


K63 Ubiquitination of P21 Can Facilitate Pellino-1 in the Context of Chronic Obstructive Pulmonary Disease and Lung Cellular Senescence
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Cell Lines
2.2. Mice Experiments
2.3. MTT Assay
2.4. Transfection of siRNA
2.5. RT-qPCR
2.6. Western Blot
2.7. Analysis of Bronchoalveolar Lavage Fluid (BALF)
2.8. Cell Cycle Distribution
2.9. Immunofluorescence Staining Assay
2.10. Immunoprecipitation
2.11. Degradation of P21
2.12. Preparation of Cigarette Smoking Extraction (CSE)
2.13. Immunohistochemistry (IHC) Staining
2.14. Adenovirus and Plasmids Construction
2.15. Quantification and Statistical Analysis
3. Results
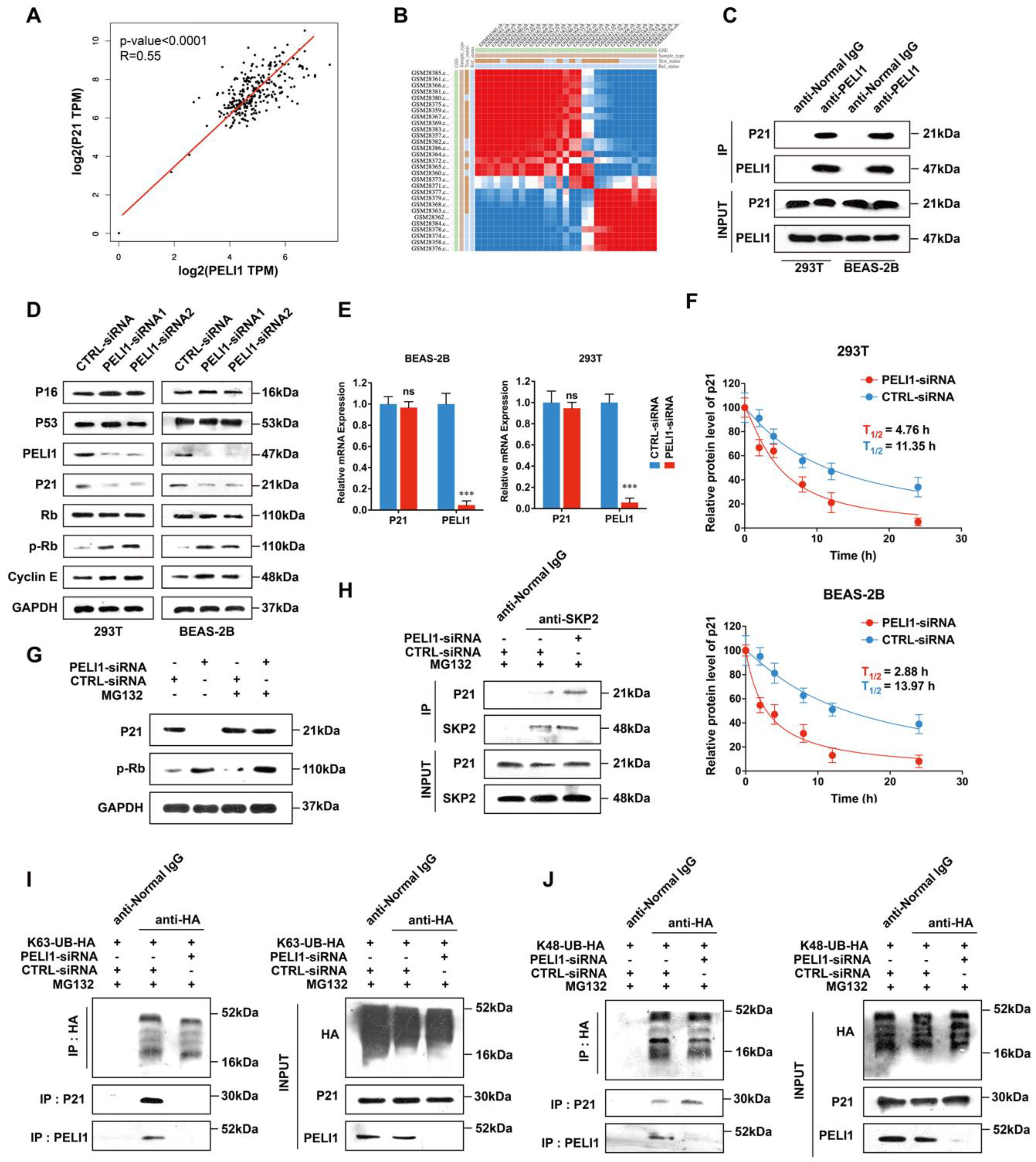
3.1. E3 Ligase Pellino-1 Ubiquitylates P21 at the K63 Site
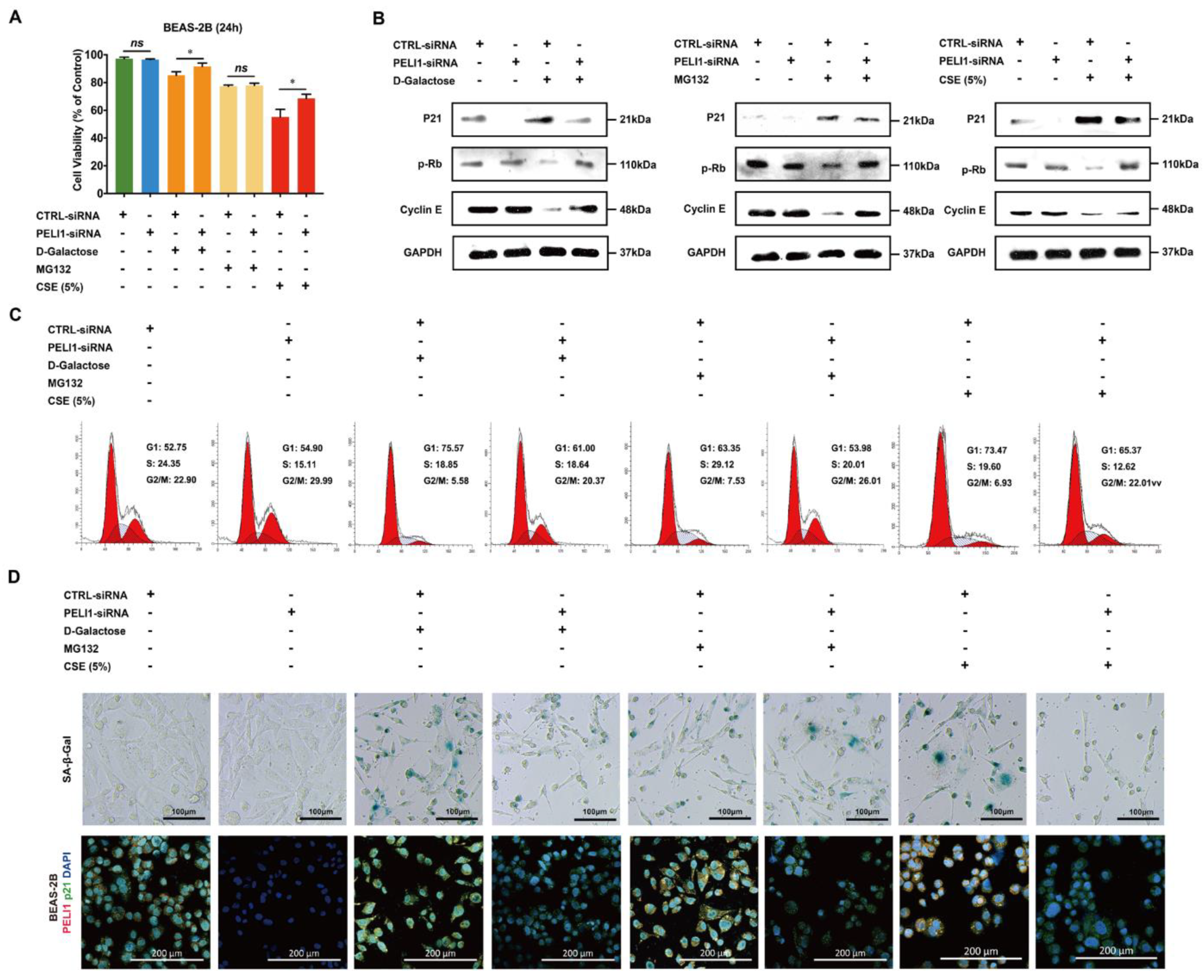
3.2. Silencing Pellino-1 Inhibits Senescence by P21-Mediated Cell-Cycle Arrest
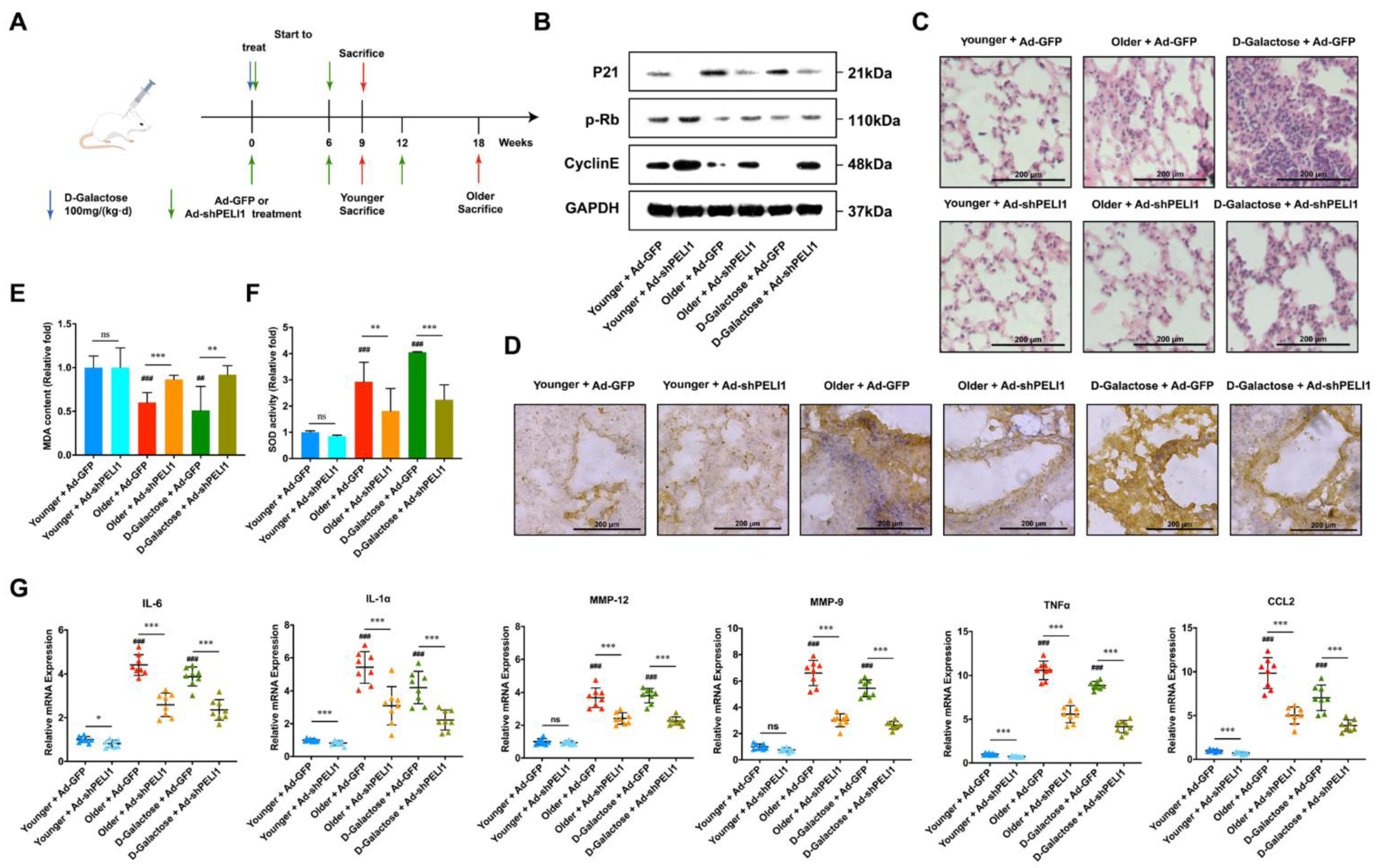
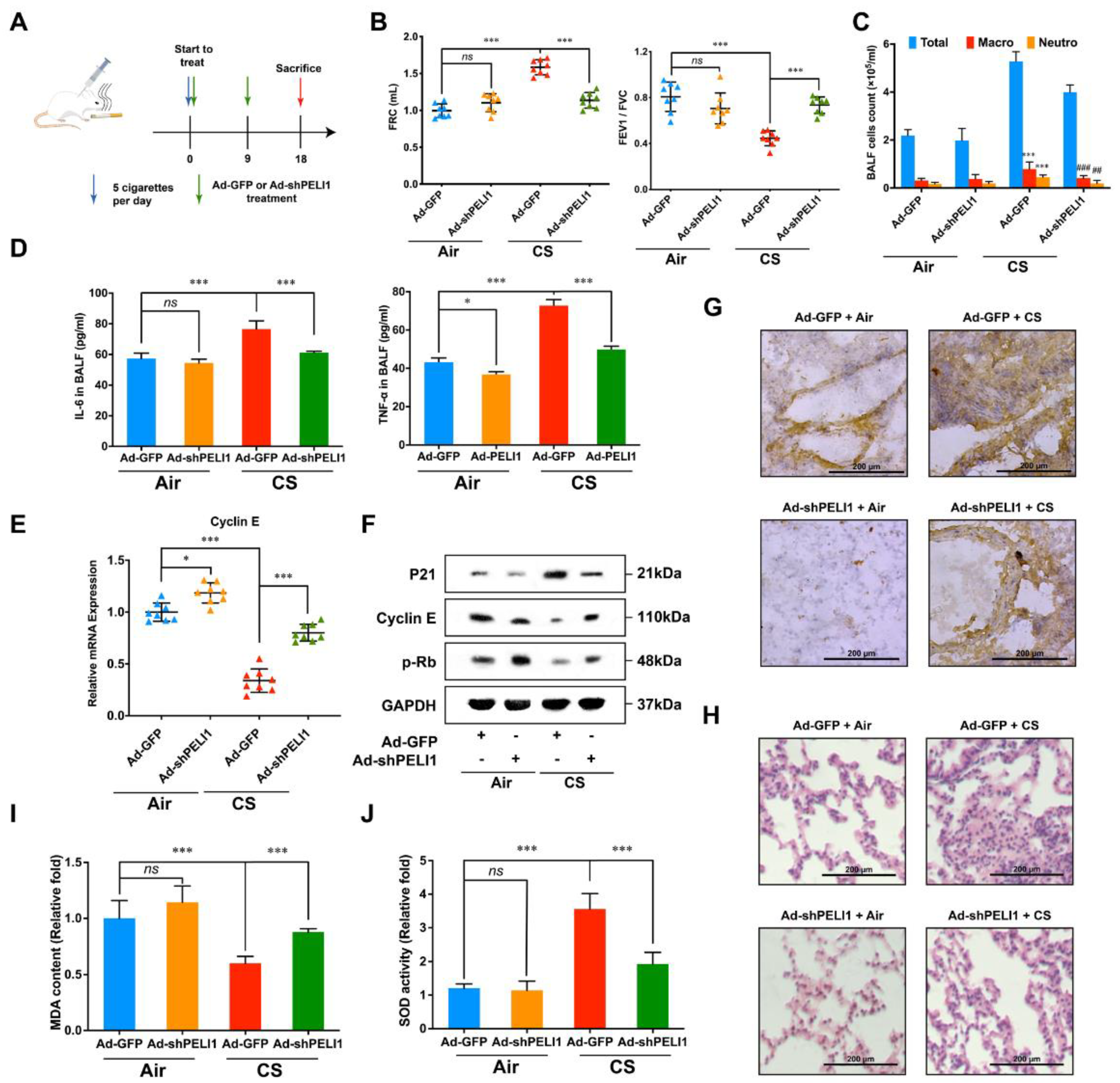
3.3. Silencing Pellino-1 Inhibits Senescence by Decreasing the P21 Level In Vivo
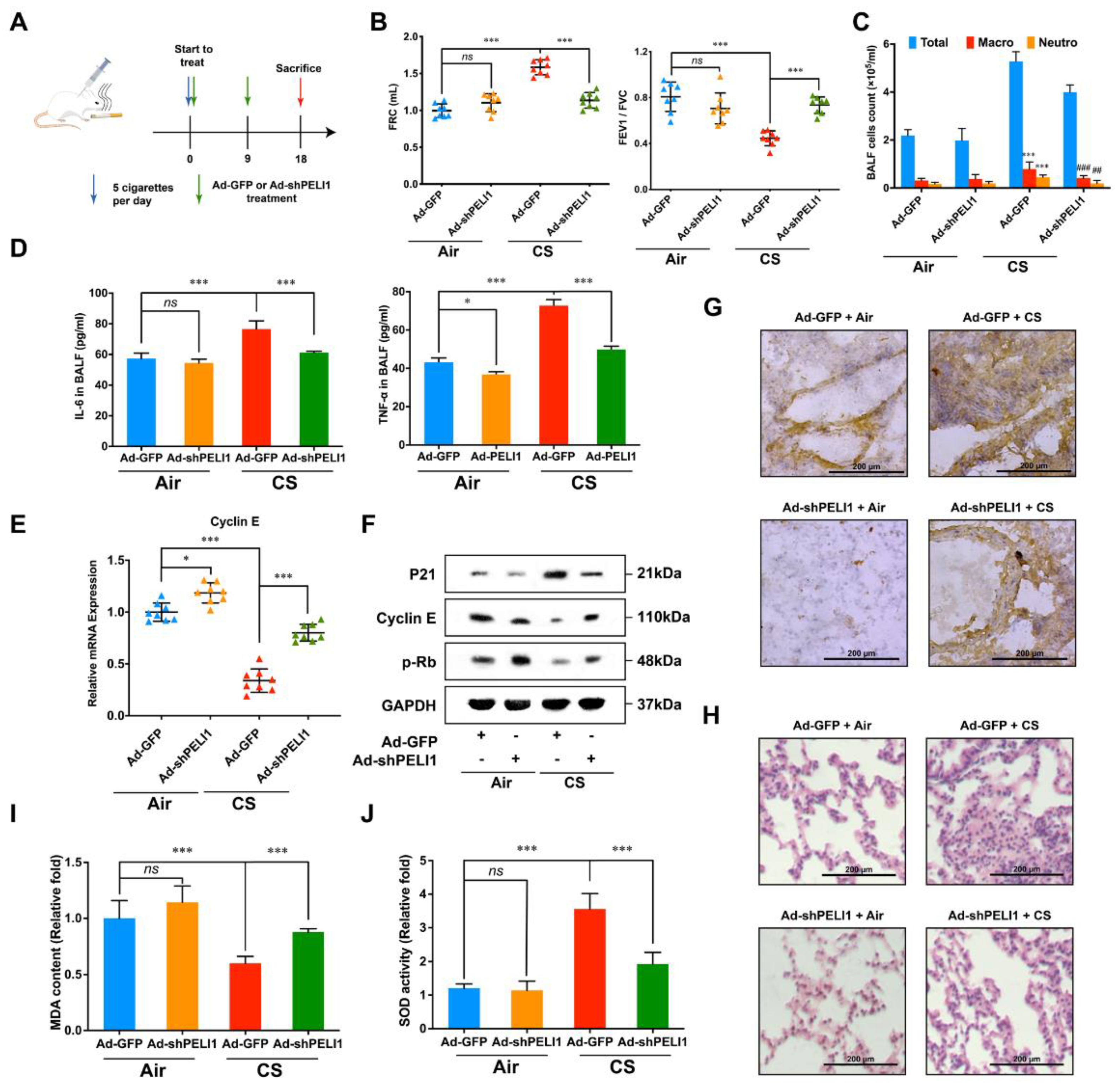
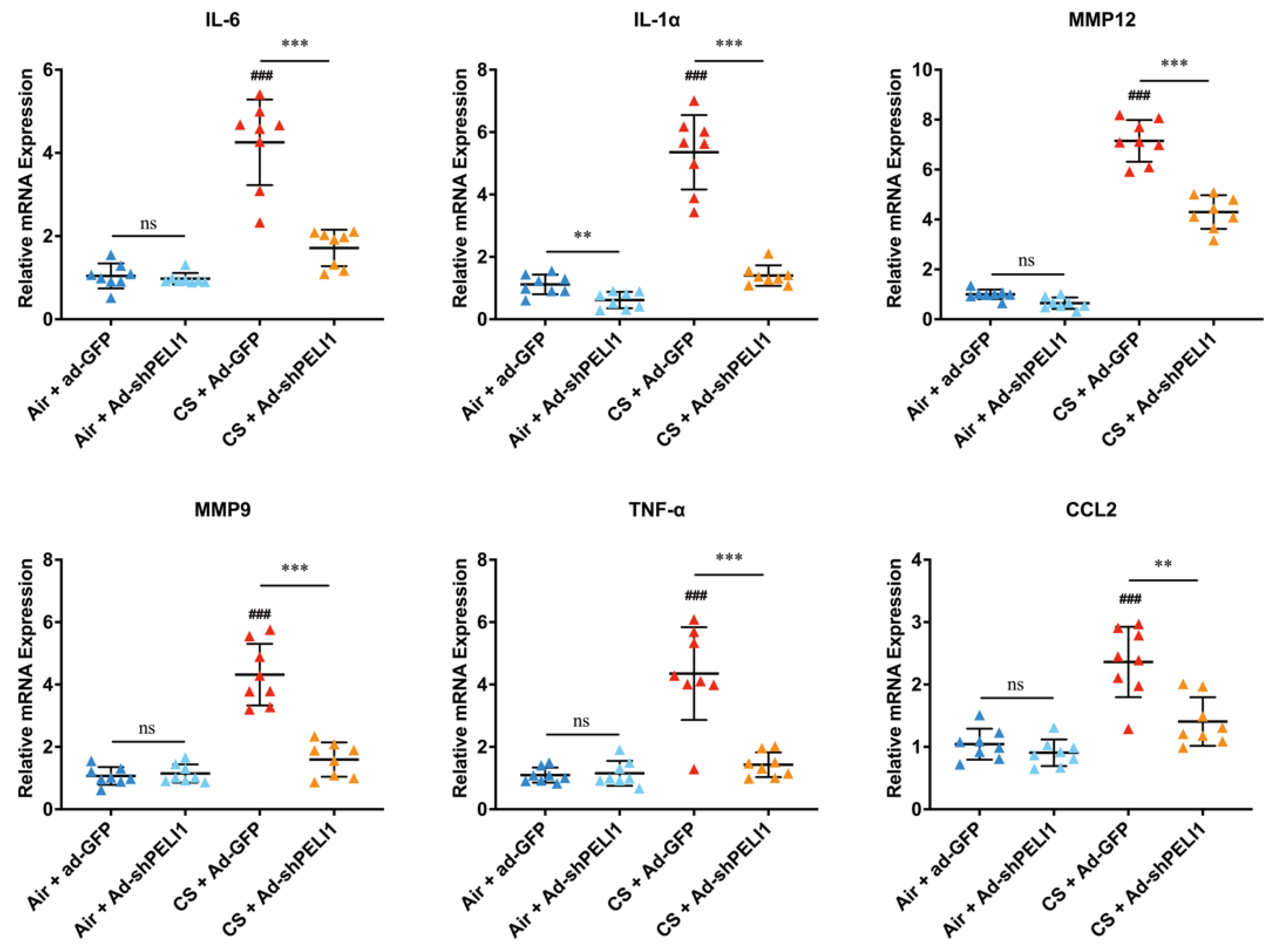
3.4. Silencing Pellino-1 Inhibits COPD and SASPs In Vivo
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Hogg, J.C.; Timens, W. The Pathology of Chronic Obstructive Pulmonary Disease. Annu. Rev. Pathol.-Mech. Dis. 2009, 4, 435–459. [Google Scholar] [CrossRef] [PubMed]
- Hong, S.; Park, E.C.; Kim, T.H.; Kwon, J.A.; Yoo, K.B.; Han, K.T.; Yoo, J.W.; Kim, S.J. Effect of pre existing respiratory conditions on survival of lung cancer patients: A nationwide population-based cohort study. Asia-Pac. J. Clin. Oncol. 2018, 14, e71–e80. [Google Scholar] [CrossRef] [PubMed]
- Keller, A.; Fehlmann, T.; Ludwig, N.; Kahraman, M.; Laufer, T.; Backes, C.; Vogelmeier, C.; Diener, C.; Biertz, F.; Herr, C.; et al. Genome-wide MicroRNA Expression Profiles in COPD: Early Predictors for Cancer Development. Genom. Proteom. Bioinform. 2018, 16, 162–171. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Y.; Zhong, N.S.; Li, X.; Chen, S.; Zheng, J.; Zhao, D.; Yao, W.; Zhi, R.; Wei, L.; He, B.; et al. Tiotropium in Early-Stage Chronic Obstructive Pulmonary Disease. N. Engl. J. Med. 2017, 377, 923–935. [Google Scholar] [CrossRef] [PubMed]
