
Mitochondrial VDAC1: A Potential Therapeutic Target of Inflammation-Related Diseases and Clinical Opportunities

{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
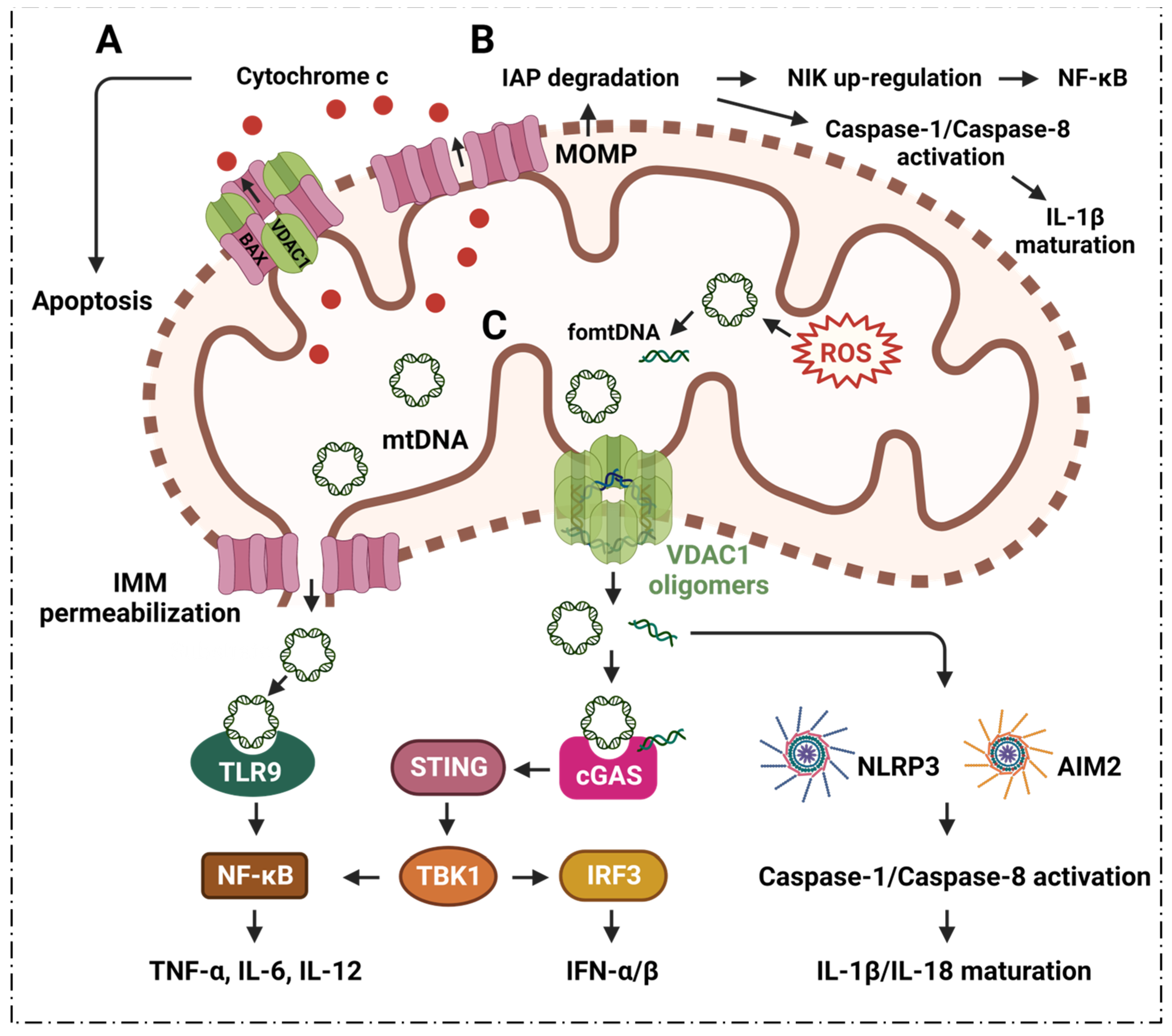
2. Inflammation, VDAC1 Mediates Apoptosis and Mitochondrial Oxidative Stress
2.1. VDAC1 Regulates Inflammation via Mediating Apoptosis
2.2. VDAC1 Mediates Mitochondrial Oxidative Stress in Immune Responses
3. Inflammation and VDAC1 Mediates Mitochondrial Ca2+ Transportation
3.1. Neutrophils
3.2. Macrophages
3.3. Dendritic Cells
4. Inflammatory Diseases and VDAC1 in Energy Metabolism
4.1. TCA Cycle
4.2. Glycolysis
5. Inflammatory Diseases and VDAC1 in Lipid Metabolism
6. Inflammatory Diseases Pathogenesis and VDAC1 in Mitophagy
6.1. Mitophagy Regulates Inflammation via VDAC1
6.2. Mitophagy Modulates mtDNA Levels in Cytoplasm
7. Summary and Conclusions, Current Clinical Conditions and Future Perspectives
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Medzhitov, R. Inflammation 2010: New adventures of an old flame. Cell 2010, 140, 771–776. [Google Scholar] [CrossRef]
- Scrivo, R.; Vasile, M.; Bartosiewicz, I.; Valesini, G. Inflammation as “common soil” of the multifactorial diseases. Autoimmun. Rev. 2011, 10, 369–374. [Google Scholar] [CrossRef]
- Medzhitov, R. Origin and physiological roles of inflammation. Nature 2008, 454, 428–435. [Google Scholar] [CrossRef] [PubMed]
- Hengartner, M.O. The biochemistry of apoptosis. Nature 2000, 407, 770–776. [Google Scholar] [CrossRef] [PubMed]
- Nunnari, J.; Suomalainen, A. Mitochondria: In sickness and in health. Cell 2012, 148, 1145–1159. [Google Scholar] [CrossRef]
- Pfanner, N.; Warscheid, B.; Wiedemann, N. Mitochondrial proteins: From biogenesis to functional networks. Nat. Rev. Mol. Cell Biol. 2019, 20, 267–284. [Google Scholar] [CrossRef]
- Bock, F.J.; Tait, S.W.G. Mitochondria as multifaceted regulators of cell death. Nat. Rev. Mol. Cell Biol. 2020, 21, 85–100. [Google Scholar] [CrossRef]
- Tait, S.W.; Green, D.R. Mitochondrial regulation of cell death. Cold Spring Harb. Perspect. Biol. 2013, 5, a008706. [Google Scholar] [CrossRef] [PubMed]
- Marchi, S.; Guilbaud, E.; Tait, S.W.G.; Yamazaki, T.; Galluzzi, L. Mitochondrial control of inflammation. Nat. Rev. Immunol. 2022, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Schapira, A.H. Mitochondrial diseases. Lancet 2012, 379, 1825–1834. [Google Scholar] [CrossRef]
- Gorman, G.S.; Chinnery, P.F.; DiMauro, S.; Hirano, M.; Koga, Y.; McFarland, R.; Suomalainen, A.; Thorburn, D.R.; Zeviani, M.; Turnbull, D.M. Mitochondrial diseases. Nat. Rev. Dis. Prim. 2016, 2, 16080. [Google Scholar] [CrossRef]
- Wallace, D.C. Mitochondrial genetic medicine. Nat. Genet. 2018, 50, 1642–1649. [Google Scholar] [CrossRef]
- Schein, S.J.; Colombini, M.; Finkelstein, A. Reconstitution in planar lipid bilayers of a voltage-dependent anion-selective channel obtained from paramecium mitochondria. J. Membr. Biol. 1976, 30, 99–120. [Google Scholar] [CrossRef] [PubMed]
- Di Rosa, M.C.; Guarino, F.; Conti Nibali, S.; Magrì, A.; De Pinto, V. Voltage-Dependent Anion Selective Channel Isoforms in Yeast: Expression, Structure, and Functions. Front. Physiol. 2021, 12, 675708. [Google Scholar] [CrossRef]
- Guardiani, C.; Magrì, A.; Karachitos, A.; Di Rosa, M.C.; Reina, S.; Bodrenko, I.; Messina, A.; Kmita, H.; Ceccarelli, M.; De Pinto, V. yVDAC2, the second mitochondrial porin isoform of Saccharomyces cerevisiae. Biochim. Biophys. Acta Bioenerg. 2018, 1859, 270–279. [Google Scholar] [CrossRef]
- De Stefani, D.; Bononi, A.; Romagnoli, A.; Messina, A.; De Pinto, V.; Pinton, P.; Rizzuto, R. VDAC1 selectively transfers apoptotic Ca2+ signals to mitochondria. Cell Death Differ. 2012, 19, 267–273. [Google Scholar] [CrossRef]
- Messina, A.; Reina, S.; Guarino, F.; De Pinto, V. VDAC isoforms in mammals. Biochim. Biophys. Acta 2012, 1818, 1466–1476. [Google Scholar] [CrossRef]
- Zinghirino, F.; Pappalardo, X.G.; Messina, A.; Guarino, F.; De Pinto, V. Is the secret of VDAC Isoforms in their gene regulation? Characterization of human VDAC genes expression profile, promoter activity, and transcriptional regulators. Int. J. Mol. Sci. 2020, 21, 7388. [Google Scholar] [CrossRef] [PubMed]
- Cheng, E.H.; Sheiko, T.V.; Fisher, J.K.; Craigen, W.J.; Korsmeyer, S.J. VDAC2 inhibits BAK activation and mitochondrial apoptosis. Science 2003, 301, 513–517. [Google Scholar] [CrossRef] [PubMed]
- Reina, S.; Nibali, S.C.; Tomasello, M.F.; Magrì, A.; Messina, A.; De Pinto, V. Voltage Dependent Anion Channel 3 (VDAC3) protects mitochondria from oxidative stress. Redox Biol. 2022, 51, 102264. [Google Scholar] [CrossRef] [PubMed]
- Zinghirino, F.; Pappalardo, X.G.; Messina, A.; Nicosia, G.; De Pinto, V.; Guarino, F. VDAC Genes Expression and Regulation in Mammals. Front. Physiol. 2021, 12, 708695. [Google Scholar] [CrossRef] [PubMed]
- Geula, S.; Ben-Hail, D.; Shoshan-Barmatz, V. Structure-based analysis of VDAC1: N-terminus location, translocation, channel gating and association with anti-apoptotic proteins. Biochem. J. 2012, 444, 475–485. [Google Scholar] [CrossRef]
- Hiller, S.; Abramson, J.; Mannella, C.; Wagner, G.; Zeth, K. The 3D structures of VDAC represent a native conformation. Trends Biochem. Sci. 2010, 35, 514–521. [Google Scholar] [CrossRef]
- Bayrhuber, M.; Meins, T.; Habeck, M.; Becker, S.; Giller, K.; Villinger, S.; Vonrhein, C.; Griesinger, C.; Zweckstetter, M.; Zeth, K. Structure of the human voltage-dependent anion channel. Proc. Natl. Acad. Sci. USA 2008, 105, 15370–15375. [Google Scholar] [CrossRef]
- Hiller, S.; Garces, R.G.; Malia, T.J.; Orekhov, V.Y.; Colombini, M.; Wagner, G. Solution structure of the integral human membrane protein VDAC-1 in detergent micelles. Science 2008, 321, 1206–1210. [Google Scholar] [CrossRef]
- Ujwal, R.; Cascio, D.; Colletier, J.P.; Faham, S.; Zhang, J.; Toro, L.; Ping, P.; Abramson, J. The crystal structure of mouse VDAC1 at 2.3 A resolution reveals mechanistic insights into metabolite gating. Proc. Natl. Acad. Sci. USA 2008, 105, 17742–17747. [Google Scholar] [CrossRef] [PubMed]
- Ham, S.J.; Lee, D.; Yoo, H.; Jun, K.; Shin, H.; Chung, J. Decision between mitophagy and apoptosis by Parkin via VDAC1 ubiquitination. Proc. Natl. Acad. Sci. USA 2020, 117, 4281–4291. [Google Scholar] [CrossRef]
- Shoshan-Barmatz, V.; Golan, M. Mitochondrial VDAC1: Function in cell life and death and a target for cancer therapy. Curr. Med. Chem. 2012, 19, 714–735. [Google Scholar] [CrossRef]
- Shoshan-Barmatz, V.; De Pinto, V.; Zweckstetter, M.; Raviv, Z.; Keinan, N.; Arbel, N. VDAC, a multi-functional mitochondrial protein regulating cell life and death. Mol. Asp. Med. 2010, 31, 227–285. [Google Scholar] [CrossRef]
- Huang, H.; Hu, X.; Eno, C.O.; Zhao, G.; Li, C.; White, C. An interaction between Bcl-xL and the voltage-dependent anion channel (VDAC) promotes mitochondrial Ca2+ uptake. J. Biol. Chem. 2013, 288, 19870–19881. [Google Scholar] [CrossRef]
- Baines, C.P.; Kaiser, R.A.; Sheiko, T.; Craigen, W.J.; Molkentin, J.D. Voltage-dependent anion channels are dispensable for mitochondrial-dependent cell death. Nat. Cell Biol. 2007, 9, 550–555. [Google Scholar] [CrossRef]
- Kim, J.; Gupta, R.; Blanco, L.P.; Yang, S.; Shteinfer-Kuzmine, A.; Wang, K.; Zhu, J.; Yoon, H.E.; Wang, X.; Kerkhofs, M.; et al. VDAC oligomers form mitochondrial pores to release mtDNA fragments and promote lupus-like disease. Science 2019, 366, 1531–1536. [Google Scholar] [CrossRef]
- Xian, H.; Watari, K.; Sanchez-Lopez, E.; Offenberger, J.; Onyuru, J.; Sampath, H.; Ying, W.; Hoffman, H.M.; Shadel, G.S.; Karin, M. Oxidized DNA fragments exit mitochondria via mPTP- and VDAC-dependent channels to activate NLRP3 inflammasome and interferon signaling. Immunity 2022, 55, 1370–1385.e1378. [Google Scholar] [CrossRef] [PubMed]
