

Pathological Role of HDAC8: Cancer and Beyond



Abstract
1. Introduction
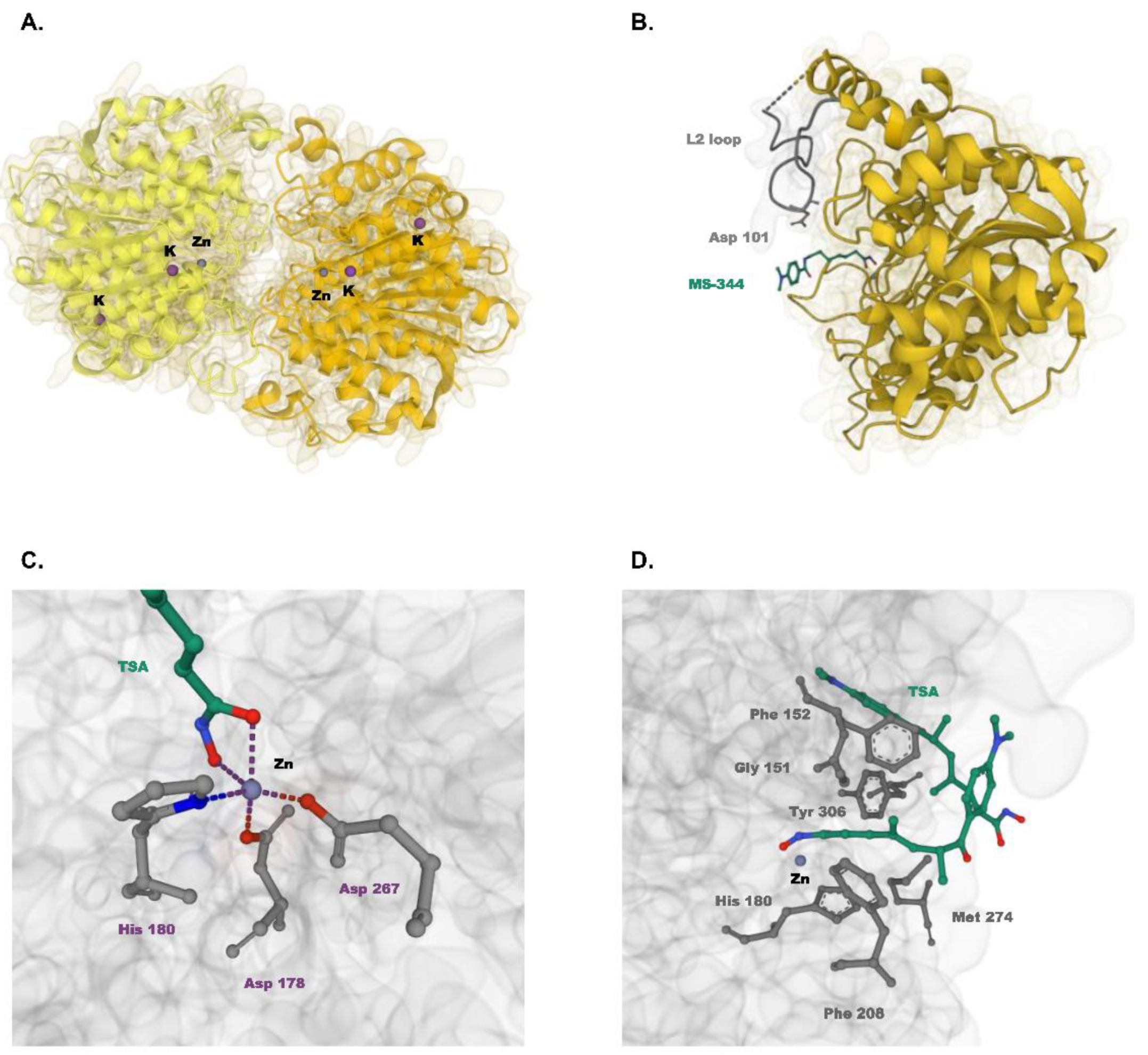
2. Subcellular Localization and Structure of HDAC8
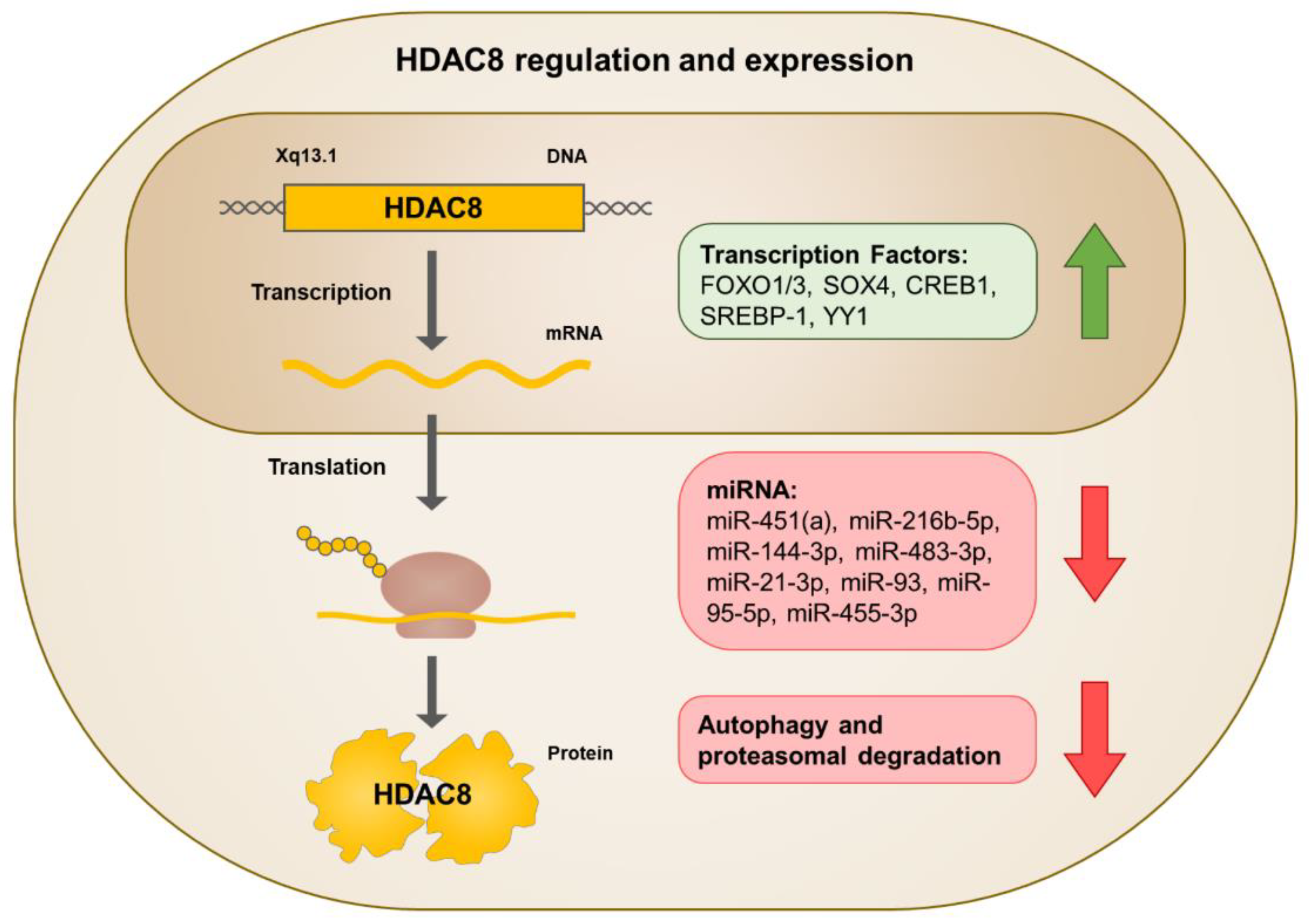
3. Regulation of HDAC8 Expression and Activity
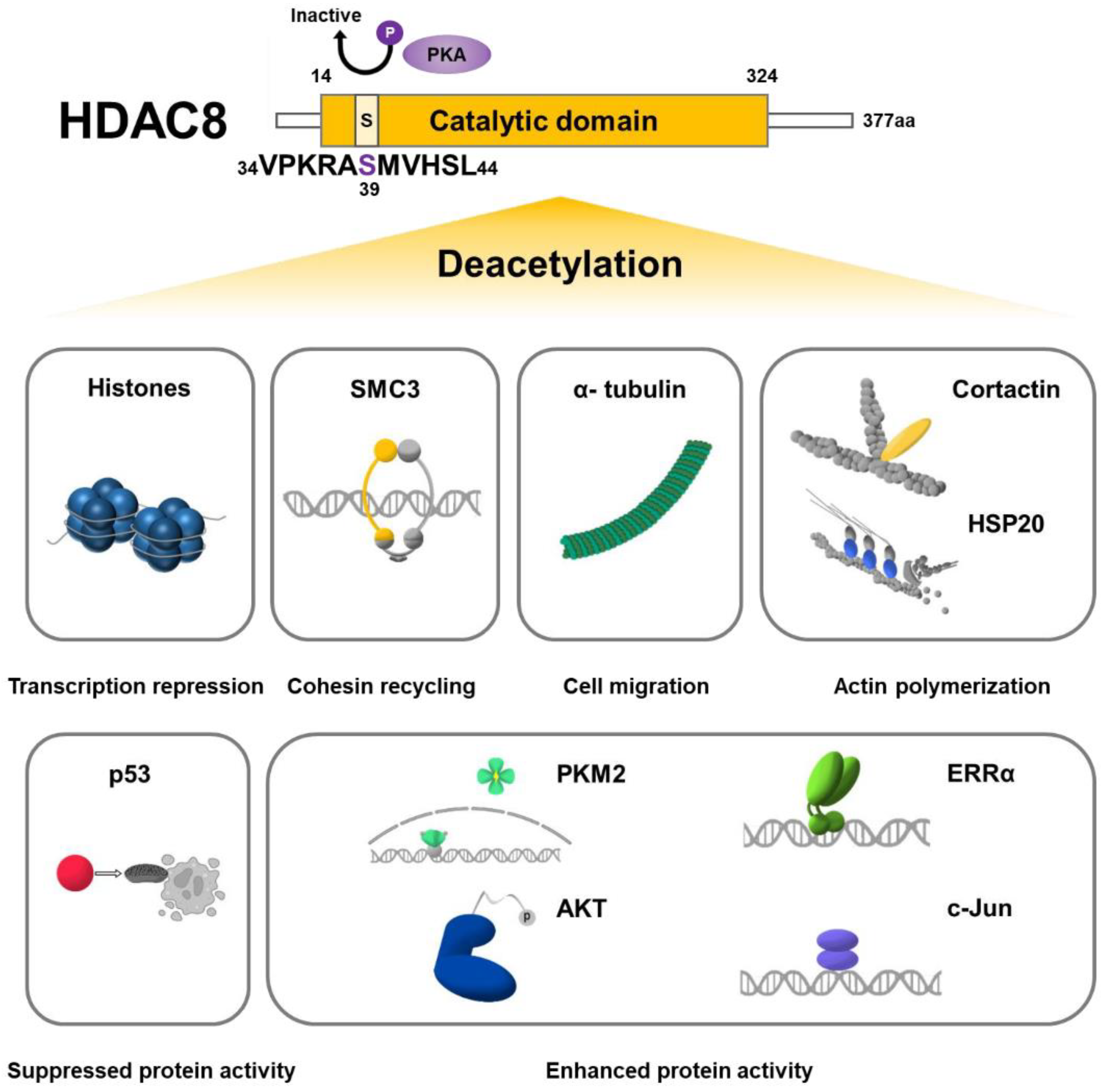
4. HDAC8 Substrates
4.1. Histone Substrates
4.2. Non-Histone Substrates
4.2.1. SMC3
4.2.2. Tumor Suppressors
4.2.3. Kinases
4.2.4. Cytoskeletal Proteins
4.2.5. Transcription Factors

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Protein | Deacetylation Site | Effect | Related Disease | Reference | |
|---|---|---|---|---|---|
| Substrate | SMC3 | Lys105 and Lys106 | Recycling of cohesin complex Chromatin organization | CdLS | [61,63,83] |
| p53 | Lys381 and Lys382 | Repressed stability and activity | Ovarian cancer HCC AML | [28,66,67,68] | |
| ARD1A | N/A | N/A | N/A | [48] | |
| PKM2 | Lys62 | Nuclear localization Glucose metabolism | HCC | [70] | |
| AKT | Lys426 | Enhanced stability and activity | BC | [71] | |
| α-Tubulin | Lys40 | Deregulation of microtubule structural organization Cell migration | Cervical cancer Glioma DMD | [76,84,85] | |
| Cortactin 1 | Nine lysines within the repeat region 1 | Actin filament polymerization Smooth muscle contraction | N/A | [77] | |
| HSP20 | Lys160 | Actin filament polymerization Smooth muscle contraction | Premature birth | [21] | |
| ERRα 1 | Four lysines within the DBD 1 | Enhanced transcriptional activity | N/A | [79]. | |
| c-Jun | Lys273 | Enhanced transcriptional activity | Melanoma | [81] | |
| Interacting Partner | CREB [82], PP1 [82], RUNX2 [50], Gal-3 [86], CM [68], Cofilin [21], HSP70 [87], EST1B [87], α-SMA [88] | ||||
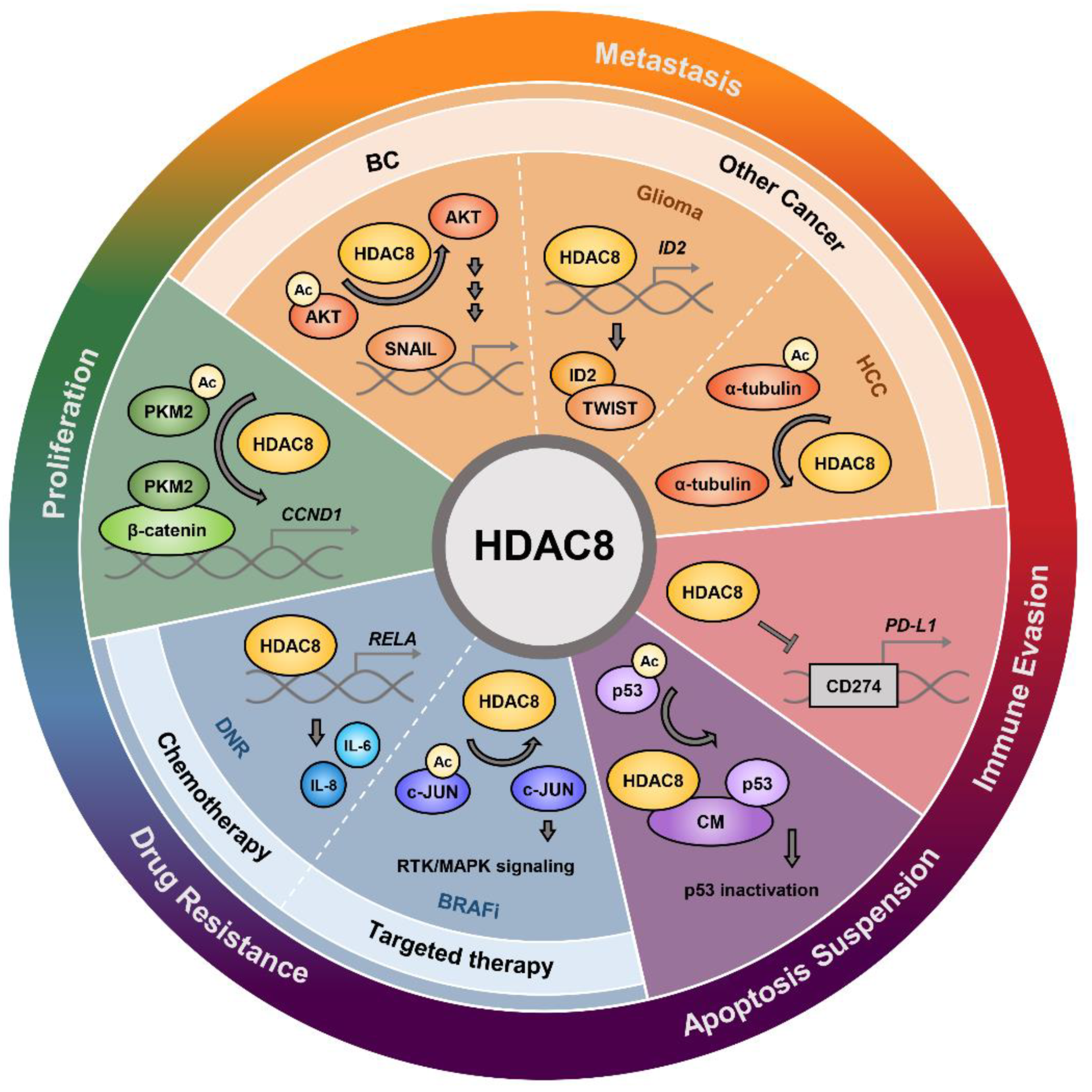
5. HDAC8 in Cancer
5.1. Cancer Cell Proliferation and Apoptosis
5.2. Metastasis
5.2.1. Breast Cancer Metastasis
5.2.2. Metastasis in Other Cancers
5.3. Immune Evasion
5.4. Drug Resistance
5.4.1. Chemotherapy
5.4.2. Targeted Therapy
6. HDAC8 in Non-Cancer Disease
6.1. CdLS and Infectious Diseases
6.2. Cardiovascular Disease
6.3. Pulmonary Disease
6.4. Hepatic Disease
6.5. Myopathy
6.6. Other Diseases
7. HDAC8-Selective Inhibitors
7.1. PCI-34051
7.2. Aryl Hydroxamic Acids
7.3. NBM-BMX (BMX)
7.4. WK2-16
7.5. Dual Inhibitors
| PCI-34051 | Aryl Hydroxamic Acids Compound 6 |
|---|---|
 |  |
| BMX | WK2-16 |
 |  |
8. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Chen, Z.; Li, S.; Subramaniam, S.; Shyy, J.Y.; Chien, S. Epigenetic Regulation: A New Frontier for Biomedical Engineers. Annu. Rev. Biomed. Eng. 2017, 19, 195–219. [Google Scholar] [CrossRef]
- Cheng, Y.; He, C.; Wang, M.; Ma, X.; Mo, F.; Yang, S.; Han, J.; Wei, X. Targeting epigenetic regulators for cancer therapy: Mechanisms and advances in clinical trials. Signal Transduct. Target. Ther. 2019, 4, 62. [Google Scholar] [CrossRef] [PubMed]
- Annunziato, A.T.; Hansen, J.C. Role of histone acetylation in the assembly and modulation of chromatin structures. Gene Expr. 2000, 9, 37–61. [Google Scholar] [CrossRef]
- Verdone, L.; Caserta, M.; Di Mauro, E. Role of histone acetylation in the control of gene expression. Biochem. Cell Biol. 2005, 83, 344–353. [Google Scholar] [CrossRef]
- Romanowska, K.; Sobecka, A.; Rawluszko-Wieczorek, A.A.; Suchorska, W.M.; Golusinski, W. Head and Neck Squamous Cell Carcinoma: Epigenetic Landscape. Diagnostics 2020, 11, 34. [Google Scholar] [CrossRef]
- Micelli, C.; Rastelli, G. Histone deacetylases: Structural determinants of inhibitor selectivity. Drug Discov. Today 2015, 20, 718–735. [Google Scholar] [CrossRef]
- Wang, P.; Wang, Z.; Liu, J. Role of HDACs in normal and malignant hematopoiesis. Mol. Cancer 2020, 19, 5. [Google Scholar] [CrossRef] [PubMed]
- Haberland, M.; Montgomery, R.L.; Olson, E.N. The many roles of histone deacetylases in development and physiology: Implications for disease and therapy. Nat. Rev. Genet. 2009, 10, 32–42. [Google Scholar] [CrossRef]
- Li, T.; Zhang, C.; Hassan, S.; Liu, X.; Song, F.; Chen, K.; Zhang, W.; Yang, J. Histone deacetylase 6 in cancer. J. Hematol. Oncol. 2018, 11, 111. [Google Scholar] [CrossRef]
- Yang, X.J.; Seto, E. HATs and HDACs: From structure, function and regulation to novel strategies for therapy and prevention. Oncogene 2007, 26, 5310–5318. [Google Scholar] [CrossRef] [PubMed]
- Buggy, J.J.; Sideris, M.L.; Mak, P.; Lorimer, D.D.; McIntosh, B.; Clark, J.M. Cloning and characterization of a novel human histone deacetylase, HDAC8. Biochem. J. 2000, 350, 199–205. [Google Scholar] [CrossRef] [PubMed]
- Hu, E.; Chen, Z.; Fredrickson, T.; Zhu, Y.; Kirkpatrick, R.; Zhang, G.F.; Johanson, K.; Sung, C.M.; Liu, R.; Winkler, J. Cloning and characterization of a novel human class I histone deacetylase that functions as a transcription repressor. J. Biol. Chem. 2000, 275, 15254–15264. [Google Scholar] [CrossRef]
- Van den Wyngaert, I.; de Vries, W.; Kremer, A.; Neefs, J.; Verhasselt, P.; Luyten, W.H.; Kass, S.U. Cloning and characterization of human histone deacetylase 8. FEBS Lett. 2000, 478, 77–83. [Google Scholar] [CrossRef]
- Chakrabarti, A.; Oehme, I.; Witt, O.; Oliveira, G.; Sippl, W.; Romier, C.; Pierce, R.J.; Jung, M. HDAC8: A multifaceted target for therapeutic interventions. Trends Pharmacol. Sci. 2015, 36, 481–492. [Google Scholar] [CrossRef] [PubMed]
