. Hypoxia as a barrier to immunotherapy in pancreatic adenocarcinoma. Clin. Transl. Med. 2019, 8, 10. [Google Scholar] [CrossRef] [PubMed]
- Erkan, M.; Adler, G.; Apte, M.V.; Bachem, M.G.; Buchholz, M.; Detlefsen, S.; Esposito, I.; Friess, H.; Gress, T.M.; Habisch, H.J.; et al. StellaTUM: Current consensus and discussion on pancreatic stellate cell research. Gut 2012, 61, 172–178. [Google Scholar] [CrossRef] [PubMed]
- Wu, Y.; Zhang, C.; Jiang, K.; Werner, J.; Bazhin, A.V.; D’Haese, J.G. The Role of Stellate Cells in Pancreatic Ductal Adenocarcinoma: Targeting Perspectives. Front. Oncol. 2020, 10, 621937. [Google Scholar] [CrossRef]
- Kuninty, P.R.; Bansal, R.; De Geus, S.W.L.; Mardhian, D.F.; Schnittert, J.; van Baarlen, J.; Storm, G.; Bijlsma, M.F.; van Laarhoven, H.W.; Metselaar, J.M.; et al. ITGA5 inhibition in pancreatic stellate cells attenuates desmoplasia and potentiates efficacy of chemotherapy in pancreatic cancer. Sci. Adv. 2019, 5, eaax2770. [Google Scholar] [CrossRef]
- Sherman, M.H.; Yu, R.T.; Engle, D.D.; Ding, N.; Atkins, A.R.; Tiriac, H.; Collisson, E.A.; Connor, F.; Van Dyke, T.; Kozlov, S.; et al. Vitamin D receptor-mediated stromal reprogramming suppresses pancreatitis and enhances pancreatic cancer therapy. Cell 2014, 159, 80–93. [Google Scholar] [CrossRef]
- Jacobetz, M.A.; Chan, D.S.; Neesse, A.; Bapiro, T.E.; Cook, N.; Frese, K.K.; Feig, C.; Nakagawa, T.; Caldwell, M.E.; Zecchini, H.I.; et al. Hyaluronan impairs vascular function and drug delivery in a mouse model of pancreatic cancer. Gut 2013, 62, 112–120. [Google Scholar] [CrossRef]
- Provenzano, P.P.; Cuevas, C.; Chang, A.E.; Goel, V.K.; Von Hoff, D.D.; Hingorani, S.R. Enzymatic targeting of the stroma ablates physical barriers to treatment of pancreatic ductal adenocarcinoma. Cancer Cell 2012, 21, 418–429. [Google Scholar] [CrossRef]
- Erkan, M.; Kleeff, J.; Gorbachevski, A.; Reiser, C.; Mitkus, T.; Esposito, I.; Giese, T.; Buchler, M.W.; Giese, N.A.; Friess, H. Periostin creates a tumor-supportive microenvironment in the pancreas by sustaining fibrogenic stellate cell activity. Gastroenterology 2007, 132, 1447–1464. [Google Scholar] [CrossRef]
- Liu, Y.; Li, F.; Gao, F.; Xing, L.; Qin, P.; Liang, X.; Zhang, J.; Qiao, X.; Lin, L.; Zhao, Q.; et al. Periostin promotes the chemotherapy resistance to gemcitabine in pancreatic cancer. Tumour. Biol. 2016, 37, 15283–15291. [Google Scholar] [CrossRef] [PubMed]
- Dangi-Garimella, S.; Krantz, S.B.; Barron, M.R.; Shields, M.A.; Heiferman, M.J.; Grippo, P.J.; Bentrem, D.J.; Munshi, H.G. Three-dimensional collagen I promotes gemcitabine resistance in pancreatic cancer through MT1-MMP-mediated expression of HMGA2. Cancer Res. 2011, 71, 1019–1028. [Google Scholar] [CrossRef] [PubMed]
- Armstrong, T.; Packham, G.; Murphy, L.B.; Bateman, A.C.; Conti, J.A.; Fine, D.R.; Johnson, C.D.; Benyon, R.C.; Iredale, J.P. Type I collagen promotes the malignant phenotype of pancreatic ductal adenocarcinoma. Clin. Cancer Res. 2004, 10, 7427–7437. [Google Scholar] [CrossRef] [PubMed]
- Amrutkar, M.; Aasrum, M.; Verbeke, C.S.; Gladhaug, I.P. Secretion of fibronectin by human pancreatic stellate cells promotes chemoresistance to gemcitabine in pancreatic cancer cells. BMC Cancer 2019, 19, 596. [Google Scholar] [CrossRef]
- Ireland, L.; Santos, A.; Ahmed, M.S.; Rainer, C.; Nielsen, S.R.; Quaranta, V.; Weyer-Czernilofsky, U.; Engle, D.D.; Perez-Mancera, P.A.; Coupland, S.E.; et al. Chemoresistance in Pancreatic Cancer Is Driven by Stroma-Derived Insulin-Like Growth Factors. Cancer Res. 2016, 76, 6851–6863. [Google Scholar] [CrossRef]
- Shi, Y.; Gao, W.; Lytle, N.K.; Huang, P.; Yuan, X.; Dann, A.M.; Ridinger-Saison, M.; DelGiorno, K.E.; Antal, C.E.; Liang, G.; et al. Targeting LIF-mediated paracrine interaction for pancreatic cancer therapy and monitoring. Nature 2019, 569, 131–135. [Google Scholar] [CrossRef]
- Xu, J.; Liu, S.; Yang, X.; Cao, S.; Zhou, Y. Paracrine HGF promotes EMT and mediates the effects of PSC on chemoresistance by activating c-Met/PI3K/Akt signaling in pancreatic cancer in vitro. Life Sci. 2020, 263, 118523. [Google Scholar] [CrossRef]
- Hesler, R.A.; Huang, J.J.; Starr, M.D.; Treboschi, V.M.; Bernanke, A.G.; Nixon, A.B.; McCall, S.J.; White, R.R.; Blobe, G.C. TGF-beta-induced stromal CYR61 promotes resistance to gemcitabine in pancreatic ductal adenocarcinoma through downregulation of the nucleoside transporters hENT1 and hCNT3. Carcinogenesis 2016, 37, 1041–1051. [Google Scholar] [CrossRef]