- Wu, J.; Ma, Z.; Raman, A.; Beckerman, P.; Dhillon, P.; Mukhi, D.; Palmer, M.; Chen, H.C.; Cohen, C.R.; Dunn, T.; et al. APOL1 risk variants in individuals of African genetic ancestry drive endothelial cell defects that exacerbate sepsis. Immunity 2021, 54, 2632–2649.e2636. [Google Scholar] [CrossRef] [PubMed]
- Onishi, M.; Yamano, K.; Sato, M.; Matsuda, N.; Okamoto, K. Molecular mechanisms and physiological functions of mitophagy. EMBO J. 2021, 40, e104705. [Google Scholar] [CrossRef]
- Marek-Iannucci, S.; Ozdemir, A.B.; Moreira, D.; Gomez, A.C.; Lane, M.; Porritt, R.A.; Lee, Y.; Shimada, K.; Abe, M.; Stotland, A.; et al. Autophagy-mitophagy induction attenuates cardiovascular inflammation in a murine model of Kawasaki disease vasculitis. JCI Insight 2021, 6, e151981. [Google Scholar] [CrossRef] [PubMed]
- Green, D.R.; Galluzzi, L.; Kroemer, G. Mitochondria and the autophagy-inflammation-cell death axis in organismal aging. Science 2011, 333, 1109–1112. [Google Scholar] [CrossRef] [PubMed]
- Sidarala, V.; Pearson, G.L.; Parekh, V.S.; Thompson, B.; Christen, L.; Gingerich, M.A.; Zhu, J.; Stromer, T.; Ren, J.; Reck, E.C.; et al. Mitophagy protects β cells from inflammatory damage in diabetes. JCI Insight 2020, 5, e141138. [Google Scholar] [CrossRef]
- Ip, W.K.E.; Hoshi, N.; Shouval, D.S.; Snapper, S.; Medzhitov, R. Anti-inflammatory effect of IL-10 mediated by metabolic reprogramming of macrophages. Science 2017, 356, 513–519. [Google Scholar] [CrossRef]
- Camara, A.K.S.; Zhou, Y.; Wen, P.C.; Tajkhorshid, E.; Kwok, W.M. Mitochondrial VDAC1: A Key Gatekeeper as Potential Therapeutic Target. Front. Physiol. 2017, 8, 460. [Google Scholar] [CrossRef] [PubMed]
- Suomalainen, A.; Battersby, B.J. Mitochondrial diseases: The contribution of organelle stress responses to pathology. Nat. Rev. Mol. Cell Biol. 2018, 19, 77–92. [Google Scholar] [CrossRef] [PubMed]
- Messer, J.S. The cellular autophagy/apoptosis checkpoint during inflammation. Cell. Mol. Life Sci. CMLS 2017, 74, 1281–1296. [Google Scholar] [CrossRef] [PubMed]
- Girardot, T.; Rimmelé, T.; Venet, F.; Monneret, G. Apoptosis-induced lymphopenia in sepsis and other severe injuries. Apoptosis Int. J. Program. Cell Death 2017, 22, 295–305. [Google Scholar] [CrossRef]
- Vignola, A.M.; Chiappara, G.; Gagliardo, R.; Gjomarkaj, M.; Merendino, A.; Siena, L.; Bousquet, J.; Bonsignore, G. Apoptosis and airway inflammation in asthma. Apoptosis 2000, 5, 473–485. [Google Scholar] [CrossRef]
- Mishra, V.; Banga, J.; Silveyra, P. Oxidative stress and cellular pathways of asthma and inflammation: Therapeutic strategies and pharmacological targets. Pharmacol. Ther. 2018, 181, 169–182. [Google Scholar] [CrossRef]
- Collison, A.; Foster, P.S.; Mattes, J. Emerging role of tumour necrosis factor-related apoptosis-inducing ligand (TRAIL) as a key regulator of inflammatory responses. Clin. Exp. Pharmacol. Physiol. 2009, 36, 1049–1053. [Google Scholar] [CrossRef]
- Roca, H.; Jones, J.D.; Purica, M.C.; Weidner, S.; Koh, A.J.; Kuo, R.; Wilkinson, J.E.; Wang, Y.; Daignault-Newton, S.; Pienta, K.J.; et al. Apoptosis-induced CXCL5 accelerates inflammation and growth of prostate tumor metastases in bone. J. Clin. Investig. 2018, 128, 248–266. [Google Scholar] [CrossRef]
- Zoller, V.; Funcke, J.B.; Roos, J.; Dahlhaus, M.; Abd El Hay, M.; Holzmann, K.; Marienfeld, R.; Kietzmann, T.; Debatin, K.M.; Wabitsch, M.; et al. Trail (TNF-related apoptosis-inducing ligand) induces an inflammatory response in human adipocytes. Sci. Rep. 2017, 7, 5691. [Google Scholar] [CrossRef] [PubMed]
- Van den Oever, I.A.; Raterman, H.G.; Nurmohamed, M.T.; Simsek, S. Endothelial dysfunction, inflammation, and apoptosis in diabetes mellitus. Mediat. Inflamm. 2010, 2010, 792393. [Google Scholar] [CrossRef]
- Giorgio, V.; Guo, L.; Bassot, C.; Petronilli, V.; Bernardi, P. Calcium and regulation of the mitochondrial permeability transition. Cell Calcium 2018, 70, 56–63. [Google Scholar] [CrossRef] [PubMed]
- Urbani, A.; Giorgio, V.; Carrer, A.; Franchin, C.; Arrigoni, G.; Jiko, C.; Abe, K.; Maeda, S.; Shinzawa-Itoh, K.; Bogers, J.F.M.; et al. Purified F-ATP synthase forms a Ca2+-dependent high-conductance channel matching the mitochondrial permeability transition pore. Nat. Commun. 2019, 10, 4341. [Google Scholar] [CrossRef] [PubMed]
- Moya, G.E.; Rivera, P.D.; Dittenhafer-Reed, K.E. Evidence for the Role of Mitochondrial DNA Release in the Inflammatory Response in Neurological Disorders. Int. J. Mol. Sci. 2021, 22, 7030. [Google Scholar] [CrossRef]
- Gutiérrez-Aguilar, M.; Baines, C.P. Structural mechanisms of cyclophilin D-dependent control of the mitochondrial permeability transition pore. Biochim. Biophys. Acta 2015, 1850, 2041–2047. [Google Scholar] [CrossRef] [PubMed]
- Jia, K.; Du, H. Mitochondrial Permeability Transition: A Pore Intertwines Brain Aging and Alzheimer’s Disease. Cells 2021, 10, 649. [Google Scholar] [CrossRef]
- Green, D.R.; Reed, J.C. Mitochondria and apoptosis. Science 1998, 281, 1309–1312. [Google Scholar] [CrossRef]
- Shimizu, S.; Narita, M.; Tsujimoto, Y. Bcl-2 family proteins regulate the release of apoptogenic cytochrome c by the mitochondrial channel VDAC. Nature 1999, 399, 483–487. [Google Scholar] [CrossRef]
- Yang, J.; Liu, X.; Bhalla, K.; Kim, C.N.; Ibrado, A.M.; Cai, J.; Peng, T.I.; Jones, D.P.; Wang, X. Prevention of apoptosis by Bcl-2: Release of cytochrome c from mitochondria blocked. Science 1997, 275, 1129–1132. [Google Scholar] [CrossRef]
- Peña-Blanco, A.; García-Sáez, A.J. Bax, Bak and beyond—Mitochondrial performance in apoptosis. FEBS J. 2018, 285, 416–431. [Google Scholar] [CrossRef]
- Banerjee, J.; Ghosh, S. Bax increases the pore size of rat brain mitochondrial voltage-dependent anion channel in the presence of tBid. Biochem. Biophys. Res. Commun. 2004, 323, 310–314. [Google Scholar] [CrossRef]
- Elmore, S. Apoptosis: A review of programmed cell death. Toxicol. Pathol. 2007, 35, 495–516. [Google Scholar] [CrossRef]
- Wang, C.; Youle, R.J. The role of mitochondria in apoptosis. Annu. Rev. Genet. 2009, 43, 95–118. [Google Scholar] [CrossRef] [PubMed]
- Riley, J.S.; Quarato, G.; Cloix, C.; Lopez, J.; O’Prey, J.; Pearson, M.; Chapman, J.; Sesaki, H.; Carlin, L.M.; Passos, J.F.; et al. Mitochondrial inner membrane permeabilisation enables mtDNA release during apoptosis. EMBO J. 2018, 37. [Google Scholar]
- Huang, L.S.; Hong, Z.; Wu, W.; Xiong, S.; Zhong, M.; Gao, X.; Rehman, J.; Malik, A.B. mtDNA Activates cGAS Signaling and Suppresses the YAP-Mediated Endothelial Cell Proliferation Program to Promote Inflammatory Injury. Immunity 2020, 52, 475–486.e475. [Google Scholar] [CrossRef]
- Sun, L.; Wu, J.; Du, F.; Chen, X.; Chen, Z.J. Cyclic GMP-AMP synthase is a cytosolic DNA sensor that activates the type I interferon pathway. Science 2013, 339, 786–791. [Google Scholar] [CrossRef] [PubMed]
- Pérez-Treviño, P.; Velásquez, M.; García, N. Mechanisms of mitochondrial DNA escape and its relationship with different metabolic diseases. Biochim. Biophys. Acta Mol. Basis Dis. 2020, 1866, 165761. [Google Scholar] [CrossRef]
- Alexeyev, M.; Shokolenko, I.; Wilson, G.; LeDoux, S. The maintenance of mitochondrial DNA integrity—critical analysis and update. Cold Spring Harb. Perspect. Biol. 2013, 5, a012641. [Google Scholar] [CrossRef]
- Moreira, P.I.; Carvalho, C.; Zhu, X.; Smith, M.A.; Perry, G. Mitochondrial dysfunction is a trigger of Alzheimer’s disease pathophysiology. Biochim. Biophys. Acta 2010, 1802, 2–10. [Google Scholar] [CrossRef]
- Schmitt, K.; Grimm, A.; Kazmierczak, A.; Strosznajder, J.B.; Götz, J.; Eckert, A. Insights into mitochondrial dysfunction: Aging, amyloid-β, and tau-A deleterious trio. Antioxid. Redox Signal. 2012, 16, 1456–1466. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.R.; Han, J. Mitochondrial Nucleoid: Shield and Switch of the Mitochondrial Genome. Oxidative Med. Cell. Longev. 2017, 2017, 8060949. [Google Scholar] [CrossRef]