- Somoza, J.R.; Skene, R.J.; Katz, B.A.; Mol, C.; Ho, J.D.; Jennings, A.J.; Luong, C.; Arvai, A.; Buggy, J.J.; Chi, E.; et al. Structural snapshots of human HDAC8 provide insights into the class I histone deacetylases. Structure 2004, 12, 1325–1334. [Google Scholar] [CrossRef]
- Dowling, D.P.; Gattis, S.G.; Fierke, C.A.; Christianson, D.W. Structures of metal-substituted human histone deacetylase 8 provide mechanistic inferences on biological function. Biochemistry 2010, 49, 5048–5056. [Google Scholar] [CrossRef]
- Nakagawa, M.; Oda, Y.; Eguchi, T.; Aishima, S.; Yao, T.; Hosoi, F.; Basaki, Y.; Ono, M.; Kuwano, M.; Tanaka, M.; et al. Expression profile of class I histone deacetylases in human cancer tissues. Oncol. Rep. 2007, 18, 769–774. [Google Scholar] [CrossRef]
- Ahn, M.Y. HDAC inhibitor apicidin suppresses murine oral squamous cell carcinoma cell growth in vitro and in vivo via inhibiting HDAC8 expression. Oncol. Lett. 2018, 16, 6552–6560. [Google Scholar] [CrossRef]
- Huang, C.; Shu, Y.; Zhu, Y.; Liu, H.; Wang, X.; Wen, H.; Liu, J.; Li, W. Discovery of non-substrate, environmentally sensitive turn-on fluorescent probes for imaging HDAC8 in tumor cells and tissue slices. Bioorg. Med. Chem. 2022, 68, 116821. [Google Scholar] [CrossRef]
- Waltregny, D.; De Leval, L.; Glénisson, W.; Tran, S.L.; North, B.J.; Bellahcène, A.; Weidle, U.; Verdin, E.; Castronovo, V. Expression of histone deacetylase 8, a class I histone deacetylase, is restricted to cells showing smooth muscle differentiation in normal human tissues. Am. J. Pathol. 2004, 165, 553–564. [Google Scholar] [CrossRef]
- Karolczak-Bayatti, M.; Sweeney, M.; Cheng, J.; Edey, L.; Robson, S.C.; Ulrich, S.M.; Treumann, A.; Taggart, M.J.; Europe-Finner, G.N. Acetylation of heat shock protein 20 (Hsp20) regulates human myometrial activity. J. Biol. Chem. 2011, 286, 34346–34355. [Google Scholar] [CrossRef] [PubMed]
- Takase, K.; Oda, S.; Kuroda, M.; Funato, H. Monoaminergic and neuropeptidergic neurons have distinct expression profiles of histone deacetylases. PLoS ONE 2013, 8, e58473. [Google Scholar] [CrossRef] [PubMed]
- Wilmott, J.S.; Colebatch, A.J.; Kakavand, H.; Shang, P.; Carlino, M.S.; Thompson, J.F.; Long, G.V.; Scolyer, R.A.; Hersey, P. Expression of the class 1 histone deacetylases HDAC8 and 3 are associated with improved survival of patients with metastatic melanoma. Mod. Pathol. 2015, 28, 884–894. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Liang, R.; Sun, M.; Li, Z.; Sheng, H.; Wang, J.; Xu, P.; Liu, S.; Yang, W.; Lu, B.; et al. AMPK-dependent phosphorylation of HDAC8 triggers PGM1 expression to promote lung cancer cell survival under glucose starvation. Cancer Lett. 2020, 478, 82–92. [Google Scholar] [CrossRef] [PubMed]
- Vannini, A.; Volpari, C.; Filocamo, G.; Casavola, E.C.; Brunetti, M.; Renzoni, D.; Chakravarty, P.; Paolini, C.; De Francesco, R.; Gallinari, P.; et al. Crystal structure of a eukaryotic zinc-dependent histone deacetylase, human HDAC8, complexed with a hydroxamic acid inhibitor. Proc. Natl. Acad. Sci. USA 2004, 101, 15064–15069. [Google Scholar] [CrossRef]
- Dowling, D.P.; Gantt, S.L.; Gattis, S.G.; Fierke, C.A.; Christianson, D.W. Structural studies of human histone deacetylase 8 and its site-specific variants complexed with substrate and inhibitors. Biochemistry 2008, 47, 13554–13563. [Google Scholar] [CrossRef]
- Vannini, A.; Volpari, C.; Gallinari, P.; Jones, P.; Mattu, M.; Carfí, A.; De Francesco, R.; Steinkühler, C.; Di Marco, S. Substrate binding to histone deacetylases as shown by the crystal structure of the HDAC8-substrate complex. EMBO Rep. 2007, 8, 879–884. [Google Scholar] [CrossRef]
- Long, J.; Jia, M.Y.; Fang, W.Y.; Chen, X.J.; Mu, L.L.; Wang, Z.Y.; Shen, Y.; Xiang, R.F.; Wang, L.N.; Wang, L.; et al. FLT3 inhibition upregulates HDAC8 via FOXO to inactivate p53 and promote maintenance of FLT3-ITD+ acute myeloid leukemia. Blood 2020, 135, 1472–1483. [Google Scholar] [CrossRef]
- Higuchi, T.; Nakayama, T.; Arao, T.; Nishio, K.; Yoshie, O. SOX4 is a direct target gene of FRA-2 and induces expression of HDAC8 in adult T-cell leukemia/lymphoma. Blood 2013, 121, 3640–3649. [Google Scholar] [CrossRef]
- Xiao, T.; Fu, Y.; Zhu, W.; Xu, R.; Xu, L.; Zhang, P.; Du, Y.; Cheng, J.; Jiang, H. HDAC8, A Potential Therapeutic Target, Regulates Proliferation and Differentiation of Bone Marrow Stromal Cells in Fibrous Dysplasia. Stem Cells Transl. Med. 2019, 8, 148–161. [Google Scholar] [CrossRef]
- Tian, Y.; Wong, V.W.; Wong, G.L.; Yang, W.; Sun, H.; Shen, J.; Tong, J.H.; Go, M.Y.; Cheung, Y.S.; Lai, P.B. Histone deacetylase HDAC8 promotes insulin resistance and β-catenin activation in NAFLD-associated hepatocellular carcinoma. Cancer Res. 2015, 75, 4803–4816. [Google Scholar] [CrossRef] [PubMed]
- Fu, W.; Liang, D.; Wu, X.; Chen, H.; Hong, X.; Wang, J.; Zhu, T.; Zeng, T.; Lin, W.; Chen, S.; et al. Long noncoding RNA LINC01435 impedes diabetic wound healing by facilitating YY1-mediated HDAC8 expression. iScience 2022, 25, 104006. [Google Scholar] [CrossRef] [PubMed]
- Gu, S.; Jin, L.; Zhang, F.; Sarnow, P.; Kay, M.A. Biological basis for restriction of microRNA targets to the 3′ untranslated region in mammalian mRNAs. Nat. Struct. Mol. Biol. 2009, 16, 144–150. [Google Scholar] [CrossRef] [PubMed]