- Barnes, P.J. Cellular and molecular mechanisms of asthma and COPD. Clin. Sci. 2017, 131, 1541–1558. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fischer, B.M.; Pavlisko, E.; Voynow, J.A. Pathogenic triad in COPD: Oxidative stress, protease-antiprotease imbalance, and inflammation. Int. J. Chronic Obstr. Pulm. Dis. 2011, 6, 413–421. [Google Scholar] [CrossRef] [Green Version]
- Fischer, B.M.; Voynow, J.A.; Ghio, A.J. COPD: Balancing oxidants and antioxidants. Int. J. Chronic Obstr. Pulm. Dis. 2015, 10, 261–276. [Google Scholar] [CrossRef] [Green Version]
- Tuder, R.M.; Kern, J.A.; Miller, Y.E. Senescence in chronic obstructive pulmonary disease. Proc. Am. Thorac. Soc. 2012, 9, 62–63. [Google Scholar] [CrossRef]
- MacNee, W. Accelerated lung aging: A novel pathogenic mechanism of chronic obstructive pulmonary disease (COPD). Biochem. Soc. Trans. 2009, 37, 819–823. [Google Scholar] [CrossRef]
- Ito, K.; Barnes, P.J. COPD as a Disease of Accelerated Lung Aging. Chest 2009, 135, 173–180. [Google Scholar] [CrossRef]
- Lange, P.; Celli, B.; Agustí, A.; Boje Jensen, G.; Divo, M.; Faner, R.; Guerra, S.; Marott, J.L.; Martinez, F.D.; Martinez-Camblor, P.; et al. Lung-Function Trajectories Leading to Chronic Obstructive Pulmonary Disease. N. Engl. J. Med. 2015, 373, 111–122. [Google Scholar] [CrossRef] [Green Version]
- Aghapour, M.; Raee, P.; Moghaddam, S.J.; Hiemstra, P.S.; Heijink, I.H. Airway Epithelial Barrier Dysfunction in Chronic Obstructive Pulmonary Disease: Role of Cigarette Smoke Exposure. Am. J. Respir. Cell Mol. Biol. 2018, 58, 157–169. [Google Scholar] [CrossRef] [Green Version]
- Hou, W.; Hu, S.; Li, C.; Ma, H.; Wang, Q.; Meng, G.; Guo, T.; Zhang, J. Cigarette Smoke Induced Lung Barrier Dysfunction, EMT, and Tissue Remodeling: A Possible Link between COPD and Lung Cancer. BioMed. Res. Int. 2019, 2019, 2025636. [Google Scholar] [CrossRef]
