
Dynamics of Actin Cytoskeleton and Their Signaling Pathways during Cellular Wound Repair

 ,
, 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Cell Culture
2.2. Plasmids and Mutants
2.3. Chamber Preparation
2.4. Wounding and Microscopy
2.5. Measurement of PI Influx
2.6. Inhibitors
2.7. Statistical Analysis
3. Results
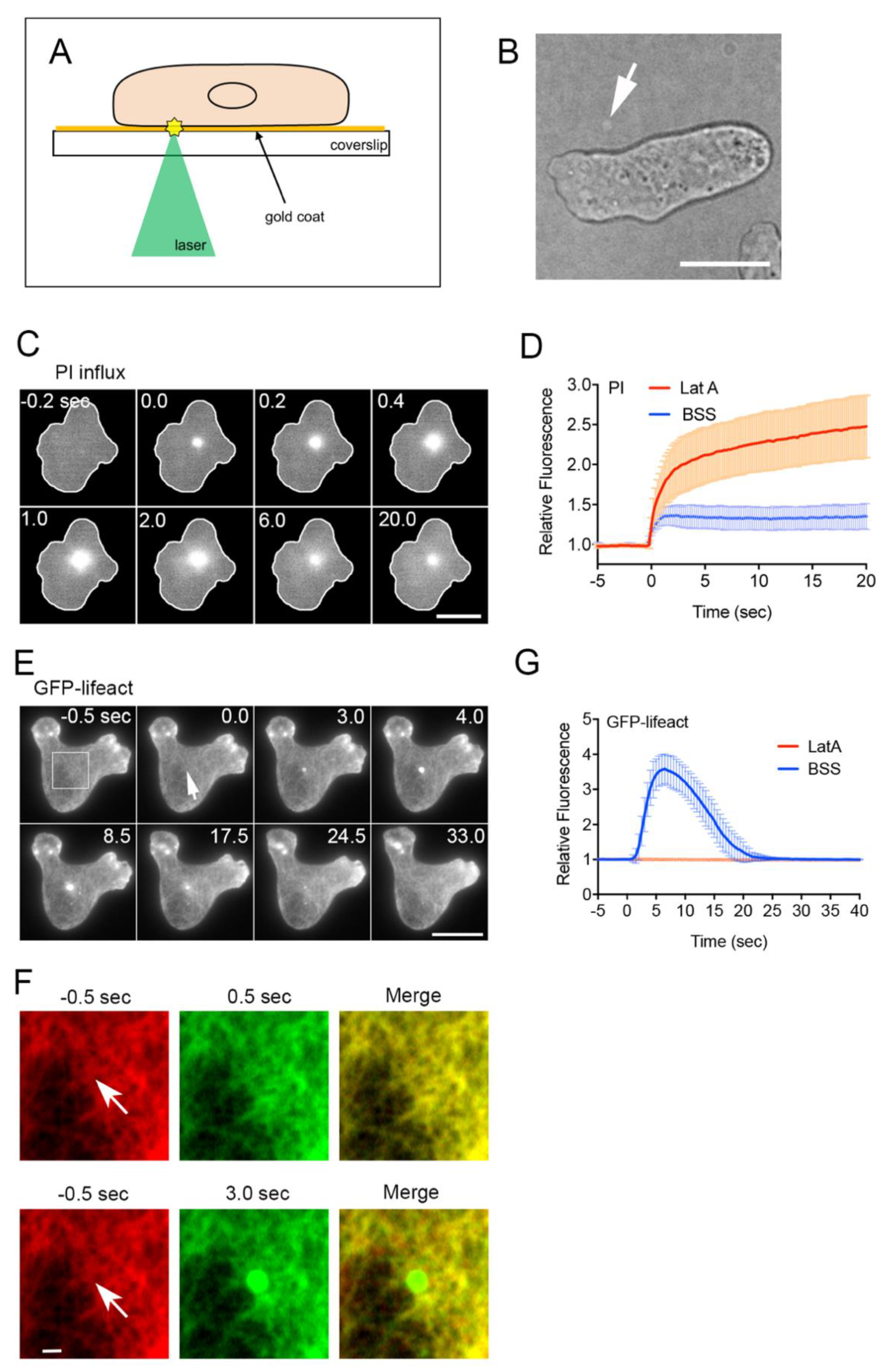
3.1. Improved Laserporation Using Gold Coating
3.2. Actin Accumulation at Wound Site Is Essential for Wound Repair
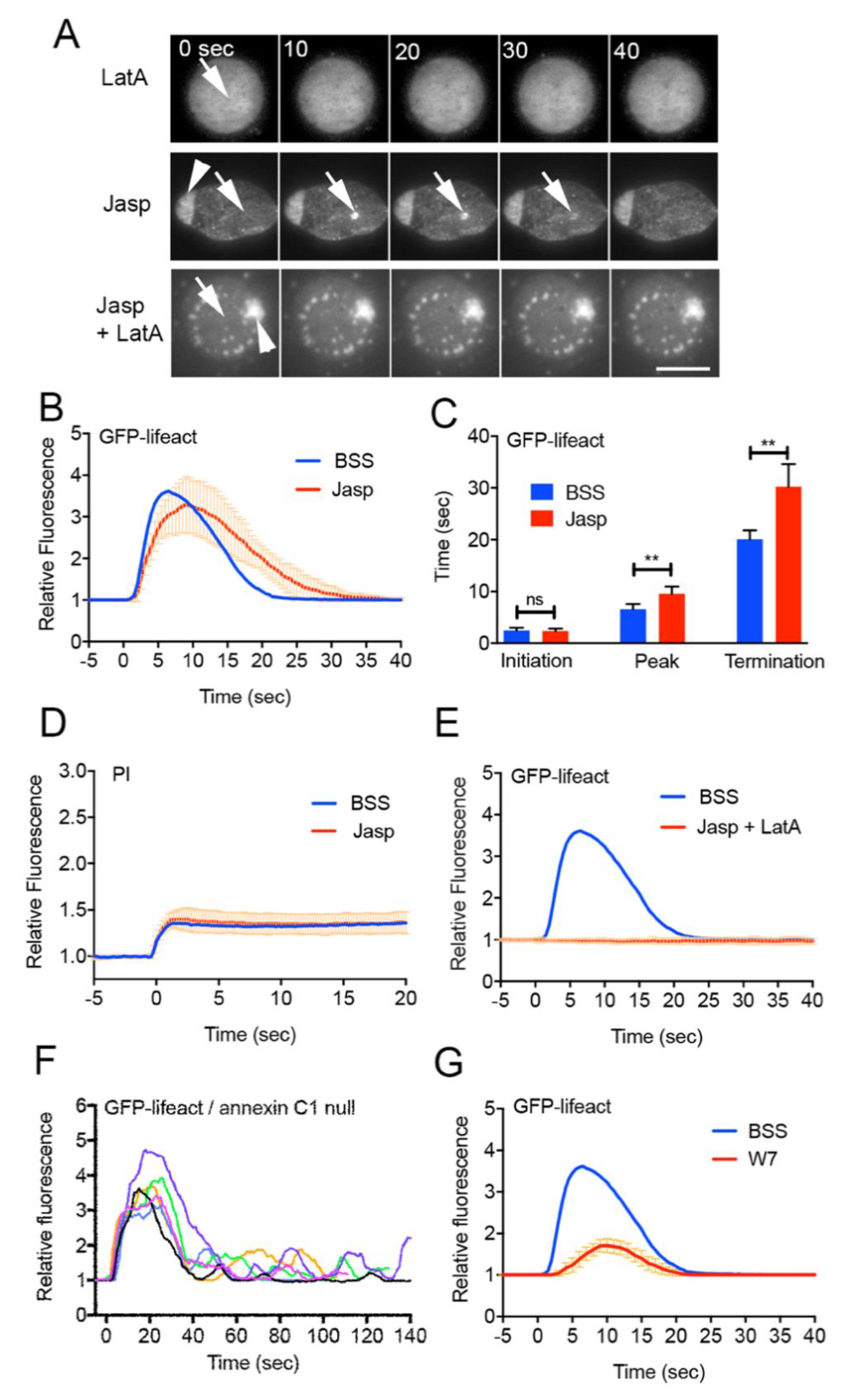
3.3. Actin Polymerizes De Novo at Wound Site
3.4. Early Signals for Wound Response
3.5. Dynamics of Actin-Related Proteins during Wound Repair
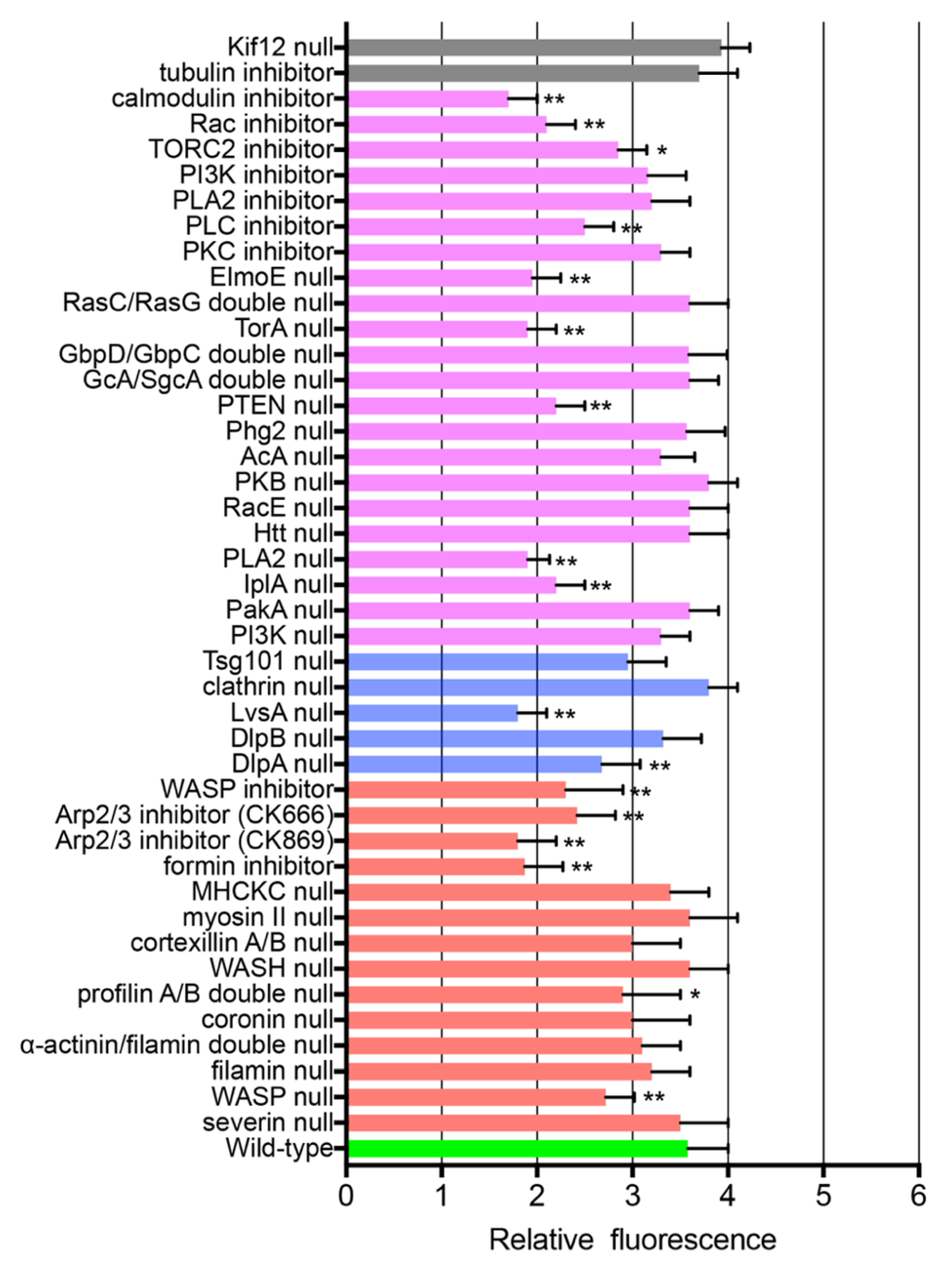
3.6. ARPs Required for Wound Repair
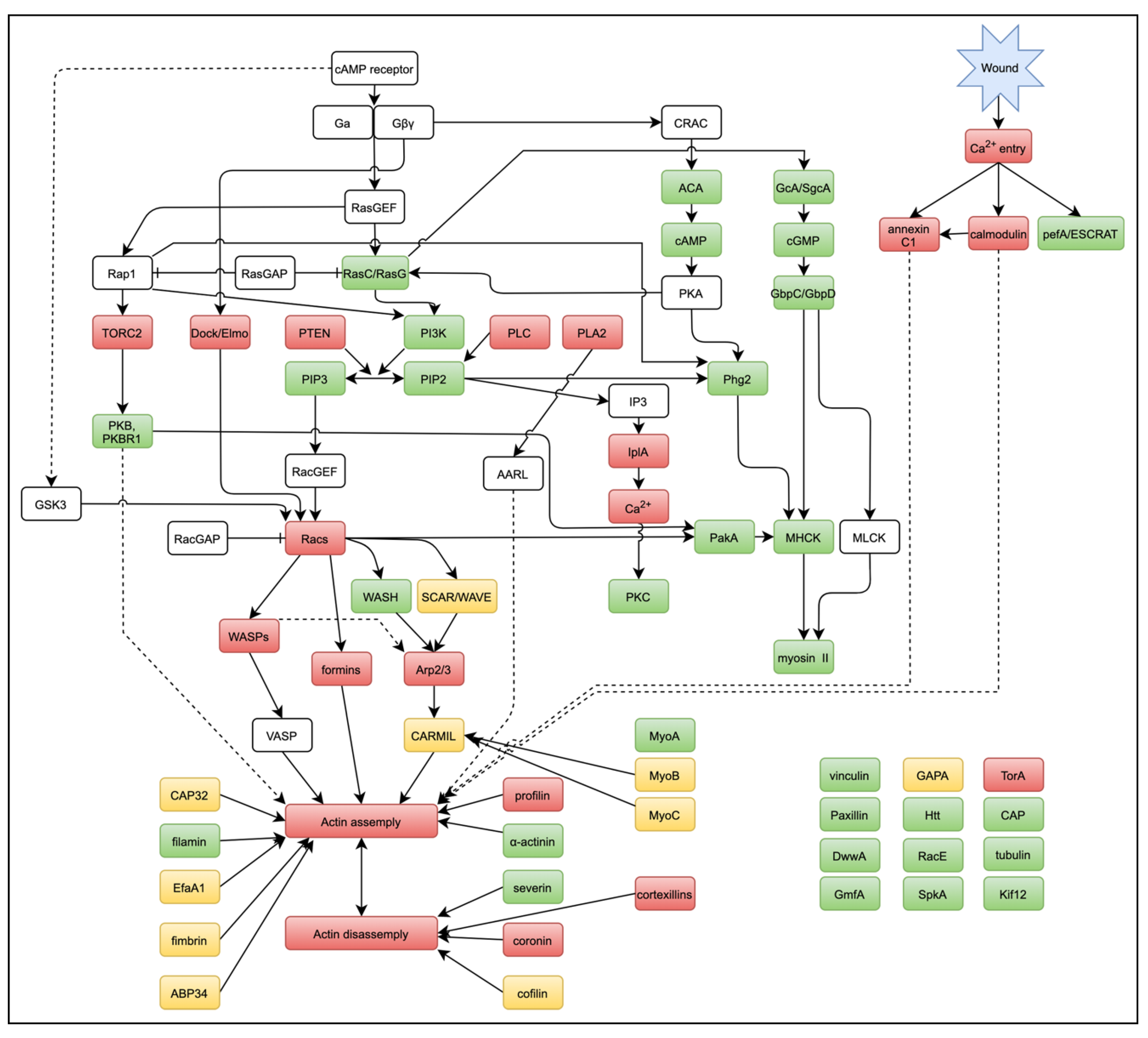
3.7. Signals for Wound-Induced Actin Dynamics
3.8. Small G Proteins for Wound-Induced Actin Dynamics
3.9. Other Signals for Wound-Induced Actin Dynamics
4. Discussion
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Bansal, D.; Miyake, K.; Vogel, S.S.; Groh, S.; Chen, C.C.; Williamson, R.; McNeil, P.L.; Campbell, K.P. Defective membrane repair in dysferlin-deficient muscular dystrophy. Nature 2003, 423, 168–172. [Google Scholar] [CrossRef] [PubMed]
- Waddell, L.B.; Lemckert, F.A.; Zheng, X.F.; Tran, J.; Evesson, F.J.; Hawkes, J.M.; Lek, A.; Street, N.E.; Lin, P.; Clarke, N.F.; et al. Dysferlin, annexin A1, and mitsugumin 53 are upregulated in muscular dystrophy and localize to longitudinal tubules of the T-system with stretch. J. Neuropathol. Exp. Neurol. 2011, 70, 302–313. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Andrews, N.W.; Corrotte, M. Plasma membrane repair. Curr. Biol. 2018, 28, R392–R397. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bement, W.M.; Capco, D.G. Analysis of inducible contractile rings suggests a role for protein kinase C in embryonic cytokinesis and wound healing. Cell Motil. Cytoskeleton 1991, 20, 145–157. [Google Scholar] [CrossRef]
- Stanisstreet, M. Calcium and wound healing in Xenopus early embryos. J. Embryol. Exp. Morphol. 1982, 67, 195–205. [Google Scholar] [CrossRef]
- McNeil, P.L.; Vogel, S.S.; Miyake, K.; Terasaki, M. Patching plasma membrane disruptions with cytoplasmic membrane. J. Cell Sci. 2000, 113, 1891–1902. [Google Scholar] [CrossRef]
- Abreu-Blanco, M.T.; Verboon, J.M.; Parkhurst, S.M. Cell wound repair in Drosophila occurs through three distinct phases of membrane and cytoskeletal remodeling. J. Cell Biol. 2011, 193, 455–464. [Google Scholar] [CrossRef] [Green Version]
- Abreu-Blanco, M.T.; Verboon, J.M.; Parkhurst, S.M. Coordination of Rho family GTPase activities to orchestrate cytoskeleton responses during cell wound repair. Curr. Biol. 2014, 24, 144–155. [Google Scholar] [CrossRef] [Green Version]
- Razzell, W.; Wood, W.; Martin, P. Swatting flies: Modelling wound healing and inflammation in Drosophila. Dis. Model. Mech. 2011, 4, 569–574. [Google Scholar] [CrossRef] [Green Version]
- Xu, S.; Chisholm, A.D. C. elegans epidermal wounding induces a mitochondrial ROS burst that promotes wound repair. Dev. Cell 2014, 31, 48–60. [Google Scholar] [CrossRef]
- Jeon, K.W.; Jeon, M.S. Cytoplasmic filaments and cellular wound healing in Amoeba proteus. J. Cell Biol 1975, 67, 243–249. [Google Scholar] [CrossRef]