- Du, B.; Tan, X.H.; Cheng, L.; Wang, F.; Zhang, H.F. MiR-451a ameliorates alcoholic hepatitis via repressing HDAC8-mediated proinflammatory response. Kaohsiung J. Med. Sci. 2020, 36, 904–910. [Google Scholar] [CrossRef]
- Pang, Y.; Li, L.; Yang, Y.; Shen, Y.; Xu, X.; Li, J. LncRNA-ANAPC2 and lncRNA-NEFM positively regulates the inflammatory response via the miR-451/npr2/hdac8 axis in grass carp. Fish Shellfish. Immunol. 2022, 128, 1–6. [Google Scholar] [CrossRef]
- Menbari, M.N.; Rahimi, K.; Ahmadi, A.; Elyasi, A.; Darvishi, N.; Hosseini, V.; Mohammadi-Yeganeh, S.; Abdi, M. MiR-216b-5p inhibits cell proliferation in human breast cancer by down-regulating HDAC8 expression. Life Sci. 2019, 237, 116945. [Google Scholar] [CrossRef]
- Wang, B.; Xu, L.; Zhang, J.; Cheng, X.; Xu, Q.; Wang, J.; Mao, F. LncRNA NORAD accelerates the progression and doxorubicin resistance of neuroblastoma through up-regulating HDAC8 via sponging miR-144-3p. Biomed. Pharmacother. 2020, 129, 110268. [Google Scholar] [CrossRef]
- Menbari, M.N.; Rahimi, K.; Ahmadi, A.; Mohammadi-Yeganeh, S.; Elyasi, A.; Darvishi, N.; Hosseini, V.; Abdi, M. miR-483-3p suppresses the proliferation and progression of human triple negative breast cancer cells by targeting the HDAC8>oncogene. J. Cell. Physiol. 2020, 235, 2631–2642. [Google Scholar] [CrossRef]
- Yan, M.; Chen, C.; Gong, W.; Yin, Z.; Zhou, L.; Chaugai, S.; Wang, D.W. miR-21-3p regulates cardiac hypertrophic response by targeting histone deacetylase-8. Cardiovasc. Res. 2015, 105, 340–352. [Google Scholar] [CrossRef]
- Xia, B.; Lu, J.; Wang, R.; Yang, Z.; Zhou, X.; Huang, P. miR-21-3p Regulates Influenza A Virus Replication by Targeting Histone Deacetylase-8. Front. Cell. Infect. Microbiol. 2018, 8, 175. [Google Scholar] [CrossRef] [PubMed]
- Yu, X.F.; Zou, J.; Bao, Z.J.; Dong, J. miR-93 suppresses proliferation and colony formation of human colon cancer stem cells. World J. Gastroenterol. 2011, 17, 4711–4717. [Google Scholar] [CrossRef]
- Mao, G.; Hu, S.; Zhang, Z.; Wu, P.; Zhao, X.; Lin, R.; Liao, W.; Kang, Y. Exosomal miR-95-5p regulates chondrogenesis and cartilage degradation via histone deacetylase 2/8. J. Cell. Mol. Med. 2018, 22, 5354–5366. [Google Scholar] [CrossRef] [PubMed]
- Chen, W.; Chen, L.; Zhang, Z.; Meng, F.; Huang, G.; Sheng, P.; Zhang, Z.; Liao, W. MicroRNA-455-3p modulates cartilage development and degeneration through modification of histone H3 acetylation. Biochim. Biophys. Acta 2016, 1863, 2881–2891. [Google Scholar] [CrossRef]
- Park, J.-Y.; Juhnn, Y.-S. cAMP signaling increases histone deacetylase 8 expression by inhibiting JNK-dependent degradation via autophagy and the proteasome system in H1299 lung cancer cells. Biochem. Biophys. Res. Commun. 2016, 470, 336–342. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.; Rezai-Zadeh, N.; Seto, E. Negative regulation of histone deacetylase 8 activity by cyclic AMP-dependent protein kinase A. Mol. Cell. Biol. 2004, 24, 765–773. [Google Scholar] [CrossRef] [PubMed]
- Alam, N.; Zimmerman, L.; Wolfson, N.A.; Joseph, C.G.; Fierke, C.A.; Schueler-Furman, O. Structure-Based Identification of HDAC8 Non-histone Substrates. Structure 2016, 24, 458–468. [Google Scholar] [CrossRef]
- Lopez, J.E.; Haynes, S.E.; Majmudar, J.D.; Martin, B.R.; Fierke, C.A. HDAC8 Substrates Identified by Genetically Encoded Active Site Photocrosslinking. J. Am. Chem. Soc. 2017, 139, 16222–16227. [Google Scholar] [CrossRef]
- Olson, D.E.; Udeshi, N.D.; Wolfson, N.A.; Pitcairn, C.A.; Sullivan, E.D.; Jaffe, J.D.; Svinkina, T.; Natoli, T.; Lu, X.; Paulk, J.; et al. An unbiased approach to identify endogenous substrates of “histone” deacetylase 8. ACS Chem. Biol. 2014, 9, 2210–2216. [Google Scholar] [CrossRef]
- Balasubramanian, S.; Ramos, J.; Luo, W.; Sirisawad, M.; Verner, E.; Buggy, J.J. A novel histone deacetylase 8 (HDAC8)-specific inhibitor PCI-34051 induces apoptosis in T-cell lymphomas. Leukemia 2008, 22, 1026–1034. [Google Scholar] [CrossRef]
- Fu, Y.; Zhang, P.; Ge, J.; Cheng, J.; Dong, W.; Yuan, H.; Du, Y.; Yang, M.; Sun, R.; Jiang, H. Histone deacetylase 8 suppresses osteogenic differentiation of bone marrow stromal cells by inhibiting histone H3K9 acetylation and RUNX2 activity. Int. J. Biochem. Cell Biol. 2014, 54, 68–77. [Google Scholar] [CrossRef]
- Pidugu, V.R.; Yarla, N.S.; Bishayee, A.; Kalle, A.M.; Satya, A.K. Novel histone deacetylase 8-selective inhibitor 1,3,4-oxadiazole-alanine hybrid induces apoptosis in breast cancer cells. Apoptosis 2017, 22, 1394–1403. [Google Scholar] [CrossRef] [PubMed]
- Biswas, S.; Reddy, N.D.; Jayashree, B.S.; Rao, C.M. Evaluation of Novel 3-Hydroxyflavone Analogues as HDAC Inhibitors against Colorectal Cancer. Adv. Pharmacol. Sci. 2018, 2018, 4751806. [Google Scholar] [CrossRef] [PubMed]
- Ha, S.-D.; Reid, C.; Meshkibaf, S.; Kim, S.O. Inhibition of interleukin 1β (IL-1β) expression by anthrax lethal toxin (LeTx) is reversed by histone deacetylase 8 (HDAC8) inhibition in murine macrophages. J. Biol. Chem. 2016, 291, 8745–8755. [Google Scholar] [CrossRef]
- Yang, W.; Feng, Y.; Zhou, J.; Cheung, O.K.-W.; Cao, J.; Wang, J.; Tang, W.; Tu, Y.; Xu, L.; Wu, F. A selective HDAC8 inhibitor potentiates antitumor immunity and efficacy of immune checkpoint blockade in hepatocellular carcinoma. Sci. Transl. Med. 2021, 13, eaaz6804. [Google Scholar] [CrossRef]
