
TIMP-1 Dependent Modulation of Metabolic Profiles Impacts Chemoresistance in NSCLC

 ,
,  , , and
, , and {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Cell Culture shRNA Transduction
2.2. Reverse Transcription Quantitative Real-Time PCR (RT-qPCR)
2.3. Western Blotting
2.4. Mitochondria Isolation
2.5. Nuclear and Cytoplasmic Protein Extraction
2.6. Liquid Chromatography-Mass Spectrometry Analysis
2.7. Mitochondrial Membrane Potential Assay
2.8. Glycolysis Cell-Based Assay
2.9. Reactive Oxygen Species (ROS) Assay
2.10. GSH/GSSG Assay
2.11. Immunofluorescence (IF)
2.12. Immunoprecipitation
2.13. Bioinformatics Evaluation
2.14. Statistical Analysis
3. Results
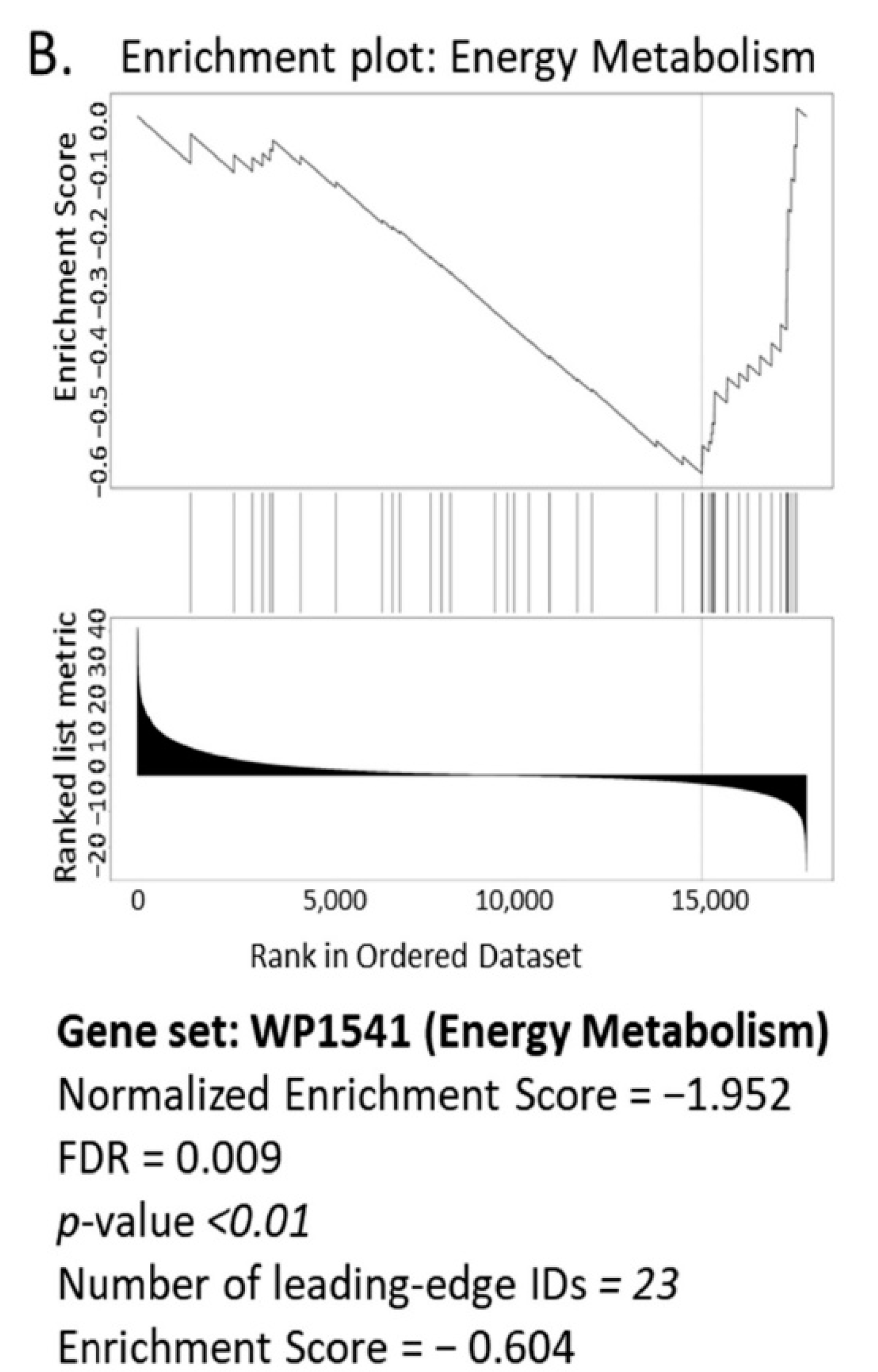
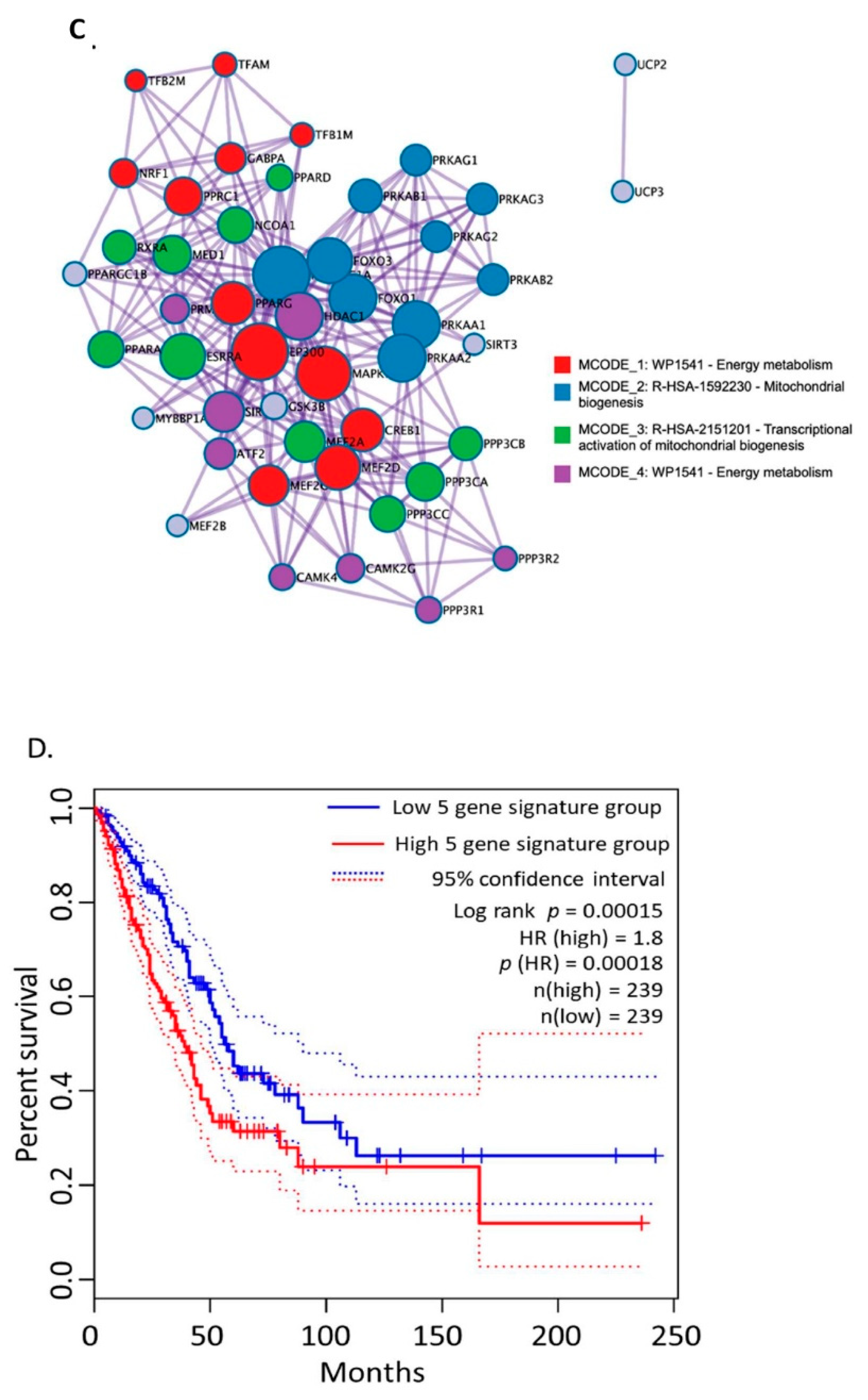
3.1. Bioinformatics Assessment Associates Mitochondrial Gene Signature with TIMP-1
3.2. TIMP-1 Modulates Metabolic Profile in NSCLC
3.3. TIMP-1 Affects STAT3 Activation via Its Acetylation
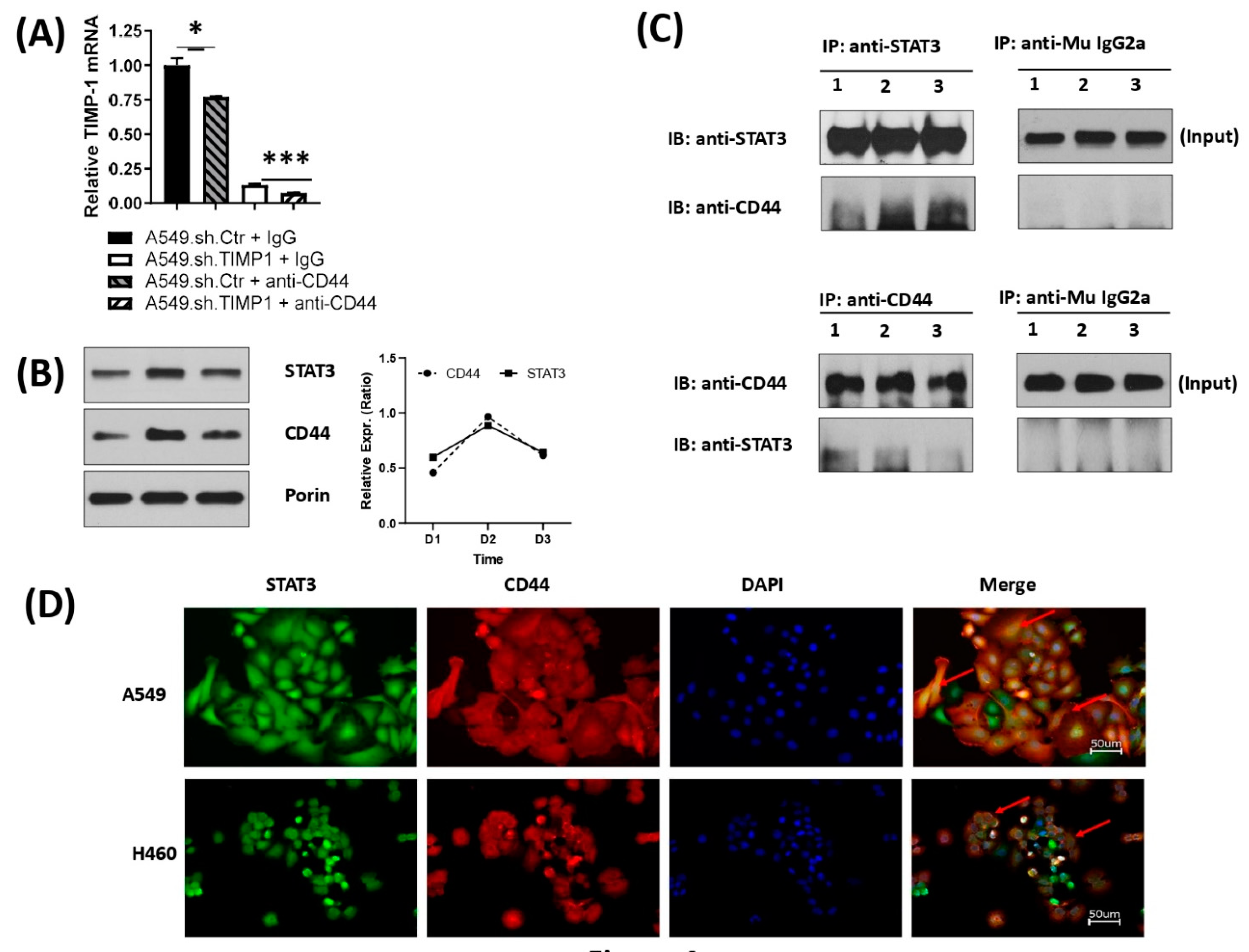
3.4. Correlation and Interaction of Mitochondrial STAT3 with CD44 upon TIMP-1 Modulation
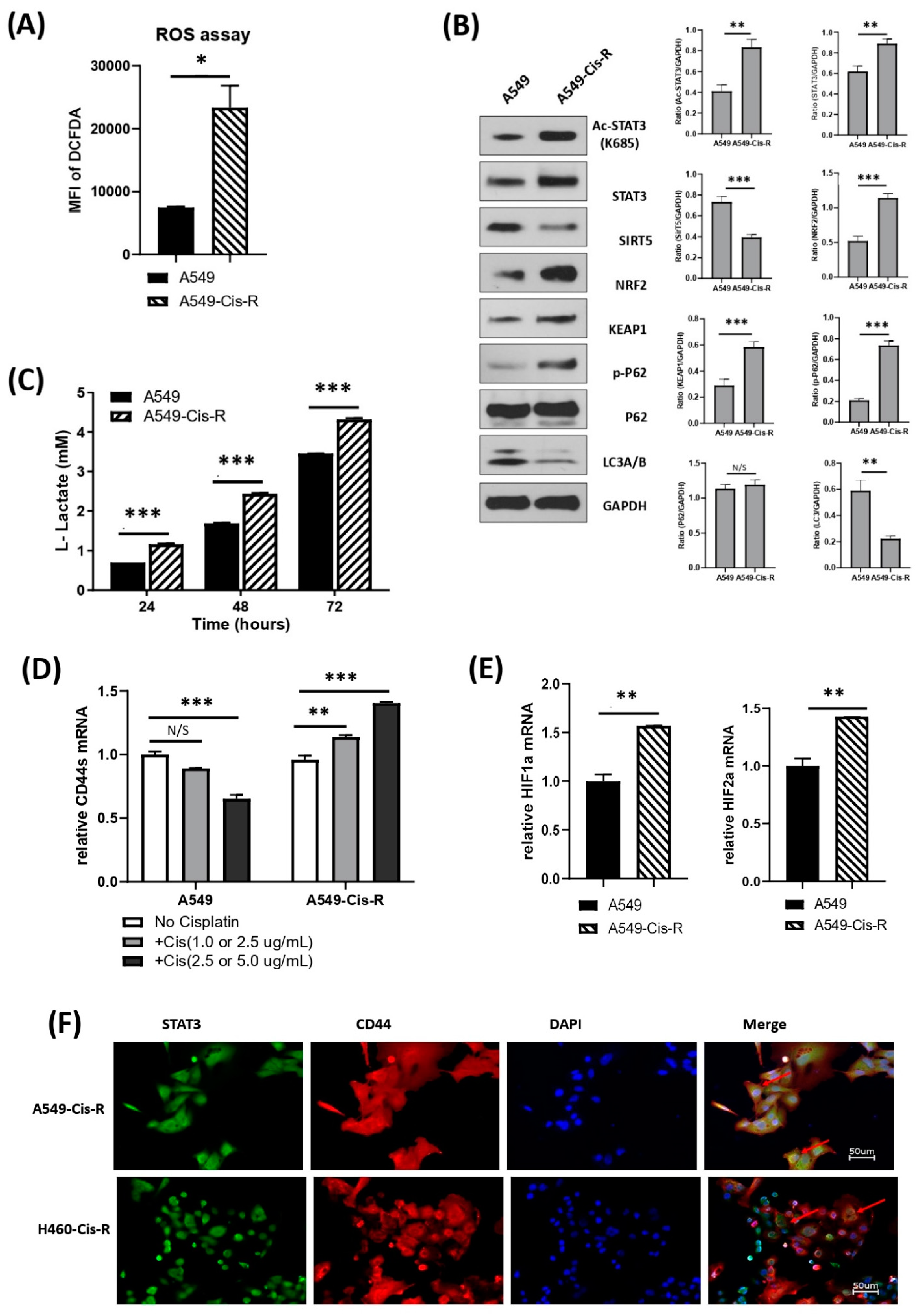
3.5. Mitochondrial Metabolic Profile Was Altered in Cisplatin-Resistant Clone of A549
3.6. CD44, STAT3 Immunohistochemistry of Archived Patient Samples of NSCLC
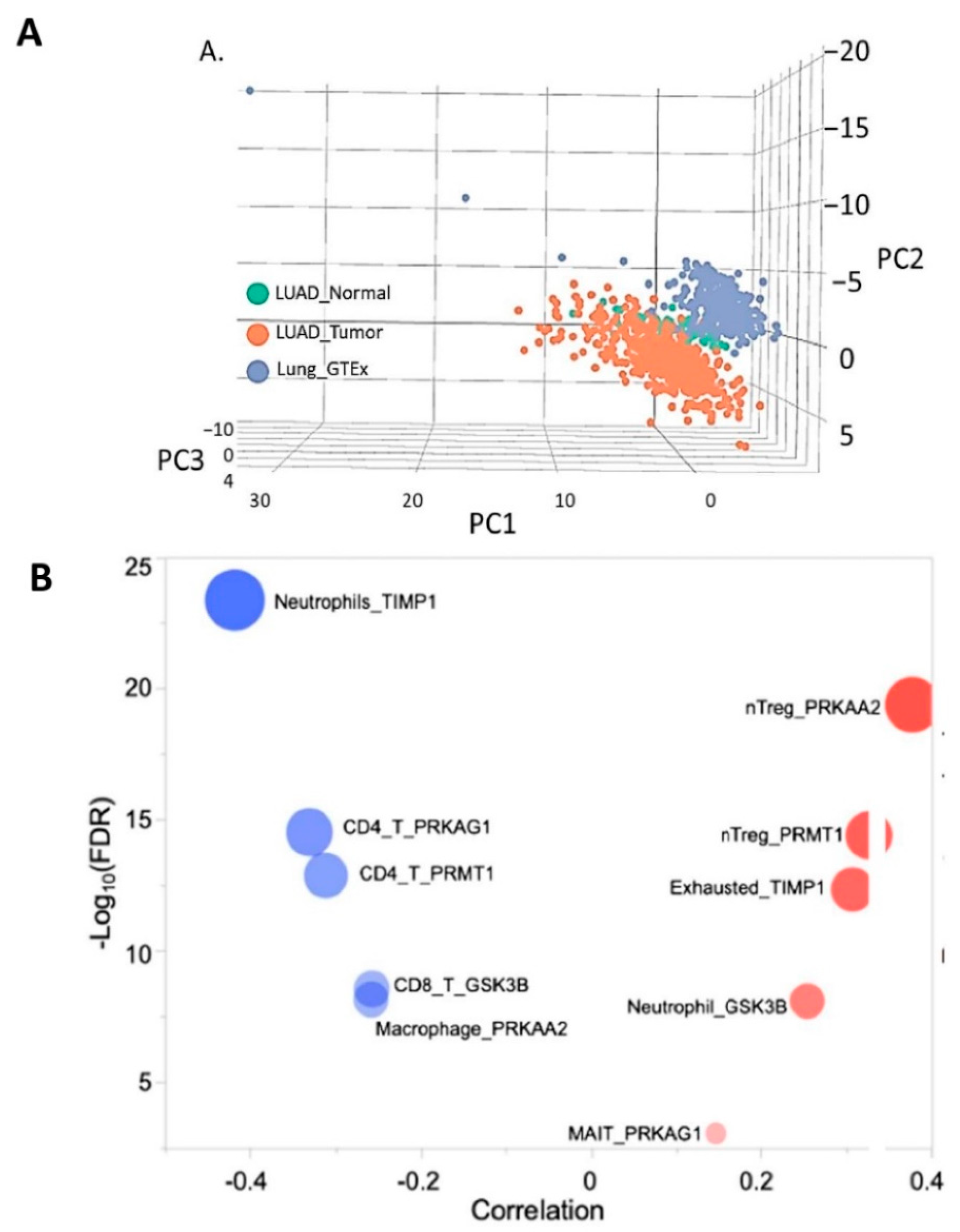
3.7. Molecular Profiling of NSCLC Tumor Based on TIMP1 Expression
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Jackson, H.W.; Defamie, V.; Waterhouse, P.; Khokha, R. TIMPs: Versatile extracellular regulators in cancer. Nat. Cancer 2017, 17, 38–53. [Google Scholar] [CrossRef]
- Xiao, W.; Wang, L.; Howard, J.; Kolhe, R.; Rojiani, A.M.; Rojiani, M.V. TIMP-1-Mediated Chemoresistance via Induction of IL-6 in NSCLC. Cancers 2019, 11, 1184. [Google Scholar] [CrossRef]
- Senthebane, D.A.; Rowe, A.; Thomford, N.E.; Shipanga, H.; Munro, D.; Al Mazeedi, M.A.M.; Almazyadi, H.A.M.; Kallmeyer, K.; Dandara, C.; Pepper, M.S.; et al. The Role of Tumor Microenvironment in Chemoresistance: To Survive, Keep Your Enemies Closer. Int. J. Mol. Sci. 2017, 18, 1586. [Google Scholar] [CrossRef]
- Yeldag, G.; Rice, A.; Del Río Hernández, A. Chemoresistance and the Self-Maintaining Tumor Microenvironment. Cancers 2018, 10, 471. [Google Scholar] [CrossRef]
- Zheng, H.-C. The molecular mechanisms of chemoresistance in cancers. Oncotarget 2017, 8, 59950–59964. [Google Scholar] [CrossRef]