- Kono, K.; Saeki, Y.; Yoshida, S.; Tanaka, K.; Pellman, D. Proteasomal degradation resolves competition between cell polarization and cellular wound healing. Cell 2012, 150, 151–164. [Google Scholar] [CrossRef] [Green Version]
- Pervin, M.S.; Itoh, G.; Talukder, M.S.U.; Fujimoto, K.; Morimoto, Y.V.; Tanaka, M.; Ueda, M.; Yumura, S. A study of wound repair in Dictyostelium cells by using novel laserporation. Sci. Rep. 2018, 8, 7969. [Google Scholar] [CrossRef] [Green Version]
- Steinhardt, R.A.; Bi, G.; Alderton, J.M. Cell membrane resealing by a vesicular mechanism similar to neurotransmitter release. Science 1994, 263, 390–393. [Google Scholar] [CrossRef]
- Terasaki, M.; Miyake, K.; McNeil, P.L. Large plasma membrane disruptions are rapidly resealed by Ca2+-dependent vesicle-vesicle fusion events. J. Cell Biol. 1997, 139, 63–74. [Google Scholar] [CrossRef] [Green Version]
- Talukder, M.S.U.; Pervin, M.S.; Tanvir, M.I.O.; Fujimoto, K.; Tanaka, M.; Itoh, G.; Yumura, S. Ca2+-calmodulin dependent wound repair in Dictyostelium cell membrane. Cells 2020, 9, 1058. [Google Scholar] [CrossRef] [Green Version]
- Moe, A.M.; Golding, A.E.; Bement, W.M. Cell healing: Calcium, repair and regeneration. Semin. Cell Dev. Biol. 2015, 45, 18–23. [Google Scholar] [CrossRef] [Green Version]
- Nakamura, M.; Dominguez, A.N.M.; Decker, J.R.; Hull, A.J.; Verboon, J.M.; Parkhurst, S.M. Into the breach: How cells cope with wounds. Open Biol. 2018, 8, 180135. [Google Scholar] [CrossRef] [Green Version]
- Bement, W.M.; Mandato, C.A.; Kirsch, M.N. Wound-induced assembly and closure of an actomyosin purse string in Xenopus oocytes. Curr. Biol. 1999, 9, 579–587. [Google Scholar] [CrossRef] [Green Version]
- Hui, J.; Stjepic, V.; Nakamura, M.; Parkhurst, S.M. Wrangling Actin Assemblies: Actin Ring Dynamics during Cell Wound Repair. Cells 2022, 11, 2777. [Google Scholar] [CrossRef]
- DeKraker, C.; Goldin-Blais, L.; Boucher, E.; Mandato, C.A. Dynamics of actin polymerisation during the mammalian single-cell wound healing response. BMC Res. Notes 2019, 12, 420. [Google Scholar] [CrossRef] [Green Version]
- Togo, T.; Steinhardt, R.A. Nonmuscle myosin IIA and IIB have distinct functions in the exocytosis-dependent process of cell membrane repair. Mol. Biol. Cell 2004, 15, 688–695. [Google Scholar] [CrossRef] [Green Version]
- Yumura, S.; Hashima, S.; Muranaka, S. Myosin II does not contribute to wound repair in Dictyostelium cells. Biol. Open 2014, 3, 966–973. [Google Scholar] [CrossRef] [Green Version]
- Jaiswal, J.K.; Lauritzen, S.P.; Scheffer, L.; Sakaguchi, M.; Bunkenborg, J.; Simon, S.M.; Kallunki, T.; Jaattela, M.; Nylandsted, J. S100A11 is required for efficient plasma membrane repair and survival of invasive cancer cells. Nat. Commun. 2014, 5, 3795. [Google Scholar] [CrossRef] [Green Version]
- McDade, J.R.; Archambeau, A.; Michele, D.E. Rapid actin-cytoskeleton-dependent recruitment of plasma membrane-derived dysferlin at wounds is critical for muscle membrane repair. FASEB J. 2014, 28, 3660–3670. [Google Scholar] [CrossRef] [Green Version]
- Shen, S.S.; Steinhardt, R.A. The mechanisms of cell membrane resealing in rabbit corneal epithelial cells. Curr. Eye Res. 2005, 30, 543–554. [Google Scholar] [CrossRef]
- Tanvir, M.I.O.; Itoh, G.; Adachi, H.; Yumura, S. Dynamics of Myosin II Filaments during Wound Repair in Dividing Cells. Cells 2021, 10, 1229. [Google Scholar] [CrossRef]