- Meng, J.; Liu, X.; Zhang, P.; Li, D.; Xu, S.; Zhou, Q.; Guo, M.; Huai, W.; Chen, X.; Wang, Q.; et al. Rb selectively inhibits innate IFN-β production by enhancing deacetylation of IFN-β promoter through HDAC1 and HDAC8. J. Autoimmun. 2016, 73, 42–53. [Google Scholar] [CrossRef]
- Gao, S.-M.; Chen, C.-Q.; Wang, L.-Y.; Hong, L.-L.; Wu, J.-B.; Dong, P.-H.; Yu, F.-J. Histone deacetylases inhibitor sodium butyrate inhibits JAK2/STAT signaling through upregulation of SOCS1 and SOCS3 mediated by HDAC8 inhibition in myeloproliferative neoplasms. Exp. Hematol. 2013, 41, 261–270. [Google Scholar] [CrossRef]
- Tang, X.; Li, G.; Su, F.; Cai, Y.; Shi, L.; Meng, Y.; Liu, Z.; Sun, J.; Wang, M.; Qian, M.; et al. HDAC8 cooperates with SMAD3/4 complex to suppress SIRT7 and promote cell survival and migration. Nucleic Acids Res. 2020, 48, 2912–2923. [Google Scholar] [CrossRef]
- Sawada, Y.; Nakatsuji, T.; Dokoshi, T.; Kulkarni, N.N.; Liggins, M.C.; Sen, G.; Gallo, R.L. Cutaneous innate immune tolerance is mediated by epigenetic control of MAP2K3 by HDAC8/9. Sci. Immunol. 2021, 6, eabe1935. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Y.; Huan, L.; Wu, Y.; Bao, C.; Chen, B.; Wang, L.; Huang, S.; Liang, L.; He, X. LncRNA ID2-AS1 suppresses tumor metastasis by activating the HDAC8/ID2 pathway in hepatocellular carcinoma. Cancer Lett. 2020, 469, 399–409. [Google Scholar] [CrossRef] [PubMed]
- Ha, S.D.; Han, C.Y.; Reid, C.; Kim, S.O. HDAC8-mediated epigenetic reprogramming plays a key role in resistance to anthrax lethal toxin-induced pyroptosis in macrophages. J. Immunol. 2014, 193, 1333–1343. [Google Scholar] [CrossRef]
- Deardorff, M.A.; Bando, M.; Nakato, R.; Watrin, E.; Itoh, T.; Minamino, M.; Saitoh, K.; Komata, M.; Katou, Y.; Clark, D.; et al. HDAC8 mutations in Cornelia de Lange syndrome affect the cohesin acetylation cycle. Nature 2012, 489, 313–317. [Google Scholar] [CrossRef]
- Dasgupta, T.; Antony, J.; Braithwaite, A.W.; Horsfield, J.A. HDAC8 Inhibition Blocks SMC3 Deacetylation and Delays Cell Cycle Progression without Affecting Cohesin-dependent Transcription in MCF7 Cancer Cells. J. Biol. Chem. 2016, 291, 12761–12770. [Google Scholar] [CrossRef]
- Van Ruiten, M.S.; van Gent, D.; Cacciatore, Á.S.; Fauster, A.; Willems, L.; Hekkelman, M.L.; Hoekman, L.; Altelaar, M.; Haarhuis, J.H.I.; Brummelkamp, T.R.; et al. The cohesin acetylation cycle controls chromatin loop length through a PDS5A brake mechanism. Nat. Struct. Mol. Biol. 2022, 29, 586–591. [Google Scholar] [CrossRef]
- Lane, D.P. p53, guardian of the genome. Nature 1992, 358, 15–16. [Google Scholar] [CrossRef]
- Liu, Y.; Tavana, O.; Gu, W. p53 modifications: Exquisite decorations of the powerful guardian. J. Mol. Cell. Biol. 2019, 11, 564–577. [Google Scholar] [CrossRef]
- Kim, J.Y.; Han, S.Y.; Yoo, J.; Kim, G.W.; Jeon, Y.H.; Lee, S.W.; Park, J.; Kwon, S.H. HDAC8-Selective Inhibition by PCI-34051 Enhances the Anticancer Effects of ACY-241 in Ovarian Cancer Cells. Int. J. Mol. Sci. 2022, 23, 8645. [Google Scholar] [CrossRef]
- Wu, J.; Du, C.; Lv, Z.; Ding, C.; Cheng, J.; Xie, H.; Zhou, L.; Zheng, S. The up-regulation of histone deacetylase 8 promotes proliferation and inhibits apoptosis in hepatocellular carcinoma. Dig. Dis. Sci. 2013, 58, 3545–3553. [Google Scholar] [CrossRef]
- Qi, J.; Singh, S.; Hua, W.-K.; Cai, Q.; Chao, S.-W.; Li, L.; Liu, H.; Ho, Y.; McDonald, T.; Lin, A.; et al. HDAC8 inhibition specifically targets Inv (16) acute myeloid leukemic stem cells by restoring p53 acetylation. Cell Stem Cell 2015, 17, 597–610. [Google Scholar] [CrossRef]
- Prakasam, G.; Iqbal, M.A.; Bamezai, R.N.K.; Mazurek, S. Posttranslational Modifications of Pyruvate Kinase M2: Tweaks that Benefit Cancer. Front. Oncol. 2018, 8, 22. [Google Scholar] [CrossRef]
- Zhang, R.; Shen, M.; Wu, C.; Chen, Y.; Lu, J.; Li, J.; Zhao, L.; Meng, H.; Zhou, X.; Huang, G.; et al. HDAC8-dependent deacetylation of PKM2 directs nuclear localization and glycolysis to promote proliferation in hepatocellular carcinoma. Cell Death Dis. 2020, 11, 1036. [Google Scholar] [CrossRef] [PubMed]
- An, P.; Chen, F.; Li, Z.; Ling, Y.; Peng, Y.; Zhang, H.; Li, J.; Chen, Z.; Wang, H. HDAC8 promotes the dissemination of breast cancer cells via AKT/GSK-3β/Snail signals. Oncogene 2020, 39, 4956–4969. [Google Scholar] [CrossRef]
- Hubbert, C.; Guardiola, A.; Shao, R.; Kawaguchi, Y.; Ito, A.; Nixon, A.; Yoshida, M.; Wang, X.F.; Yao, T.P. HDAC6 is a microtubule-associated deacetylase. Nature 2002, 417, 455–458. [Google Scholar] [CrossRef]
- Zhang, X.; Yuan, Z.; Zhang, Y.; Yong, S.; Salas-Burgos, A.; Koomen, J.; Olashaw, N.; Parsons, J.T.; Yang, X.-J.; Dent, S.R.; et al. HDAC6 modulates cell motility by altering the acetylation level of cortactin. Mol. Cell 2007, 27, 197–213. [Google Scholar] [CrossRef] [PubMed]
- Shida, T.; Cueva, J.G.; Xu, Z.; Goodman, M.B.; Nachury, M.V. The major α-tubulin K40 acetyltransferase αTAT1 promotes rapid ciliogenesis and efficient mechanosensation. Proc. Natl. Acad. Sci. USA 2010, 107, 21517–21522. [Google Scholar] [CrossRef] [PubMed]