- MacNee, W. Is Chronic Obstructive Pulmonary Disease an Accelerated Aging Disease? Ann. Am. Thorac. Soc. 2016, 13 (Suppl. 5), S429–S437. [Google Scholar] [CrossRef]
- Bowdish, D.M.E. The Aging Lung Is Lung Health Good Health for Older Adults? Chest 2019, 155, 391–400. [Google Scholar] [CrossRef]
- Hecker, L. Mechanisms and consequences of oxidative stress in lung disease: Therapeutic implications for an aging populace. Am. J. Physiol.-Lung Cell. Mol. Physiol. 2018, 314, 1642–1653. [Google Scholar] [CrossRef]
- Ascher, K.; Elliot, S.J.; Rubio, G.A.; Glassberg, M.K. Lung Diseases of the Elderly: Cellular Mechanisms. Clin. Geriatr. Med. 2017, 33, 473–490. [Google Scholar] [CrossRef]
- Tchkonia, T.; Zhu, Y.; van Deursen, J.; Campisi, J.; Kirkland, J.L. Cellular senescence and the senescent secretory phenotype: Therapeutic opportunities. J. Clin. Investig. 2013, 123, 966–972. [Google Scholar] [CrossRef] [Green Version]
- Beghe, B.; Cerri, S.; Fabbri, L.M.; Marchioni, A. COPD, Pulmonary Fibrosis and ILAs in Aging Smokers: The Paradox of Striking Different Responses to the Major Risk Factors. Int. J. Mol. Sci. 2021, 22, 9292. [Google Scholar] [CrossRef]
- Salama, R.; Sadaie, M.; Hoare, M.; Narita, M. Cellular senescence and its effector programs. Genes Dev. 2014, 28, 99–114. [Google Scholar] [CrossRef]
- Hacievliyagil, S.S.; Mutlu, L.C.; Temel, İ. Airway inflammatory markers in chronic obstructive pulmonary disease patients and healthy smokers. Niger. J. Clin. Pract. 2013, 16, 76–81. [Google Scholar] [CrossRef] [Green Version]
- Barnes, P.J.; Baker, J.; Donnelly, L.E. Cellular Senescence as a Mechanism and Target in Chronic Lung Diseases. Am. J. Respir. Crit. Care Med. 2019, 200, 556–564. [Google Scholar] [CrossRef]
- Hughes, B.M.; Burton, C.S.; Reese, A.; Jabeen, M.F.; Wright, C.; Willis, J.; Khoshaein, N.; Marsh, E.K.; Peachell, P.; Sun, S.C.; et al. Pellino-1 Regulates Immune Responses to Haemophilus influenzae in Models of Inflammatory Lung Disease. Front. Immunol. 2019, 10, 1721. [Google Scholar] [CrossRef] [Green Version]