- Ben-Hail, D.; Begas-Shvartz, R.; Shalev, M.; Shteinfer-Kuzmine, A.; Gruzman, A.; Reina, S.; De Pinto, V.; Shoshan-Barmatz, V. Novel Compounds Targeting the Mitochondrial Protein VDAC1 Inhibit Apoptosis and Protect against Mitochondrial Dysfunction. J. Biol. Chem. 2016, 291, 24986–25003. [Google Scholar] [CrossRef] [PubMed]
- Shteinfer-Kuzmine, A.; Argueti-Ostrovsky, S.; Leyton-Jaimes, M.F.; Anand, U.; Abu-Hamad, S.; Zalk, R.; Shoshan-Barmatz, V.; Israelson, A. Targeting the Mitochondrial Protein VDAC1 as a Potential Therapeutic Strategy in ALS. Int. J. Mol. Sci. 2022, 23, 9946. [Google Scholar] [CrossRef]
- Zhang, E.; Mohammed Al-Amily, I.; Mohammed, S.; Luan, C.; Asplund, O.; Ahmed, M.; Ye, Y.; Ben-Hail, D.; Soni, A.; Vishnu, N.; et al. Preserving Insulin Secretion in Diabetes by Inhibiting VDAC1 Overexpression and Surface Translocation in β Cells. Cell Metab. 2019, 29, 64–77.e66. [Google Scholar] [CrossRef]
- Klapper-Goldstein, H.; Verma, A.; Elyagon, S.; Gillis, R.; Murninkas, M.; Pittala, S.; Paul, A.; Shoshan-Barmatz, V.; Etzion, Y. VDAC1 in the diseased myocardium and the effect of VDAC1-interacting compound on atrial fibrosis induced by hyperaldosteronism. Sci. Rep. 2020, 10, 22101. [Google Scholar] [CrossRef] [PubMed]
- Verma, A.; Pittala, S.; Alhozeel, B.; Shteinfer-Kuzmine, A.; Ohana, E.; Gupta, R.; Chung, J.H.; Shoshan-Barmatz, V. The role of the mitochondrial protein VDAC1 in inflammatory bowel disease: A potential therapeutic target. Mol. Ther. J. Am. Soc. Gene Ther. 2022, 30, 726–744. [Google Scholar] [CrossRef]
- Shoshan-Barmatz, V.; Ben-Hail, D.; Admoni, L.; Krelin, Y.; Tripathi, S.S. The mitochondrial voltage-dependent anion channel 1 in tumor cells. Biochim. Biophys. Acta 2015, 1848, 2547–2575. [Google Scholar] [CrossRef] [PubMed]
- Arif, T.; Vasilkovsky, L.; Refaely, Y.; Konson, A.; Shoshan-Barmatz, V. Silencing VDAC1 Expression by siRNA Inhibits Cancer Cell Proliferation and Tumor Growth In Vivo. Mol. Ther. Nucleic Acids 2017, 8, 493. [Google Scholar] [CrossRef] [PubMed]
- Manczak, M.; Reddy, P.H. RNA silencing of genes involved in Alzheimer’s disease enhances mitochondrial function and synaptic activity. Biochim. Biophys. Acta 2013, 1832, 2368–2378. [Google Scholar] [CrossRef] [PubMed]
- Smilansky, A.; Dangoor, L.; Nakdimon, I.; Ben-Hail, D.; Mizrachi, D.; Shoshan-Barmatz, V. The Voltage-dependent Anion Channel 1 Mediates Amyloid β Toxicity and Represents a Potential Target for Alzheimer Disease Therapy. J. Biol. Chem. 2015, 290, 30670–30683. [Google Scholar] [CrossRef]
- Wu, J.; Sun, L.; Chen, X.; Du, F.; Shi, H.; Chen, C.; Chen, Z.J. Cyclic GMP-AMP is an endogenous second messenger in innate immune signaling by cytosolic DNA. Science 2013, 339, 826–830. [Google Scholar] [CrossRef]
- Wan, D.; Jiang, W.; Hao, J. Research Advances in How the cGAS-STING Pathway Controls the Cellular Inflammatory Response. Front. Immunol. 2020, 11, 615. [Google Scholar] [CrossRef]
- Liu, S.; Cai, X.; Wu, J.; Cong, Q.; Chen, X.; Li, T.; Du, F.; Ren, J.; Wu, Y.T.; Grishin, N.V.; et al. Phosphorylation of innate immune adaptor proteins MAVS, STING, and TRIF induces IRF3 activation. Science 2015, 347, aaa2630. [Google Scholar] [CrossRef] [PubMed]
- Müller, T.; Hamm, S.; Bauer, S. TLR9-mediated recognition of DNA. In Toll-Like Receptors (TLRs) and Innate Immunity; Springer: Berlin/Heidelberg, Germany, 2008; pp. 51–70. [Google Scholar]
- De Gaetano, A.; Solodka, K.; Zanini, G.; Selleri, V.; Mattioli, A.V.; Nasi, M.; Pinti, M. Molecular Mechanisms of mtDNA-Mediated Inflammation. Cells 2021, 10, 2898. [Google Scholar] [CrossRef]
- Riley, J.S.; Tait, S.W. Mitochondrial DNA in inflammation and immunity. EMBO Rep. 2020, 21, e49799. [Google Scholar] [CrossRef]
- Li, Z.; Guo, J.; Bi, L. Role of the NLRP3 inflammasome in autoimmune diseases. Biomed. Pharmacother. 2020, 130, 110542. [Google Scholar] [CrossRef]
- Shimada, K.; Crother, T.R.; Karlin, J.; Dagvadorj, J.; Chiba, N.; Chen, S.; Ramanujan, V.K.; Wolf, A.J.; Vergnes, L.; Ojcius, D.M.; et al. Oxidized mitochondrial DNA activates the NLRP3 inflammasome during apoptosis. Immunity 2012, 36, 401–414. [Google Scholar] [CrossRef]
- Zhong, Z.; Sanchez-Lopez, E.; Karin, M. Autophagy, NLRP3 inflammasome and auto-inflammatory/immune diseases. Clin. Exp. Rheumatol. 2016, 34 (Suppl. S98), 12–16. [Google Scholar]
- Nakahira, K.; Haspel, J.A.; Rathinam, V.A.; Lee, S.J.; Dolinay, T.; Lam, H.C.; Englert, J.A.; Rabinovitch, M.; Cernadas, M.; Kim, H.P.; et al. Autophagy proteins regulate innate immune responses by inhibiting the release of mitochondrial DNA mediated by the NALP3 inflammasome. Nat. Immunol. 2011, 12, 222–230. [Google Scholar] [CrossRef]
- Man, S.M.; Karki, R.; Kanneganti, T.D. AIM2 inflammasome in infection, cancer, and autoimmunity: Role in DNA sensing, inflammation, and innate immunity. Eur. J. Immunol. 2016, 46, 269–280. [Google Scholar] [CrossRef] [PubMed]
- Lugrin, J.; Martinon, F. The AIM2 inflammasome: Sensor of pathogens and cellular perturbations. Immunol. Rev. 2018, 281, 99–114. [Google Scholar] [CrossRef]
- Man, S.M.; Karki, R.; Sasai, M.; Place, D.E.; Kesavardhana, S.; Temirov, J.; Frase, S.; Zhu, Q.; Malireddi, R.K.S.; Kuriakose, T.; et al. IRGB10 Liberates Bacterial Ligands for Sensing by the AIM2 and Caspase-11-NLRP3 Inflammasomes. Cell 2016, 167, 382–396.e317. [Google Scholar] [CrossRef]
- Lammert, C.R.; Frost, E.L.; Bellinger, C.E.; Bolte, A.C.; McKee, C.A.; Hurt, M.E.; Paysour, M.J.; Ennerfelt, H.E.; Lukens, J.R. AIM2 inflammasome surveillance of DNA damage shapes neurodevelopment. Nature 2020, 580, 647–652. [Google Scholar]
- Bravo-Sagua, R.; Parra, V.; López-Crisosto, C.; Díaz, P.; Quest, A.F.; Lavandero, S. Calcium Transport and Signaling in Mitochondria. Compr. Physiol. 2017, 7, 623–634. [Google Scholar] [PubMed]
- Shoshan-Barmatz, V.; De, S. Mitochondrial VDAC, the Na+/Ca2+ Exchanger, and the Ca2+ Uniporter in Ca2+ Dynamics and Signaling. Adv. Exp. Med. Biol. 2017, 981, 323–347. [Google Scholar]
- Ben-Hail, D.; Palty, R.; Shoshan-Barmatz, V. Measurement of mitochondrial Ca2+ transport mediated by three transport proteins: VDAC1, the Na+/Ca2+ exchanger, and the Ca2+ uniporter. Cold Spring Harb. Protoc. 2014, 2014, 161–166. [Google Scholar] [CrossRef]
- Gincel, D.; Zaid, H.; Shoshan-Barmatz, V. Calcium binding and translocation by the voltage-dependent anion channel: A possible regulatory mechanism in mitochondrial function. Biochem. J. 2001, 358, 147–155. [Google Scholar] [CrossRef]
- Abu-Hamad, S.; Sivan, S.; Shoshan-Barmatz, V. The expression level of the voltage-dependent anion channel controls life and death of the cell. Proc. Natl. Acad. Sci. USA 2006, 103, 5787–5792. [Google Scholar] [CrossRef]
- Griesbeck, O.; Baird, G.S.; Campbell, R.E.; Zacharias, D.A.; Tsien, R.Y. Reducing the environmental sensitivity of yellow fluorescent protein. Mechanism and applications. J. Biol. Chem. 2001, 276, 29188–29194. [Google Scholar] [CrossRef]
- Dalal, P.J.; Muller, W.A.; Sullivan, D.P. Endothelial Cell Calcium Signaling during Barrier Function and Inflammation. Am. J. Pathol. 2020, 190, 535–542. [Google Scholar] [CrossRef]
- Berridge, M.J.; Bootman, M.D.; Lipp, P. Calcium—A life and death signal. Nature 1998, 395, 645–648. [Google Scholar] [CrossRef] [PubMed]
- Yang, R.; Lirussi, D.; Thornton, T.M.; Jelley-Gibbs, D.M.; Diehl, S.A.; Case, L.K.; Madesh, M.; Taatjes, D.J.; Teuscher, C.; Haynes, L.; et al. Mitochondrial Ca2+ and membrane potential, an alternative pathway for Interleukin 6 to regulate CD4 cell effector function. eLife 2015, 4, e06376. [Google Scholar] [CrossRef] [PubMed]
- Valença-Pereira, F.; Fang, Q.; Marié, I.J.; Giddings, E.L.; Fortner, K.A.; Yang, R.; Villarino, A.V.; Huang, Y.H.; Frank, D.A.; Wen, H.; et al. IL-6 enhances CD4 cell motility by sustaining mitochondrial Ca2+ through the noncanonical STAT3 pathway. Proc. Natl. Acad. Sci. USA 2021, 118, e2103444118. [Google Scholar] [CrossRef]