- Likus, W.; Siemianowicz, K.; Bieńk, K.; Pakuła, M.; Pathak, H.; Dutta, C.; Wang, Q.; Shojaei, S.; Assaraf, Y.G.; Ghavami, S.; et al. Could drugs inhibiting the mevalonate pathway also target cancer stem cells? Drug Resist. Updat. 2016, 25, 13–25. [Google Scholar] [CrossRef]
- Schon, E.A.; DiMauro, S.; Hirano, M. Human mitochondrial DNA: Roles of inherited and somatic mutations. Nat. Rev. Genet. 2012, 13, 878–890. [Google Scholar] [CrossRef]
- Koh, Y.W.; Han, J.-H.; Park, S.Y.; Yoon, D.H.; Suh, C.; Huh, J. GLUT1 as a Prognostic Factor for Classical Hodgkin’s Lymphoma: Correlation with PD-L1 and PD-L2 Expression. J. Pathol. Transl. Med. 2017, 51, 152–158. [Google Scholar] [CrossRef]
- Cruz-Bermúdez, A.; Vicente-Blanco, R.J.; Briviesca, R.L.; García-Grande, A.; Laine-Menéndez, S.; Gutiérrez, L.; Calvo, V.; Romero, A.; Martin-Acosta, P.; García, J.M.; et al. PGC-1alpha levels correlate with survival in patients with stage III NSCLC and may define a new biomarker to metabolism-targeted therapy. Sci. Rep. 2017, 7, 16661. [Google Scholar] [CrossRef]
- Makinoshima, H.; Takita, M.; Matsumoto, S.; Yagishita, A.; Owada, S.; Esumi, H.; Tsuchihara, K. Epidermal Growth Factor Receptor (EGFR) Signaling Regulates Global Metabolic Pathways in EGFR-mutated Lung Adenocarcinoma. J. Biol. Chem. 2014, 289, 20813–20823. [Google Scholar] [CrossRef] [Green Version]
- Liao, Z.-X.; Kempson, I.M.; Hsieh, C.-C.; Tseng, S.-J.; Yang, P.-C. Potential therapeutics using tumor-secreted lactate in nonsmall cell lung cancer. Drug Discov. Today 2021, 26, 2508–2514. [Google Scholar] [CrossRef]
- Kerr, E.M.; Martins, C.P. Metabolic rewiring in mutant Kras lung cancer. FEBS J. 2018, 285, 28–41. [Google Scholar] [CrossRef]
- Ghoshal-Gupta, S.; Kutiyanawalla, A.; Lee, B.R.; Ojha, J.; Nurani, A.; Mondal, A.K.; Kolhe, R.; Rojiani, A.M.; Rojiani, M.V. TIMP-1 downregulation modulates miR-125a-5p expression and triggers the apoptotic pathway. Oncotarget 2018, 9, 8941–8956. [Google Scholar] [CrossRef]
- Kolhe, R.; Owens, V.; Sharma, A.; Lee, T.J.; Zhi, W.; Ghilzai, U.; Mondal, A.K.; Liu, Y.; Isales, C.M.; Hamrick, M.W.; et al. Sex-Specific Differences in Extracellular Vesicle Protein Cargo in Synovial Fluid of Patients with Osteoarthritis. Life 2020, 10, 337. [Google Scholar] [CrossRef]
- Martens, M.; Ammar, A.; Riutta, A.; Waagmeester, A.; Slenter, D.N.; Hanspers, K.; Miller, R.A.; Digles, D.; Lopes, E.N.; Ehrhart, F.; et al. WikiPathways: Connecting communities. Nucleic Acids Res. 2021, 49, D613–D621. [Google Scholar] [CrossRef]