- Lim, R.; Barker, G.; Lappas, M. Pellino 1 is a novel regulator of TNF and TLR signalling in human myometrial and amnion cells. J. Reprod. Immunol. 2018, 127, 24–35. [Google Scholar] [CrossRef]
- Medvedev, A.E.; Murphy, M.; Zhou, H.; Li, X.X. E3 ubiquitin ligases Pellinos as regulators of pattern recognition receptor signaling and immune responses. Immunol. Rev. 2015, 266, 109–122. [Google Scholar] [CrossRef] [Green Version]
- Marsh, E.K.; Prestwich, E.C.; Williams, L.; Hart, A.R.; Muir, C.F.; Parker, L.C.; Jonker, M.R.; Heijink, I.H.; Timens, W.; Fife, M.; et al. Pellino-1 Regulates the Responses of the Airway to Viral Infection. Front. Cell. Infect. Microbiol. 2020, 10, 456. [Google Scholar] [CrossRef]
- Jeon, Y.K.; Kim, C.K.; Hwang, K.R.; Park, H.-Y.; Koh, J.; Chung, D.H.; Lee, C.-W.; Ha, G.-H. Pellino-1 promotes lung carcinogenesis via the stabilization of Slug and Snail through K63-mediated polyubiquitination. Cell Death Differ. 2017, 24, 469–480. [Google Scholar] [CrossRef] [Green Version]
- Jeon, Y.K.; Kim, C.K.; Koh, J.; Chung, D.H.; Ha, G.-H. Pellino-1 confers chemoresistance in lung cancer cells by upregulating cIAP2 through Lys63-mediated polyubiquitination. Oncotarget 2016, 7, 41811–41824. [Google Scholar] [CrossRef] [Green Version]
- Bennett, J.A.; Prince, L.R.; Parker, L.C.; Stokes, C.A.; de Bruin, H.G.; van den Berge, M.; Heijink, I.H.; Whyte, M.K.; Sabroe, I. Pellino-1 Selectively Regulates Epithelial Cell Responses to Rhinovirus. J. Virol. 2012, 86, 6595–6604. [Google Scholar] [CrossRef] [Green Version]
- Wang, Y.Q.; Wang, X.Q.; Zhang, H.; Han, B.; Ye, Y.M.; Zhang, M.J.; Wang, Y.B.; Xue, J.J.; Wang, C.A. Transforming Growth Factor-beta 1 Promotes M1 Alveolar Macrophage Polarization in Acute Lung Injury by Up-Regulating DNMT1 to Mediate the microRNA-124/PELI1/IRF5 Axis. Front. Cell. Infect. Microbiol. 2021, 11, 693981. [Google Scholar] [CrossRef]