- Lokuta, M.A.; Nuzzi, P.A.; Huttenlocher, A. Calpain regulates neutrophil chemotaxis. Proc. Natl. Acad. Sci. USA 2003, 100, 4006–4011. [Google Scholar] [CrossRef] [PubMed]
- Kolaczkowska, E.; Kubes, P. Neutrophil recruitment and function in health and inflammation. Nat. Rev. Immunol. 2013, 13, 159–175. [Google Scholar] [CrossRef]
- Immler, R.; Simon, S.I.; Sperandio, M. Calcium signalling and related ion channels in neutrophil recruitment and function. Eur. J. Clin. Investig. 2018, 48 (Suppl. S2), e12964. [Google Scholar] [CrossRef] [PubMed]
- Dewitt, S.; Francis, R.J.; Hallett, M.B. Ca²⁺ and calpain control membrane expansion during the rapid cell spreading of neutrophils. J. Cell Sci. 2013, 126 Pt 20, 4627–4635. [Google Scholar] [PubMed]
- Nguyen, G.T.; Green, E.R.; Mecsas, J. Neutrophils to the ROScue: Mechanisms of NADPH Oxidase Activation and Bacterial Resistance. Front. Cell. Infect. Microbiol. 2017, 7, 373. [Google Scholar] [CrossRef] [PubMed]
- Bouti, P.; Webbers, S.D.S.; Fagerholm, S.C.; Alon, R.; Moser, M.; Matlung, H.L.; Kuijpers, T.W. β2 Integrin Signaling Cascade in Neutrophils: More Than a Single Function. Front. Immunol. 2021, 11, 619925. [Google Scholar] [CrossRef]
- Chen, H.; Gao, W.; Yang, Y.; Guo, S.; Wang, H.; Wang, W.; Zhang, S.; Zhou, Q.; Xu, H.; Yao, J.; et al. Inhibition of VDAC1 prevents Ca²⁺-mediated oxidative stress and apoptosis induced by 5-aminolevulinic acid mediated sonodynamic therapy in THP-1 macrophages. Apoptosis Int. J. Program. Cell Death 2014, 19, 1712–1726. [Google Scholar] [CrossRef] [PubMed]
- Danelishvili, L.; Chinison, J.J.J.; Pham, T.; Gupta, R.; Bermudez, L.E. The Voltage-Dependent Anion Channels (VDAC) of Mycobacterium avium phagosome are associated with bacterial survival and lipid export in macrophages. Sci. Rep. 2017, 7, 7007. [Google Scholar] [CrossRef]
- Hirayama, D.; Iida, T.; Nakase, H. The Phagocytic Function of Macrophage-Enforcing Innate Immunity and Tissue Homeostasis. Int. J. Mol. Sci. 2017, 19, 92. [Google Scholar] [CrossRef] [PubMed]
- Arango Duque, G.; Descoteaux, A. Macrophage cytokines: Involvement in immunity and infectious diseases. Front. Immunol. 2014, 5, 491. [Google Scholar] [CrossRef]
- Nunes, P.; Demaurex, N. The role of calcium signaling in phagocytosis. J. Leukoc. Biol. 2010, 88, 57–68. [Google Scholar]
- Tedesco, S.; Scattolini, V.; Albiero, M.; Bortolozzi, M.; Avogaro, A.; Cignarella, A.; Fadini, G.P. Mitochondrial Calcium Uptake Is Instrumental to Alternative Macrophage Polarization and Phagocytic Activity. Int. J. Mol. Sci. 2019, 20, 4966. [Google Scholar]
- Paredes-Gamero, E.J.; Leon, C.M.; Borojevic, R.; Oshiro, M.E.; Ferreira, A.T. Changes in intracellular Ca2+ levels induced by cytokines and P2 agonists differentially modulate proliferation or commitment with macrophage differentiation in murine hematopoietic cells. J. Biol. Chem. 2008, 283, 31909–31919. [Google Scholar] [CrossRef] [PubMed]
- Kang, H.; Zhang, K.; Wong, D.S.H.; Han, F.; Li, B.; Bian, L. Near-infrared light-controlled regulation of intracellular calcium to modulate macrophage polarization. Biomaterials 2018, 178, 681–696. [Google Scholar] [CrossRef] [PubMed]
- Hilligan, K.L.; Ronchese, F. Antigen presentation by dendritic cells and their instruction of CD4+ T helper cell responses. Cell. Mol. Immunol. 2020, 17, 587–599. [Google Scholar] [CrossRef]
- Sumoza-Toledo, A.; Lange, I.; Cortado, H.; Bhagat, H.; Mori, Y.; Fleig, A.; Penner, R.; Partida-Sánchez, S. Dendritic cell maturation and chemotaxis is regulated by TRPM2-mediated lysosomal Ca2+ release. FASEB J. 2011, 25, 3529–3542. [Google Scholar] [CrossRef]
- Worbs, T.; Hammerschmidt, S.I.; Förster, R. Dendritic cell migration in health and disease. Nat. Rev. Immunol. 2017, 17, 30–48. [Google Scholar]
- Braun, A.; Worbs, T.; Moschovakis, G.L.; Halle, S.; Hoffmann, K.; Bölter, J.; Münk, A.; Förster, R. Afferent lymph-derived T cells and DCs use different chemokine receptor CCR7-dependent routes for entry into the lymph node and intranodal migration. Nat. Immunol. 2011, 12, 879–887. [Google Scholar] [CrossRef]
- Shao, Z.; Makinde, T.O.; Agrawal, D.K. Calcium-activated potassium channel KCa3.1 in lung dendritic cell migration. Am. J. Respir. Cell Mol. Biol. 2011, 45, 962–968. [Google Scholar] [CrossRef]
- Rodríguez-Fernández, J.L.; Criado-García, O. The Chemokine Receptor CCR7 Uses Distinct Signaling Modules With Biased Functionality to Regulate Dendritic Cells. Front. Immunol. 2020, 11, 528. [Google Scholar] [CrossRef]
- Riol-Blanco, L.; Sánchez-Sánchez, N.; Torres, A.; Tejedor, A.; Narumiya, S.; Corbí, A.L.; Sánchez-Mateos, P.; Rodríguez-Fernández, J.L. The chemokine receptor CCR7 activates in dendritic cells two signaling modules that independently regulate chemotaxis and migratory speed. J. Immunol. 2005, 174, 4070–4080. [Google Scholar] [CrossRef]
- Rosencrans, W.M.; Rajendran, M.; Bezrukov, S.M.; Rostovtseva, T.K. VDAC regulation of mitochondrial calcium flux: From channel biophysics to disease. Cell Calcium 2021, 94, 102356. [Google Scholar] [CrossRef] [PubMed]
- Shoshan-Barmatz, V.; Pittala, S.; Mizrachi, D. VDAC1 and the TSPO: Expression, Interactions, and Associated Functions in Health and Disease States. Int. J. Mol. Sci. 2019, 20, 3348. [Google Scholar] [CrossRef]
- Shoshan-Barmatz, V.; Shteinfer-Kuzmine, A.; Verma, A. VDAC1 at the Intersection of Cell Metabolism, Apoptosis, and Diseases. Biomolecules 2020, 10, 1485. [Google Scholar] [CrossRef]
- Mills, E.L.; Kelly, B.; O’Neill, L.A.J. Mitochondria are the powerhouses of immunity. Nat. Immunol. 2017, 18, 488–498. [Google Scholar] [CrossRef] [PubMed]
- Cruzat, V.; Macedo Rogero, M.; Noel Keane, K.; Curi, R.; Newsholme, P. Glutamine: Metabolism and Immune Function, Supplementation and Clinical Translation. Nutrients 2018, 10, 1564. [Google Scholar] [CrossRef]
- Judge A, Dodd MS: Metabolism. Essays Biochem. 2020, 64, 607–647. [CrossRef]
- Mergenthaler, P.; Lindauer, U.; Dienel, G.A.; Meisel, A. Sugar for the brain: The role of glucose in physiological and pathological brain function. Trends Neurosci. 2013, 36, 587–597. [Google Scholar] [CrossRef]
- Zhu, J.; Thompson, C.B. Metabolic regulation of cell growth and proliferation. Nat. Rev. Mol. Cell Biol. 2019, 20, 436–450. [Google Scholar] [CrossRef]
- Denton, R.M. Regulation of mitochondrial dehydrogenases by calcium ions. Biochim. Biophys. Acta 2009, 1787, 1309–1316. [Google Scholar] [CrossRef]
- Cárdenas, C.; Miller, R.A.; Smith, I.; Bui, T.; Molgó, J.; Müller, M.; Vais, H.; Cheung, K.H.; Yang, J.; Parker, I.; et al. Essential regulation of cell bioenergetics by constitutive InsP3 receptor Ca2+ transfer to mitochondria. Cell 2010, 142, 270–283. [Google Scholar] [CrossRef] [PubMed]
- Szado, T.; Vanderheyden, V.; Parys, J.B.; De Smedt, H.; Rietdorf, K.; Kotelevets, L.; Chastre, E.; Khan, F.; Landegren, U.; Söderberg, O.; et al. Phosphorylation of inositol 1,4,5-trisphosphate receptors by protein kinase B/Akt inhibits Ca2+ release and apoptosis. Proc. Natl. Acad. Sci. USA 2008, 105, 2427–2432. [Google Scholar] [CrossRef]
- Gellerich, F.N.; Gizatullina, Z.; Arandarcikaite, O.; Jerzembek, D.; Vielhaber, S.; Seppet, E.; Striggow, F. Extramitochondrial Ca2+ in the nanomolar range regulates glutamate-dependent oxidative phosphorylation on demand. PLoS ONE 2009, 4, e8181. [Google Scholar] [CrossRef] [PubMed]
- Pérez-Liébana, I.; Juaristi, I.; González-Sánchez, P.; González-Moreno, L.; Rial, E.; Podunavac, M.; Zakarian, A.; Molgó, J.; Vallejo-Illarramendi, A.; Mosqueira-Martín, L.; et al. A Ca2+-Dependent Mechanism Boosting Glycolysis and OXPHOS by Activating Aralar-Malate-Aspartate Shuttle, upon Neuronal Stimulation. J. Neurosci. 2022, 42, 3879–3895. [Google Scholar] [PubMed]