- Akella, J.S.; Wloga, D.; Kim, J.; Starostina, N.G.; Lyons-Abbott, S.; Morrissette, N.S.; Dougan, S.T.; Kipreos, E.T.; Gaertig, J. MEC-17 is an α-tubulin acetyltransferase. Nature 2010, 467, 218–222. [Google Scholar] [CrossRef] [PubMed]
- Vanaja, G.R.; Ramulu, H.G.; Kalle, A.M. Overexpressed HDAC8 in cervical cancer cells shows functional redundancy of tubulin deacetylation with HDAC6. Cell Commun. Signal. 2018, 16, 20. [Google Scholar] [CrossRef]
- Li, J.; Chen, S.; Cleary, R.A.; Wang, R.; Gannon, O.J.; Seto, E.; Tang, D.D. Histone deacetylase 8 regulates cortactin deacetylation and contraction in smooth muscle tissues. Am. J. Physiol. Cell Physiol. 2014, 307, C288–C295. [Google Scholar] [CrossRef]
- Tran, A.; Scholtes, C.; Songane, M.; Champagne, C.; Galarneau, L.; Levasseur, M.-P.; Fodil, N.; Dufour, C.R.; Giguère, V.; Saleh, M. Estrogen-related receptor alpha (ERRα) is a key regulator of intestinal homeostasis and protects against colitis. Sci. Rep. 2021, 11, 15073. [Google Scholar] [CrossRef]
- Wilson, B.J.; Tremblay, A.M.; Deblois, G.; Sylvain-Drolet, G.; Giguère, V. An acetylation switch modulates the transcriptional activity of estrogen-related receptor alpha. Mol. Endocrinol. 2010, 24, 1349–1358. [Google Scholar] [CrossRef]
- Wisdom, R.; Johnson, R.S.; Moore, C. c-Jun regulates cell cycle progression and apoptosis by distinct mechanisms. Embo. J. 1999, 18, 188–197. [Google Scholar] [CrossRef]
- Emmons, M.F.; Faiao-Flores, F.; Sharma, R.; Thapa, R.; Messina, J.L.; Becker, J.C.; Schadendorf, D.; Seto, E.; Sondak, V.K.; Koomen, J.M.; et al. HDAC8 Regulates a Stress Response Pathway in Melanoma to Mediate Escape from BRAF Inhibitor Therapy. Cancer Res. 2019, 79, 2947–2961. [Google Scholar] [CrossRef] [PubMed]
- Gao, J.; Siddoway, B.; Huang, Q.; Xia, H. Inactivation of CREB mediated gene transcription by HDAC8 bound protein phosphatase. Biochem. Biophys. Res. Commun. 2009, 379, 1–5. [Google Scholar] [CrossRef] [PubMed]
- Kaiser, F.J.; Ansari, M.; Braunholz, D.; Gil-Rodríguez, M.C.; Decroos, C.; Wilde, J.J.; Fincher, C.T.; Kaur, M.; Bando, M.; Amor, D.J.; et al. Loss-of-function HDAC8 mutations cause a phenotypic spectrum of Cornelia de Lange syndrome-like features, ocular hypertelorism, large fontanelle and X-linked inheritance. Hum. Mol. Genet. 2014, 23, 2888–2900. [Google Scholar] [CrossRef] [PubMed]
- Mormino, A.; Cocozza, G.; Fontemaggi, G.; Valente, S.; Esposito, V.; Santoro, A.; Bernardini, G.; Santoni, A.; Fazi, F.; Mai, A.; et al. Histone-deacetylase 8 drives the immune response and the growth of glioma. Glia 2021, 69, 2682–2698. [Google Scholar] [CrossRef]
- Spreafico, M.; Cafora, M.; Bragato, C.; Capitanio, D.; Marasca, F.; Bodega, B.; De Palma, C.; Mora, M.; Gelfi, C.; Marozzi, A.; et al. Targeting HDAC8 to ameliorate skeletal muscle differentiation in Duchenne muscular dystrophy. Pharmacol. Res. 2021, 170, 105750. [Google Scholar] [CrossRef]
- Li, M.-L.; Su, X.-M.; Ren, Y.; Zhao, X.; Kong, L.-F.; Kang, J. HDAC8 inhibitor attenuates airway responses to antigen stimulus through synchronously suppressing galectin-3 expression and reducing macrophage-2 polarization. Respir. Res. 2020, 21, 62. [Google Scholar] [CrossRef]
- Lee, H.; Sengupta, N.; Villagra, A.; Rezai-Zadeh, N.; Seto, E. Histone deacetylase 8 safeguards the human ever-shorter telomeres 1B (hEST1B) protein from ubiquitin-mediated degradation. Mol. Cell. Biol. 2006, 26, 5259–5269. [Google Scholar] [CrossRef]
- Waltregny, D.; Glenisson, W.; Tran, S.L.; North, B.J.; Verdin, E.; Colige, A.; Castronovo, V. Histone deacetylase HDAC8 associates with smooth muscle alpha-actin and is essential for smooth muscle cell contractility. FASEB J. 2005, 19, 966–968. [Google Scholar] [CrossRef]
- Song, S.; Wang, Y.; Xu, P.; Yang, R.; Ma, Z.; Liang, S.; Zhang, G. The inhibition of histone deacetylase 8 suppresses proliferation and inhibits apoptosis in gastric adenocarcinoma. Int. J. Oncol. 2015, 47, 1819–1828. [Google Scholar] [CrossRef]
- Ahn, M.Y.; Yoon, J.H. Histone deacetylase 8 as a novel therapeutic target in oral squamous cell carcinoma. Oncol. Rep. 2017, 37, 540–546. [Google Scholar] [CrossRef]
- Moreno, D.A.; Scrideli, C.A.; Cortez, M.A.A.; De Paula Queiroz, R.; Valera, E.T.; Da Silva Silveira, V.; Yunes, J.A.; Brandalise, S.R.; Tone, L.G. Differential expression of HDAC3, HDAC7 and HDAC9 is associated with prognosis and survival in childhood acute lymphoblastic leukaemia. Br. J. Haematol. 2010, 150, 665–673. [Google Scholar] [CrossRef] [PubMed]
- Oehme, I.; Deubzer, H.E.; Wegener, D.; Pickert, D.; Linke, J.-P.; Hero, B.; Kopp-Schneider, A.; Westermann, F.; Ulrich, S.M.; Von Deimling, A. Histone deacetylase 8 in neuroblastoma tumorigenesis. Clin. Cancer Res. 2009, 15, 91–99. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Y.J.; Xu, Q.; Shao, M.Y.; Cao, X.Y.; Wu, Z.R.; Chen, Y.W.; Bu, H.; Shi, Y.J. Decreased expression of HDAC8 indicates poor prognosis in patients with intrahepatic cholangiocarcinoma. Hepatobiliary Pancreat. Dis. Int. 2019, 18, 464–470. [Google Scholar] [CrossRef] [PubMed]
- Tilekar, K.; Upadhyay, N.; Jänsch, N.; Schweipert, M.; Mrowka, P.; Meyer-Almes, F.J.; Ramaa, C.S. Discovery of 5-naphthylidene-2,4-thiazolidinedione derivatives as selective HDAC8 inhibitors and evaluation of their cytotoxic effects in leukemic cell lines. Bioorg. Chem. 2020, 95, 103522. [Google Scholar] [CrossRef] [PubMed]