- Dalin, S.; Sullivan, M.R.; Lau, A.N.; Grauman-Boss, B.; Mueller, H.S.; Kreidl, E.; Fenoglio, S.; Luengo, A.; Lees, J.A.; Vander Heiden, M.G.; et al. Deoxycytidine Release from Pancreatic Stellate Cells Promotes Gemcitabine Resistance. Cancer Res. 2019, 79, 5723–5733. [Google Scholar] [CrossRef]
- Ene-Obong, A.; Clear, A.J.; Watt, J.; Wang, J.; Fatah, R.; Riches, J.C.; Marshall, J.F.; Chin-Aleong, J.; Chelala, C.; Gribben, J.G.; et al. Activated pancreatic stellate cells sequester CD8+ T cells to reduce their infiltration of the juxtatumoral compartment of pancreatic ductal adenocarcinoma. Gastroenterology 2013, 145, 1121–1132. [Google Scholar] [CrossRef] [Green Version]
- Tang, D.; Yuan, Z.; Xue, X.; Lu, Z.; Zhang, Y.; Wang, H.; Chen, M.; An, Y.; Wei, J.; Zhu, Y.; et al. High expression of Galectin-1 in pancreatic stellate cells plays a role in the development and maintenance of an immunosuppressive microenvironment in pancreatic cancer. Int. J. Cancer 2012, 130, 2337–2348. [Google Scholar] [CrossRef] [PubMed]
- Mace, T.A.; Ameen, Z.; Collins, A.; Wojcik, S.; Mair, M.; Young, G.S.; Fuchs, J.R.; Eubank, T.D.; Frankel, W.L.; Bekaii-Saab, T.; et al. Pancreatic cancer-associated stellate cells promote differentiation of myeloid-derived suppressor cells in a STAT3-dependent manner. Cancer Res. 2013, 73, 3007–3018. [Google Scholar] [CrossRef] [PubMed]
- Chronopoulos, A.; Robinson, B.; Sarper, M.; Cortes, E.; Auernheimer, V.; Lachowski, D.; Attwood, S.; Garcia, R.; Ghassemi, S.; Fabry, B.; et al. ATRA mechanically reprograms pancreatic stellate cells to suppress matrix remodelling and inhibit cancer cell invasion. Nat. Commun. 2016, 7, 12630. [Google Scholar] [CrossRef] [PubMed]
- Froeling, F.E.; Feig, C.; Chelala, C.; Dobson, R.; Mein, C.E.; Tuveson, D.A.; Clevers, H.; Hart, I.R.; Kocher, H.M. Retinoic acid-induced pancreatic stellate cell quiescence reduces paracrine Wnt-beta-catenin signaling to slow tumor progression. Gastroenterology 2011, 141, 1486–1497.e14. [Google Scholar] [CrossRef]
- Hessmann, E.; Patzak, M.S.; Klein, L.; Chen, N.; Kari, V.; Ramu, I.; Bapiro, T.E.; Frese, K.K.; Gopinathan, A.; Richards, F.M.; et al. Fibroblast drug scavenging increases intratumoural gemcitabine accumulation in murine pancreas cancer. Gut 2018, 67, 497–507. [Google Scholar] [CrossRef]
- Richards, K.E.; Zeleniak, A.E.; Fishel, M.L.; Wu, J.; Littlepage, L.E.; Hill, R. Cancer-associated fibroblast exosomes regulate survival and proliferation of pancreatic cancer cells. Oncogene 2017, 36, 1770–1778. [Google Scholar] [CrossRef]
- Hu, C.; Xia, R.; Zhang, X.; Li, T.; Ye, Y.; Li, G.; He, R.; Li, Z.; Lin, Q.; Zheng, S.; et al. circFARP1 enables cancer-associated fibroblasts to promote gemcitabine resistance in pancreatic cancer via the LIF/STAT3 axis. Mol. Cancer 2022, 21, 24. [Google Scholar] [CrossRef]
- Wei, L.; Lin, Q.; Lu, Y.; Li, G.; Huang, L.; Fu, Z.; Chen, R.; Zhou, Q. Cancer-associated fibroblasts-mediated ATF4 expression promotes malignancy and gemcitabine resistance in pancreatic cancer via the TGF-beta1/SMAD2/3 pathway and ABCC1 transactivation. Cell Death Dis. 2021, 12, 334. [Google Scholar] [CrossRef]
- Wei, L.; Ye, H.; Li, G.; Lu, Y.; Zhou, Q.; Zheng, S.; Lin, Q.; Liu, Y.; Li, Z.; Chen, R. Cancer-associated fibroblasts promote progression and gemcitabine resistance via the SDF-1/SATB-1 pathway in pancreatic cancer. Cell Death Dis. 2018, 9, 1065. [Google Scholar] [CrossRef]
- Long, K.B.; Tooker, G.; Tooker, E.; Luque, S.L.; Lee, J.W.; Pan, X.; Beatty, G.L. IL6 Receptor Blockade Enhances Chemotherapy Efficacy in Pancreatic Ductal Adenocarcinoma. Mol. Cancer Ther. 2017, 16, 1898–1908. [Google Scholar] [CrossRef]
- Singh, S.; Srivastava, S.K.; Bhardwaj, A.; Owen, L.B.; Singh, A.P. CXCL12-CXCR4 signalling axis confers gemcitabine resistance to pancreatic cancer cells: A novel target for therapy. Br. J. Cancer 2010, 103, 1671–1679. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Zheng, S.; Hu, C.; Li, G.; Lin, H.; Xia, R.; Ye, Y.; He, R.; Li, Z.; Lin, Q.; et al. Cancer-associated fibroblast-induced lncRNA UPK1A-AS1 confers platinum resistance in pancreatic cancer via efficient double-strand break repair. Oncogene 2022, 41, 2372–2389. [Google Scholar] [CrossRef] [PubMed]
- Muerkoster, S.; Wegehenkel, K.; Arlt, A.; Witt, M.; Sipos, B.; Kruse, M.L.; Sebens, T.; Kloppel, G.; Kalthoff, H.; Folsch, U.R.; et al. Tumor stroma interactions induce chemoresistance in pancreatic ductal carcinoma cells involving increased secretion and paracrine effects of nitric oxide and interleukin-1beta. Cancer Res. 2004, 64, 1331–1337. [Google Scholar] [CrossRef] [PubMed]