- Mi, H.; Ebert, D.; Muruganujan, A.; Mills, C.; Albou, L.-P.; Mushayamaha, T.; Thomas, P.D. PANTHER version 16: A revised family classification, tree-based classification tool, enhancer regions and extensive API. Nucleic Acids Res. 2021, 49, D394–D403. [Google Scholar] [CrossRef]
- Vasaikar, S.; Straub, P.; Wang, J.; Zhang, B. Abstract 5112: LinkedOmics: Analyzing multi-omics data within and across 32 cancer types. Nucleic Acids Res. 2019, 46, D956–D963. [Google Scholar] [CrossRef]
- Slenter, D.N.; Kutmon, M.; Hanspers, K.; Riutta, A.; Windsor, J.; Nunes, N.; Mélius, J.; Cirillo, E.; Coort, S.L.; Digles, D.; et al. WikiPathways: A multifaceted pathway database bridging metabolomics to other omics research. Nucleic Acids Res. 2018, 46, D661–D667. [Google Scholar] [CrossRef]
- Tang, Z.; Li, C.; Kang, B.; Gao, G.; Li, C.; Zhang, Z. GEPIA: A web server for cancer and normal gene expression profiling and interactive analyses. Nucleic Acids Res. 2017, 45, W98–W102. [Google Scholar] [CrossRef]
- Bader, G.D.; Hogue, C.W.V. An automated method for finding molecular complexes in large protein interaction networks. BMC Bioinform. 2003, 4, 2. [Google Scholar] [CrossRef] [Green Version]
- Shannon, P.; Markiel, A.; Ozier, O.; Baliga, N.S.; Wang, J.T.; Ramage, D.; Amin, N.; Schwikowski, B.; Ideker, T. Cytoscape: A software environment for integrated models of Biomolecular Interaction Networks. Genome Res. 2003, 13, 2498–2504. [Google Scholar] [CrossRef] [PubMed]
- Choi, Y.; Kendziorski, C. Statistical methods for gene set co-expression analysis. Bioinformatics 2009, 25, 2780–2786. [Google Scholar] [CrossRef] [PubMed]
- Tang, Z.; Kang, B.; Li, C.; Chen, T.; Zhang, Z. GEPIA2: An enhanced web server for large-scale expression profiling and interactive analysis. Nucleic Acids Res. 2019, 47, W556–W560. [Google Scholar] [CrossRef] [PubMed]
- Bonnet, S.; Archer, S.L.; Allalunis-Turner, J.; Haromy, A.; Beaulieu, C.; Thompson, R.; Lee, C.T.; Lopaschuk, G.D.; Puttagunta, L.; Bonnet, S. A mitochondria-K+ channel axis is suppressed in cancer and its normalization promotes apoptosis and inhibits cancer growth. Cancer Cell. 2007, 11, 37–51. [Google Scholar] [CrossRef] [PubMed]
- Demaria, M.; Camporeale, A.; Poli, V. STAT3 and metabolism: How many ways to use a single molecule? Int. J. Cancer 2014, 135, 1997–2003. [Google Scholar] [CrossRef]