- Lei, L.; Bandola-Simon, J.; Roche, P.A. Ubiquitin-conjugating enzyme E2 D1 (Ube2D1) mediates lysine-independent ubiquitination of the E3 ubiquitin ligase March-I. J. Biol. Chem. 2018, 293, 3904–3912. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, W.; Yang, X.; Chen, L.; Liu, Y.Y.; Venkatarangan, V.; Reist, L.; Hanson, P.; Xu, H.; Wang, Y.; Li, M. A conserved ubiquitin- and ESCRT-dependent pathway internalizes human lysosomal membrane proteins for degradation. PLoS Biol. 2021, 19, e3001361. [Google Scholar] [CrossRef] [PubMed]
- Sun, X.; Dong, Z.; Li, N.; Feng, X.; Liu, Y.; Li, A.; Zhu, X.; Li, C.; Zhao, Z. Nucleosides isolated from Ophiocordyceps sinensis inhibit cigarette smoke extract-induced inflammation via the SIRT1-nuclear factor-κB/p65 pathway in RAW264.7 macrophages and in COPD mice. Int. J. Chronic Obstr. Pulm. Dis. 2018, 13, 2821–2832. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lim, K.L.; Chew, K.C.; Tan, J.M.; Wang, C.; Chung, K.K.; Zhang, Y.; Tanaka, Y.; Smith, W.; Engelender, S.; Ross, C.A.; et al. Parkin mediates nonclassical, proteasomal-independent ubiquitination of synphilin-1: Implications for Lewy body formation. J. Neurosci. Off. J. Soc. Neurosci. 2005, 25, 2002–2009. [Google Scholar] [CrossRef] [Green Version]
- Smith, H.; Peggie, M.; Campbell, D.G.; Vandermoere, F.; Carrick, E.; Cohen, P. Identification of the phosphorylation sites on the E3 ubiquitin ligase Pellino that are critical for activation by IRAK1 and IRAK4. Proc. Natl. Acad. Sci. USA 2009, 106, 4584–4590. [Google Scholar] [CrossRef] [Green Version]
- Choi, K.C.; Lee, Y.S.; Lim, S.; Choi, H.K.; Lee, C.H.; Lee, E.K.; Hong, S.; Kim, I.H.; Kim, S.J.; Park, S.H. Smad6 negatively regulates interleukin 1-receptor Toll-like receptor signaling through direct interaction with the adaptor Pellino-1. Nat. Immunol. 2006, 7, 1057–1065. [Google Scholar] [CrossRef]
