
Mitochondrion-Mediated Cell Death through Erk1-Alox5 Independent of Caspase-9 Signaling


Abstract
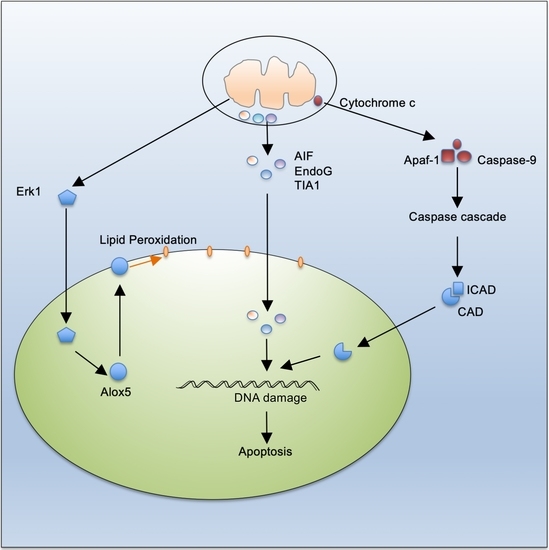
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
2.1. Mice
2.2. siRNA Screening
2.3. Cell Death Assay
2.4. Measurement of ROS and Lipid Peroxidation
2.5. Immunocytochemistry
2.6. Immunoprecipitation and Western Blot
2.7. Statistic Analyses
3. Results
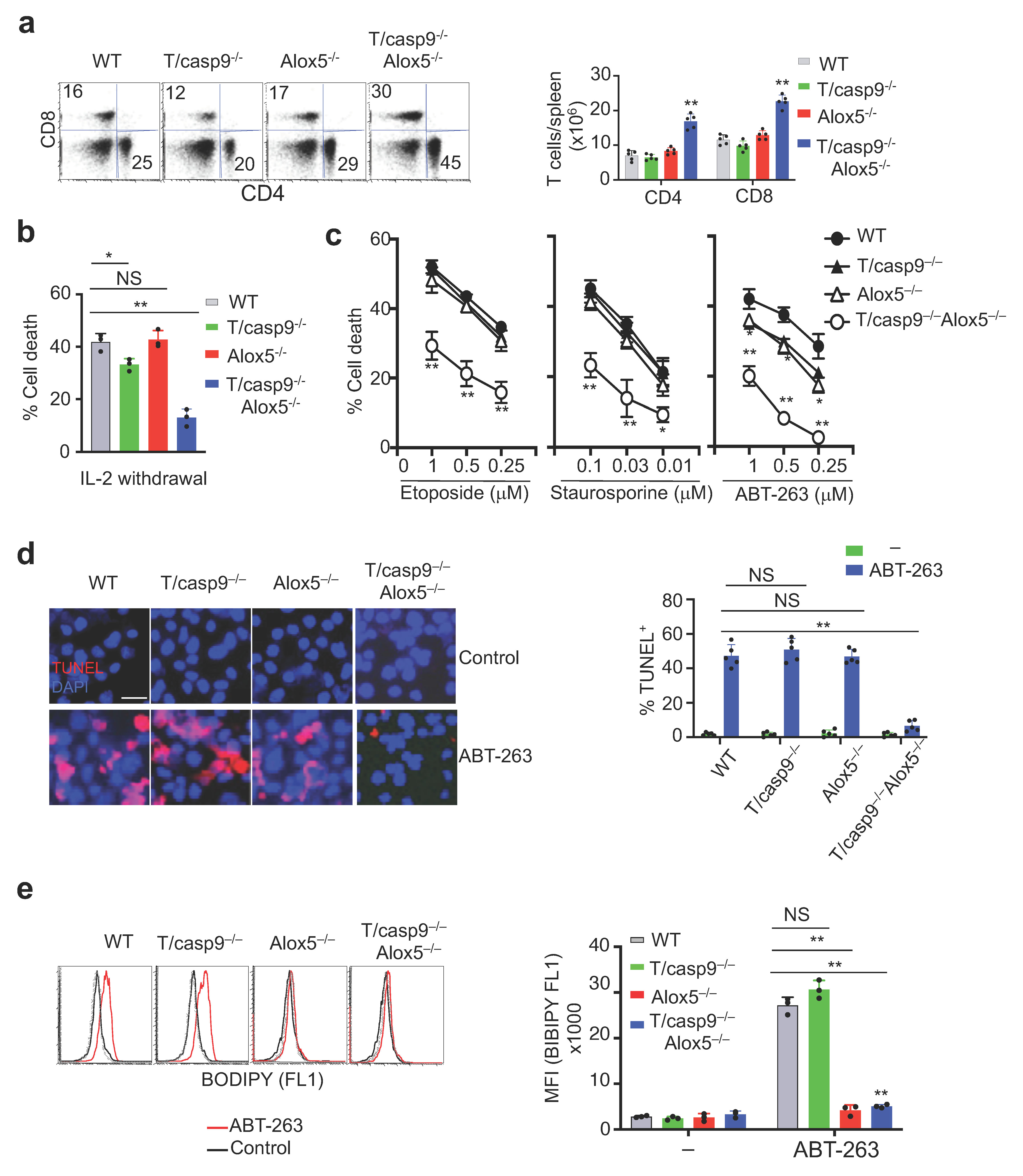
3.1. Caspase-9-Independent Mechanisms Efficiently Mediate Mitochondrion-Dependent Cell Death and Maintain T Cell Homeostasis
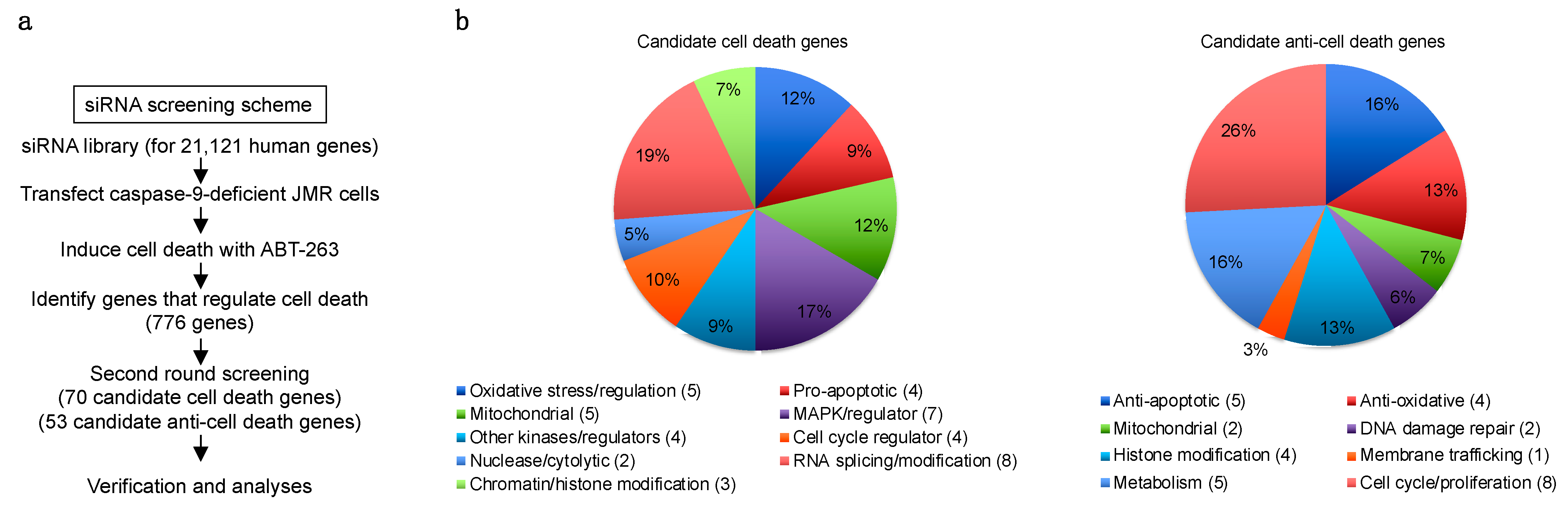
3.2. Genome-Wide siRNA Screening for Genes That Regulate Caspase-9-Independent Cell Death
3.3. Mitochondrial Proteins Essential for ROS Production during Caspase-9-Independent Cell Death
3.4. Protection against Caspase-9-Independent Cell Death by Genes That Inhibit Oxidative Stress
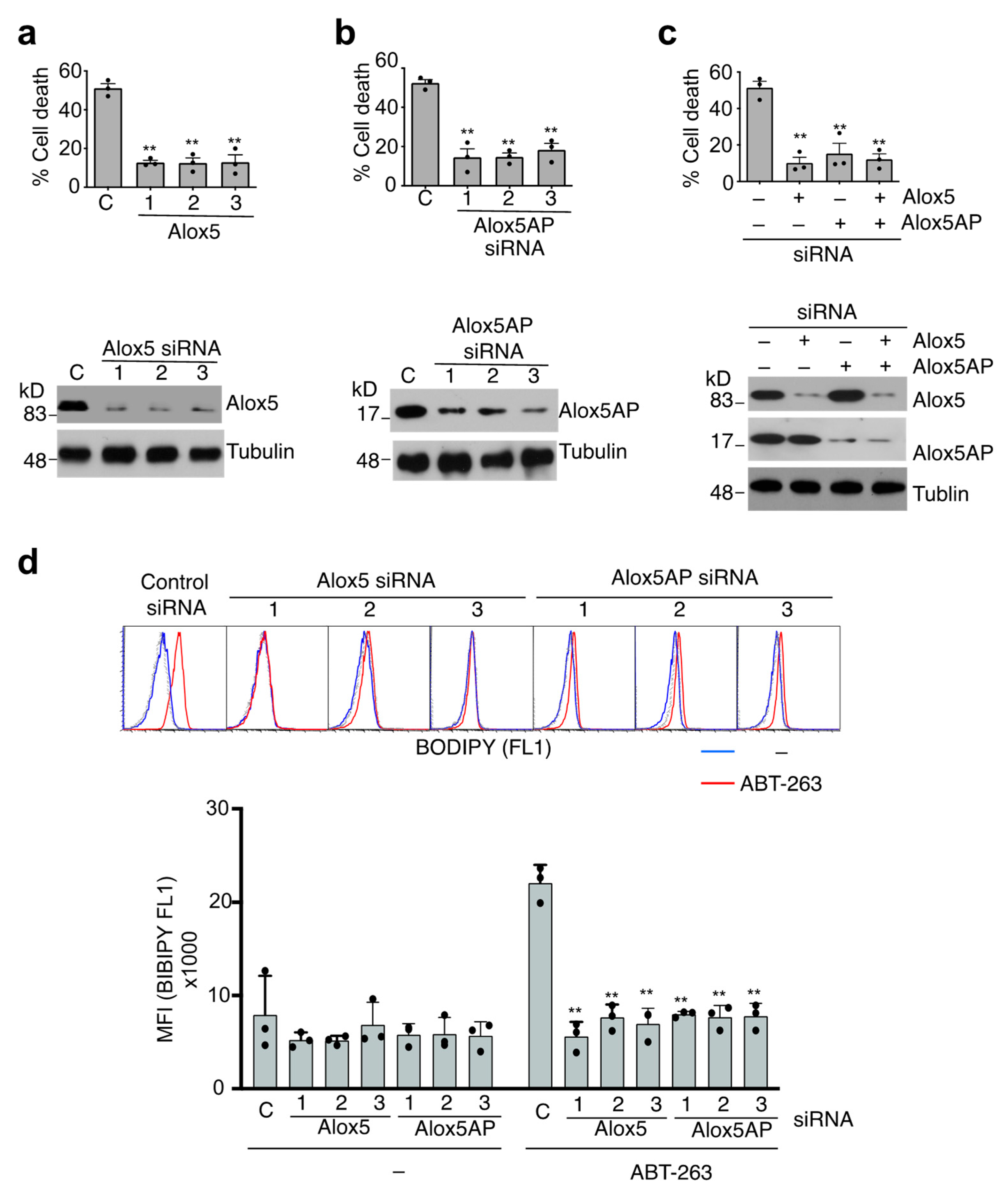
3.5. Alox5-Mediated Lipid Peroxidation in the Induction of Caspase-9-Independent Cell Death
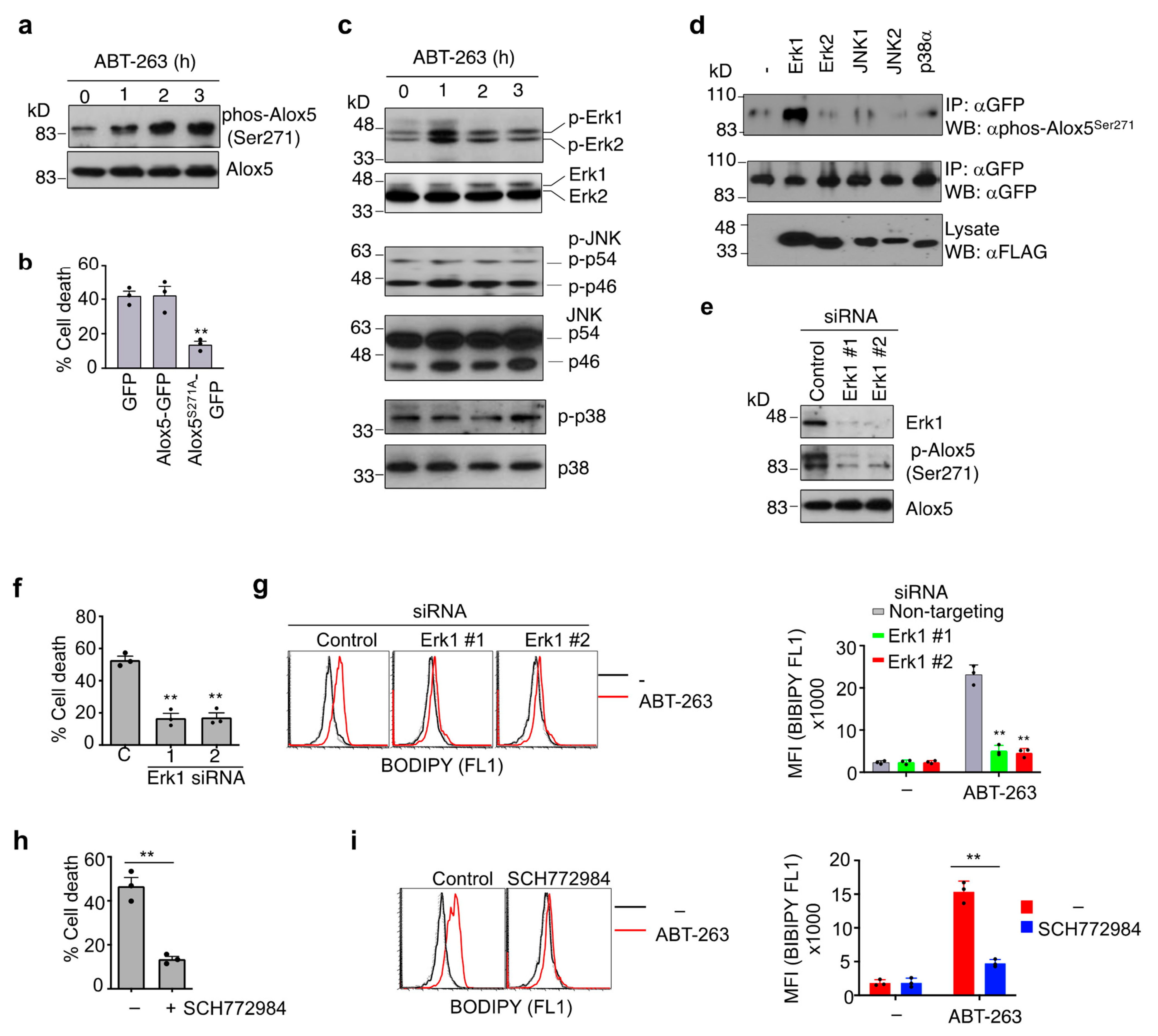
3.6. Phosphorylation of Alox5 Serine 271 by Erk1 Is Critical for Its Nuclear Membrane Localization and Cell Death Function
3.7. Loss of Alox5 Inhibits Membrane Lipid Peroxidation and Cell Death in Caspase-9−/− T Cells
3.8. Defects in Nuclear Translocation of TIA-1 and EndoG in the Absence of Caspase-9 and Alox5
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Kluck, R.M.; Bossy-Wetzel, E.; Green, D.R.; Newmeyer, D.D. The release of cytochrome c from mitochondria: A primary site for Bcl-2 regulation of apoptosis. Science 1997, 275, 1132–1136. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Kim, C.N.; Yang, J.; Jemmerson, R.; Wang, X. Induction of apoptotic program in cell-free extracts: Requirement for dATP and cytochrome c. Cell 1996, 86, 147–157. [Google Scholar] [CrossRef]
- Yang, J.; Liu, X.; Bhalla, K.; Kim, C.N.; Ibrado, A.M.; Cai, J.; Peng, T.I.; Jones, D.P.; Wang, X. Prevention of apoptosis by Bcl-2: Release of cytochrome c from mitochondria blocked. Science 1997, 275, 1129–1132. [Google Scholar] [CrossRef] [PubMed]
- Li, P.; Nijhawan, D.; Budihardjo, I.; Srinivasula, S.M.; Ahmad, M.; Alnemri, E.S.; Wang, X. Cytochrome c and dATP-dependent formation of Apaf-1/caspase-9 complex initiates an apoptotic protease cascade. Cell 1997, 91, 479–489. [Google Scholar] [CrossRef]
- Thornberry, N.A.; Lazebnik, Y. Caspases: Enemies within. Science 1998, 281, 1312–1316. [Google Scholar] [CrossRef]
- Enari, M.; Sakahira, H.; Yokoyama, H.; Okawa, K.; Iwamatsu, A.; Nagata, S. A caspase-activated DNase that degrades DNA during apoptosis, and its inhibitor ICAD. Nature 1998, 391, 43–50. [Google Scholar] [CrossRef]
- Sakahira, H.; Enari, M.; Nagata, S. Cleavage of CAD inhibitor in CAD activation and DNA degradation during apoptosis. Nature 1998, 391, 96–99. [Google Scholar] [CrossRef]
- Liu, X.; Zou, H.; Slaughter, C.; Wang, X. DFF, a heterodimeric protein that functions downstream of caspase-3 to trigger DNA fragmentation during apoptosis. Cell 1997, 89, 175–184. [Google Scholar] [CrossRef]