- Mahajan, M.; Suryavanshi, S.; Bhowmick, S.; Alasmary, F.A.; Almutairi, T.M.; Islam, M.A.; Kaul-Ghanekar, R. Matairesinol, an active constituent of HC9 polyherbal formulation, exhibits HDAC8 inhibitory and anticancer activity. Biophys. Chem. 2021, 273, 106588. [Google Scholar] [CrossRef] [PubMed]
- Ramos, T.L.; Sánchez-Abarca, L.I.; Redondo, A.; Hernández-Hernández, Á.; Almeida, A.M.; Puig, N.; Rodríguez, C.; Ortega, R.; Preciado, S.; Rico, A.; et al. HDAC8 overexpression in mesenchymal stromal cells from JAK2+ myeloproliferative neoplasms: A new therapeutic target? Oncotarget 2017, 8, 28187–28202. [Google Scholar] [CrossRef]
- Lopez, G.; Bill, K.L.; Bid, H.K.; Braggio, D.; Constantino, D.; Prudner, B.; Zewdu, A.; Batte, K.; Lev, D.; Pollock, R.E. HDAC8, A Potential Therapeutic Target for the Treatment of Malignant Peripheral Nerve Sheath Tumors (MPNST). PLoS ONE 2015, 10, e0133302. [Google Scholar] [CrossRef] [PubMed]
- Martínez-Pacheco, H.; Ramírez-Galicia, G.; Vergara-Arias, M.; Gertsch, J.; Fragoso-Vazquez, J.M.; Mendez-Luna, D.; Abujamra, A.L.; Cristina, C.L.; Cecilia, R.M.; Mendoza-Lujambio, I.; et al. Docking and QSAR Studies of Aryl-valproic Acid Derivatives to Identify Antiproliferative Agents Targeting the HDAC8. Anti Cancer Agents Med. Chem. 2017, 17, 927–940. [Google Scholar] [CrossRef]
- Watters, J.M.; Wright, G.; Smith, M.A.; Shah, B.; Wright, K.L. Histone deacetylase 8 inhibition suppresses mantle cell lymphoma viability while preserving natural killer cell function. Biochem. Biophys. Res. Commun. 2021, 534, 773–779. [Google Scholar] [CrossRef]
- Kang, Y.; Nian, H.; Rajendran, P.; Kim, E.; Dashwood, W.; Pinto, J.; Boardman, L.; Thibodeau, S.; Limburg, P.; Löhr, C. HDAC8 and STAT3 repress BMF gene activity in colon cancer cells. Cell Death Dis. 2014, 5, e1476. [Google Scholar] [CrossRef]
- Wang, Z.T.; Chen, Z.J.; Jiang, G.M.; Wu, Y.M.; Liu, T.; Yi, Y.M.; Zeng, J.; Du, J.; Wang, H.S. Histone deacetylase inhibitors suppress mutant p53 transcription via HDAC8/YY1 signals in triple negative breast cancer cells. Cell. Signal. 2016, 28, 506–515. [Google Scholar] [CrossRef] [PubMed]
- Yan, W.; Liu, S.; Xu, E.; Zhang, J.; Zhang, Y.; Chen, X. Histone deacetylase inhibitors suppress mutant p53 transcription via histone deacetylase 8. Oncogene 2013, 32, 599–609. [Google Scholar] [CrossRef] [PubMed]
- Singh, R.K.; Cho, K.; Padi, S.K.; Yu, J.; Haldar, M.; Mandal, T.; Yan, C.; Cook, G.; Guo, B.; Mallik, S.; et al. Mechanism of N-Acylthiourea-mediated activation of human histone deacetylase 8 (HDAC8) at molecular and cellular levels. J. Biol. Chem. 2015, 290, 6607–6619. [Google Scholar] [CrossRef] [PubMed]
- Park, S.Y.; Jun, J.A.; Jeong, K.J.; Heo, H.J.; Sohn, J.S.; Lee, H.Y.; Park, C.G.; Kang, J. Histone deacetylases 1, 6 and 8 are critical for invasion in breast cancer. Oncol. Rep. 2011, 25, 1677–1681. [Google Scholar] [CrossRef] [PubMed]
- An, P.; Li, J.; Lu, L.; Wu, Y.; Ling, Y.; Du, J.; Chen, Z.; Wang, H. Histone deacetylase 8 triggers the migration of triple negative breast cancer cells via regulation of YAP signals. Eur. J. Pharmacol. 2019, 845, 16–23. [Google Scholar] [CrossRef] [PubMed]
- Hsieh, C.L.; Ma, H.P.; Su, C.M.; Chang, Y.J.; Hung, W.Y.; Ho, Y.S.; Huang, W.J.; Lin, R.K. Alterations in histone deacetylase 8 lead to cell migration and poor prognosis in breast cancer. Life Sci. 2016, 151, 7–14. [Google Scholar] [CrossRef]
- Shankar, E.; Pandey, M.; Verma, S.; Abbas, A.; Candamo, M.; Kanwal, R.; Shukla, S.; MacLennan, G.T.; Gupta, S. Role of class I histone deacetylases in the regulation of maspin expression in prostate cancer. Mol. Carcinog. 2020, 59, 955–966. [Google Scholar] [CrossRef]
- Wang, Y.F.; Liu, F.; Sherwin, S.; Farrelly, M.; Yan, X.G.; Croft, A.; Liu, T.; Jin, L.; Zhang, X.D.; Jiang, C.C. Cooperativity of HOXA5 and STAT3 Is Critical for HDAC8 Inhibition-Mediated Transcriptional Activation of PD-L1 in Human Melanoma Cells. J. Investig. Dermatol. 2018, 138, 922–932. [Google Scholar] [CrossRef]
- Tsai, C.Y.; Ko, H.J.; Chiou, S.J.; Lai, Y.L.; Hou, C.C.; Javaria, T.; Huang, Z.Y.; Cheng, T.S.; Hsu, T.I.; Chuang, J.Y.; et al. NBM-BMX, an HDAC8 Inhibitor, Overcomes Temozolomide Resistance in Glioblastoma Multiforme by Downregulating the β-Catenin/c-Myc/SOX2 Pathway and Upregulating p53-Mediated MGMT Inhibition. Int. J. Mol. Sci. 2021, 22, 5907. [Google Scholar] [CrossRef]
- Zhao, G.; Wang, G.; Bai, H.; Li, T.; Gong, F.; Yang, H.; Wen, J.; Wang, W. Targeted inhibition of HDAC8 increases the doxorubicin sensitivity of neuroblastoma cells via up regulation of miR-137. Eur. J. Pharmacol. 2017, 802, 20–26. [Google Scholar] [CrossRef]
- Wu, J.; Zhang, L.; Feng, Y.; Khadka, B.; Fang, Z.; Liu, J. HDAC8 promotes daunorubicin resistance of human acute myeloid leukemia cells via regulation of IL-6 and IL-8. Biol. Chem. 2021, 402, 461–468. [Google Scholar] [CrossRef] [PubMed]
- McCubrey, J.A.; Steelman, L.S.; Chappell, W.H.; Abrams, S.L.; Franklin, R.A.; Montalto, G.; Cervello, M.; Libra, M.; Candido, S.; Malaponte, G.; et al. Ras/Raf/MEK/ERK and PI3K/PTEN/Akt/mTOR cascade inhibitors: How mutations can result in therapy resistance and how to overcome resistance. Oncotarget 2012, 3, 1068–1111. [Google Scholar] [CrossRef] [PubMed]