- Liles, J.S.; Arnoletti, J.P.; Kossenkov, A.V.; Mikhaylina, A.; Frost, A.R.; Kulesza, P.; Heslin, M.J.; Frolov, A. Targeting ErbB3-mediated stromal-epithelial interactions in pancreatic ductal adenocarcinoma. Br. J. Cancer 2011, 105, 523–533. [Google Scholar] [CrossRef] [PubMed]
- Ogier, C.; Colombo, P.E.; Bousquet, C.; Canterel-Thouennon, L.; Sicard, P.; Garambois, V.; Thomas, G.; Gaborit, N.; Jarlier, M.; Pirot, N.; et al. Targeting the NRG1/HER3 pathway in tumor cells and cancer-associated fibroblasts with an anti-neuregulin 1 antibody inhibits tumor growth in pre-clinical models of pancreatic cancer. Cancer Lett. 2018, 432, 227–236. [Google Scholar] [CrossRef]
- Hartmann, N.; Giese, N.A.; Giese, T.; Poschke, I.; Offringa, R.; Werner, J.; Ryschich, E. Prevailing role of contact guidance in intrastromal T-cell trapping in human pancreatic cancer. Clin. Cancer Res. 2014, 20, 3422–3433. [Google Scholar] [CrossRef]
- Zhang, A.; Qian, Y.; Ye, Z.; Chen, H.; Xie, H.; Zhou, L.; Shen, Y.; Zheng, S. Cancer-associated fibroblasts promote M2 polarization of macrophages in pancreatic ductal adenocarcinoma. Cancer Med. 2017, 6, 463–470. [Google Scholar] [CrossRef]
- Kraman, M.; Bambrough, P.J.; Arnold, J.N.; Roberts, E.W.; Magiera, L.; Jones, J.O.; Gopinathan, A.; Tuveson, D.A.; Fearon, D.T. Suppression of antitumor immunity by stromal cells expressing fibroblast activation protein-alpha. Science 2010, 330, 827–830. [Google Scholar] [CrossRef]
- Feig, C.; Jones, J.O.; Kraman, M.; Wells, R.J.; Deonarine, A.; Chan, D.S.; Connell, C.M.; Roberts, E.W.; Zhao, Q.; Caballero, O.L.; et al. Targeting CXCL12 from FAP-expressing carcinoma-associated fibroblasts synergizes with anti-PD-L1 immunotherapy in pancreatic cancer. Proc. Natl. Acad. Sci. USA 2013, 110, 20212–20217. [Google Scholar] [CrossRef]
- Gorchs, L.; Fernandez Moro, C.; Bankhead, P.; Kern, K.P.; Sadeak, I.; Meng, Q.; Rangelova, E.; Kaipe, H. Human Pancreatic Carcinoma-Associated Fibroblasts Promote Expression of Co-inhibitory Markers on CD4(+) and CD8(+) T-Cells. Front. Immunol. 2019, 10, 847. [Google Scholar] [CrossRef]
- Kerk, S.A.; Lin, L.; Myers, A.L.; Sutton, D.J.; Andren, A.; Sajjakulnukit, P.; Zhang, L.; Zhang, Y.; Jimenez, J.A.; Nelson, B.S.; et al. Metabolic requirement for GOT2 in pancreatic cancer depends on environmental context. Elife 2022, 11, e73245. [Google Scholar] [CrossRef] [PubMed]
- Datta, R.; Sivanand, S.; Lau, A.N.; Florek, L.V.; Barbeau, A.M.; Wyckoff, J.; Skala, M.C.; Vander Heiden, M.G. Interactions with stromal cells promote a more oxidized cancer cell redox state in pancreatic tumors. Sci. Adv. 2022, 8, eabg6383. [Google Scholar] [CrossRef] [PubMed]
- Spek, C.A.; Aberson, H.L.; Duitman, J. Macrophage C/EBPdelta Drives Gemcitabine, but Not 5-FU or Paclitaxel, Resistance of Pancreatic Cancer Cells in a Deoxycytidine-Dependent Manner. Biomedicines 2022, 10, 219. [Google Scholar] [CrossRef] [PubMed]
- Weizman, N.; Krelin, Y.; Shabtay-Orbach, A.; Amit, M.; Binenbaum, Y.; Wong, R.J.; Gil, Z. Macrophages mediate gemcitabine resistance of pancreatic adenocarcinoma by upregulating cytidine deaminase. Oncogene 2014, 33, 3812–3819. [Google Scholar] [CrossRef]
- Hou, P.; Kapoor, A.; Zhang, Q.; Li, J.; Wu, C.J.; Li, J.; Lan, Z.; Tang, M.; Ma, X.; Ackroyd, J.J.; et al. Tumor Microenvironment Remodeling Enables Bypass of Oncogenic KRAS Dependency in Pancreatic Cancer. Cancer Discov. 2020, 10, 1058–1077. [Google Scholar] [CrossRef]
- Quaranta, V.; Rainer, C.; Nielsen, S.R.; Raymant, M.L.; Ahmed, M.S.; Engle, D.D.; Taylor, A.; Murray, T.; Campbell, F.; Palmer, D.H.; et al. Macrophage-Derived Granulin Drives Resistance to Immune Checkpoint Inhibition in Metastatic Pancreatic Cancer. Cancer Res. 2018, 78, 4253–4269. [Google Scholar] [CrossRef]
- Seifert, L.; Werba, G.; Tiwari, S.; Giao Ly, N.N.; Alothman, S.; Alqunaibit, D.; Avanzi, A.; Barilla, R.; Daley, D.; Greco, S.H.; et al. The necrosome promotes pancreatic oncogenesis via CXCL1 and Mincle-induced immune suppression. Nature 2016, 532, 245–249. [Google Scholar] [CrossRef]
- Wang, W.; Marinis, J.M.; Beal, A.M.; Savadkar, S.; Wu, Y.; Khan, M.; Taunk, P.S.; Wu, N.; Su, W.; Wu, J.; et al. RIP1 Kinase Drives Macrophage-Mediated Adaptive Immune Tolerance in Pancreatic Cancer. Cancer Cell 2018, 34, 757–774.e7. [Google Scholar] [CrossRef]
- Kalbasi, A.; Komar, C.; Tooker, G.M.; Liu, M.; Lee, J.W.; Gladney, W.L.; Ben-Josef, E.; Beatty, G.L. Tumor-Derived CCL2 Mediates Resistance to Radiotherapy in Pancreatic Ductal Adenocarcinoma. Clin. Cancer Res. 2017, 23, 137–148. [Google Scholar] [CrossRef]