- Liu, X.; Li, P.; Widlak, P.; Zou, H.; Luo, X.; Garrard, W.T.; Wang, X. The 40-kDa subunit of DNA fragmentation factor induces DNA fragmentation and chromatin condensation during apoptosis. Proc. Natl. Acad. Sci. USA 1998, 95, 8461–8466. [Google Scholar] [CrossRef]
- Du, C.; Fang, M.; Li, Y.; Li, L.; Wang, X. Smac, a mitochondrial protein that promotes cytochrome c-dependent caspase activation by eliminating IAP inhibition. Cell 2000, 102, 33–42. [Google Scholar] [CrossRef] [Green Version]
- Verhagen, A.M.; Ekert, P.G.; Pakusch, M.; Silke, J.; Connolly, L.M.; Reid, G.E.; Moritz, R.L.; Simpson, R.J.; Vaux, D.L. Identification of DIABLO, a mammalian protein that promotes apoptosis by binding to and antagonizing IAP proteins. Cell 2000, 102, 43–53. [Google Scholar] [CrossRef]
- Susin, S.A.; Zamzami, N.; Castedo, M.; Hirsch, T.; Marchetti, P.; Macho, A.; Daugas, E.; Geuskens, M.; Kroemer, G. Bcl-2 inhibits the mitochondrial release of an apoptogenic protease. J. Exp. Med. 1996, 184, 1331–1341. [Google Scholar] [CrossRef] [PubMed]
- Li, L.Y.; Luo, X.; Wang, X. Endonuclease G is an apoptotic DNase when released from mitochondria. Nature 2001, 412, 95–99. [Google Scholar] [CrossRef] [PubMed]
- Tait, S.W.; Ichim, G.; Green, D.R. Die another way--non-apoptotic mechanisms of cell death. J. Cell Sci. 2014, 127, 2135–2144. [Google Scholar] [CrossRef] [PubMed]
- He, S.; Wang, L.; Miao, L.; Wang, T.; Du, F.; Zhao, L.; Wang, X. Receptor interacting protein kinase-3 determines cellular necrotic response to TNF-alpha. Cell 2009, 137, 1100–1111. [Google Scholar] [CrossRef]
- Cho, Y.S.; Challa, S.; Moquin, D.; Genga, R.; Ray, T.D.; Guildford, M.; Chan, F.K. Phosphorylation-driven assembly of the RIP1-RIP3 complex regulates programmed necrosis and virus-induced inflammation. Cell 2009, 137, 1112–1123. [Google Scholar] [CrossRef]
- Sun, L.; Wang, H.; Wang, Z.; He, S.; Chen, S.; Liao, D.; Wang, L.; Yan, J.; Liu, W.; Lei, X.; et al. Mixed lineage kinase domain-like protein mediates necrosis signaling downstream of RIP3 kinase. Cell 2012, 148, 213–227. [Google Scholar] [CrossRef]
- Molnar, T.; Mazlo, A.; Tslaf, V.; Szollosi, A.G.; Emri, G.; Koncz, G. Current translational potential and underlying molecular mechanisms of necroptosis. Cell Death Dis. 2019, 10, 860. [Google Scholar] [CrossRef]
- Tan, Y.; Chen, Q.; Li, X.; Zeng, Z.; Xiong, W.; Li, G.; Li, X.; Yang, J.; Xiang, B.; Yi, M. Pyroptosis: A new paradigm of cell death for fighting against cancer. J. Exp. Clin. Cancer Res. 2021, 40, 153. [Google Scholar] [CrossRef]
- Stockwell, B.R.; Angeli, J.P.F.; Bayir, H.; Bush, A.I.; Conrad, M.; Dixon, S.J.; Fulda, S.; Gascon, S.; Hatzios, S.K.; Kagan, V.E.; et al. Ferroptosis: A Regulated Cell Death Nexus Linking Metabolism, Redox Biology, and Disease. Cell 2017, 171, 273–285. [Google Scholar] [CrossRef] [Green Version]
- Yao, Y.; Chen, Z.; Zhang, H.; Chen, C.; Zeng, M.; Yunis, J.; Wei, Y.; Wan, Y.; Wang, N.; Zhou, M.; et al. Selenium-GPX4 axis protects follicular helper T cells from ferroptosis. Nat. Immunol. 2021, 22, 1127–1139. [Google Scholar] [CrossRef] [PubMed]