- Ha, S.D.; Lewin, N.; Li, S.S.C.; Kim, S.O. HDAC8 Activates AKT through Upregulating PLCB1 and Suppressing DESC1 Expression in MEK1/2 Inhibition-Resistant Cells. Cells 2021, 10, 1101. [Google Scholar] [CrossRef] [PubMed]
- Kindler, T.; Lipka, D.B.; Fischer, T. FLT3 as a therapeutic target in AML: Still challenging after all these years. Blood 2010, 116, 5089–5102. [Google Scholar] [CrossRef]
- Yamauchi, Y.; Boukari, H.; Banerjee, I.; Sbalzarini, I.F.; Horvath, P.; Helenius, A. Histone deacetylase 8 is required for centrosome cohesion and influenza A virus entry. PLoS Pathog. 2011, 7, e1002316. [Google Scholar] [CrossRef]
- Chelladurai, P.; Dabral, S.; Basineni, S.R.; Chen, C.N.; Schmoranzer, M.; Bender, N.; Feld, C.; Nötzold, R.R.; Dobreva, G.; Wilhelm, J.; et al. Isoform-specific characterization of class I histone deacetylases and their therapeutic modulation in pulmonary hypertension. Sci. Rep. 2020, 10, 12864. [Google Scholar] [CrossRef]
- Wedel, T.; Van Eys, G.J.; Waltregny, D.; Glenisson, W.; Castronovo, V.; Vanderwinden, J.M. Novel smooth muscle markers reveal abnormalities of the intestinal musculature in severe colorectal motility disorders. Neurogastroenterol. Motil. 2006, 18, 526–538. [Google Scholar] [CrossRef]
- Long, K.; Vaughn, Z.; McDaniels, M.D.; Joyasawal, S.; Przepiorski, A.; Parasky, E.; Sander, V.; Close, D.; Johnston, P.A.; Davidson, A.J.; et al. Validation of HDAC8 Inhibitors as Drug Discovery Starting Points to Treat Acute Kidney Injury. ACS Pharmacol. Transl. Sci. 2022, 5, 207–215. [Google Scholar] [CrossRef]
- Lin, F.L.; Yen, J.L.; Kuo, Y.C.; Kang, J.J.; Cheng, Y.W.; Huang, W.J.; Hsiao, G. HADC8 Inhibitor WK2-16 Therapeutically Targets Lipopolysaccharide-Induced Mouse Model of Neuroinflammation and Microglial Activation. Int. J. Mol. Sci. 2019, 20, 410. [Google Scholar] [CrossRef]
- Marek, M.; Kannan, S.; Hauser, A.-T.; Mourão, M.M.; Caby, S.; Cura, V.; Stolfa, D.A.; Schmidtkunz, K.; Lancelot, J.; Andrade, L. Structural basis for the inhibition of histone deacetylase 8 (HDAC8), a key epigenetic player in the blood fluke Schistosoma mansoni. PLoS Pathog. 2013, 9, e1003645. [Google Scholar] [CrossRef]
- Vaca, H.R.; Celentano, A.M.; Macchiaroli, N.; Kamenetzky, L.; Camicia, F.; Rosenzvit, M.C. Histone deacetylase enzymes as potential drug targets of Neglected Tropical Diseases caused by cestodes. PLoS Pathog. 2019, 9, 120–132. [Google Scholar] [CrossRef]
- Zhao, T.; Kee, H.J.; Bai, L.; Kim, M.K.; Kee, S.J.; Jeong, M.H. Selective HDAC8 Inhibition Attenuates Isoproterenol-Induced Cardiac Hypertrophy and Fibrosis via p38 MAPK Pathway. Front. Pharmacol. 2021, 12, 677757. [Google Scholar] [CrossRef]
- Zhao, T.; Kee, H.J.; Kee, S.J.; Jeong, M.H. Hdac8 Inhibitor Alleviates Transverse Aortic Constriction-Induced Heart Failure in Mice by Downregulating Ace1. Oxidative Med. Cell. Longev. 2022, 2022, 6227330. [Google Scholar] [CrossRef]
- Kee, H.J.; Ryu, Y.; Seok, Y.M.; Choi, S.Y.; Sun, S.; Kim, G.R.; Jeong, M.H. Selective inhibition of histone deacetylase 8 improves vascular hypertrophy, relaxation, and inflammation in angiotensin II hypertensive mice. Clin. Hypertens. 2019, 25, 13. [Google Scholar] [CrossRef] [PubMed]
- Saito, S.; Zhuang, Y.; Suzuki, T.; Ota, Y.; Bateman, M.E.; Alkhatib, A.L.; Morris, G.F.; Lasky, J.A. HDAC8 inhibition ameliorates pulmonary fibrosis. Am. J. Physiol. Lung Cell. Mol. Physiol. 2019, 316, L175–L186. [Google Scholar] [CrossRef]
- Lee, C.H.; Choi, Y.; Cho, H.; Bang, I.H.; Hao, L.; Lee, S.O.; Jeon, R.; Bae, E.J.; Park, B.H. Histone deacetylase 8 inhibition alleviates cholestatic liver injury and fibrosis. Biochem. Pharmacol. 2021, 183, 114312. [Google Scholar] [CrossRef] [PubMed]
- Kim, D.S.; Kwon, J.E.; Lee, S.H.; Kim, E.K.; Ryu, J.G.; Jung, K.A.; Choi, J.W.; Park, M.J.; Moon, Y.M.; Park, S.H.; et al. Attenuation of Rheumatoid Inflammation by Sodium Butyrate Through Reciprocal Targeting of HDAC2 in Osteoclasts and HDAC8 in T Cells. Front. Immunol. 2018, 9, 1525. [Google Scholar] [CrossRef]
- Wang, P.; Mao, Z.; Pan, Q.; Lu, R.; Huang, X.; Shang, X.; Zhang, R.; You, H. Histone deacetylase-4 and histone deacetylase-8 regulate interleukin-1β-induced cartilage catabolic degradation through MAPK/JNK and ERK pathways. Int. J. Mol. Med. 2018, 41, 2117–2127. [Google Scholar] [CrossRef]
- Huang, W.J.; Wang, Y.C.; Chao, S.W.; Yang, C.Y.; Chen, L.C.; Lin, M.H.; Hou, W.C.; Chen, M.Y.; Lee, T.L.; Yang, P.; et al. Synthesis and biological evaluation of ortho-aryl N-hydroxycinnamides as potent histone deacetylase (HDAC) 8 isoform-selective inhibitors. ChemMedChem 2012, 7, 1815–1824. [Google Scholar] [CrossRef]
- Spreafico, M.; Gruszka, A.M.; Valli, D.; Mazzola, M.; Deflorian, G.; Quinte, A.; Totaro, M.G.; Battaglia, C.; Alcalay, M.; Marozzi, A.; et al. HDAC8: A Promising Therapeutic Target for Acute Myeloid Leukemia. Front. Cell Dev. Biol. 2020, 8, 844. [Google Scholar] [CrossRef]
- Shen, J.; Najafi, S.; Stäble, S.; Fabian, J.; Koeneke, E.; Kolbinger, F.R.; Wrobel, J.K.; Meder, B.; Distel, M.; Heimburg, T.; et al. A kinome-wide RNAi screen identifies ALK as a target to sensitize neuroblastoma cells for HDAC8-inhibitor treatment. Cell Death Differ. 2018, 25, 2053–2070. [Google Scholar] [CrossRef] [PubMed]