- Jimenez, A.J.; Maiuri, P.; Lafaurie-Janvore, J.; Divoux, S.; Piel, M.; Perez, F. ESCRT machinery is required for plasma membrane repair. Science 2014, 343, 1247136. [Google Scholar] [CrossRef]
- Scheffer, L.L.; Sreetama, S.C.; Sharma, N.; Medikayala, S.; Brown, K.J.; Defour, A.; Jaiswal, J.K. Mechanism of Ca2+-triggered ESCRT assembly and regulation of cell membrane repair. Nat. Commun. 2014, 5, 5646. [Google Scholar] [CrossRef] [Green Version]
- Pollard, T.D. Regulation of actin filament assembly by Arp2/3 complex and formins. Annu. Rev. Biophys. Biomol. Struct. 2007, 36, 451–477. [Google Scholar] [CrossRef]
- Goode, B.L.; Eck, M.J. Mechanism and function of formins in the control of actin assembly. Annu. Rev. Biochem. 2007, 76, 593–627. [Google Scholar] [CrossRef] [PubMed]
- Winder, S.J.; Ayscough, K.R. Actin-binding proteins. J. Cell Sci. 2005, 118, 651–654. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ebstrup, M.L.; Dias, C.; Heitmann, A.S.B.; Sonder, S.L.; Nylandsted, J. Actin Cytoskeletal Dynamics in Single-Cell Wound Repair. Int. J. Mol. Sci 2021, 22, 10886. [Google Scholar] [CrossRef] [PubMed]
- Henson, J.H.; Nazarian, R.; Schulberg, K.L.; Trabosh, V.A.; Kolnik, S.E.; Burns, A.R.; McPartland, K.J. Wound Closure in the Lamellipodia of Single Cells: Mediation by Actin Polymerization in the Absence of an Actomyosin Purse String. Mol. Biol. Cell 2002, 13, 1001–1014. [Google Scholar] [CrossRef] [Green Version]
- Mandato, C.A.; Bement, W.M. Actomyosin transports microtubules and microtubules control actomyosin recruitment during Xenopus oocyte wound healing. Curr. Biol. 2003, 13, 1096–1105. [Google Scholar] [CrossRef] [Green Version]
- Wales, P.; Schuberth, C.E.; Aufschnaiter, R.; Fels, J.; García-Aguilar, I.; Janning, A.; Dlugos, C.P.; Sch√§fer-Herte, M.; Klingner, C.; Wälte, M.; et al. Calcium-mediated actin reset (CaAR) mediates acute cell adaptations. eLife 2016, 5, e19850. [Google Scholar] [CrossRef]
- Vaughan, E.M.; You, J.S.; Elsie Yu, H.Y.; Lasek, A.; Vitale, N.; Hornberger, T.A.; Bement, W.M. Lipid domain-dependent regulation of single-cell wound repair. Mol. Biol. Cell 2014, 25, 1867–1876. [Google Scholar] [CrossRef]
- Vaughan, E.M.; Miller, A.L.; Yu, H.Y.; Bement, W.M. Control of local Rho GTPase crosstalk by Abr. Curr. Biol. 2011, 21, 270–277. [Google Scholar] [CrossRef]
- Hayes, M.J.; Shao, D.; Bailly, M.; Moss, S.E. Regulation of actin dynamics by annexin 2. EMBO J. 2006, 25, 1816–1826. [Google Scholar] [CrossRef] [Green Version]
- Tang, M.; Wang, M.; Shi, C.; Iglesias, P.A.; Devreotes, P.N.; Huang, C.H. Evolutionarily conserved coupling of adaptive and excitable networks mediates eukaryotic chemotaxis. Nat. Commun. 2014, 5, 5175. [Google Scholar] [CrossRef]
- Wang, Y.; Chen, C.L.; Iijima, M. Signaling mechanisms for chemotaxis. Dev. Growth Differ. 2011, 53, 495–502. [Google Scholar] [CrossRef] [Green Version]
- Affolter, M.; Weijer, C.J. Signaling to cytoskeletal dynamics during chemotaxis. Dev. Cell 2005, 9, 19–34. [Google Scholar] [CrossRef] [Green Version]
- van Haastert, P.J.M.; Keizer-Gunnink, I.; Kortholt, A. Essential role of PI3-kinase and phospholipase A2 in Dictyostelium discoideum chemotaxis. J. Cell Biol. 2007, 177, 809–816. [Google Scholar] [CrossRef] [Green Version]
- Yan, J.; Jin, T. Signaling network from GPCR to the actin cytoskeleton during chemotaxis. BioArchitecture 2012, 2, 15–18. [Google Scholar] [CrossRef] [Green Version]