- Matsushita, M.; Freigang, S.; Schneider, C.; Conrad, M.; Bornkamm, G.W.; Kopf, M. T cell lipid peroxidation induces ferroptosis and prevents immunity to infection. J. Exp. Med. 2015, 212, 555–568. [Google Scholar] [CrossRef] [PubMed]
- Kuida, K.; Haydar, T.F.; Kuan, C.Y.; Gu, Y.; Taya, C.; Karasuyama, H.; Su, M.S.; Rakic, P.; Flavell, R.A. Reduced apoptosis and cytochrome c-mediated caspase activation in mice lacking caspase 9. Cell 1998, 94, 325–337. [Google Scholar] [CrossRef]
- Hakem, R.; Hakem, A.; Duncan, G.S.; Henderson, J.T.; Woo, M.; Soengas, M.S.; Elia, A.; de la Pompa, J.L.; Kagi, D.; Khoo, W.; et al. Differential requirement for caspase 9 in apoptotic pathways in vivo. Cell 1998, 94, 339–352. [Google Scholar] [CrossRef]
- Ekert, P.G.; Read, S.H.; Silke, J.; Marsden, V.S.; Kaufmann, H.; Hawkins, C.J.; Gerl, R.; Kumar, S.; Vaux, D.L. Apaf-1 and caspase-9 accelerate apoptosis, but do not determine whether factor-deprived or drug-treated cells die. J. Cell Biol. 2004, 165, 835–842. [Google Scholar] [CrossRef]
- Marsden, V.S.; O’Connor, L.; O’Reilly, L.A.; Silke, J.; Metcalf, D.; Ekert, P.G.; Huang, D.C.; Cecconi, F.; Kuida, K.; Tomaselli, K.J.; et al. Apoptosis initiated by Bcl-2-regulated caspase activation independently of the cytochrome c/Apaf-1/caspase-9 apoptosome. Nature 2002, 419, 634–637. [Google Scholar] [CrossRef]
- Tait, S.W.; Green, D.R. Caspase-independent cell death: Leaving the set without the final cut. Oncogene 2008, 27, 6452–6461. [Google Scholar] [CrossRef]
- Zhang, J.; Kodali, S.; Chen, M.; Wang, J. Maintenance of Germinal Center B Cells by Caspase-9 through Promotion of Apoptosis and Inhibition of Necroptosis. J. Immunol. 2020, 205, 113–120. [Google Scholar] [CrossRef]
- Subramaniam, S.; Zirrgiebel, U.; von Halbach, O.B.U.; Strelau, J.; Laliberte, C.; Kaplan, D.R.; Unsicker, K. ERK activation promotes neuronal degeneration predominantly through plasma membrane damage and independently of caspase-3. J. Cell Biol. 2004, 165, 357–369. [Google Scholar] [CrossRef]
- Bosch, R.; Dieguez-Gonzalez, R.; Cespedes, M.V.; Parreno, M.; Pavon, M.A.; Granena, A.; Sierra, J.; Mangues, R.; Casanova, I. A novel inhibitor of focal adhesion signaling induces caspase-independent cell death in diffuse large B-cell lymphoma. Blood 2011, 118, 4411–4420. [Google Scholar] [CrossRef] [Green Version]
- Lavallard, V.J.; Pradelli, L.A.; Paul, A.; Beneteau, M.; Jacquel, A.; Auberger, P.; Ricci, J.E. Modulation of caspase-independent cell death leads to resensitization of imatinib mesylate-resistant cells. Cancer Res. 2009, 69, 3013–3020. [Google Scholar] [CrossRef] [PubMed]
- Kim, R.H.; Coates, J.M.; Bowles, T.L.; McNerney, G.P.; Sutcliffe, J.; Jung, J.U.; Gandour-Edwards, R.; Chuang, F.Y.; Bold, R.J.; Kung, H.J. Arginine deiminase as a novel therapy for prostate cancer induces autophagy and caspase-independent apoptosis. Cancer Res. 2009, 69, 700–708. [Google Scholar] [CrossRef] [PubMed]