- Poli, V.; Camporeale, A. STAT3-Mediated Metabolic Reprograming in Cellular Transformation and Implications for Drug Resistance. Front. Oncol. 2015, 5, 121. [Google Scholar] [CrossRef] [PubMed]
- Xu, Y.; Liang, J.; Wang, Y.; Zhao, X.; Xu, L.; Xu, Y.; Zou, Q.; Zhang, J.; Tu, C.; Cui, Y.; et al. STAT3 Undergoes Acetylation-dependent Mitochondrial Translocation to Regulate Pyruvate Metabolism. Sci. Rep. 2016, 6, 39517. [Google Scholar] [CrossRef]
- Horvath, M.; Petrvalska, O.; Herman, P.; Obsilova, V.; Obsil, T. 14-3-3 proteins inactivate DAPK2 by promoting its dimerization and protecting key regulatory phosphosites. Commun. Biol. 2021, 4, 986. [Google Scholar] [CrossRef]
- Zhong, M.; Henriksen, M.A.; Takeuchi, K.; Schaefer, O.; Liu, B.; Hoeve, J.T.; Ren, Z.; Mao, X.; Chen, X.; Shuai, K.; et al. Implications of an antiparallel dimeric structure of nonphosphorylated STAT1 for the activation–inactivation cycle. Proc. Natl. Acad. Sci. USA 2005, 102, 3966–3971. [Google Scholar] [CrossRef]
- Wu, C.-T.; Huang, Y.-C.; Chen, W.-C.; Chen, M.-F. Effect of Tumor Burden on Tumor Aggressiveness and Immune Modulation in Prostate Cancer: Association with IL-6 Signaling. Cancers 2019, 11, 992. [Google Scholar] [CrossRef] [Green Version]
- Chang, M.T.; Lee, S.P.; Fang, C.Y.; Hsieh, P.L.; Liao, Y.W.; Lu, M.Y.; Tsai, L.L.; Yu, C.C.; Liu, C.M. Chemosensitizing effect of honokiol in oral carcinoma stem cells via regulation of IL-6/Stat3 signaling. Environ. Toxicol. 2018, 33, 1105–1112. [Google Scholar] [CrossRef]
- Chung, S.S.; Aroh, C.; Vadgama, J.V. Constitutive Activation of STAT3 Signaling Regulates hTERT and Promotes Stem Cell-Like Traits in Human Breast Cancer Cells. PLoS ONE 2013, 8, e83971. [Google Scholar] [CrossRef] [PubMed]
- Khurana, S.S.; Riehl, T.E.; Moore, B.D.; Fassan, M.; Rugge, M.; Romero-Gallo, J.; Noto, J.; Peek, R.M.; Stenson, W.F.; Mills, J.C. The Hyaluronic Acid Receptor CD44 Coordinates Normal and Metaplastic Gastric Epithelial Progenitor Cell Proliferation. J. Biol. Chem. 2013, 288, 16085–16097. [Google Scholar] [CrossRef]
- So, J.Y.; Smolarek, A.K.; Salerno, D.M.; Maehr, H.; Uskokovic, M.; Liu, F.; Suh, N. Targeting CD44-STAT3 Signaling by Gemini Vitamin D Analog Leads to Inhibition of Invasion in Basal-Like Breast Cancer. PLoS ONE 2013, 8, e54020. [Google Scholar] [CrossRef]