- Yumura, S. Myosin II dynamics and cortical flow during contractile ring formation in Dictyostelium cells. J. Cell Biol. 2001, 154, 137–146. [Google Scholar] [CrossRef] [Green Version]
- Yumura, S.; Matsuzaki, R.; Kitanishi-Yumura, T. Introduction of macromolecules into living Dictyostelium cells by electroporation. Cell Struct. Funct. 1995, 20, 185–190. [Google Scholar] [CrossRef] [Green Version]
- Yumura, S. A novel low-power laser-mediated transfer of foreign molecules into cells. Sci. Rep. 2016, 6, 22055. [Google Scholar] [CrossRef] [Green Version]
- Nakatoh, T.; Osaki, T.; Tanimoto, S.; Jahan, M.G.S.; Kawakami, T.; Chihara, K.; Sakai, N.; Yumura, S. Cell behaviors within a confined adhesive area fabricated using novel micropatterning methods. PLoS ONE 2022, 17, e0262632. [Google Scholar] [CrossRef]
- Pervin, M.S.; Yumura, S. Manipulation of cell migration by laserporation-induced local wounding. Sci. Rep. 2019, 9, 4291. [Google Scholar] [CrossRef] [Green Version]
- Fukui, Y.; Yumura, S.; Yumura, T.K.; Mori, H. Agar overlay method: High-resolution immunofluorescence for the study of the contractile apparatus. Methods Enzymol. 1986, 134, 573–580. [Google Scholar]
- Yumura, S.; Itoh, G.; Kikuta, Y.; Kikuchi, T.; Kitanishi-Yumura, T.; Tsujioka, M. Cell-scale dynamic recycling and cortical flow of the actin-myosin cytoskeleton for rapid cell migration. Biol. Open 2013, 2, 200–209. [Google Scholar] [CrossRef]
- Williams, T.D.; Kay, R.R. The physiological regulation of macropinocytosis during Dictyostelium growth and development. J. Cell Sci. 2018, 131, jcs213736. [Google Scholar]
- Ashraf, A.P.K.; Gerke, V. Plasma membrane wound repair is characterized by extensive membrane lipid and protein rearrangements in vascular endothelial cells. Biochim. Biophys. Acta Mol. Cell Res. 2021, 1868, 118991. [Google Scholar] [CrossRef]
- Mellgren, R.L.; Zhang, W.; Miyake, K.; McNeil, P.L. Calpain is required for the rapid, calcium-dependent repair of wounded plasma membrane. J. Biol. Chem. 2007, 282, 2567–2575. [Google Scholar] [CrossRef] [Green Version]
- Miyake, K.; McNeil, P.L.; Suzuki, K.; Tsunoda, R.; Sugai, N. An actin barrier to resealing. J. Cell Sci. 2001, 114, 3487–3494. [Google Scholar] [CrossRef]
- Bretschneider, T.; Diez, S.; Anderson, K.; Heuser, J.; Clarke, M.; Müller-Taubenberger, A.; Köhler, J.; Gerisch, G. Dynamic actin patterns and Arp2/3 assembly at the substrate-attached surface of motile cells. Curr. Biol. 2004, 14, 1–10. [Google Scholar] [CrossRef]
- Mandato, C.A.; Bement, W.M. Contraction and polymerization cooperate to assemble and close actomyosin rings around Xenopus oocyte wounds. J. Cell Biol. 2001, 154, 785–797. [Google Scholar] [CrossRef]
- Lee, E.; Shelden, E.A.; Knecht, D.A. Formation of F-actin aggregates in cells treated with actin stabilizing drugs. Cell Motil. Cytoskeleton 1998, 39, 122–133. [Google Scholar] [CrossRef]
- Tanaka, Y.; Jahan, M.G.S.; Kondo, T.; Nakano, M.; Yumura, S. Cytokinesis D is mediated by cortical flow of dividing cells Instead of chemotaxis. Cells 2019, 8, 473. [Google Scholar] [CrossRef] [Green Version]
- Bouter, A.; Gounou, C.; Berat, R.; Tan, S.; Gallois, B.; Granier, T.; d’Estaintot, B.L.; Poschl, E.; Brachvogel, B.; Brisson, A.R. Annexin-A5 assembled into two-dimensional arrays promotes cell membrane repair. Nat. Commun. 2011, 2, 270. [Google Scholar] [CrossRef] [Green Version]