- Jiang, Z.F.; Johnson, H.J.; Nie, H.Q.; Qin, J.Z.; Bird, T.A.; Li, X.X. Pellino 1 is required for interleukin-1 (IL-1)-mediated signaling through its interaction with the IL-1 receptor-associated kinase 4 (IRAK4)-IRAK-tumor necrosis factor receptor-associated factor 6 (TRAF6) complex. J. Biol. Chem. 2003, 278, 10952–10956. [Google Scholar] [CrossRef] [Green Version]
- Bornstein, G.; Bloom, J.; Sitry-Shevah, D.; Nakayama, K.; Pagano, M.; Hershko, A. Role of the SCFSkp2 ubiquitin ligase in the degradation of p21(Cip1) in S phase. J. Biol. Chem. 2003, 278, 25752–25757. [Google Scholar] [CrossRef] [Green Version]
- Yu, Z.K.; Gervais, J.L.M.; Zhang, H. Human CUL-1 associates with the SKP1/SKP2 complex and regulates p21(CIP1/WAF1) and cyclin D proteins. Proc. Natl. Acad. Sci. USA 1998, 95, 11324–11329. [Google Scholar] [CrossRef] [Green Version]
- Chen, P.; Chen, F.C.; Zhou, B.H. Leonurine ameliorates D-galactose-induced aging in mice through activation of the Nrf2 signalling pathway. Aging 2019, 11, 7339–7356. [Google Scholar] [CrossRef]
- Azman, K.F.; Zakaria, R. D-Galactose-induced accelerated aging model: An overview. Biogerontology 2019, 20, 763–782. [Google Scholar] [CrossRef]
- Miller, M.R. Structural and physiological age-associated changes in aging lungs. Semin. Respir. Crit. Care Med. 2010, 31, 521–527. [Google Scholar] [CrossRef]
- Yeh, S.L.; Wu, T.C.; Chan, S.T.; Hong, M.J.; Chen, H.L. Fructo-oligosaccharide attenuates the production of pro-inflammatory cytokines and the activation of JNK/Jun pathway in the lungs of D-galactose-treated Balb/cJ mice. Eur. J. Nutr. 2014, 53, 449–456. [Google Scholar] [CrossRef]
- Lozano, R.; Naghavi, M.; Foreman, K.; Lim, S.; Shibuya, K.; Aboyans, V.; Abraham, J.; Adair, T.; Aggarwal, R.; Ahn, S.Y.; et al. Global and regional mortality from 235 causes of death for 20 age groups in 1990 and 2010: A systematic analysis for the Global Burden of Disease Study 2010. Lancet 2012, 380, 2095–2128. [Google Scholar] [CrossRef]
- Halpin, D.M.G.; Criner, G.J.; Papi, A.; Singh, D.; Anzueto, A.; Martinez, F.J.; Agusti, A.A.; Vogelmeier, C.F. Global Initiative for the Diagnosis, Management, and Prevention of Chronic Obstructive Lung Disease. The 2020 GOLD Science Committee Report on COVID-19 and Chronic Obstructive Pulmonary Disease. Am. J. Respir. Crit. Care Med. 2021, 203, 24–36. [Google Scholar] [CrossRef]