- Hurren, R.; Zavareh, R.B.; Dalili, S.; Wood, T.; Rose, D.; Chang, H.; Jamal, N.; Messner, H.; Batey, R.A.; Schimmer, A.D. A novel diquinolonium displays preclinical anti-cancer activity and induces caspase-independent cell death. Apoptosis 2008, 13, 748–755. [Google Scholar] [CrossRef] [PubMed]
- Strauss, G.; Westhoff, M.A.; Fischer-Posovszky, P.; Fulda, S.; Schanbacher, M.; Eckhoff, S.M.; Stahnke, K.; Vahsen, N.; Kroemer, G.; Debatin, K.M. 4-hydroperoxy-cyclophosphamide mediates caspase-independent T-cell apoptosis involving oxidative stress-induced nuclear relocation of mitochondrial apoptogenic factors AIF and EndoG. Cell Death Differ. 2008, 15, 332–343. [Google Scholar] [CrossRef]
- de Milito, A.; Iessi, E.; Logozzi, M.; Lozupone, F.; Spada, M.; Marino, M.L.; Federici, C.; Perdicchio, M.; Matarrese, P.; Lugini, L.; et al. Proton pump inhibitors induce apoptosis of human B-cell tumors through a caspase-independent mechanism involving reactive oxygen species. Cancer Res. 2007, 67, 5408–5417. [Google Scholar] [CrossRef]
- Parreno, M.; Vaque, J.P.; Casanova, I.; Frade, P.; Cespedes, M.V.; Pavon, M.A.; Molins, A.; Camacho, M.; Vila, L.; Nomdedeu, J.F.; et al. Novel triiodophenol derivatives induce caspase-independent mitochondrial cell death in leukemia cells inhibited by Myc. Mol. Cancer Ther. 2006, 5, 1166–1175. [Google Scholar] [CrossRef]
- Daniels, I.; Abulayha, A.M.; Thomson, B.J.; Haynes, A.P. Caspase-independent killing of Burkitt lymphoma cell lines by rituximab. Apoptosis 2006, 11, 1013–1023. [Google Scholar] [CrossRef]
- Torre, L.D.; Nebbioso, A.; Stunnenberg, H.G.; Martens, J.H.A.; Carafa, V.; Altucci, L. The Role of Necroptosis: Biological Relevance and Its Involvement in Cancer. Cancers 2021, 13, 684. [Google Scholar] [CrossRef]
- Hitomi, J.; Christofferson, D.E.; Ng, A.; Yao, J.; Degterev, A.; Xavier, R.J.; Yuan, J. Identification of a molecular signaling network that regulates a cellular necrotic cell death pathway. Cell 2008, 135, 1311–1323. [Google Scholar] [CrossRef]
- Zhang, D.W.; Shao, J.; Lin, J.; Zhang, N.; Lu, B.J.; Lin, S.C.; Dong, M.Q.; Han, J. RIP3, an energy metabolism regulator that switches TNF-induced cell death from apoptosis to necrosis. Science 2009, 325, 332–336. [Google Scholar] [CrossRef]
- Wang, Z.; Jiang, H.; Chen, S.; Du, F.; Wang, X. The mitochondrial phosphatase PGAM5 functions at the convergence point of multiple necrotic death pathways. Cell 2012, 148, 228–243. [Google Scholar] [CrossRef]
- Green, D.R.; Galluzzi, L.; Kroemer, G. Mitochondria and the autophagy-inflammation-cell death axis in organismal aging. Science 2011, 333, 1109–1112. [Google Scholar] [CrossRef] [PubMed]
- Rathmell, J.C.; Lindsten, T.; Zong, W.X.; Cinalli, R.M.; Thompson, C.B. Deficiency in Bak and Bax perturbs thymic selection and lymphoid homeostasis. Nat. Immunol. 2002, 3, 932–939. [Google Scholar] [CrossRef] [PubMed]
- Samraj, A.K.; Keil, E.; Ueffing, N.; Schulze-Osthoff, K.; Schmitz, I. Loss of caspase-9 provides genetic evidence for the type I/II concept of CD95-mediated apoptosis. J. Biol. Chem. 2006, 281, 29652–29659. [Google Scholar] [CrossRef]