- Kim, Y.-S.; Seo, D.-W.; Kong, S.-K.; Lee, J.-H.; Lee, E.-S.; Stetler-Stevenson, M.; Stetler-Stevenson, W.G. TIMP1 induces CD44 expression and the activation and nuclear translocation of SHP1 during the late centrocyte/post-germinal center B cell differentiation. Cancer Lett. 2008, 269, 37–45. [Google Scholar] [CrossRef]
- Guedez, L.; Martinez, A.; Zhao, S.; Vivero, A.; Pittaluga, S.; Stetler-Stevenson, M.; Raffeld, M.; Stetler-Stevenson, W.G. Tissue inhibitor of metalloproteinase 1 (TIMP-1) promotes plasmablastic differentiation of a Burkitt lymphoma cell line: Implications in the pathogenesis of plasmacytic/plasmablastic tumors. Blood 2005, 105, 1660–1668. [Google Scholar] [CrossRef]
- Kolenda, J.; Jensen, S.S.; Aaberg-Jessen, C.; Christensen, K.; Andersen, C.; Brünner, N.; Kristensen, B.W. Effects of hypoxia on expression of a panel of stem cell and chemoresistance markers in glioblastoma-derived spheroids. J. Neuro-Oncol. 2011, 103, 43–58. [Google Scholar] [CrossRef]
- Kuo, Y.-L.; Jou, I.-M.; Jeng, S.-F.; Chu, C.-H.; Huang, J.-S.; Hsu, T.-I.; Chang, L.-R.; Huang, P.-W.; Chen, J.-A.; Chou, T.-M. Hypoxia-induced epithelial-mesenchymal transition and fibrosis for the development of breast capsular contracture. Sci. Rep. 2019, 9, 10269. [Google Scholar] [CrossRef] [PubMed]
- Ilie, M.I.; Lassalle, S.; Long-Mira, E.; Hofman, V.; Zangari, J.; Bénaim, G.; Bozec, A.; Guevara, N.; Haudebourg, J.; Birtwisle-Peyrottes, I.; et al. In papillary thyroid carcinoma, TIMP-1 expression correlates with BRAF (V600E) mutation status and together with hypoxia-related proteins predicts aggressive behavior. Virchows Arch. 2013, 463, 437–444. [Google Scholar] [CrossRef] [PubMed]
- Cui, H.; Seubert, B.; Stahl, E.; Dietz, H.; Reuning, U.; Moreno-Leon, L.; Ilie, M.; Hofman, P.; Nagase, H.; Mari, B.; et al. Tissue inhibitor of metalloproteinases-1 induces a pro-tumourigenic increase of miR-210 in lung adenocarcinoma cells and their exosomes. Oncogene 2015, 34, 3640–3650. [Google Scholar] [CrossRef] [PubMed]
- Carlsson, R.; Özen, I.; Barbariga, M.; Gaceb, A.; Roth, M.; Paul, G. STAT3 precedes HIF1α transcriptional responses to oxygen and oxygen and glucose deprivation in human brain pericytes. PLoS ONE 2018, 13, e0194146. [Google Scholar] [CrossRef] [PubMed]