- Hogg, J.C.; Chu, F.; Utokaparch, S.; Woods, R.; Elliott, W.M.; Buzatu, L.; Cherniack, R.M.; Rogers, R.M.; Sciurba, F.C.; Coxson, H.O.; et al. The nature of small-airway obstruction in chronic obstructive pulmonary disease. N. Engl. J. Med. 2004, 350, 2645–2653. [Google Scholar] [CrossRef]
- Cohen, A.J.; Brauer, M.; Burnett, R.; Anderson, H.R.; Frostad, J.; Estep, K.; Balakrishnan, K.; Brunekreef, B.; Dandona, L.; Dandona, R.; et al. Estimates and 25-year trends of the global burden of disease attributable to ambient air pollution: An analysis of data from the Global Burden of Diseases Study 2015. Lancet 2017, 389, 1907–1918. [Google Scholar] [CrossRef] [Green Version]
- Karrasch, S.; Holz, O.; Jörres, R.A. Aging and induced senescence as factors in the pathogenesis of lung emphysema. Respir. Med. 2008, 102, 1215–1230. [Google Scholar] [CrossRef] [Green Version]
- Sherr, C.J.; Roberts, J.M. CDK inhibitors: Positive and negative regulators of G(1)-phase progression. Genes Dev. 1999, 13, 1501–1512. [Google Scholar] [CrossRef] [Green Version]
- Sherr, C.J.; Roberts, J.M. Inhibitors of Mammalian G(1) Cyclin-Dependent Kinases. Genes Dev. 1995, 9, 1149–1163. [Google Scholar] [CrossRef]
- Abbas, T.; Dutta, A. p21 in cancer: Intricate networks and multiple activities. Nat. Rev. Cancer 2009, 9, 400–414. [Google Scholar] [CrossRef]
- Xing, L.; Tang, X.; Wu, K.; Huang, X.; Yi, Y.; Huan, J. TRIM27 Functions as a Novel Oncogene in Non-Triple-Negative Breast Cancer by Blocking Cellular Senescence through p21 Ubiquitination. Mol. Ther. Nucleic Acids 2020, 22, 910–923. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Chen, J.; Ning, D.; Liu, Q.; Wang, C.; Zhang, Z.; Chu, L.; Yu, C.; Liang, H.F.; Zhang, B.; et al. FBXO22 promotes the development of hepatocellular carcinoma by regulating the ubiquitination and degradation of p21. J. Exp. Clin. Cancer Res. CR 2019, 38, 101. [Google Scholar] [CrossRef] [Green Version]