- Rueda, C.B.; Llorente-Folch, I.; Traba, J.; Amigo, I.; Gonzalez-Sanchez, P.; Contreras, L.; Juaristi, I.; Martinez-Valero, P.; Pardo, B.; Del Arco, A.; et al. Glutamate excitotoxicity and Ca2+-regulation of respiration: Role of the Ca2+ activated mitochondrial transporters (CaMCs). Biochim. Biophys. Acta 2016, 1857, 1158–1166. [Google Scholar] [CrossRef]
- Vander Heiden, M.G.; Chandel, N.S.; Li, X.X.; Schumacker, P.T.; Colombini, M.; Thompson, C.B. Outer mitochondrial membrane permeability can regulate coupled respiration and cell survival. Proc. Natl. Acad. Sci. USA 2000, 97, 4666–4671. [Google Scholar] [CrossRef] [PubMed]
- Magrì, A.; Di Rosa, M.C.; Orlandi, I.; Guarino, F.; Reina, S.; Guarnaccia, M.; Morello, G.; Spampinato, A.; Cavallaro, S.; Messina, A.; et al. Deletion of Voltage-Dependent Anion Channel 1 knocks mitochondria down triggering metabolic rewiring in yeast. Cell. Mol. Life Sci. CMLS 2020, 77, 3195–3213. [Google Scholar] [CrossRef]
- Soto-Heredero, G.; Gómez de Las Heras, M.M.; Gabandé-Rodríguez, E.; Oller, J.; Mittelbrunn, M. Glycolysis—A key player in the inflammatory response. FEBS J. 2020, 287, 3350–3369. [Google Scholar] [CrossRef]
- Ko, C.W.; Counihan, D.; Wu, J.; Hatzoglou, M.; Puchowicz, M.A.; Croniger, C.M. Macrophages with a deletion of the phosphoenolpyruvate carboxykinase 1 (Pck1) gene have a more proinflammatory phenotype. J. Biol. Chem. 2018, 293, 3399–3409. [Google Scholar] [CrossRef] [PubMed]
- Satapati, S.; Kucejova, B.; Duarte, J.A.; Fletcher, J.A.; Reynolds, L.; Sunny, N.E.; He, T.; Nair, L.A.; Livingston, K.A.; Fu, X.; et al. Mitochondrial metabolism mediates oxidative stress and inflammation in fatty liver. J. Clin. Investig. 2015, 125, 4447–4462. [Google Scholar] [CrossRef]
- Viola, A.; Munari, F.; Sánchez-Rodríguez, R.; Scolaro, T.; Castegna, A. The Metabolic Signature of Macrophage Responses. Front. Immunol. 2019, 10, 1462. [Google Scholar] [CrossRef] [PubMed]
- Tannahill, G.M.; Curtis, A.M.; Adamik, J.; Palsson-McDermott, E.M.; McGettrick, A.F.; Goel, G.; Frezza, C.; Bernard, N.J.; Kelly, B.; Foley, N.H.; et al. Succinate is an inflammatory signal that induces IL-1β through HIF-1α. Nature 2013, 496, 238–242. [Google Scholar] [CrossRef] [PubMed]
- Littlewood-Evans, A.; Sarret, S.; Apfel, V.; Loesle, P.; Dawson, J.; Zhang, J.; Muller, A.; Tigani, B.; Kneuer, R.; Patel, S.; et al. GPR91 senses extracellular succinate released from inflammatory macrophages and exacerbates rheumatoid arthritis. J. Exp. Med. 2016, 213, 1655–1662. [Google Scholar] [CrossRef]
- Mills, E.; O’Neill, L.A. Succinate: A metabolic signal in inflammation. Trends Cell Biol. 2014, 24, 313–320. [Google Scholar] [CrossRef] [PubMed]
- De Souza, D.P.; Achuthan, A.; Lee, M.K.; Binger, K.J.; Lee, M.C.; Davidson, S.; Tull, D.L.; McConville, M.J.; Cook, A.D.; Murphy, A.J.; et al. Autocrine IFN-I inhibits isocitrate dehydrogenase in the TCA cycle of LPS-stimulated macrophages. J. Clin. Investig. 2019, 129, 4239–4244. [Google Scholar] [CrossRef]
- Seim, G.L.; Britt, E.C.; John, S.V.; Yeo, F.J.; Johnson, A.R.; Eisenstein, R.S.; Pagliarini, D.J.; Fan, J. Two-stage metabolic remodelling in macrophages in response to lipopolysaccharide and interferon-γ stimulation. Nat. Metab. 2019, 1, 731–742. [Google Scholar] [CrossRef] [PubMed]
- Infantino, V.; Convertini, P.; Cucci, L.; Panaro, M.A.; Di Noia, M.A.; Calvello, R.; Palmieri, F.; Iacobazzi, V. The mitochondrial citrate carrier: A new player in inflammation. Biochem. J. 2011, 438, 433–436. [Google Scholar] [CrossRef] [PubMed]
- Bryland, A.; Wieslander, A.; Carlsson, O.; Hellmark, T.; Godaly, G. Citrate treatment reduces endothelial death and inflammation under hyperglycaemic conditions. Diabetes Vasc. Dis. Res. 2012, 9, 42–51. [Google Scholar] [CrossRef]
- Williams, N.C.; O’Neill, L.A.J. A Role for the Krebs Cycle Intermediate Citrate in Metabolic Reprogramming in Innate Immunity and Inflammation. Front. Immunol. 2018, 9, 141. [Google Scholar] [CrossRef]
- Choi, E.Y.; Kim, H.J.; Han, J.S. Anti-inflammatory effects of calcium citrate in RAW 264.7cells via suppression of NF-κB activation. Environ. Toxicol. Pharmacol. 2015, 39, 27–34. [Google Scholar] [CrossRef]
- Jha, A.K.; Huang, S.C.; Sergushichev, A.; Lampropoulou, V.; Ivanova, Y.; Loginicheva, E.; Chmielewski, K.; Stewart, K.M.; Ashall, J.; Everts, B.; et al. Network integration of parallel metabolic and transcriptional data reveals metabolic modules that regulate macrophage polarization. Immunity 2015, 42, 419–430. [Google Scholar] [CrossRef]
- Langston, P.K.; Nambu, A.; Jung, J.; Shibata, M.; Aksoylar, H.I.; Lei, J.; Xu, P.; Doan, M.T.; Jiang, H.; MacArthur, M.R.; et al. Glycerol phosphate shuttle enzyme GPD2 regulates macrophage inflammatory responses. Nat. Immunol. 2019, 20, 1186–1195. [Google Scholar] [CrossRef] [PubMed]
- Wilson, J.L.; Nägele, T.; Linke, M.; Demel, F.; Fritsch, S.D.; Mayr, H.K.; Cai, Z.; Katholnig, K.; Sun, X.; Fragner, L.; et al. Inverse Data-Driven Modeling and Multiomics Analysis Reveals Phgdh as a Metabolic Checkpoint of Macrophage Polarization and Proliferation. Cell Rep. 2020, 30, 1542–1552.e1547. [Google Scholar] [CrossRef]
- Brucklacher-Waldert, V.; Ferreira, C.; Stebegg, M.; Fesneau, O.; Innocentin, S.; Marie, J.C.; Veldhoen, M. Cellular Stress in the Context of an Inflammatory Environment Supports TGF-β-Independent T Helper-17 Differentiation. Cell Rep. 2017, 19, 2357–2370. [Google Scholar] [CrossRef]
- Nagy, E.; Rigby, W.F. Glyceraldehyde-3-phosphate dehydrogenase selectively binds AU-rich RNA in the NAD(+)-binding region (Rossmann fold). J. Biol. Chem. 1995, 270, 2755–2763. [Google Scholar] [CrossRef]
- Chang, C.H.; Curtis, J.D.; Maggi, L.B., Jr.; Faubert, B.; Villarino, A.V.; O’Sullivan, D.; Huang, S.C.; van der Windt, G.J.; Blagih, J.; Qiu, J.; et al. Posttranscriptional control of T cell effector function by aerobic glycolysis. Cell 2013, 153, 1239–1251. [Google Scholar] [CrossRef]
- Everts, B.; Amiel, E.; Huang, S.C.; Smith, A.M.; Chang, C.H.; Lam, W.Y.; Redmann, V.; Freitas, T.C.; Blagih, J.; van der Windt, G.J.; et al. TLR-driven early glycolytic reprogramming via the kinases TBK1-IKKɛ supports the anabolic demands of dendritic cell activation. Nat. Immunol. 2014, 15, 323–332. [Google Scholar] [CrossRef] [PubMed]
- Du, X.; Chapman, N.M.; Chi, H. Emerging Roles of Cellular Metabolism in Regulating Dendritic Cell Subsets and Function. Front. Cell Dev. Biol. 2018, 6, 152. [Google Scholar] [CrossRef]
- Wculek, S.K.; Khouili, S.C.; Priego, E.; Heras-Murillo, I.; Sancho, D. Metabolic Control of Dendritic Cell Functions: Digesting Information. Front. Immunol. 2019, 10, 775. [Google Scholar] [CrossRef]
- Malinarich, F.; Duan, K.; Hamid, R.A.; Bijin, A.; Lin, W.X.; Poidinger, M.; Fairhurst, A.M.; Connolly, J.E. High mitochondrial respiration and glycolytic capacity represent a metabolic phenotype of human tolerogenic dendritic cells. J. Immunol. 2015, 194, 5174–5186. [Google Scholar] [CrossRef] [PubMed]
- Vander Heiden, M.G.; Locasale, J.W.; Swanson, K.D.; Sharfi, H.; Heffron, G.J.; Amador-Noguez, D.; Christofk, H.R.; Wagner, G.; Rabinowitz, J.D.; Asara, J.M.; et al. Evidence for an alternative glycolytic pathway in rapidly proliferating cells. Science 2010, 329, 1492–1499. [Google Scholar] [CrossRef]
- Ho, P.C.; Bihuniak, J.D.; Macintyre, A.N.; Staron, M.; Liu, X.; Amezquita, R.; Tsui, Y.C.; Cui, G.; Micevic, G.; Perales, J.C.; et al. Phosphoenolpyruvate Is a Metabolic Checkpoint of Anti-tumor T Cell Responses. Cell 2015, 162, 1217–1228. [Google Scholar] [CrossRef] [PubMed]
- Brooks, G.A. The Science and Translation of Lactate Shuttle Theory. Cell Metab. 2018, 27, 757–785. [Google Scholar] [CrossRef]
- Pearce, E.J.; Everts, B. Dendritic cell metabolism. Nat. Rev. Immunol. 2015, 15, 18–29. [Google Scholar] [CrossRef]