- Demaria, M.; Giorgi, C.; Lebiedzinska, M.; Esposito, G.; D’Angeli, L.; Bartoli, A.; Gough, D.J.; Turkson, J.; Levy, D.E.; Watson, C.J.; et al. A STAT3-mediated metabolic switch is involved in tumour transformation and STAT3 addiction. Aging 2010, 2, 823–842. [Google Scholar] [CrossRef]
- Semenza, G.L. Hypoxia-inducible factors: Coupling glucose metabolism and redox regulation with induction of the breast cancer stem cell phenotype. EMBO J. 2016, 36, 252–259. [Google Scholar] [CrossRef]
- Elfawy, H.A.; Das, B. Crosstalk between mitochondrial dysfunction, oxidative stress, and age related neurodegenerative disease: Etiologies and therapeutic strategies. Life Sci. 2019, 218, 165–184. [Google Scholar] [CrossRef] [PubMed]
- Sergi, D.; Naumovski, N.N.; Heilbronn, L.H.K.; Abeywardena, M.; O’Callaghan, N.; Lionetti, L.; Luscombe-Marsh, N.L.-M. Mitochondrial (Dys)function and Insulin Resistance: From Pathophysiological Molecular Mechanisms to the Impact of Diet. Front. Physiol. 2019, 10, 532. [Google Scholar] [CrossRef] [PubMed]
- Vyas, S.; Zaganjor, E.; Haigis, M.C. Mitochondria and Cancer. Cell 2016, 166, 555–566. [Google Scholar] [CrossRef] [PubMed]
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef] [PubMed]
- Garama, D.J.; Harris, T.J.; White, C.L.; Rossello, F.; Abdul-Hay, M.; Gough, D.; Levy, D.E. A Synthetic Lethal Interaction between Glutathione Synthesis and Mitochondrial Reactive Oxygen Species Provides a Tumor-Specific Vulnerability Dependent on STAT3. Mol. Cell. Biol. 2015, 35, 3646–3656. [Google Scholar] [CrossRef]
- Li, L.; Shaw, P.E. A STAT3 dimer formed by inter-chain disulphide bridging during oxidative stress. Biochem. Biophys. Res. Commun. 2004, 322, 1005–1011. [Google Scholar] [CrossRef]
- Morath, I.; Hartmann, T.; Orian-Rousseau, V. CD44: More than a mere stem cell marker. Int. J. Biochem. Cell Biol. 2016, 81, 166–173. [Google Scholar] [CrossRef] [PubMed]
- Nagano, O.; Okazaki, S.; Saya, H. Redox regulation in stem-like cancer cells by CD44 variant isoforms. Oncogene 2013, 32, 5191–5198. [Google Scholar] [CrossRef]
- Nam, K.; Oh, S.; Shin, I. Ablation of CD44 induces glycolysis-to-oxidative phosphorylation transition via modulation of the c-Src-Akt-LKB1-AMPKalpha pathway. Biochem. J. 2016, 473, 3013–3030. [Google Scholar] [CrossRef] [PubMed]