- Demonbreun, A.R.; Quattrocelli, M.; Barefield, D.Y.; Allen, M.V.; Swanson, K.E.; McNally, E.M. An actin-dependent annexin complex mediates plasma membrane repair in muscle. J. Cell Biol. 2016, 213, 705–718. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McNeil, A.K.; Rescher, U.; Gerke, V.; McNeil, P.L. Requirement for annexin A1 in plasma membrane repair. J. Biol. Chem. 2006, 281, 35202–35207. [Google Scholar] [CrossRef] [PubMed]
- Cai, L.; Marshall, T.W.; Uetrecht, A.C.; Schafer, D.A.; Bear, J.E. Coronin 1B coordinates Arp2/3 complex and cofilin activities at the leading edge. Cell 2007, 128, 915–929. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pollard, T.D.; Borisy, G.G. Cellular Motility Driven by Assembly and Disassembly of Actin Filaments. Cell 2003, 112, 453–465. [Google Scholar] [CrossRef] [Green Version]
- Devreotes, P.; Horwitz, A.R. Signaling networks that regulate cell migration. Cold Spring Harb. Perspect. Biol. 2015, 7, a005959. [Google Scholar] [CrossRef] [Green Version]
- Rivero, F.; Xiong, H. Rho Signaling in Dictyostelium discoideum. Int Rev. Cell Mol. Biol. 2016, 322, 61–181. [Google Scholar]
- Veltman, D.M.; Keizer-Gunnik, I.; Van Haastert, P.J.M. Four key signaling pathways mediating chemotaxis in Dictyostelium discoideum. J. Cell Biol. 2008, 180, 747–753. [Google Scholar] [CrossRef] [Green Version]
- Xu, X.; Jin, T. ELMO proteins transduce G protein-coupled receptor signal to control reorganization of actin cytoskeleton in chemotaxis of eukaryotic cells. Small GTPases 2019, 10, 271–279. [Google Scholar] [CrossRef]
- Malchow, D.; Lusche, D.F.; De Lozanne, A.; Schlatterer, C. A fast Ca2+-induced Ca2+-release mechanism in Dictyostelium discoideum. Cell Calcium 2008, 43, 521–530. [Google Scholar] [CrossRef]
- Funamoto, S.; Milan, K.; Meili, R.; Firtel, R.A. Role of phosphatidylinositol 3’ kinase and a downstream pleckstrin homology domain-containing protein in controlling chemotaxis in dictyostelium. J. Cell Biol. 2001, 153, 795–810. [Google Scholar] [CrossRef] [Green Version]
- Zhou, K.; Pandol, S.; Bokoch, G.; Traynor-Kaplan, A.E. Disruption of Dictyostelium PI3K genes reduces [32P]phosphatidylinositol 3,4 bisphosphate and [32P]phosphatidylinositol trisphosphate levels, alters F-actin distribution and impairs pinocytosis. J. Cell Sci. 1998, 111, 283–294. [Google Scholar] [CrossRef]
- Chen, L.; Iijima, M.; Tang, M.; Landree, M.A.; Huang, Y.E.; Xiong, Y.; Iglesias, P.A.; Devreotes, P.N. PLA2 and PI3K/PTEN pathways act in parallel to mediate chemotaxis. Dev. Cell 2007, 12, 603–614. [Google Scholar] [CrossRef] [Green Version]
- van Haastert, P.J.M.; Keizer-Gunnink, I.; Kortholt, A. The cytoskeleton regulates symmetry transitions in moving amoeboid cells. J. Cell Sci. 2018, 131, jcs208892. [Google Scholar] [CrossRef]
- Lee, S.; Comer, F.I.; Sasaki, A.; McLeod, I.X.; Duong, Y.; Okumura, K.; Yates, J.R.; Parent, C.A.; Firtel, R.A. TOR complex 2 integrates cell movement during chemotaxis and signal relay in Dictyostelium. Mol. Biol. Cell 2005, 16, 4572–4583. [Google Scholar] [CrossRef] [Green Version]
- Cai, H.; Das, S.; Kamimura, Y.; Long, Y.; Parent, C.A.; Devreotes, P.N. Ras-mediated activation of the TORC2-PKB pathway is critical for chemotaxis. J. Cell Biol. 2010, 190, 233–245. [Google Scholar] [CrossRef] [Green Version]