- Errea, A.; Cayet, D.; Marchetti, P.; Tang, C.; Kluza, J.; Offermanns, S.; Sirard, J.C.; Rumbo, M. Lactate Inhibits the Pro-Inflammatory Response and Metabolic Reprogramming in Murine Macrophages in a GPR81-Independent Manner. PLoS ONE 2016, 11, e0163694. [Google Scholar] [CrossRef]
- Nasi, A.; Fekete, T.; Krishnamurthy, A.; Snowden, S.; Rajnavölgyi, E.; Catrina, A.I.; Wheelock, C.E.; Vivar, N.; Rethi, B. Dendritic cell reprogramming by endogenously produced lactic acid. J. Immunol. 2013, 191, 3090–3099. [Google Scholar] [CrossRef]
- Haas, R.; Smith, J.; Rocher-Ros, V.; Nadkarni, S.; Montero-Melendez, T.; D’Acquisto, F.; Bland, E.J.; Bombardieri, M.; Pitzalis, C.; Perretti, M.; et al. Lactate Regulates Metabolic and Pro-inflammatory Circuits in Control of T Cell Migration and Effector Functions. PLoS Biol. 2015, 13, e1002202. [Google Scholar] [CrossRef]
- Samuvel, D.J.; Sundararaj, K.P.; Nareika, A.; Lopes-Virella, M.F.; Huang, Y. Lactate boosts TLR4 signaling and NF-kappaB pathway-mediated gene transcription in macrophages via monocarboxylate transporters and MD-2 up-regulation. J. Immunol. 2009, 182, 2476–2484. [Google Scholar] [CrossRef]
- Végran, F.; Boidot, R.; Michiels, C.; Sonveaux, P.; Feron, O. Lactate influx through the endothelial cell monocarboxylate transporter MCT1 supports an NF-κB/IL-8 pathway that drives tumor angiogenesis. Cancer Res. 2011, 71, 2550–2560. [Google Scholar] [CrossRef] [PubMed]
- Ivashkiv, L.B. The hypoxia-lactate axis tempers inflammation. Nat. Rev. Immunol. 2020, 20, 85–86. [Google Scholar] [CrossRef] [PubMed]
- Colegio, O.R.; Chu, N.Q.; Szabo, A.L.; Chu, T.; Rhebergen, A.M.; Jairam, V.; Cyrus, N.; Brokowski, C.E.; Eisenbarth, S.C.; Phillips, G.M.; et al. Functional polarization of tumour-associated macrophages by tumour-derived lactic acid. Nature 2014, 513, 559–563. [Google Scholar] [CrossRef] [PubMed]
- Peng, M.; Yin, N.; Chhangawala, S.; Xu, K.; Leslie, C.S.; Li, M.O. Aerobic glycolysis promotes T helper 1 cell differentiation through an epigenetic mechanism. Science 2016, 354, 481–484. [Google Scholar] [CrossRef]
- Manosalva, C.; Quiroga, J.; Hidalgo, A.I.; Alarcón, P.; Anseoleaga, N.; Hidalgo, M.A.; Burgos, R.A. Role of Lactate in Inflammatory Processes: Friend or Foe. Front. Immunol. 2021, 12, 808799. [Google Scholar] [CrossRef] [PubMed]
- Roberts, D.J.; Miyamoto, S. Hexokinase II integrates energy metabolism and cellular protection: Akting on mitochondria and TORCing to autophagy. Cell Death Differ. 2015, 22, 248–257. [Google Scholar] [CrossRef]
- Zhang, D.; Yip, Y.M.; Li, L. In silico construction of HK2-VDAC1 complex and investigating the HK2 binding-induced molecular gating mechanism of VDAC1. Mitochondrion 2016, 30, 222–228. [Google Scholar] [CrossRef]
- Xu, D.; Jin, J.; Yu, H.; Zhao, Z.; Ma, D.; Zhang, C.; Jiang, H. Chrysin inhibited tumor glycolysis and induced apoptosis in hepatocellular carcinoma by targeting hexokinase-2. J. Exp. Clin. Cancer Res. CR 2017, 36, 44. [Google Scholar] [CrossRef]
- Yoon, H.; Shaw, J.L.; Haigis, M.C.; Greka, A. Lipid metabolism in sickness and in health: Emerging regulators of lipotoxicity. Mol. Cell 2021, 81, 3708–3730. [Google Scholar] [CrossRef] [PubMed]
- Snaebjornsson, M.T.; Janaki-Raman, S.; Schulze, A. Greasing the Wheels of the Cancer Machine: The Role of Lipid Metabolism in Cancer. Cell Metab. 2020, 31, 62–76. [Google Scholar] [CrossRef]
- Shimano, H.; Sato, R. SREBP-regulated lipid metabolism: Convergent physiology—Divergent pathophysiology. Nat. Rev. Endocrinol. 2017, 13, 710–730. [Google Scholar] [CrossRef] [PubMed]
- Currie, E.; Schulze, A.; Zechner, R.; Walther, T.C.; Farese, R.V., Jr. Cellular fatty acid metabolism and cancer. Cell Metab. 2013, 18, 153–161. [Google Scholar] [CrossRef]
- Lee, K.; Kerner, J.; Hoppel, C.L. Mitochondrial carnitine palmitoyltransferase 1a (CPT1a) is part of an outer membrane fatty acid transfer complex. J. Biol. Chem. 2011, 286, 25655–25662. [Google Scholar] [CrossRef]
- Martel, C.; Allouche, M.; Esposti, D.D.; Fanelli, E.; Boursier, C.; Henry, C.; Chopineau, J.; Calamita, G.; Kroemer, G.; Lemoine, A.; et al. Glycogen synthase kinase 3-mediated voltage-dependent anion channel phosphorylation controls outer mitochondrial membrane permeability during lipid accumulation. Hepatology 2013, 57, 93–102. [Google Scholar] [CrossRef] [PubMed]
- Pittala, S.; Krelin, Y.; Kuperman, Y.; Shoshan-Barmatz, V. A Mitochondrial VDAC1-Based Peptide Greatly Suppresses Steatosis and NASH-Associated Pathologies in a Mouse Model. Mol. Ther. J. Am. Soc. Gene Ther. 2019, 27, 1848–1862. [Google Scholar] [CrossRef]
- Turkaly, P.; Kerner, J.; Hoppel, C. A 22 kDa polyanion inhibits carnitine-dependent fatty acid oxidation in rat liver mitochondria. FEBS Lett. 1999, 460, 241–245. [Google Scholar] [CrossRef]
- Ashrafi, G.; Schwarz, T.L. The pathways of mitophagy for quality control and clearance of mitochondria. Cell Death Differ. 2013, 20, 31–42. [Google Scholar] [CrossRef] [PubMed]
- Xu, Y.; Shen, J.; Ran, Z. Emerging views of mitophagy in immunity and autoimmune diseases. Autophagy 2020, 16, 3–17. [Google Scholar] [CrossRef]
- Ziegler, P.K.; Bollrath, J.; Pallangyo, C.K.; Matsutani, T.; Canli, Ö.; De Oliveira, T.; Diamanti, M.A.; Müller, N.; Gamrekelashvili, J.; Putoczki, T.; et al. Mitophagy in Intestinal Epithelial Cells Triggers Adaptive Immunity during Tumorigenesis. Cell 2018, 174, 88–101.e116. [Google Scholar] [CrossRef] [PubMed]
- Ma, K.; Chen, G.; Li, W.; Kepp, O.; Zhu, Y.; Chen, Q. Mitophagy, Mitochondrial Homeostasis, and Cell Fate. Front. Cell Dev. Biol. 2020, 8, 467. [Google Scholar] [CrossRef] [PubMed]
- Matsuda, N.; Sato, S.; Shiba, K.; Okatsu, K.; Saisho, K.; Gautier, C.A.; Sou, Y.S.; Saiki, S.; Kawajiri, S.; Sato, F.; et al. PINK1 stabilized by mitochondrial depolarization recruits Parkin to damaged mitochondria and activates latent Parkin for mitophagy. J. Cell Biol. 2010, 189, 211–221. [Google Scholar] [CrossRef] [PubMed]
- Vives-Bauza, C.; Zhou, C.; Huang, Y.; Cui, M.; de Vries, R.L.; Kim, J.; May, J.; Tocilescu, M.A.; Liu, W.; Ko, H.S.; et al. PINK1-dependent recruitment of Parkin to mitochondria in mitophagy. Proc. Natl. Acad. Sci. USA 2010, 107, 378–383. [Google Scholar] [CrossRef] [PubMed]
- Shiba-Fukushima, K.; Imai, Y.; Yoshida, S.; Ishihama, Y.; Kanao, T.; Sato, S.; Hattori, N. PINK1-mediated phosphorylation of the Parkin ubiquitin-like domain primes mitochondrial translocation of Parkin and regulates mitophagy. Sci. Rep. 2012, 2, 1002. [Google Scholar] [CrossRef]
- Narendra, D.; Tanaka, A.; Suen, D.F.; Youle, R.J. Parkin is recruited selectively to impaired mitochondria and promotes their autophagy. J. Cell Biol. 2008, 183, 795–803. [Google Scholar] [CrossRef]
- Cho, D.H.; Kim, J.K.; Jo, E.K. Mitophagy and Innate Immunity in Infection. Mol. Cells 2020, 43, 10–22. [Google Scholar]
- Ham, S.J.; Lee, S.Y.; Song, S.; Chung, J.R.; Choi, S.; Chung, J. Interaction between RING1 (R1) and the Ubiquitin-like (UBL) Domains Is Critical for the Regulation of Parkin Activity. J. Biol. Chem. 2016, 291, 1803–1816. [Google Scholar] [CrossRef]
- Geisler, S.; Holmström, K.M.; Skujat, D.; Fiesel, F.C.; Rothfuss, O.C.; Kahle, P.J.; Springer, W. PINK1/Parkin-mediated mitophagy is dependent on VDAC1 and p62/SQSTM1. Nat. Cell Biol. 2010, 12, 119–131. [Google Scholar] [CrossRef]
- Yang, X.; Zhou, Y.; Liang, H.; Meng, Y.; Liu, H.; Zhou, Y.; Huang, C.; An, B.; Mao, H.; Liao, Z. VDAC1 promotes cardiomyocyte autophagy in anoxia/reoxygenation injury via the PINK1/Parkin pathway. Cell Biol. Int. 2021, 45, 1448–1458. [Google Scholar] [CrossRef]
- Sun, Y.; Vashisht, A.A.; Tchieu, J.; Wohlschlegel, J.A.; Dreier, L. Voltage-dependent anion channels (VDACs) recruit Parkin to defective mitochondria to promote mitochondrial autophagy. J. Biol. Chem. 2012, 287, 40652–40660. [Google Scholar] [CrossRef]
- Fang, E.F.; Hou, Y.; Palikaras, K.; Adriaanse, B.A.; Kerr, J.S.; Yang, B.; Lautrup, S.; Hasan-Olive, M.M.; Caponio, D.; Dan, X.; et al. Mitophagy inhibits amyloid-β and tau pathology and reverses cognitive deficits in models of Alzheimer’s disease. Nat. Neurosci. 2019, 22, 401–412. [Google Scholar] [CrossRef]