- Zhang, P.; Brinton, L.T.; Williams, K.; Sher, S.; Orwick, S.; Tzung-Huei, L.; Mims, A.S.; Coss, C.C.; Kulp, S.K.; Youssef, Y.; et al. Targeting DNA Damage Repair Functions of Two Histone Deacetylases, HDAC8 and SIRT6, Sensitizes Acute Myeloid Leukemia to NAMPT Inhibition. Clin. Cancer Res. 2021, 27, 2352–2366. [Google Scholar] [CrossRef]
- Zhang, B.; Liu, B.; Chen, D.; Setroikromo, R.; Haisma, H.J.; Quax, W.J. Histone Deacetylase Inhibitors Sensitize TRAIL-Induced Apoptosis in Colon Cancer Cells. Cancers 2019, 11, 645. [Google Scholar] [CrossRef]
- Krennhrubec, K.; Marshall, B.L.; Hedglin, M.; Verdin, E.; Ulrich, S.M. Design and evaluation of ‘Linkerless’ hydroxamic acids as selective HDAC8 inhibitors. Bioorg. Med. Chem. Lett. 2007, 17, 2874–2878. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.C.; Chen, C.N.; Wu, C.I.; Huang, W.J.; Kuo, T.Y.; Kuan, M.C.; Tsai, T.H.; Huang, J.S.; Huang, C.Y. NBM-T-L-BMX-OS01, Semisynthesized from Osthole, Is a Novel Inhibitor of Histone Deacetylase and Enhances Learning and Memory in Rats. Evid. Based Complement. Altern. Med. 2013, 2013, 514908. [Google Scholar] [CrossRef]
- Kampa-Schittenhelm, K.M.; Heinrich, M.C.; Akmut, F.; Dohner, H.; Dohner, K.; Schittenhelm, M.M. Quizartinib (AC220) is a potent second generation class III tyrosine kinase inhibitor that displays a distinct inhibition profile against mutant-FLT3, -PDGFRA and -KIT isoforms. Mol. Cancer 2013, 12, 19. [Google Scholar] [CrossRef]
- Halder, A.K.; Mallick, S.; Shikha, D.; Saha, A.; Saha, K.D.; Jha, T. Design of dual MMP-2/HDAC-8 inhibitors by pharmacophore mapping, molecular docking, synthesis and biological activity. RSC Adv. 2015, 5, 72373–72386. [Google Scholar] [CrossRef]
- Amin, S.A.; Adhikari, N.; Jha, T. Is dual inhibition of metalloenzymes HDAC-8 and MMP-2 a potential pharmacological target to combat hematological malignancies? Pharmacol. Res. 2017, 122, 8–19. [Google Scholar] [CrossRef]
- Cheng, X.C.; Wang, R.L.; Dong, Z.K.; Li, J.; Li, Y.Y.; Li, R.R. Design, synthesis and evaluation of novel metalloproteinase inhibitors based on L-tyrosine scaffold. Bioorg. Med. Chem. 2012, 20, 5738–5744. [Google Scholar] [CrossRef] [PubMed]
- Banerjee, S.; Adhikari, N.; Amin, S.A.; Jha, T. Histone deacetylase 8 (HDAC8) and its inhibitors with selectivity to other isoforms: An overview. Eur. J. Med. Chem. 2019, 164, 214–240. [Google Scholar] [CrossRef] [PubMed]
- Chotitumnavee, J.; Yamashita, Y.; Takahashi, Y.; Takada, Y.; Iida, T.; Oba, M.; Itoh, Y.; Suzuki, T. Selective degradation of histone deacetylase 8 mediated by a proteolysis targeting chimera (PROTAC). Chem. Commun. 2022, 58, 4635–4638. [Google Scholar] [CrossRef]
- Sun, Z.; Deng, B.; Yang, Z.; Mai, R.; Huang, J.; Ma, Z.; Chen, T.; Chen, J. Discovery of pomalidomide-based PROTACs for selective degradation of histone deacetylase 8. Eur. J. Med. Chem. 2022, 239, 114544. [Google Scholar] [CrossRef]
- Ferrari, L.; Bragato, C.; Brioschi, L.; Spreafico, M.; Esposito, S.; Pezzotta, A.; Pizzetti, F.; Moreno-Fortuny, A.; Bellipanni, G.; Giordano, A.; et al. HDAC8 regulates canonical Wnt pathway to promote differentiation in skeletal muscles. J. Cell. Physiol. 2019, 234, 6067–6076. [Google Scholar] [CrossRef] [PubMed]
- Katayama, S.; Morii, A.; Makanga, J.O.; Suzuki, T.; Miyata, N.; Inazu, T. HDAC8 regulates neural differentiation through embryoid body formation in P19cells. Biochem. Biophys. Res. Commun. 2018, 498, 45–51. [Google Scholar] [CrossRef]
- Morii, A.; Katayama, S.; Inazu, T. Establishment of a Simple Method for Inducing Neuronal Differentiation of P19 EC Cells without Embryoid Body Formation and Analysis of the Role of Histone Deacetylase 8 Activity in This Differentiation. Biol. Pharm. Bull. 2020, 43, 1096–1103. [Google Scholar] [CrossRef]
- Hua, W.K.; Qi, J.; Cai, Q.; Carnahan, E.; Ramirez, M.A.; Li, L.; Marcucci, G.; Kuo, Y.H. HDAC8 regulates long-term hematopoietic stem-cell maintenance under stress by modulating p53 activity. Blood 2017, 130, 2619–2630. [Google Scholar] [CrossRef]




| Gene | Region | Histone Substrate | Cells | Reference |
|---|---|---|---|---|
| IFNB1 | promoter | H3/H4 | Macrophages | [55] |
| SOCS1/3 | promoter | H3/H4 | Erythroleukemia cells | [56] |
| SIRT7 | promoter | H4 | BC cells | [57] |
| MAP2K3 | promoter | H3K9 and H3K27 | Keratinocytes | [58] |
| CCL4 | enhancer | H3K27 | HCC cells | [54] |
| ID2 | enhancer | H3K27 | HCC cells | [59] |
| BNIP3 | N/A | H3K27 | TIR cells | [60] |
| MLN64 | N/A | H3K27 | TIR cells | [60] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kim, J.Y.; Cho, H.; Yoo, J.; Kim, G.W.; Jeon, Y.H.; Lee, S.W.; Kwon, S.H. Pathological Role of HDAC8: Cancer and Beyond. Cells 2022, 11, 3161. https://doi.org/10.3390/cells11193161
Kim JY, Cho H, Yoo J, Kim GW, Jeon YH, Lee SW, Kwon SH. Pathological Role of HDAC8: Cancer and Beyond. Cells. 2022; 11(19):3161. https://doi.org/10.3390/cells11193161
Chicago/Turabian StyleKim, Ji Yoon, Hayoung Cho, Jung Yoo, Go Woon Kim, Yu Hyun Jeon, Sang Wu Lee, and So Hee Kwon. 2022. "Pathological Role of HDAC8: Cancer and Beyond" Cells 11, no. 19: 3161. https://doi.org/10.3390/cells11193161
APA StyleKim, J. Y., Cho, H., Yoo, J., Kim, G. W., Jeon, Y. H., Lee, S. W., & Kwon, S. H. (2022). Pathological Role of HDAC8: Cancer and Beyond. Cells, 11(19), 3161. https://doi.org/10.3390/cells11193161