- Beatty, G.L.; Winograd, R.; Evans, R.A.; Long, K.B.; Luque, S.L.; Lee, J.W.; Clendenin, C.; Gladney, W.L.; Knoblock, D.M.; Guirnalda, P.D.; et al. Exclusion of T Cells From Pancreatic Carcinomas in Mice Is Regulated by Ly6C(low) F4/80(+) Extratumoral Macrophages. Gastroenterology 2015, 149, 201–210. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Y.; Chandra, V.; Riquelme Sanchez, E.; Dutta, P.; Quesada, P.R.; Rakoski, A.; Zoltan, M.; Arora, N.; Baydogan, S.; Horne, W.; et al. Interleukin-17-induced neutrophil extracellular traps mediate resistance to checkpoint blockade in pancreatic cancer. J. Exp. Med. 2020, 217, e20190354. [Google Scholar] [CrossRef] [PubMed]
- Nywening, T.M.; Belt, B.A.; Cullinan, D.R.; Panni, R.Z.; Han, B.J.; Sanford, D.E.; Jacobs, R.C.; Ye, J.; Patel, A.A.; Gillanders, W.E.; et al. Targeting both tumour-associated CXCR2(+) neutrophils and CCR2(+) macrophages disrupts myeloid recruitment and improves chemotherapeutic responses in pancreatic ductal adenocarcinoma. Gut 2018, 67, 1112–1123. [Google Scholar] [CrossRef] [PubMed]
- Si, Y.; Merz, S.F.; Jansen, P.; Wang, B.; Bruderek, K.; Altenhoff, P.; Mattheis, S.; Lang, S.; Gunzer, M.; Klode, J.; et al. Multidimensional imaging provides evidence for down-regulation of T cell effector function by MDSC in human cancer tissue. Sci. Immunol. 2019, 4, eaaw9159. [Google Scholar] [CrossRef] [PubMed]
- Wisdom, A.J.; Hong, C.S.; Lin, A.J.; Xiang, Y.; Cooper, D.E.; Zhang, J.; Xu, E.S.; Kuo, H.C.; Mowery, Y.M.; Carpenter, D.J.; et al. Neutrophils promote tumor resistance to radiation therapy. Proc. Natl. Acad. Sci. USA 2019, 116, 18584–18589. [Google Scholar] [CrossRef] [PubMed]
- Siolas, D.; Vucic, E.; Kurz, E.; Hajdu, C.; Bar-Sagi, D. Gain-of-function p53(R172H) mutation drives accumulation of neutrophils in pancreatic tumors, promoting resistance to immunotherapy. Cell Rep. 2021, 36, 109578. [Google Scholar] [CrossRef]
- Li, J.; Byrne, K.T.; Yan, F.; Yamazoe, T.; Chen, Z.; Baslan, T.; Richman, L.P.; Lin, J.H.; Sun, Y.H.; Rech, A.J.; et al. Tumor Cell-Intrinsic Factors Underlie Heterogeneity of Immune Cell Infiltration and Response to Immunotherapy. Immunity 2018, 49, 178–193.e7. [Google Scholar] [CrossRef]
- Steele, C.W.; Karim, S.A.; Leach, J.D.G.; Bailey, P.; Upstill-Goddard, R.; Rishi, L.; Foth, M.; Bryson, S.; McDaid, K.; Wilson, Z.; et al. CXCR2 Inhibition Profoundly Suppresses Metastases and Augments Immunotherapy in Pancreatic Ductal Adenocarcinoma. Cancer Cell 2016, 29, 832–845. [Google Scholar] [CrossRef]
- Vaish, U.; Jain, T.; Are, A.C.; Dudeja, V. Cancer-Associated Fibroblasts in Pancreatic Ductal Adenocarcinoma: An Update on Heterogeneity and Therapeutic Targeting. Int. J. Mol. Sci. 2021, 22, 13408. [Google Scholar] [CrossRef]
- Ohlund, D.; Handly-Santana, A.; Biffi, G.; Elyada, E.; Almeida, A.S.; Ponz-Sarvise, M.; Corbo, V.; Oni, T.E.; Hearn, S.A.; Lee, E.J.; et al. Distinct populations of inflammatory fibroblasts and myofibroblasts in pancreatic cancer. J. Exp. Med. 2017, 214, 579–596. [Google Scholar] [CrossRef]
- Elyada, E.; Bolisetty, M.; Laise, P.; Flynn, W.F.; Courtois, E.T.; Burkhart, R.A.; Teinor, J.A.; Belleau, P.; Biffi, G.; Lucito, M.S.; et al. Cross-Species Single-Cell Analysis of Pancreatic Ductal Adenocarcinoma Reveals Antigen-Presenting Cancer-Associated Fibroblasts. Cancer Discov. 2019, 9, 1102–1123. [Google Scholar] [CrossRef] [Green Version]
- Hutton, C.; Heider, F.; Blanco-Gomez, A.; Banyard, A.; Kononov, A.; Zhang, X.; Karim, S.; Paulus-Hock, V.; Watt, D.; Steele, N.; et al. Single-cell analysis defines a pancreatic fibroblast lineage that supports anti-tumor immunity. Cancer Cell 2021, 39, 1227–1244.e20. [Google Scholar] [CrossRef] [PubMed]
- Huang, H.; Wang, Z.; Zhang, Y.; Pradhan, R.N.; Ganguly, D.; Chandra, R.; Murimwa, G.; Wright, S.; Gu, X.; Maddipati, R.; et al. Mesothelial cell-derived antigen-presenting cancer-associated fibroblasts induce expansion of regulatory T cells in pancreatic cancer. Cancer Cell 2022, 40, 656–673.e7. [Google Scholar] [CrossRef]
- Yokoi, K.; Fidler, I.J. Hypoxia increases resistance of human pancreatic cancer cells to apoptosis induced by gemcitabine. Clin. Cancer Res. 2004, 10, 2299–2306. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.; Han, H.; Rong, Y.; Zhu, K.; Zhu, Z.; Tang, Z.; Xiong, C.; Tao, J. Hypoxia potentiates gemcitabine-induced stemness in pancreatic cancer cells through AKT/Notch1 signaling. J. Exp. Clin. Cancer Res. 2018, 37, 291. [Google Scholar] [CrossRef] [PubMed]
- He, X.; Wang, J.; Wei, W.; Shi, M.; Xin, B.; Zhang, T.; Shen, X. Hypoxia regulates ABCG2 activity through the activivation of ERK1/2/HIF-1alpha and contributes to chemoresistance in pancreatic cancer cells. Cancer Biol. Ther. 2016, 17, 188–198. [Google Scholar] [CrossRef]