- Bu, Y.; Yang, Z.; Li, Q.; Song, F. Silencing of polo-like kinase (Plk) 1 via siRNA causes inhibition of growth and induction of apoptosis in human esophageal cancer cells. Oncology 2008, 74, 198–206. [Google Scholar] [CrossRef] [PubMed]
- Tse, C.; Shoemaker, A.R.; Adickes, J.; Anderson, M.G.; Chen, J.; Jin, S.; Johnson, E.F.; Marsh, K.C.; Mitten, M.J.; Nimmer, P.; et al. ABT-263: A potent and orally bioavailable Bcl-2 family inhibitor. Cancer Res. 2008, 68, 3421–3428. [Google Scholar] [CrossRef]
- Kim, H.J.; Koo, S.Y.; Ahn, B.H.; Park, O.; Park, D.H.; Seo, D.O.; Won, J.H.; Yim, H.J.; Kwak, H.S.; Park, H.S.; et al. NecroX as a novel class of mitochondrial reactive oxygen species and ONOO(-) scavenger. Arch. Pharm. Res. 2010, 33, 1813–1823. [Google Scholar] [CrossRef]
- Degterev, A.; Hitomi, J.; Germscheid, M.; Ch’en, I.L.; Korkina, O.; Teng, X.; Abbott, D.; Cuny, G.D.; Yuan, C.; Wagner, G.; et al. Identification of RIP1 kinase as a specific cellular target of necrostatins. Nat. Chem. Biol. 2008, 4, 313–321. [Google Scholar] [CrossRef]
- Hail, N., Jr.; Chen, P.; Kepa, J.J.; Bushman, L.R.; Shearn, C. Dihydroorotate dehydrogenase is required for N-(4-hydroxyphenyl)retinamide-induced reactive oxygen species production and apoptosis. Free Radic. Biol. Med. 2010, 49, 109–116. [Google Scholar] [CrossRef]
- Mamtani, M.; Kulkarni, H. Association of HADHA expression with the risk of breast cancer: Targeted subset analysis and meta-analysis of microarray data. BMC Res. Notes 2012, 5, 25. [Google Scholar] [CrossRef] [Green Version]
- Karpova, T.; Danchuk, S.; Huang, B.; Popov, K.M. Probing a putative active site of the catalytic subunit of pyruvate dehydrogenase phosphatase 1 (PDP1c) by site-directed mutagenesis. Biochim. Biophys. Acta 2004, 1700, 43–51. [Google Scholar] [CrossRef] [PubMed]
- Khutornenko, A.A.; Roudko, V.V.; Chernyak, B.V.; Vartapetian, A.B.; Chumakov, P.M.; Evstafieva, A.G. Pyrimidine biosynthesis links mitochondrial respiration to the p53 pathway. Proc. Natl. Acad. Sci. USA 2010, 107, 12828–12833. [Google Scholar] [CrossRef] [PubMed]
- Casares, C.; Ramirez-Camacho, R.; Trinidad, A.; Roldan, A.; Jorge, E.; Garcia-Berrocal, J.R. Reactive oxygen species in apoptosis induced by cisplatin: Review of physiopathological mechanisms in animal models. Eur. Arch. Otorhinolaryngol. 2012, 269, 2455–2459. [Google Scholar] [CrossRef]
- Seiler, A.; Schneider, M.; Forster, H.; Roth, S.; Wirth, E.K.; Culmsee, C.; Plesnila, N.; Kremmer, E.; Radmark, O.; Wurst, W.; et al. Glutathione peroxidase 4 senses and translates oxidative stress into 12/15-lipoxygenase dependent- and AIF-mediated cell death. Cell Metab. 2008, 8, 237–248. [Google Scholar] [CrossRef]
- Drummen, G.P.; van Liebergen, L.C.; den Kamp, J.A.F.O.; Post, J.A. C11-BODIPY(581/591), an oxidation-sensitive fluorescent lipid peroxidation probe: (micro)spectroscopic characterization and validation of methodology. Free Radic. Biol. Med. 2002, 33, 473–490. [Google Scholar] [CrossRef]
- Maeda, A.; Crabb, J.W.; Palczewski, K. Microsomal glutathione S-transferase 1 in the retinal pigment epithelium: Protection against oxidative stress and a potential role in aging. Biochemistry 2005, 44, 480–489. [Google Scholar] [CrossRef]