- Tamada, M.; Nagano, O.; Tateyama, S.; Ohmura, M.; Yae, T.; Ishimoto, T.; Sugihara, E.; Onishi, N. Modulation of glucose metabolism by CD44 contributes to antioxidant status and drug resistance in cancer cells. Cancer Res. 2012, 72, 1438–1448. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Liu, Y.; Ma, H.; Guan, Y.; Cao, Y.; Tian, Y.; Zhang, Y. Enhancement of Glucose Metabolism via PGC-1α Participates in the Cardioprotection of Chronic Intermittent Hypobaric Hypoxia. Front. Physiol. 2016, 7, 219. [Google Scholar] [CrossRef] [PubMed]
- Dhar, D.; Antonucci, L.; Nakagawa, H.; Kim, J.Y.; Glitzner, E.; Caruso, S.; Shalapour, S.; Yang, L.; Valasek, M.A.; Lee, S.; et al. Liver Cancer Initiation Requires p53 Inhibition by CD44-Enhanced Growth Factor Signaling. Cancer Cell 2018, 33, 1061–1077.e6. [Google Scholar] [CrossRef] [PubMed]
- Janiszewska, M.; De Vito, C.; Le Bitoux, M.-A.; Fusco, C.; Stamenkovic, I. Transportin Regulates Nuclear Import of CD44. J. Biol. Chem. 2010, 285, 30548–30557. [Google Scholar] [CrossRef] [PubMed]
- Su, M.; Wang, P.; Wang, X.; Zhang, M.; Wei, S.; Liu, K.; Han, S.; Han, X.; Deng, Y.; Shen, L. Nuclear CD44 Mediated by Importin beta Participated in Naive Genes Transcriptional Regulation in C3A-iCSCs. Int. J. Biol. Sci. 2019, 15, 1252–1260. [Google Scholar] [CrossRef]
- Nolan, M.J.; Koga, T.; Walker, L.; Mccarty, R.; Grybauskas, A.; Giovingo, M.C.; Skuran, K.; Kuprys, P.V.; Knepper, P.A. sCD44 Internalization in Human Trabecular Meshwork Cells. Investig. Opthalmol. Vis. Sci. 2013, 54, 592–601. [Google Scholar] [CrossRef] [PubMed]
- Gorrini, C.; Baniasadi, P.S.; Harris, I.S.; Silvester, J.; Inoue, S.; Snow, B.; Joshi, P.A.; Wakeham, A.; Molyneux, S.D.; Martin, B.; et al. BRCA1 interacts with Nrf2 to regulate antioxidant signaling and cell survival. J. Exp. Med. 2013, 210, 1529–1544. [Google Scholar] [CrossRef]
- Lu, W.; Zuo, Y.; Feng, Y.; Zhang, M. SIRT5 facilitates cancer cell growth and drug resistance in non-small cell lung cancer. Tumor Biol. 2014, 35, 10699–10705. [Google Scholar] [CrossRef]
- Chen, X.; Tian, M.; Sun, R.; Zhang, M.; Zhou, L.; Jin, L.; Chen, L.; Zhou, W.; Duan, K.; Chen, Y.; et al. SIRT 5 inhibits peroxisomal ACOX 1 to prevent oxidative damage and is downregulated in liver cancer. EMBO Rep. 2018, 19, e45124. [Google Scholar] [CrossRef]
- Chen, X.; Xu, Z.; Zeng, S.; Wang, X.; Liu, W.; Qian, L.; Wei, J.; Yang, X.; Shen, Q.; Gong, Z.; et al. SIRT5 downregulation is associated with poor prognosis in glioblastoma. Cancer Biomarkers 2019, 24, 449–459. [Google Scholar] [CrossRef]