- Jiang, B.; Guan, Y.; Shen, H.J.; Zhang, L.H.; Jiang, J.X.; Dong, X.W.; Shen, H.H.; Xie, Q.M. Akt/PKB signaling regulates cigarette smoke-induced pulmonary epithelial-mesenchymal transition. Lung Cancer 2018, 122, 44–53. [Google Scholar] [CrossRef]
- Eapen, M.S.; Sharma, P.; Gaikwad, A.V.; Lu, W.; Myers, S.; Hansbro, P.M.; Sohal, S.S. Epithelial-mesenchymal transition is driven by transcriptional and post transcriptional modulations in COPD: Implications for disease progression and new therapeutics. Int. J. Chronic Obstr. Pulm. Dis. 2019, 14, 1603–1610. [Google Scholar] [CrossRef]

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ma, J.-H.; Zhang, Y.-T.; Wang, L.-P.; Sun, Q.-Y.; Zhang, H.; Li, J.-J.; Han, N.-N.; Zhu, Y.-Y.; Xie, X.-Y.; Li, X. K63 Ubiquitination of P21 Can Facilitate Pellino-1 in the Context of Chronic Obstructive Pulmonary Disease and Lung Cellular Senescence. Cells 2022, 11, 3115. https://doi.org/10.3390/cells11193115
Ma J-H, Zhang Y-T, Wang L-P, Sun Q-Y, Zhang H, Li J-J, Han N-N, Zhu Y-Y, Xie X-Y, Li X. K63 Ubiquitination of P21 Can Facilitate Pellino-1 in the Context of Chronic Obstructive Pulmonary Disease and Lung Cellular Senescence. Cells. 2022; 11(19):3115. https://doi.org/10.3390/cells11193115
Chicago/Turabian StyleMa, Jia-Hui, Yi-Ting Zhang, Lu-Ping Wang, Qing-Yu Sun, Hao Zhang, Jian-Jiang Li, Ning-Ning Han, Yao-Yao Zhu, Xiao-Yu Xie, and Xia Li. 2022. "K63 Ubiquitination of P21 Can Facilitate Pellino-1 in the Context of Chronic Obstructive Pulmonary Disease and Lung Cellular Senescence" Cells 11, no. 19: 3115. https://doi.org/10.3390/cells11193115
APA StyleMa, J.-H., Zhang, Y.-T., Wang, L.-P., Sun, Q.-Y., Zhang, H., Li, J.-J., Han, N.-N., Zhu, Y.-Y., Xie, X.-Y., & Li, X. (2022). K63 Ubiquitination of P21 Can Facilitate Pellino-1 in the Context of Chronic Obstructive Pulmonary Disease and Lung Cellular Senescence. Cells, 11(19), 3115. https://doi.org/10.3390/cells11193115