- Ben-Sahra, I.; Dirat, B.; Laurent, K.; Puissant, A.; Auberger, P.; Budanov, A.; Tanti, J.F.; Bost, F. Sestrin2 integrates Akt and mTOR signaling to protect cells against energetic stress-induced death. Cell Death Differ. 2013, 20, 611–619. [Google Scholar] [CrossRef]
- Liu, S.Y.; Lee, Y.J.; Lee, T.C. Association of platelet-derived growth factor receptor beta accumulation with increased oxidative stress and cellular injury in sestrin 2 silenced human glioblastoma cells. FEBS Lett. 2011, 585, 1853–1858. [Google Scholar] [CrossRef]
- Oliver, P.L.; Finelli, M.J.; Edwards, B.; Bitoun, E.; Butts, D.L.; Becker, E.B.; Cheeseman, M.T.; Davies, B.; Davies, K.E. Oxr1 is essential for protection against oxidative stress-induced neurodegeneration. PLoS Genet. 2011, 7, e1002338. [Google Scholar] [CrossRef]
- Elliott, N.A.; Volkert, M.R. Stress induction and mitochondrial localization of Oxr1 proteins in yeast and humans. Mol. Cell. Biol. 2004, 24, 3180–3187. [Google Scholar] [CrossRef] [Green Version]
- Riendeau, D.; Denis, D.; Choo, L.Y.; Nathaniel, D.J. Stimulation of 5-lipoxygenase activity under conditions which promote lipid peroxidation. Biochem. J. 1989, 263, 565–572. [Google Scholar] [CrossRef] [PubMed]
- Jian, W.; Lee, S.H.; Williams, M.V.; Blair, I.A. 5-Lipoxygenase-mediated endogenous DNA damage. J. Biol. Chem. 2009, 284, 16799–16807. [Google Scholar] [CrossRef]
- Sun, Q.Y.; Zhou, H.H.; Mao, X.Y. Emerging Roles of 5-Lipoxygenase Phosphorylation in Inflammation and Cell Death. Oxid. Med. Cell. Longev. 2019, 2019, 2749173. [Google Scholar] [CrossRef]
- Abramovitz, M.; Wong, E.; Cox, M.E.; Richardson, C.D.; Li, C.; Vickers, P.J. 5-lipoxygenase-activating protein stimulates the utilization of arachidonic acid by 5-lipoxygenase. Eur. J. Biochem. 1993, 215, 105–111. [Google Scholar] [CrossRef]
- Flamand, N.; Luo, M.; Peters-Golden, M.; Brock, T.G. Phosphorylation of serine 271 on 5-lipoxygenase and its role in nuclear export. J. Biol. Chem. 2009, 284, 306–313. [Google Scholar] [CrossRef] [PubMed]
- Radmark, O.; Werz, O.; Steinhilber, D.; Samuelsson, B. 5-Lipoxygenase: Regulation of expression and enzyme activity. Trends Biochem. Sci. 2007, 32, 332–341. [Google Scholar] [CrossRef] [PubMed]
- Wada, T.; Penninger, J.M. Mitogen-activated protein kinases in apoptosis regulation. Oncogene 2004, 23, 2838–2849. [Google Scholar] [CrossRef] [PubMed]
- Cheung, E.C.; Slack, R.S. Emerging role for ERK as a key regulator of neuronal apoptosis. Sci. STKE 2004, 2004, PE45. [Google Scholar] [CrossRef]
- Morris, E.J.; Jha, S.; Restaino, C.R.; Dayananth, P.; Zhu, H.; Cooper, A.; Carr, D.; Deng, Y.; Jin, W.; Black, S.; et al. Discovery of a novel ERK inhibitor with activity in models of acquired resistance to BRAF and MEK inhibitors. Cancer Discov. 2013, 3, 742–750. [Google Scholar] [CrossRef]
- Woods, J.W.; Coffey, M.J.; Brock, T.G.; Singer, I.I.; Peters-Golden, M. 5-Lipoxygenase is located in the euchromatin of the nucleus in resting human alveolar macrophages and translocates to the nuclear envelope upon cell activation. J. Clin. Investig. 1995, 95, 2035–2046. [Google Scholar] [CrossRef] [Green Version]