- Kamimura, Y.; Xiong, Y.; Iglesias, P.A.; Hoeller, O.; Bolourani, P.; Devreotes, P.N. PIP3-independent activation of TorC2 and PKB at the cell’s leading edge mediates chemotaxis. Curr. Biol. 2008, 18, 1034–1043. [Google Scholar] [CrossRef] [Green Version]
- Charest, P.G.; Shen, Z.; Lakoduk, A.; Sasaki, A.T.; Briggs, S.P.; Firtel, R.A. A Ras signaling complex controls the RasC-TORC2 pathway and directed cell migration. Dev. Cell 2010, 18, 737–749. [Google Scholar] [CrossRef] [Green Version]
- Sumagin, R.; Robin, A.Z.; Nusrat, A.; Parkos, C.A. Activation of PKCbetaII by PMA facilitates enhanced epithelial wound repair through increased cell spreading and migration. PLoS ONE 2013, 8, e55775. [Google Scholar] [CrossRef] [Green Version]
- Togo, T.; Alderton, J.M.; Bi, G.Q.; Steinhardt, R.A. The mechanism of facilitated cell membrane resealing. J. Cell Sci. 1999, 112, 719–731. [Google Scholar] [CrossRef]
- Mohamed, W.; Ray, S.; Brazill, D.; Baskar, R. Absence of catalytic domain in a putative protein kinase C (PkcA) suppresses tip dominance in Dictyostelium discoideum. Dev. Biol. 2015, 405, 10–20. [Google Scholar] [CrossRef] [Green Version]
- Singh, S.; Mohamed, W.; Aguessy, A.; Dyett, E.; Shah, S.; Khan, M.; Baskar, R.; Brazill, D. Functional interaction of PkcA and PldB regulate aggregation and development in Dictyostelium discoideum. Cell Signal. 2017, 34, 47–54. [Google Scholar] [CrossRef] [Green Version]
- Bolourani, P.; Spiegelman, G.B.; Weeks, G. Delineation of the roles played by RasG and RasC in cAMP-dependent signal transduction during the early development of Dictyostelium discoideum. Mol. Biol. Cell 2006, 17, 4543–4550. [Google Scholar] [CrossRef] [Green Version]
- Wang, Y.; Steimle, P.A.; Ren, Y.; Ross, C.A.; Robinson, D.N.; Egelhoff, T.T.; Sesaki, H.; Iijima, M. Dictyostelium huntingtin controls chemotaxis and cytokinesis through the regulation of myosin II phosphorylation. Mol. Biol. Cell 2011, 22, 2270–2281. [Google Scholar] [CrossRef]
- Wessels, D.; Lusche, D.F.; Scherer, A.; Kuhl, S.; Myre, M.A.; Soll, D.R. Huntingtin regulates Ca(2+) chemotaxis and K(+)-facilitated cAMP chemotaxis, in conjunction with the monovalent cation/H(+) exchanger Nhe1, in a model developmental system: Insights into its possible role in Huntington’s disease. Dev. Biol. 2014, 394, 24–38. [Google Scholar] [CrossRef] [Green Version]
- Blanc, C.; Charette, S.; Cherix, N.; Lefkir, Y.; Cosson, P.; Letourneur, F. A novel phosphatidylinositol 4,5-bisphosphate-binding domain targeting the Phg2 kinase to the membrane in Dictyostelium cells. Eur. J. Cell Biol. 2005, 84, 951–960. [Google Scholar] [CrossRef]
- Gebbie, L.; Benghezal, M.; Cornillon, S.; Froquet, R.; Cherix, N.; Malbouyres, M.; Lefkir, Y.; Grangeasse, C.; Fache, S.; Dalous, J.; et al. Phg2, a kinase involved in adhesion and focal site modeling in Dictyostelium. Mol. Biol. Cell 2004, 15, 3915–3925. [Google Scholar] [CrossRef] [Green Version]
- van Es, S.; Wessels, D.; Soll, D.R.; Borleis, J.; Devreotes, P.N. Tortoise, a novel mitochondrial protein, is required for directional responses of Dictyostelium in chemotactic gradients. J. Cell Biol. 2001, 152, 621–632. [Google Scholar] [CrossRef] [Green Version]