- Yoo, H.C.; Park, S.J.; Nam, M.; Kang, J.; Kim, K.; Yeo, J.H.; Kim, J.K.; Heo, Y.; Lee, H.S.; Lee, M.Y.; et al. A Variant of SLC1A5 Is a Mitochondrial Glutamine Transporter for Metabolic Reprogramming in Cancer Cells. Cell Metab. 2020, 31, 267–283.e12. [Google Scholar] [CrossRef]
- Garcia Garcia, C.J.; Huang, Y.; Fuentes, N.R.; Turner, M.C.; Monberg, M.E.; Lin, D.; Nguyen, N.D.; Fujimoto, T.N.; Zhao, J.; Lee, J.J.; et al. Stromal HIF2 Regulates Immune Suppression in the Pancreatic Cancer Microenvironment. Gastroenterology 2022, 162, 2018–2031. [Google Scholar] [CrossRef]
- Domen, A.; Quatannens, D.; Zanivan, S.; Deben, C.; Van Audenaerde, J.; Smits, E.; Wouters, A.; Lardon, F.; Roeyen, G.; Verhoeven, Y.; et al. Cancer-Associated Fibroblasts as a Common Orchestrator of Therapy Resistance in Lung and Pancreatic Cancer. Cancers 2021, 13, 987. [Google Scholar] [CrossRef]
- Han, X.; Zhang, W.H.; Wang, W.Q.; Yu, X.J.; Liu, L. Cancer-associated fibroblasts in therapeutic resistance of pancreatic cancer: Present situation, predicaments, and perspectives. Biochim. Biophys. Acta Rev. Cancer 2020, 1874, 188444. [Google Scholar] [CrossRef]
- Beatty, G.L.; Werba, G.; Lyssiotis, C.A.; Simeone, D.M. The biological underpinnings of therapeutic resistance in pancreatic cancer. Genes Dev. 2021, 35, 940–962. [Google Scholar] [CrossRef]
- Geng, X.; Chen, H.; Zhao, L.; Hu, J.; Yang, W.; Li, G.; Cheng, C.; Zhao, Z.; Zhang, T.; Li, L.; et al. Cancer-Associated Fibroblast (CAF) Heterogeneity and Targeting Therapy of CAFs in Pancreatic Cancer. Front. Cell Dev. Biol. 2021, 9, 655152. [Google Scholar] [CrossRef] [PubMed]
- Fabre, M.; Ferrer, C.; Dominguez-Hormaetxe, S.; Bockorny, B.; Murias, L.; Seifert, O.; Eisler, S.A.; Kontermann, R.E.; Pfizenmaier, K.; Lee, S.Y.; et al. OMTX705, a Novel FAP-Targeting ADC Demonstrates Activity in Chemotherapy and Pembrolizumab-Resistant Solid Tumor Models. Clin. Cancer Res. 2020, 26, 3420–3430. [Google Scholar] [CrossRef]
- Wang, L.C.; Lo, A.; Scholler, J.; Sun, J.; Majumdar, R.S.; Kapoor, V.; Antzis, M.; Cotner, C.E.; Johnson, L.A.; Durham, A.C.; et al. Targeting fibroblast activation protein in tumor stroma with chimeric antigen receptor T cells can inhibit tumor growth and augment host immunity without severe toxicity. Cancer Immunol. Res. 2014, 2, 154–166. [Google Scholar] [CrossRef] [PubMed]
- Cox, N.; Pokrovskii, M.; Vicario, R.; Geissmann, F. Origins, Biology, and Diseases of Tissue Macrophages. Annu. Rev. Immunol. 2021, 39, 313–344. [Google Scholar] [CrossRef] [PubMed]
- Yu, M.; Guan, R.; Hong, W.; Zhou, Y.; Lin, Y.; Jin, H.; Hou, B.; Jian, Z. Prognostic value of tumor-associated macrophages in pancreatic cancer: A meta-analysis. Cancer Manag. Res. 2019, 11, 4041–4058. [Google Scholar] [CrossRef]
- Yang, S.; Liu, Q.; Liao, Q. Tumor-Associated Macrophages in Pancreatic Ductal Adenocarcinoma: Origin, Polarization, Function, and Reprogramming. Front. Cell Dev. Biol. 2020, 8, 607209. [Google Scholar] [CrossRef]
- Atanasov, G.; Potner, C.; Aust, G.; Schierle, K.; Dietel, C.; Benzing, C.; Krenzien, F.; Bartels, M.; Eichfeld, U.; Schmelzle, M.; et al. TIE2-expressing monocytes and M2-polarized macrophages impact survival and correlate with angiogenesis in adenocarcinoma of the pancreas. Oncotarget 2018, 9, 29715–29726. [Google Scholar] [CrossRef]
- Zhu, Y.; Herndon, J.M.; Sojka, D.K.; Kim, K.W.; Knolhoff, B.L.; Zuo, C.; Cullinan, D.R.; Luo, J.; Bearden, A.R.; Lavine, K.J.; et al. Tissue-Resident Macrophages in Pancreatic Ductal Adenocarcinoma Originate from Embryonic Hematopoiesis and Promote Tumor Progression. Immunity 2017, 47, 323–338.e6. [Google Scholar] [CrossRef]
- Ruffell, B.; Coussens, L.M. Macrophages and therapeutic resistance in cancer. Cancer Cell 2015, 27, 462–472. [Google Scholar] [CrossRef]
- Mitchem, J.B.; Brennan, D.J.; Knolhoff, B.L.; Belt, B.A.; Zhu, Y.; Sanford, D.E.; Belaygorod, L.; Carpenter, D.; Collins, L.; Piwnica-Worms, D.; et al. Targeting tumor-infiltrating macrophages decreases tumor-initiating cells, relieves immunosuppression, and improves chemotherapeutic responses. Cancer Res. 2013, 73, 1128–1141. [Google Scholar] [CrossRef] [Green Version]
- Larionova, I.; Cherdyntseva, N.; Liu, T.; Patysheva, M.; Rakina, M.; Kzhyshkowska, J. Interaction of tumor-associated macrophages and cancer chemotherapy. Oncoimmunology 2019, 8, 1596004. [Google Scholar] [CrossRef]
- Hughes, R.; Qian, B.Z.; Rowan, C.; Muthana, M.; Keklikoglou, I.; Olson, O.C.; Tazzyman, S.; Danson, S.; Addison, C.; Clemons, M.; et al. Perivascular M2 Macrophages Stimulate Tumor Relapse after Chemotherapy. Cancer Res. 2015, 75, 3479–3491. [Google Scholar] [CrossRef] [PubMed]