- Henning, W.; Sturzbecher, H.W. Homologous recombination and cell cycle checkpoints: Rad51 in tumour progression and therapy resistance. Toxicology 2003, 193, 91–109. [Google Scholar] [CrossRef]
- Jilani, A.; Ramotar, D.; Slack, C.; Ong, C.; Yang, X.M.; Scherer, S.W.; Lasko, D.D. Molecular cloning of the human gene, PNKP, encoding a polynucleotide kinase 3′-phosphatase and evidence for its role in repair of DNA strand breaks caused by oxidative damage. J. Biol. Chem. 1999, 274, 24176–24186. [Google Scholar] [CrossRef] [PubMed]
- Rigou, P.; Piddubnyak, V.; Faye, A.; Rain, J.C.; Michel, L.; Calvo, F.; Poyet, J.L. The antiapoptotic protein AAC-11 interacts with and regulates Acinus-mediated DNA fragmentation. EMBO J. 2009, 28, 1576–1588. [Google Scholar] [CrossRef] [PubMed]
- Tewari, M.; Yu, M.; Ross, B.; Dean, C.; Giordano, A.; Rubin, R. AAC-11, a novel cDNA that inhibits apoptosis after growth factor withdrawal. Cancer Res. 1997, 57, 4063–4069. [Google Scholar]
- Wu, M.; Xu, L.G.; Li, X.; Zhai, Z.; Shu, H.B. AMID, an apoptosis-inducing factor-homologous mitochondrion-associated protein, induces caspase-independent apoptosis. J. Biol. Chem. 2002, 277, 25617–25623. [Google Scholar] [CrossRef] [PubMed]
- Tian, Q.; Streuli, M.; Saito, H.; Schlossman, S.F.; Anderson, P. A polyadenylate binding protein localized to the granules of cytolytic lymphocytes induces DNA fragmentation in target cells. Cell 1991, 67, 629–639. [Google Scholar] [CrossRef]
- Masuda, K.; Marasa, B.; Martindale, J.L.; Halushka, M.K.; Gorospe, M. Tissue- and age-dependent expression of RNA-binding proteins that influence mRNA turnover and translation. Aging 2009, 1, 681–698. [Google Scholar] [CrossRef]
- Fernandez-Gomez, A.; Izquierdo, J.M. The Multifunctional Faces of T-Cell Intracellular Antigen 1 in Health and Disease. Int. J. Mol. Sci. 2022, 23, 1400. [Google Scholar] [CrossRef]
- Izquierdo, J.M.; Alcalde, J.; Carrascoso, I.; Reyes, R.; Ludena, M.D. Knockdown of T-cell intracellular antigens triggers cell proliferation, invasion and tumour growth. Biochem. J. 2011, 435, 337–344. [Google Scholar] [CrossRef]
- Mebratu, Y.; Tesfaigzi, Y. How ERK1/2 activation controls cell proliferation and cell death: Is subcellular localization the answer? Cell Cycle 2009, 8, 1168–1175. [Google Scholar] [CrossRef]
- Lesuisse, C.; Martin, L.J. Immature and mature cortical neurons engage different apoptotic mechanisms involving caspase-3 and the mitogen-activated protein kinase pathway. J. Cereb. Blood Flow Metab. 2002, 22, 935–950. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.J.; Lee, N.K.; Lee, N.Y.; Lee, J.W.; Park, S.J. Cell death mediated by Vibrio parahaemolyticus type III secretion system 1 is dependent on ERK1/2 MAPK, but independent of caspases. J. Microbiol. Biotechnol. 2011, 21, 903–913. [Google Scholar] [CrossRef] [PubMed]