- Zhong, Z.; Umemura, A.; Sanchez-Lopez, E.; Liang, S.; Shalapour, S.; Wong, J.; He, F.; Boassa, D.; Perkins, G.; Ali, S.R.; et al. NF-κB Restricts Inflammasome Activation via Elimination of Damaged Mitochondria. Cell 2016, 164, 896–910. [Google Scholar] [CrossRef]
- Lupfer, C.; Thomas, P.G.; Anand, P.K.; Vogel, P.; Milasta, S.; Martinez, J.; Huang, G.; Green, M.; Kundu, M.; Chi, H.; et al. Receptor interacting protein kinase 2-mediated mitophagy regulates inflammasome activation during virus infection. Nat. Immunol. 2013, 14, 480–488. [Google Scholar] [CrossRef]
- Ivashkiv, L.B.; Donlin, L.T. Regulation of type I interferon responses. Nat. Rev. Immunol. 2014, 14, 36–49. [Google Scholar] [CrossRef] [PubMed]
- Tal, M.C.; Sasai, M.; Lee, H.K.; Yordy, B.; Shadel, G.S.; Iwasaki, A. Absence of autophagy results in reactive oxygen species-dependent amplification of RLR signaling. Proc. Natl. Acad. Sci. USA 2009, 106, 2770–2775. [Google Scholar] [CrossRef]
- Xia, M.; Gonzalez, P.; Li, C.; Meng, G.; Jiang, A.; Wang, H.; Gao, Q.; Debatin, K.M.; Beltinger, C.; Wei, J. Mitophagy enhances oncolytic measles virus replication by mitigating DDX58/RIG-I-like receptor signaling. J. Virol. 2014, 88, 5152–5164. [Google Scholar] [CrossRef]
- García-Sastre, A. Ten Strategies of Interferon Evasion by Viruses. Cell Host Microbe 2017, 22, 176–184. [Google Scholar] [CrossRef]
- Zhang, L.; Qin, Y.; Chen, M. Viral strategies for triggering and manipulating mitophagy. Autophagy 2018, 14, 1665–1673. [Google Scholar] [CrossRef] [PubMed]
- He, X.; Zhu, Y.; Zhang, Y.; Geng, Y.; Gong, J.; Geng, J.; Zhang, P.; Zhang, X.; Liu, N.; Peng, Y.; et al. RNF34 functions in immunity and selective mitophagy by targeting MAVS for autophagic degradation. EBMO J. 2019, 38, e100978. [Google Scholar]
- Seth, R.B.; Sun, L.; Ea, C.K.; Chen, Z.J. Identification and characterization of MAVS, a mitochondrial antiviral signaling protein that activates NF-kappaB and IRF 3. Cell 2005, 122, 669–682. [Google Scholar] [CrossRef]
- Jena, K.K.; Mehto, S.; Nath, P.; Chauhan, N.R.; Sahu, R.; Dhar, K.; Das, S.K.; Kolapalli, S.P.; Murmu, K.C.; Jain, A.; et al. Autoimmunity gene IRGM suppresses cGAS-STING and RIG-I-MAVS signaling to control interferon response. EMBO Rep. 2020, 21, e50051. [Google Scholar] [CrossRef]
- Wang, K.; Ma, H.; Liu, H.; Ye, W.; Li, Z.; Cheng, L.; Zhang, L.; Lei, Y.; Shen, L.; Zhang, F. The Glycoprotein and Nucleocapsid Protein of Hantaviruses Manipulate Autophagy Flux to Restrain Host Innate Immune Responses. Cell Rep. 2019, 27, 2075–2091.e2075. [Google Scholar] [CrossRef]
- Tseng, C.C.; Liao, W.T.; Wong, M.C.; Chen, C.J.; Lee, S.C.; Yen, J.H.; Chang, S.J. Cell lineage-specific methylome and genome alterations in gout. Aging 2021, 13, 3843–3865. [Google Scholar] [CrossRef]
- Joo, H.K.; Lee, Y.R.; Lim, S.Y.; Lee, E.J.; Choi, S.; Cho, E.J.; Park, M.S.; Ryoo, S.; Park, J.B.; Jeon, B.H. Peripheral benzodiazepine receptor regulates vascular endothelial activations via suppression of the voltage-dependent anion channel-1. FEBS Lett. 2012, 586, 1349–1355. [Google Scholar] [CrossRef]
- Srisomboon, Y.; Squillace, D.L.; Maniak, P.J.; Kita, H.; O’Grady, S.M. Fungal allergen-induced IL-33 secretion involves cholesterol-dependent, VDAC-1-mediated ATP release from the airway epithelium. J. Physiol. 2020, 598, 1829–1845. [Google Scholar] [CrossRef]
- Lin, Y.C.; Lin, Y.C.; Tsai, M.L.; Tsai, Y.G.; Kuo, C.H.; Hung, C.H. IL-33 regulates M1/M2 chemokine expression via mitochondrial redox-related mitophagy in human monocytes. Chem. Biol. Interact. 2022, 359, 109915. [Google Scholar] [CrossRef] [PubMed]
- Biasizzo, M.; Kopitar-Jerala, N. Interplay between NLRP3 Inflammasome and Autophagy. Front. Immunol. 2020, 11, 591803. [Google Scholar] [CrossRef]
- Zhou, R.; Yazdi, A.S.; Menu, P.; Tschopp, J. A role for mitochondria in NLRP3 inflammasome activation. Nature 2011, 469, 221–225. [Google Scholar] [CrossRef]
- Harris, J.; Hartman, M.; Roche, C.; Zeng, S.G.; O’Shea, A.; Sharp, F.A.; Lambe, E.M.; Creagh, E.M.; Golenbock, D.T.; Tschopp, J.; et al. Autophagy controls IL-1beta secretion by targeting pro-IL-1beta for degradation. J. Biol. Chem. 2011, 286, 9587–9597. [Google Scholar] [CrossRef]
- Li, W.; Zeng, H.; Xu, M.; Huang, C.; Tao, L.; Li, J.; Zhang, T.; Chen, H.; Xia, J.; Li, C.; et al. Oleanolic Acid Improves Obesity-Related Inflammation and Insulin Resistance by Regulating Macrophages Activation. Front. Pharmacol. 2021, 12, 697483. [Google Scholar] [CrossRef]
- Oka, T.; Hikoso, S.; Yamaguchi, O.; Taneike, M.; Takeda, T.; Tamai, T.; Oyabu, J.; Murakawa, T.; Nakayama, H.; Nishida, K.; et al. Mitochondrial DNA that escapes from autophagy causes inflammation and heart failure. Nature 2012, 485, 251–255. [Google Scholar] [CrossRef]
- Wu, K.K.L.; Long, K.; Lin, H.; Siu, P.M.F.; Hoo, R.L.C.; Ye, D.; Xu, A.; Cheng, K.K.Y. The APPL1-Rab5 axis restricts NLRP3 inflammasome activation through early endosomal-dependent mitophagy in macrophages. Nat. Commun. 2021, 12, 6637. [Google Scholar] [CrossRef]
- Sims, J.E.; Smith, D.E. The IL-1 family: Regulators of immunity. Nat. Rev. Immunol. 2010, 10, 89–102. [Google Scholar] [CrossRef] [PubMed]
- Dinarello, C.A. Introduction to the interleukin-1 family of cytokines and receptors: Drivers of innate inflammation and acquired immunity. Immunol. Rev. 2018, 281, 5–7. [Google Scholar] [CrossRef] [PubMed]
- Mantovani, A.; Dinarello, C.A.; Molgora, M.; Garlanda, C. Interleukin-1 and Related Cytokines in the Regulation of Inflammation and Immunity. Immunity 2019, 50, 778–795. [Google Scholar] [CrossRef] [PubMed]
- Wang, B.; Zhou, C.; Wu, Q.; Lin, P.; Pu, Q.; Qin, S.; Gao, P.; Wang, Z.; Liu, Y.; Arel, J.; et al. cGAS modulates cytokine secretion and bacterial burdens by altering the release of mitochondrial DNA in pseudomonas pulmonary infection. Immunology 2022, 166, 408–423. [Google Scholar] [CrossRef]
- Zhong, W.; Rao, Z.; Xu, J.; Sun, Y.; Hu, H.; Wang, P.; Xia, Y.; Pan, X.; Tang, W.; Chen, Z.; et al. Defective mitophagy in aged macrophages promotes mitochondrial DNA cytosolic leakage to activate STING signaling during liver sterile inflammation. Aging Cell 2022, 21, e13622. [Google Scholar] [CrossRef]
- Zhao, X.; Yu, M.; Zhao, Y.; Zheng, Y.; Meng, L.; Du, K.; Xie, Z.; Lv, H.; Zhang, W.; Liu, J.; et al. Circulating cell-free mtDNA release is associated with the activation of cGAS-STING pathway and inflammation in mitochondrial diseases. J. Neurol. 2022, 269, 4985–4996. [Google Scholar] [CrossRef]
- Willemsen, J.; Neuhoff, M.T.; Hoyler, T.; Noir, E.; Tessier, C.; Sarret, S.; Thorsen, T.N.; Littlewood-Evans, A.; Zhang, J.; Hasan, M.; et al. TNF leads to mtDNA release and cGAS/STING-dependent interferon responses that support inflammatory arthritis. Cell Rep. 2021, 37, 109977. [Google Scholar] [CrossRef]
- Borsche, M.; König, I.R.; Delcambre, S.; Petrucci, S.; Balck, A.; Brüggemann, N.; Zimprich, A.; Wasner, K.; Pereira, S.L.; Avenali, M.; et al. Mitochondrial damage-associated inflammation highlights biomarkers in PRKN/PINK1 parkinsonism. Brain J. Neurol. 2020, 143, 3041–3051. [Google Scholar] [CrossRef]
- Choudhuri, S.; Chowdhury, I.H.; Garg, N.J. Mitochondrial Regulation of Macrophage Response against Pathogens. Front. Immunol. 2020, 11, 622602. [Google Scholar] [CrossRef]
- Lin, J.Y.; Jing, R.; Lin, F.; Ge, W.Y.; Dai, H.J.; Pan, L. High Tidal Volume Induces Mitochondria Damage and Releases Mitochondrial DNA to Aggravate the Ventilator-Induced Lung Injury. Front. Immunol. 2018, 9, 1477. [Google Scholar] [CrossRef]
- Sliter, D.A.; Martinez, J.; Hao, L.; Chen, X.; Sun, N.; Fischer, T.D.; Burman, J.L.; Li, Y.; Zhang, Z.; Narendra, D.P.; et al. Parkin and PINK1 mitigate STING-induced inflammation. Nature 2018, 561, 258–262. [Google Scholar] [CrossRef]