- Halbrook, C.J.; Pontious, C.; Kovalenko, I.; Lapienyte, L.; Dreyer, S.; Lee, H.J.; Thurston, G.; Zhang, Y.; Lazarus, J.; Sajjakulnukit, P.; et al. Macrophage-Released Pyrimidines Inhibit Gemcitabine Therapy in Pancreatic Cancer. Cell Metab. 2019, 29, 1390–1399.e6. [Google Scholar] [CrossRef] [PubMed]
- Kapoor, A.; Yao, W.; Ying, H.; Hua, S.; Liewen, A.; Wang, Q.; Zhong, Y.; Wu, C.J.; Sadanandam, A.; Hu, B.; et al. Yap1 activation enables bypass of oncogenic Kras addiction in pancreatic cancer. Cell 2014, 158, 185–197. [Google Scholar] [CrossRef] [PubMed]
- Shao, D.D.; Xue, W.; Krall, E.B.; Bhutkar, A.; Piccioni, F.; Wang, X.; Schinzel, A.C.; Sood, S.; Rosenbluh, J.; Kim, J.W.; et al. KRAS and YAP1 converge to regulate EMT and tumor survival. Cell 2014, 158, 171–184. [Google Scholar] [CrossRef]
- Adachi, Y.; Ito, K.; Hayashi, Y.; Kimura, R.; Tan, T.Z.; Yamaguchi, R.; Ebi, H. Epithelial-to-Mesenchymal Transition is a Cause of Both Intrinsic and Acquired Resistance to KRAS G12C Inhibitor in KRAS G12C-Mutant Non-Small Cell Lung Cancer. Clin. Cancer Res. 2020, 26, 5962–5973. [Google Scholar] [CrossRef] [PubMed]
- Tekin, C.; Aberson, H.L.; Waasdorp, C.; Hooijer, G.K.J.; de Boer, O.J.; Dijk, F.; Bijlsma, M.F.; Spek, C.A. Macrophage-secreted MMP9 induces mesenchymal transition in pancreatic cancer cells via PAR1 activation. Cell Oncol. 2020, 43, 1161–1174. [Google Scholar] [CrossRef]
- Zuo, C.; Baer, J.M.; Knolhoff, B.L.; Belle, J.I.; Liu, X.; Hogg, G.D.; Fu, C.; Kingston, N.L.; Brenden, M.A.; De La Lastra, A.A.; et al. Macrophage proliferation machinery leads to PDAC progression, but susceptibility to innate immunotherapy. bioRxiv 2021. [Google Scholar] [CrossRef]
- Uribe-Querol, E.; Rosales, C. Neutrophils in Cancer: Two Sides of the Same Coin. J. Immunol. Res. 2015, 2015, 983698. [Google Scholar] [CrossRef]
- McFarlane, A.J.; Fercoq, F.; Coffelt, S.B.; Carlin, L.M. Neutrophil dynamics in the tumor microenvironment. J. Clin. Investig. 2021, 131, e143759. [Google Scholar] [CrossRef]
- Jin, L.; Kim, H.S.; Shi, J. Neutrophil in the Pancreatic Tumor Microenvironment. Biomolecules 2021, 11, 1170. [Google Scholar] [CrossRef] [PubMed]
- Faget, J.; Peters, S.; Quantin, X.; Meylan, E.; Bonnefoy, N. Neutrophils in the era of immune checkpoint blockade. J. Immunother. Cancer 2021, 9, e002242. [Google Scholar] [CrossRef] [PubMed]
- Jaillon, S.; Ponzetta, A.; Di Mitri, D.; Santoni, A.; Bonecchi, R.; Mantovani, A. Neutrophil diversity and plasticity in tumour progression and therapy. Nat. Rev. Cancer 2020, 20, 485–503. [Google Scholar] [CrossRef] [PubMed]
- Fridlender, Z.G.; Sun, J.; Kim, S.; Kapoor, V.; Cheng, G.; Ling, L.; Worthen, G.S.; Albelda, S.M. Polarization of tumor-associated neutrophil phenotype by TGF-beta: “N1” versus “N2” TAN. Cancer Cell 2009, 16, 183–194. [Google Scholar] [CrossRef] [PubMed]
- Andzinski, L.; Kasnitz, N.; Stahnke, S.; Wu, C.F.; Gereke, M.; von Kockritz-Blickwede, M.; Schilling, B.; Brandau, S.; Weiss, S.; Jablonska, J. Type I IFNs induce anti-tumor polarization of tumor associated neutrophils in mice and human. Int. J. Cancer 2016, 138, 1982–1993. [Google Scholar] [CrossRef]
- Singhal, S.; Bhojnagarwala, P.S.; O’Brien, S.; Moon, E.K.; Garfall, A.L.; Rao, A.S.; Quatromoni, J.G.; Stephen, T.L.; Litzky, L.; Deshpande, C.; et al. Origin and Role of a Subset of Tumor-Associated Neutrophils with Antigen-Presenting Cell Features in Early-Stage Human Lung Cancer. Cancer Cell 2016, 30, 120–135. [Google Scholar] [CrossRef]
- Evrard, M.; Kwok, I.W.H.; Chong, S.Z.; Teng, K.W.W.; Becht, E.; Chen, J.; Sieow, J.L.; Penny, H.L.; Ching, G.C.; Devi, S.; et al. Developmental Analysis of Bone Marrow Neutrophils Reveals Populations Specialized in Expansion, Trafficking, and Effector Functions. Immunity 2018, 48, 364–379.e8. [Google Scholar] [CrossRef]
- Engblom, C.; Pfirschke, C.; Zilionis, R.; Da Silva Martins, J.; Bos, S.A.; Courties, G.; Rickelt, S.; Severe, N.; Baryawno, N.; Faget, J.; et al. Osteoblasts remotely supply lung tumors with cancer-promoting SiglecF(high) neutrophils. Science 2017, 358, eaal5081. [Google Scholar] [CrossRef]
- Wang, W.Q.; Liu, L.; Xu, H.X.; Wu, C.T.; Xiang, J.F.; Xu, J.; Liu, C.; Long, J.; Ni, Q.X.; Yu, X.J. Infiltrating immune cells and gene mutations in pancreatic ductal adenocarcinoma. Br. J. Surg. 2016, 103, 1189–1199. [Google Scholar] [CrossRef]
- Nielsen, S.R.; Strobech, J.E.; Horton, E.R.; Jackstadt, R.; Laitala, A.; Bravo, M.C.; Maltese, G.; Jensen, A.R.D.; Reuten, R.; Rafaeva, M.; et al. Suppression of tumor-associated neutrophils by lorlatinib attenuates pancreatic cancer growth and improves treatment with immune checkpoint blockade. Nat. Commun. 2021, 12, 3414. [Google Scholar] [CrossRef]