- Noegel, A.A.; Blau-Wasser, R.; Sultana, H.; Müller, R.; Israel, L.; Schleicher, M.; Patel, H.; Weijer, C.J. The cyclase-associated protein CAP as regulator of cell polarity and cAMP signaling in Dictyostelium. Mol. Biol. Cell 2004, 15, 934–945. [Google Scholar] [CrossRef] [Green Version]
- Sun, B.; Ma, H.; Firtel, R.A. Dictyostelium stress-activated protein kinase alpha, a novel stress-activated mitogen-activated protein kinase kinase kinase-like kinase, is important for the proper regulation of the cytoskeleton. Mol. Biol. Cell 2003, 14, 4526–4540. [Google Scholar] [CrossRef] [Green Version]
- Abreu-Blanco, M.T.; Verboon, J.M.; Liu, R.; Watts, J.J.; Parkhurst, S.M. Drosophila embryos close epithelial wounds using a combination of cellular protrusions and an actomyosin purse string. J. Cell Sci. 2012, 125, 5984–5997. [Google Scholar] [CrossRef] [Green Version]
- Togo, T. Disruption of the plasma membrane stimulates rearrangement of microtubules and lipid traffic toward the wound site. J. Cell Sci. 2006, 119, 2780–2786. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fujimoto, K.; Tanaka, M.; Rana, A.Y.K.M.M.; Jahan, M.G.S.; Itoh, G.; Tsujioka, M.; Uyeda, T.Q.P.; Miyagishima, S.Y.; Yumura, S. Dynamin-like protein B of Dictyostelium contributes to cytokinesis cooperatively with other dynamins. Cells 2019, 8, 781. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Masud Rana, A.Y.; Tsujioka, M.; Miyagishima, S.; Ueda, M.; Yumura, S. Dynamin contributes to cytokinesis by stabilizing actin filaments in the contractile ring. Genes Cells 2013, 18, 621–635. [Google Scholar] [CrossRef] [PubMed]
- Wu, W.I.; Yajnik, J.; Siano, M.; De Lozanne, A. Structure-function analysis of the BEACH protein LvsA. Traffic 2004, 5, 346–355. [Google Scholar] [CrossRef]
- Yumura, S.; Fukui, Y. Filopodelike projections induced with dimethyl sulfoxide and their relevance to cellular polarity in Dictyostelium. J. Cell Biol. 1983, 96, 857–865. [Google Scholar] [CrossRef] [Green Version]
- Brzeska, H.; Pridham, K.; Chery, G.; Titus, M.A.; Korn, E.D. The association of myosin IB with actin waves in dictyostelium requires both the plasma membrane-binding site and actin-binding region in the myosin tail. PLoS ONE 2014, 9, e94306. [Google Scholar] [CrossRef] [Green Version]
- McRobbie, S.J.; Newell, P.C. Chemoattractant-mediated changes in cytoskeletal actin of cellular slime moulds. J. Cell Sci. 1984, 68, 139–151. [Google Scholar] [CrossRef]
- Yumura, S. Reorganization of actin and myosin II in Dictyostelium amoeba during stimulation by cAMP. Cell Struct. Funct. 1993, 18, 379–388. [Google Scholar] [CrossRef] [Green Version]
- Yumura, S. Rapid translocation of myosin II in vegetative Dictyostelium amoebae during chemotactic stimulation by folic acid. Cell Struct. Funct. 1994, 19, 143–151. [Google Scholar] [CrossRef] [Green Version]
- Junemann, A.; Filić, V.; Winterhoff, M.; Nordholz, B.; Litschko, C.; Schwellenbach, H.; Stephan, T.; Weber, I.; Faix, J. A Diaphanous-related formin links Ras signaling directly to actin assembly in macropinocytosis and phagocytosis. Proc. Natl. Acad. Sci. USA 2016, 113, E7464–E7473. [Google Scholar] [CrossRef] [Green Version]
- Litschko, C.; Brühmann, S.; Csiször, A.; Stephan, T.; Dimchev, V.; Damiano-Guercio, J.; Junemann, A.; Körber, S.; Winterhoff, M.; Nordholz, B.; et al. Functional integrity of the contractile actin cortex is safeguarded by multiple Diaphanous-related formins. Proc. Natl. Acad. Sci. USA 2019, 116, 3594–3603. [Google Scholar] [CrossRef] [Green Version]