- Liu, Z.; Wang, M.; Wang, X.; Bu, Q.; Wang, Q.; Su, W.; Li, L.; Zhou, H.; Lu, L. XBP1 deficiency promotes hepatocyte pyroptosis by impairing mitophagy to activate mtDNA-cGAS-STING signaling in macrophages during acute liver injury. Redox Biol. 2022, 52, 102305. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.; Wang, L.; Qin, X.; Turdi, S.; Sun, D.; Culver, B.; Reiter, R.J.; Wang, X.; Zhou, H.; Ren, J. ALDH2 contributes to melatonin-induced protection against APP/PS1 mutation-prompted cardiac anomalies through cGAS-STING-TBK1-mediated regulation of mitophagy. Signal Transduct. Target. Ther. 2020, 5, 119. [Google Scholar] [CrossRef] [PubMed]
- Caielli, S.; Athale, S.; Domic, B.; Murat, E.; Chandra, M.; Banchereau, R.; Baisch, J.; Phelps, K.; Clayton, S.; Gong, M.; et al. Oxidized mitochondrial nucleoids released by neutrophils drive type I interferon production in human lupus. J. Exp. Med. 2016, 213, 697–713. [Google Scholar] [CrossRef] [PubMed]
- Chen, G.; Kroemer, G.; Kepp, O. Mitophagy: An Emerging Role in Aging and Age-Associated Diseases. Front. Cell Dev. Biol. 2020, 8, 200. [Google Scholar] [CrossRef]
- Jing, R.; Hu, Z.K.; Lin, F.; He, S.; Zhang, S.S.; Ge, W.Y.; Dai, H.J.; Du, X.K.; Lin, J.Y.; Pan, L.H. Mitophagy-Mediated mtDNA Release Aggravates Stretching-Induced Inflammation and Lung Epithelial Cell Injury via the TLR9/MyD88/NF-κB Pathway. Front. Cell Dev. Biol. 2020, 8, 819. [Google Scholar] [CrossRef]
- Sasaki, M.; Miyakoshi, M.; Sato, Y.; Nakanuma, Y. Increased expression of mitochondrial proteins associated with autophagy in biliary epithelial lesions in primary biliary cirrhosis. Liver Int. 2013, 33, 312–320. [Google Scholar] [CrossRef]
- Caza, T.N.; Fernandez, D.R.; Talaber, G.; Oaks, Z.; Haas, M.; Madaio, M.P.; Lai, Z.W.; Miklossy, G.; Singh, R.R.; Chudakov, D.M.; et al. HRES-1/Rab4-mediated depletion of Drp1 impairs mitochondrial homeostasis and represents a target for treatment in SLE. Ann. Rheum. Dis. 2014, 73, 1888–1897. [Google Scholar] [CrossRef]
- Sachdeva, K.; Do, D.C.; Zhang, Y.; Hu, X.; Chen, J.; Gao, P. Environmental Exposures and Asthma Development: Autophagy, Mitophagy, and Cellular Senescence. Front. Immunol. 2019, 10, 2787. [Google Scholar] [CrossRef]
- McAlinden, K.D.; Deshpande, D.A.; Ghavami, S.; Xenaki, D.; Sohal, S.S.; Oliver, B.G.; Haghi, M.; Sharma, P. Autophagy Activation in Asthma Airways Remodeling. Am. J. Respir. Cell Mol. Biol. 2019, 60, 541–553. [Google Scholar] [CrossRef]
- Lee, J.; Kim, H.S. The Role of Autophagy in Eosinophilic Airway Inflammation. Immune Netw. 2019, 19, e5. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, Y.; Aono, Y.; Akiyama, N.; Horiike, Y.; Naoi, H.; Horiguchi, R.; Shibata, K.; Hozumi, H.; Karayama, M.; Furuhashi, K.; et al. Involvement of autophagy in exacerbation of eosinophilic airway inflammation in a murine model of obese asthma. Autophagy 2022, 18, 2216–2228. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.N.; Suh, D.H.; Trinh, H.K.; Chwae, Y.J.; Park, H.S.; Shin, Y.S. The role of autophagy in allergic inflammation: A new target for severe asthma. Exp. Mol. Med. 2016, 48, e243. [Google Scholar] [CrossRef] [PubMed]
- Liu, T.; Liu, Y.; Miller, M.; Cao, L.; Zhao, J.; Wu, J.; Wang, J.; Liu, L.; Li, S.; Zou, M.; et al. Autophagy plays a role in FSTL1-induced epithelial mesenchymal transition and airway remodeling in asthma. Am. J. Physiol. Lung Cell. Mol. Physiol. 2017, 313, L27–L40. [Google Scholar] [CrossRef]
- Zeki, A.A.; Yeganeh, B.; Kenyon, N.J.; Post, M.; Ghavami, S. Autophagy in airway diseases: A new frontier in human asthma? Allergy 2016, 71, 5–14. [Google Scholar] [CrossRef] [PubMed]
- Picca, A.; Lezza, A.M.S.; Leeuwenburgh, C.; Pesce, V.; Calvani, R.; Landi, F.; Bernabei, R.; Marzetti, E. Fueling Inflamm-Aging through Mitochondrial Dysfunction: Mechanisms and Molecular Targets. Int. J. Mol. Sci. 2017, 18, 933. [Google Scholar] [CrossRef]
- Picca, A.; Calvani, R.; Coelho-Junior, H.J.; Marzetti, E. Cell Death and Inflammation: The Role of Mitochondria in Health and Disease. Cells 2021, 10, 537. [Google Scholar] [CrossRef]
- Missiroli, S.; Genovese, I.; Perrone, M.; Vezzani, B.; Vitto, V.A.M.; Giorgi, C. The Role of Mitochondria in Inflammation: From Cancer to Neurodegenerative Disorders. J. Clin. Med. 2020, 9, 740. [Google Scholar] [CrossRef]
- Shoshan-Barmatz, V.; Maldonado, E.N.; Krelin, Y. VDAC1 at the crossroads of cell metabolism, apoptosis and cell stress. Cell Stress 2017, 1, 11–36. [Google Scholar] [CrossRef]
- Thinnes, F.P. Opening up of plasmalemma type-1 VDAC to form apoptotic “find me signal” pathways is essential in early apoptosis—Evidence from the pathogenesis of cystic fibrosis resulting from failure of apoptotic cell clearance followed by sterile inflammation. Mol. Genet. Metab. 2014, 111, 439–444. [Google Scholar] [CrossRef]
- Karachitos, A.; Jordan, J.; Kmita, H. VDAC-Targeted Drugs Affecting Cytoprotection and Mitochondrial Physiology in Cerebrovascular and Cardiovascular Diseases. Curr. Med. Chem. 2017, 24, 4419–4434. [Google Scholar] [CrossRef] [PubMed]
- Shoshan-Barmatz, V.; Mizrachi, D. VDAC1: From structure to cancer therapy. Front. Oncol. 2012, 2, 164. [Google Scholar] [CrossRef] [PubMed]
- Shoshan-Barmatz, V.; Nahon-Crystal, E.; Shteinfer-Kuzmine, A.; Gupta, R. VDAC1, mitochondrial dysfunction, and Alzheimer’s disease. Pharmacol. Res. 2018, 131, 87–101. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Ma, X.; Fujioka, H.; Liu, J.; Chen, S.; Zhu, X. DJ-1 regulates the integrity and function of ER-mitochondria association through interaction with IP3R3-Grp75-VDAC1. Proc. Natl. Acad. Sci. USA 2019, 116, 25322–25328. [Google Scholar] [CrossRef]
- Sasaki, K.; Donthamsetty, R.; Heldak, M.; Cho, Y.E.; Scott, B.T.; Makino, A. VDAC: Old protein with new roles in diabetes. Am. J. Physiol. Cell Physiol. 2012, 303, C1055–C1060. [Google Scholar] [CrossRef]
- Li, X.; Pan, J.; Li, H.; Li, G.; Liu, B.; Tang, X.; Liu, X.; He, Z.; Peng, Z.; Zhang, H.; et al. DsbA-L interacts with VDAC1 in mitochondrion-mediated tubular cell apoptosis and contributes to the progression of acute kidney disease. EBioMedicine 2022, 76, 103859. [Google Scholar] [CrossRef]
- Nowak, G.; Megyesi, J.; Craigen, W.J. Deletion of VDAC1 Hinders Recovery of Mitochondrial and Renal Functions After Acute Kidney Injury. Biomolecules 2020, 10, 585. [Google Scholar] [CrossRef]
- Shoshan-Barmatz, V.; Anand, U.; Nahon-Crystal, E.; Di Carlo, M.; Shteinfer-Kuzmine, A. Adverse Effects of Metformin From Diabetes to COVID-19, Cancer, Neurodegenerative Diseases, and Aging: Is VDAC1 a Common Target? Front. Physiol. 2021, 12, 730048. [Google Scholar] [CrossRef]
- Geula, S.; Naveed, H.; Liang, J.; Shoshan-Barmatz, V. Structure-based analysis of VDAC1 protein: Defining oligomer contact sites. J. Biol. Chem. 2012, 287, 2179–2190. [Google Scholar] [CrossRef]




Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hu, H.; Guo, L.; Overholser, J.; Wang, X. Mitochondrial VDAC1: A Potential Therapeutic Target of Inflammation-Related Diseases and Clinical Opportunities. Cells 2022, 11, 3174. https://doi.org/10.3390/cells11193174
Hu H, Guo L, Overholser J, Wang X. Mitochondrial VDAC1: A Potential Therapeutic Target of Inflammation-Related Diseases and Clinical Opportunities. Cells. 2022; 11(19):3174. https://doi.org/10.3390/cells11193174
Chicago/Turabian StyleHu, Hang, Linlin Guo, Jay Overholser, and Xing Wang. 2022. "Mitochondrial VDAC1: A Potential Therapeutic Target of Inflammation-Related Diseases and Clinical Opportunities" Cells 11, no. 19: 3174. https://doi.org/10.3390/cells11193174
APA StyleHu, H., Guo, L., Overholser, J., & Wang, X. (2022). Mitochondrial VDAC1: A Potential Therapeutic Target of Inflammation-Related Diseases and Clinical Opportunities. Cells, 11(19), 3174. https://doi.org/10.3390/cells11193174