- Apostolov, E.O.; Ray, D.; Alobuia, W.M.; Mikhailova, M.V.; Wang, X.; Basnakian, A.G.; Shah, S.V. Endonuclease G mediates endothelial cell death induced by carbamylated LDL. Am. J. Physiol. Heart Circ. Physiol. 2011, 300, H1997–H2004. [Google Scholar] [CrossRef] [PubMed]
- Chu, C.T.; Levinthal, D.J.; Kulich, S.M.; Chalovich, E.M.; DeFranco, D.B. Oxidative neuronal injury. The dark side of ERK1/2. Eur. J. Biochem. 2004, 271, 2060–2066. [Google Scholar] [CrossRef] [PubMed]
- Su, L.J.; Zhang, J.H.; Gomez, H.; Murugan, R.; Hong, X.; Xu, D.; Jiang, F.; Peng, Z.Y. Reactive Oxygen Species-Induced Lipid Peroxidation in Apoptosis, Autophagy, and Ferroptosis. Oxid. Med. Cell. Longev. 2019, 2019, 5080843. [Google Scholar] [CrossRef]
- Yamasaki, S.; Stoecklin, G.; Kedersha, N.; Simarro, M.; Anderson, P. T-cell intracellular antigen-1 (TIA-1)-induced translational silencing promotes the decay of selected mRNAs. J. Biol. Chem. 2007, 282, 30070–30077. [Google Scholar] [CrossRef]
- Forch, P.; Puig, O.; Kedersha, N.; Martinez, C.; Granneman, S.; Seraphin, B.; Anderson, P.; Valcarcel, J. The apoptosis-promoting factor TIA-1 is a regulator of alternative pre-mRNA splicing. Mol. Cell 2000, 6, 1089–1098. [Google Scholar] [CrossRef]
- Kawakami, A.; Tian, Q.; Duan, X.; Streuli, M.; Schlossman, S.F.; Anderson, P. Identification and functional characterization of a TIA-1-related nucleolysin. Proc. Natl. Acad. Sci. USA 1992, 89, 8681–8685. [Google Scholar] [CrossRef]
- McAlinden, A.; Liang, L.; Mukudai, Y.; Imamura, T.; Sandell, L.J. Nuclear protein TIA-1 regulates COL2A1 alternative splicing and interacts with precursor mRNA and genomic DNA. J. Biol. Chem. 2007, 282, 24444–24454. [Google Scholar] [CrossRef]
- Yang, F.; Peng, Y.; Murray, E.L.; Otsuka, Y.; Kedersha, N.; Schoenberg, D.R. Polysome-bound endonuclease PMR1 is targeted to stress granules via stress-specific binding to TIA-1. Mol. Cell Biol. 2006, 26, 8803–8813. [Google Scholar] [CrossRef]
- Wu, L.; Miao, S.; Zou, L.B.; Wu, P.; Hao, H.; Tang, K.; Zeng, P.; Xiong, J.; Li, H.H.; Wu, Q.; et al. Lipoxin A4 inhibits 5-lipoxygenase translocation and leukotrienes biosynthesis to exert a neuroprotective effect in cerebral ischemia/reperfusion injury. J. Mol. Neurosci. 2012, 48, 185–200. [Google Scholar] [CrossRef] [PubMed]





Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chen, M.; Wang, L.; Li, M.; Budai, M.M.; Wang, J. Mitochondrion-Mediated Cell Death through Erk1-Alox5 Independent of Caspase-9 Signaling. Cells 2022, 11, 3053. https://doi.org/10.3390/cells11193053
Chen M, Wang L, Li M, Budai MM, Wang J. Mitochondrion-Mediated Cell Death through Erk1-Alox5 Independent of Caspase-9 Signaling. Cells. 2022; 11(19):3053. https://doi.org/10.3390/cells11193053
Chicago/Turabian StyleChen, Min, Lei Wang, Min Li, Marietta M. Budai, and Jin Wang. 2022. "Mitochondrion-Mediated Cell Death through Erk1-Alox5 Independent of Caspase-9 Signaling" Cells 11, no. 19: 3053. https://doi.org/10.3390/cells11193053
APA StyleChen, M., Wang, L., Li, M., Budai, M. M., & Wang, J. (2022). Mitochondrion-Mediated Cell Death through Erk1-Alox5 Independent of Caspase-9 Signaling. Cells, 11(19), 3053. https://doi.org/10.3390/cells11193053