- Schirenbeck, A.; Bretschneider, T.; Arasada, R.; Schleicher, M.; Faix, J. The Diaphanous-related formin dDia2 is required for the formation and maintenance of filopodia. Nat. Cell Biol. 2005, 7, 619–625. [Google Scholar] [CrossRef]
- Aizawa, H.; Sutoh, K.; Tsubuki, S.; Kawashima, S.; Ishii, A.; Yahara, I. Identification, characterization, and intracellular distribution of cofilin in Dictyostelium discoideum. J. Biol. Chem. 1995, 270, 10923–10932. [Google Scholar] [CrossRef]
- Mikati, M.A.; Breitsprecher, D.; Jansen, S.; Reisler, E.; Goode, B.L. Coronin Enhances Actin Filament Severing by Recruiting Cofilin to Filament Sides and Altering F-Actin Conformation. J. Mol. Biol. 2015, 427, 3137–3147. [Google Scholar] [CrossRef] [Green Version]
- Rivero, F.; Furukawa, R.; Fechheimer, M.; Noegel, A.A. Three actin cross-linking proteins, the 34 kDa actin-bundling protein, alpha-actinin and gelation factor (ABP-120), have both unique and redundant roles in the growth and development of Dictyostelium. J. Cell Sci. 1999, 112, 2737–2751. [Google Scholar] [CrossRef]
- Sasaki, A.T.; Chun, C.; Takeda, K.; Firtel, R.A. Localized Ras signaling at the leading edge regulates PI3K, cell polarity, and directional cell movement. J. Cell Biol. 2004, 167, 505–518. [Google Scholar] [CrossRef]
- Rozelle, A.L.; Machesky, L.M.; Yamamoto, M.; Driessens, M.H.; Insall, R.H.; Roth, M.G.; Luby-Phelps, K.; Marriott, G.; Hall, A.; Yin, H.L. Phosphatidylinositol 4,5-bisphosphate induces actin-based movement of raft-enriched vesicles through WASP-Arp2/3. Curr. Biol. 2000, 10, 311–320. [Google Scholar] [CrossRef] [Green Version]
- Zhang, L.; Mao, Y.S.; Janmey, P.A.; Yin, H.L. Phosphatidylinositol 4, 5 bisphosphate and the actin cytoskeleton. Subcell. Biochem. 2012, 59, 177–215. [Google Scholar]
- Schaloske, R.; Malchow, D. Mechanism of cAMP-induced Ca2+ influx in Dictyostelium: Role of phospholipase A2. Biochem. J. 1997, 327, 233–238. [Google Scholar] [CrossRef]
- O’Day, D.H.; Taylor, R.J.; Myre, M.A. Calmodulin and Calmodulin Binding Proteins in Dictyostelium: A Primer. Int. J. Mol. Sci. 2020, 21, 1210. [Google Scholar] [CrossRef]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yumura, S.; Talukder, M.S.U.; Pervin, M.S.; Tanvir, M.I.O.; Matsumura, T.; Fujimoto, K.; Tanaka, M.; Itoh, G. Dynamics of Actin Cytoskeleton and Their Signaling Pathways during Cellular Wound Repair. Cells 2022, 11, 3166. https://doi.org/10.3390/cells11193166
Yumura S, Talukder MSU, Pervin MS, Tanvir MIO, Matsumura T, Fujimoto K, Tanaka M, Itoh G. Dynamics of Actin Cytoskeleton and Their Signaling Pathways during Cellular Wound Repair. Cells. 2022; 11(19):3166. https://doi.org/10.3390/cells11193166
Chicago/Turabian StyleYumura, Shigehiko, Md. Shahabe Uddin Talukder, Mst. Shaela Pervin, Md. Istiaq Obaidi Tanvir, Takashi Matsumura, Koushiro Fujimoto, Masahito Tanaka, and Go Itoh. 2022. "Dynamics of Actin Cytoskeleton and Their Signaling Pathways during Cellular Wound Repair" Cells 11, no. 19: 3166. https://doi.org/10.3390/cells11193166
APA StyleYumura, S., Talukder, M. S. U., Pervin, M. S., Tanvir, M. I. O., Matsumura, T., Fujimoto, K., Tanaka, M., & Itoh, G. (2022). Dynamics of Actin Cytoskeleton and Their Signaling Pathways during Cellular Wound Repair. Cells, 11(19), 3166. https://doi.org/10.3390/cells11193166