- Liao, W.; Overman, M.J.; Boutin, A.T.; Shang, X.; Zhao, D.; Dey, P.; Li, J.; Wang, G.; Lan, Z.; Li, J.; et al. KRAS-IRF2 Axis Drives Immune Suppression and Immune Therapy Resistance in Colorectal Cancer. Cancer Cell 2019, 35, 559–572.e7. [Google Scholar] [CrossRef] [PubMed]
- Lu, X.; Horner, J.W.; Paul, E.; Shang, X.; Troncoso, P.; Deng, P.; Jiang, S.; Chang, Q.; Spring, D.J.; Sharma, P.; et al. Effective combinatorial immunotherapy for castration-resistant prostate cancer. Nature 2017, 543, 728–732. [Google Scholar] [CrossRef] [PubMed]
- Patnaik, A.; Eisenberg, P.D.; Sachdev, J.C.; Weise, A.M.; Tse, A.N.; Hutchinson, M.; Aromin, I.; West, B.; Tong, S.; Ribas, A.; et al. Phase 1/2a study of double immune suppression blockade by combining a CSF1R inhibitor (Pexidartinib/PLX3397) with an anti PD-1 antibody (Pembrolizumab) to treat advanced, solid tumors. J. Clin. Oncol. 2016, 34, TPS11618-TPS. [Google Scholar] [CrossRef]
- Wang-Gillam, A.; O’Reilly, E.M.; Bendell, J.C.; Wainberg, Z.A.; Borazanci, E.H.; Bahary, N.; O’Hara, M.H.; Beatty, G.L.; Pant, S.; Cohen, D.J.; et al. A randomized phase II study of cabiralizumab (cabira) + nivolumab (nivo) ± chemotherapy (chemo) in advanced pancreatic ductal adenocarcinoma (PDAC). J. Clin. Oncol. 2019, 37, TPS465-TPS. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
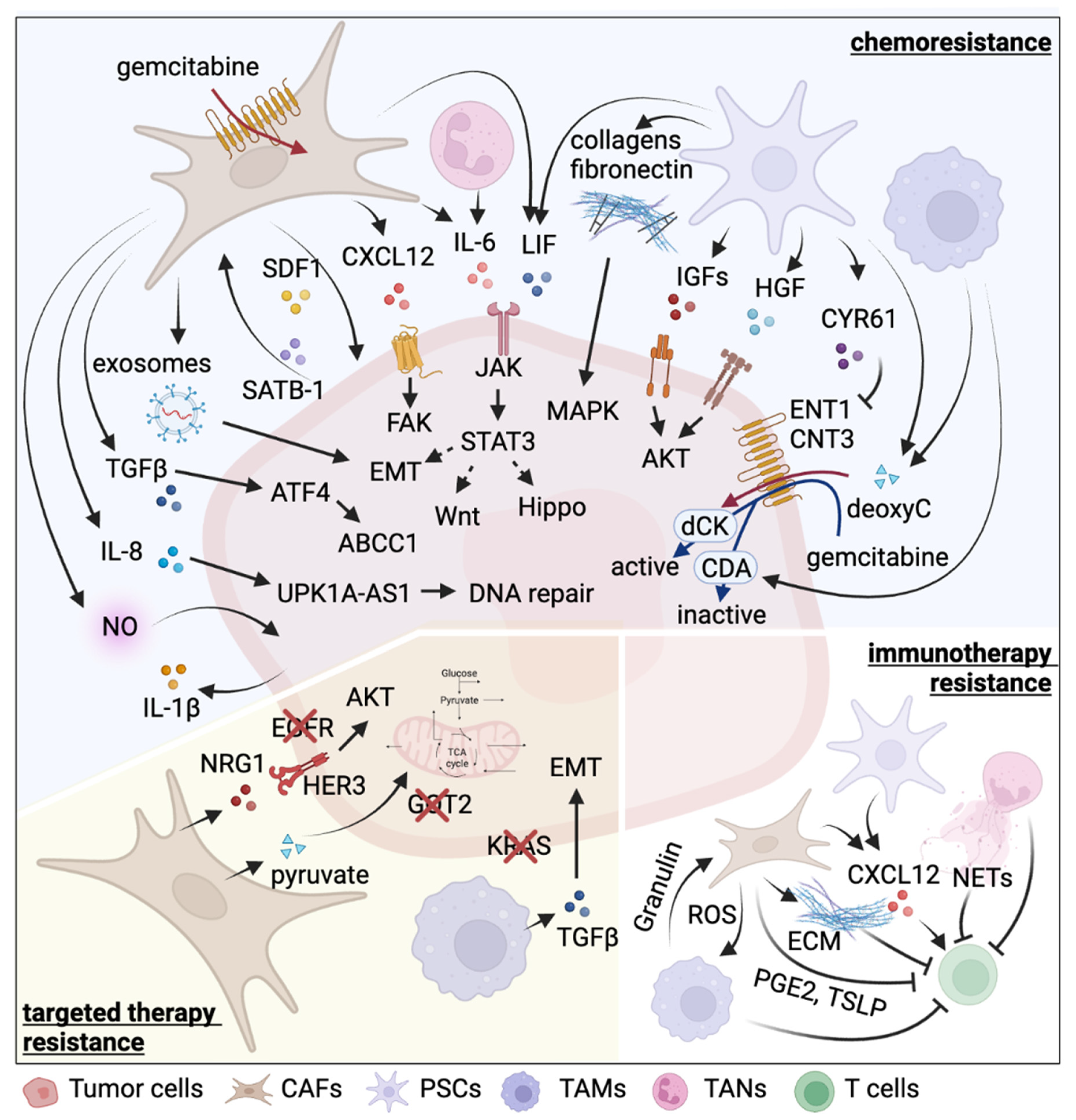
| Cell Type | Therapy | Resistance Inducer | Detailed Mechanism | Reference |
|---|---|---|---|---|
| PSCs | chemotherapy (gemcitabine) | Collagen I | Promote proliferation by MAPK pathway activation and chromatin remodeling | [62,63] |
| Periostin | Induce ECM molecules, including collagen I | [60,61] | ||
| fibronectin | Promote proliferation by MAPK pathway activation | [64] | ||
| IGF1, IGF2 | Activate IGFR-PI3K-AKT pathway | [65] | ||
| LIF | Activate Wnt and Hippo signaling pathways and induce EMT | [66] | ||
| HGF | Activate c-Met-PI3K-Akt pathway and induce EMT | [67] | ||
| CYR61 | Downregulate nucleoside transporters ENT1 and CNT3 | [68] | ||
| Deoxycytidine | Compete with gemcitabine for deoxycytidine kinase-mediated phosphorylation | [69] | ||
| immunotherapy | CXCL12 | Chemoattract CD8+ T cells via CXCL12-CXCR4 axis to sequester them in the panstromal compartment | [70] | |
| Galectin-1 | Induce T cell apoptosis and Th2 differentiation | [71] | ||
| IL-6 | Promote MDSC differentiation via STAT3 activation and suppress T cell proliferation | [72] | ||
| CAFs | chemotherapy (gemcitabine) | 5′-nucleotidases | Entrap active gemcitabine intracellularly via downregulation of Nt5c1A, Nt5c3 | [75] |
| Exosomes | Deliver SNAI1 and miR-146a to tumor cells via exosomes | [76] | ||
| circFARP1 | Enhance LIF expression and secretion | [77] | ||
| TGF-β | Upregulate ATF4 in tumor cells to activate ABCC1 expression | [78] | ||
| SDF-1 | Form a reciprocal feedback loop with tumor cells via SDF-1/SATB-1 axis | [79] | ||
| IL-6 | Activate JAK-STAT3 signaling pathway | [80] | ||
| CXCL12 | Bind to CXCR4 to activate FAK, AKT, and ERK pathways | [81] | ||
| chemotherapy (oxaliplatin) | IL-8 | Upregulate UPK1A-AS1 to facilitate DNA repair | [82] | |
| chemotherapy (etoposide) | NO | Elevate IL-1β production in tumor cells | [83] | |
| targeted therapy (EGFRi erlotinib) | NRG-1 | Activate ERBB3-AKT signaling pathway | [84,85] | |
| immunotherapy | ECM | Form a physical barrier to impede T cell-tumor cell contact | [86] | |
| ROS | Induce M2 TAM polarization | [87] | ||
| / | Suppress immunogenic activities | [88] | ||
| CXCL12 | Exclude T cells from tumor region by binding to CXCR4 | [89] | ||
| PGE2 | Induce expression of immune checkpoints on CD4+ and CD8+ T cells | [90] | ||
| TSLP | Induce Th2 cell polarization through dendritic cell conditioning | [41] | ||
| targeted therapy (GOT2i) | Pyruvate | Provide tumor cells with pyruvate to maintain redox balance | [91,92] | |
| TAMs | chemotherapy (gemcitabine) | Deoxycytidine | Interfere the uptake and metabolism of gemcitabine | [93] |
| Cytidine deaminase | Elevate cytidine deaminase expression in tumor cells to inactivate gemcitabine | [94] | ||
| targeted therapy (KRASi) | TGFβ | Activate canonical SMAD3/4 pathway and promote EMT | [95] | |
| immunotherapy | Granulin | Induce fibrosis to prevent T cell infiltration | [96] | |
| Mincle | Ligate to SAP130 expressed by tumor cells to suppress cancer immunity | [97] | ||
| RIP1 | Regulate M2 TAM polarization | [98] | ||