- Quinlan, C.L.; Treberg, J.R.; Perevoshchikova, I.V.; Orr, A.L.; Brand, M.D. Native rates of superoxide production from multiple sites in isolated mitochondria measured using endogenous reporters. Free Radic. Biol. Med. 2012, 53, 1807–1817. [Google Scholar] [CrossRef]
- Sullivan, L.B.; Chandel, N.S. Mitochondrial reactive oxygen species and cancer. Cancer Metab. 2014, 2, 17. [Google Scholar] [CrossRef]
- Genova, C.; Dellepiane, C.; Carrega, P.; Sommariva, S.; Ferlazzo, G.; Pronzato, P.; Gangemi, R.; Filaci, G.; Coco, S.; Croce, M. Therapeutic Implications of Tumor Microenvironment in Lung Cancer: Focus on Immune Checkpoint Blockade. Front. Immunol. 2022, 12, 799455. [Google Scholar] [CrossRef] [PubMed]
- Principe, D.R.; Chiec, L.; Mohindra, N.A.; Munshi, H.G. Regulatory T-Cells as an Emerging Barrier to Immune Checkpoint Inhibition in Lung Cancer. Front. Oncol. 2021, 11, 684098. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.; Liu, S.; Zhang, B.; Qiao, L.; Zhang, Y. T Cell Dysfunction and Exhaustion in Cancer. Front. Cell Dev. Biol. 2020, 8, 17. [Google Scholar] [CrossRef]
- Kim, K.; Park, S.; Park, S.Y.; Kim, G.; Park, S.M.; Cho, J.-W.; Kim, D.H.; Park, Y.M.; Koh, Y.W.; Kim, H.R.; et al. Single-cell transcriptome analysis reveals TOX as a promoting factor for T cell exhaustion and a predictor for anti-PD-1 responses in human cancer. Genome Med. 2020, 12, 22. [Google Scholar] [CrossRef] [PubMed] [Green Version]






Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Xiao, W.; Ahluwalia, P.; Wang, L.; Howard, J.; Kolhe, R.; Rojiani, A.M.; Rojiani, M.V. TIMP-1 Dependent Modulation of Metabolic Profiles Impacts Chemoresistance in NSCLC. Cells 2022, 11, 3036. https://doi.org/10.3390/cells11193036
Xiao W, Ahluwalia P, Wang L, Howard J, Kolhe R, Rojiani AM, Rojiani MV. TIMP-1 Dependent Modulation of Metabolic Profiles Impacts Chemoresistance in NSCLC. Cells. 2022; 11(19):3036. https://doi.org/10.3390/cells11193036
Chicago/Turabian StyleXiao, Wei, Pankaj Ahluwalia, Lan Wang, John Howard, Ravindra Kolhe, Amyn M. Rojiani, and Mumtaz V. Rojiani. 2022. "TIMP-1 Dependent Modulation of Metabolic Profiles Impacts Chemoresistance in NSCLC" Cells 11, no. 19: 3036. https://doi.org/10.3390/cells11193036
APA StyleXiao, W., Ahluwalia, P., Wang, L., Howard, J., Kolhe, R., Rojiani, A. M., & Rojiani, M. V. (2022). TIMP-1 Dependent Modulation of Metabolic Profiles Impacts Chemoresistance in NSCLC. Cells, 11(19), 3036. https://doi.org/10.3390/cells11193036