| radiotherapy, immunotherapy | / | n/a | [99,100] | |
| TANs | chemotherapy (gemcitabine) | IL-6 | Activate JAK-STAT3 signaling pathway | [80] |
| immunotherapy | NETs | Cause tumor CD8+ T cell inactivation and spatial exclusion | [101] | |
| chemotherapy (FOLFIRINOX, gemcitabine, nab-paclitaxel), radiotherapy, immunotherapy | / | n/a | [102,103,104,105,106,107] |
| Target | Agent | Combined Agent | Selected Clinical Trials |
|---|---|---|---|
| ECM or membrane proteins | |||
| Hyaluronic acid | PEGPH20 | Avelumab, chemotherapy, pembrolizumab | NCT03481920, NCT01453153, NCT01839487, NCT04058964 |
| Plectin | ZB131 | NCT05074472 | |
| Galectin-9 | LYT-200 | NCT04666688 | |
| CTLA-4 | Zalifrelimab | NCT04827953 | |
| RARα/β | Am80 | NCT05064618 | |
| Receptors | |||
| IGF1R | MK-0646 | Chemotherapy + TKI | NCT00769483 |
| Cixutumumab | NCT00617708 | ||
| AMG 479 | Chemotherapy, radiotherapy, AMG 655 | NCT00630552, NCT01298401, NCT00819169, NCT01231347 | |
| Metformin | Everolimus, octreotide LAR | NCT01971034, NCT02431676 | |
| MM-141 | Chemotherapy | NCT02399137 | |
| HER3 | Seribantumab | NCT04790695, NCT04383210 | |
| HMBD-001 | NCT05057013 | ||
| HER2/3 | Zenocutuzumab (MCLA-128) | NCT02912949 | |
| IL6R | Tocilizumab | Chemotherapy | NCT02767557, NCT04258150 |
| CNTO 328 | NCT00841191 | ||
| CXCR4 | MB1707 | NCT05465590 | |
| Plerixafor | Cemiplimab | NCT03277209, NCT02179970 | |
| IL1RAP | CAN04 | FOLFIRINOX | NCT04990037 |
| TGFβR | PF-06952229 | NCT03685591 | |
| SHR-1701 | Chemotherapy | NCT04624217 | |
| CSF1R | Cabiralizumab | Nivolumab, chemotherapy | NCT02526017, NCT03697564 |
| Pexidartinib | Durvalumab | NCT02777710 | |
| IMC-CS4 | Pembrolizumab, GVAX | NCT03153410 | |
| CXCR2 | SX-682 | Nivolumab | NCT04477343 |
| Enzymes | |||
| COX | Etodolac | NCT03838029 | |
| Celecoxib | Chemotherapy, irinotecan, interferon α-2b, DC vaccine | NCT00198081, NCT00068432, NCT00177853, NCT01111591 | |
| RIPK1 | GSK3145095 | NCT03681951 | |
| Cytokines, chemokines, or growth factors | |||
| LIF | MSC-1 | NCT03490669 | |
| HGF | Ficlatuzumab | NCT03316599 | |
| CXCL12 | Olaptesed pegol (NOX-A12) | Pembrolizumab | NCT03168139, NCT04901741 |
| IL-6 | Siltuximab | Spartalizumab | NCT04191421 |
| IL-12 | VG161 | Nivolumab | NCT05162118 |
| IL-15 | ALT-803 | NCT02559674 | |
| IL-1β | Canakinumab | Spartalizumab, nab-paclitaxel, gemcitabine | NCT04581343, NCT04229004 |
| IL-2 | Aldesleukin | chemotherapy, anti-KRAS G12D mTCR PBL, anti-KRAS G12V mTCR PBL, pembrolizumab, anti-hCD70 CAR-transduced PBL, HER2Bi-armed T cells, sargramostim, ALVAC-CEA vaccine, neoantigen-specific TCR-T | NCT05194735, NCT02620865, NCT01583686, NCT01212887, NCT03745326, NCT01174121, NCT03190941, NCT02830724, NCT02662348, NCT00003125, NCT05194735, NCT04426669 |
| IL-8 | BMS-986253 | Nivolumab | NCT02451982 |
| VEGF | Bevacizumab | Chemotherapy, radiotherapy, TKI, cetuximab, ALT-803, cancer vaccine, immunotherapy, pembrolizumab, ZN-c3, PEGPH20, durvalumab, TGR-1202 | NCT00047710, NCT00417976, NCT00614653, NCT00460174, NCT00365144, NCT00602602, NCT00410774, NCT00126633 |
| Bevacizumab-800CW | NCT02743975 | ||
| Avastin | Chemotherapy, NANT-008, radiotherapy | NCT03127124, NCT00735306, NCT00609765 | |
| rhuMAB-VEGF | Chemotherapy | NCT00066677 | |
| TGF-β | HCW9218 | NCT05304936 | |
| BCA101 | NCT04429542 | ||
| NIS793 | PDR001, chemotherapy | NCT02947165, NCT05417386 | |
| AP 12009 | NCT00844064 | ||
| M-CSF | MCS110 | Spartalizumab | NCT02807844 |
| GM-CSF | Sargramostim | Carcinoembryonic antigen peptide 1-6D | NCT00669734, NCT00012246 |
| GM-CSF | iNeo-Vac-P01, TG-01 | NCT04810910, NCT03645148 | |
| OH2 injection | NCT04637698 | ||
| PANC 10.05 pcDNA-1/GM-Neo | NCT01088789 | ||
| PANVAC™-VF | NCT00088660 | ||
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Deng, D.; Patel, R.; Chiang, C.-Y.; Hou, P. Role of the Tumor Microenvironment in Regulating Pancreatic Cancer Therapy Resistance. Cells 2022, 11, 2952. https://doi.org/10.3390/cells11192952
Deng D, Patel R, Chiang C-Y, Hou P. Role of the Tumor Microenvironment in Regulating Pancreatic Cancer Therapy Resistance. Cells. 2022; 11(19):2952. https://doi.org/10.3390/cells11192952
Chicago/Turabian StyleDeng, Daiyong, Riya Patel, Cheng-Yao Chiang, and Pingping Hou. 2022. "Role of the Tumor Microenvironment in Regulating Pancreatic Cancer Therapy Resistance" Cells 11, no. 19: 2952. https://doi.org/10.3390/cells11192952
APA StyleDeng, D., Patel, R., Chiang, C. -Y., & Hou, P. (2022). Role of the Tumor Microenvironment in Regulating Pancreatic Cancer Therapy Resistance. Cells, 11(19), 2952. https://doi.org/10.3390/cells11192952