
Mitophagy Regulation Following Myocardial Infarction


Abstract
:1. Introduction
2. Role and Regulation of Cardiac Mitophagy in Physiological Condition
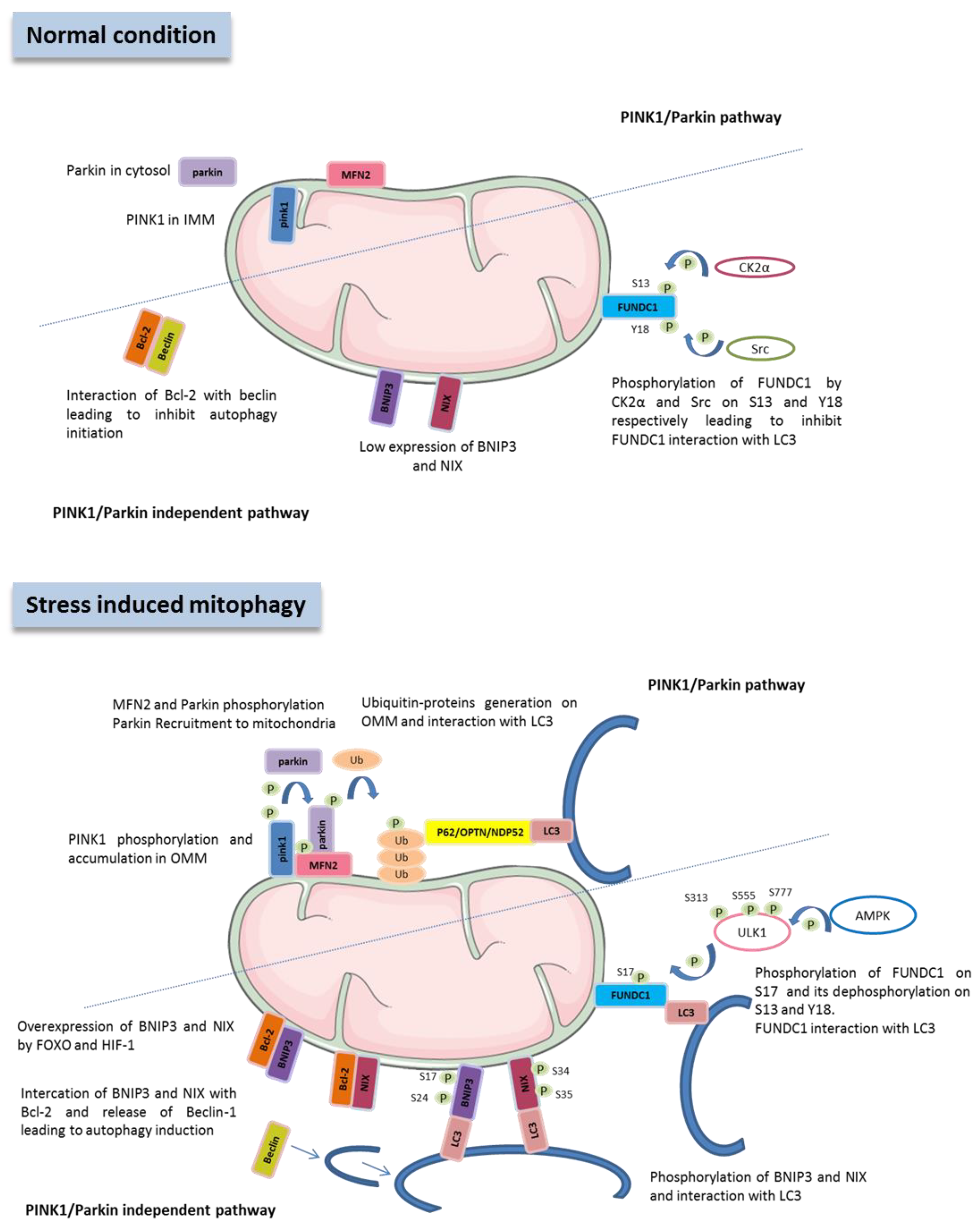
2.1. Machinery of Mitophagy
2.2. Regulation of Mitophagy by Non-Coding RNAs
2.3. Physiological Role of Cardiac Mitophagy
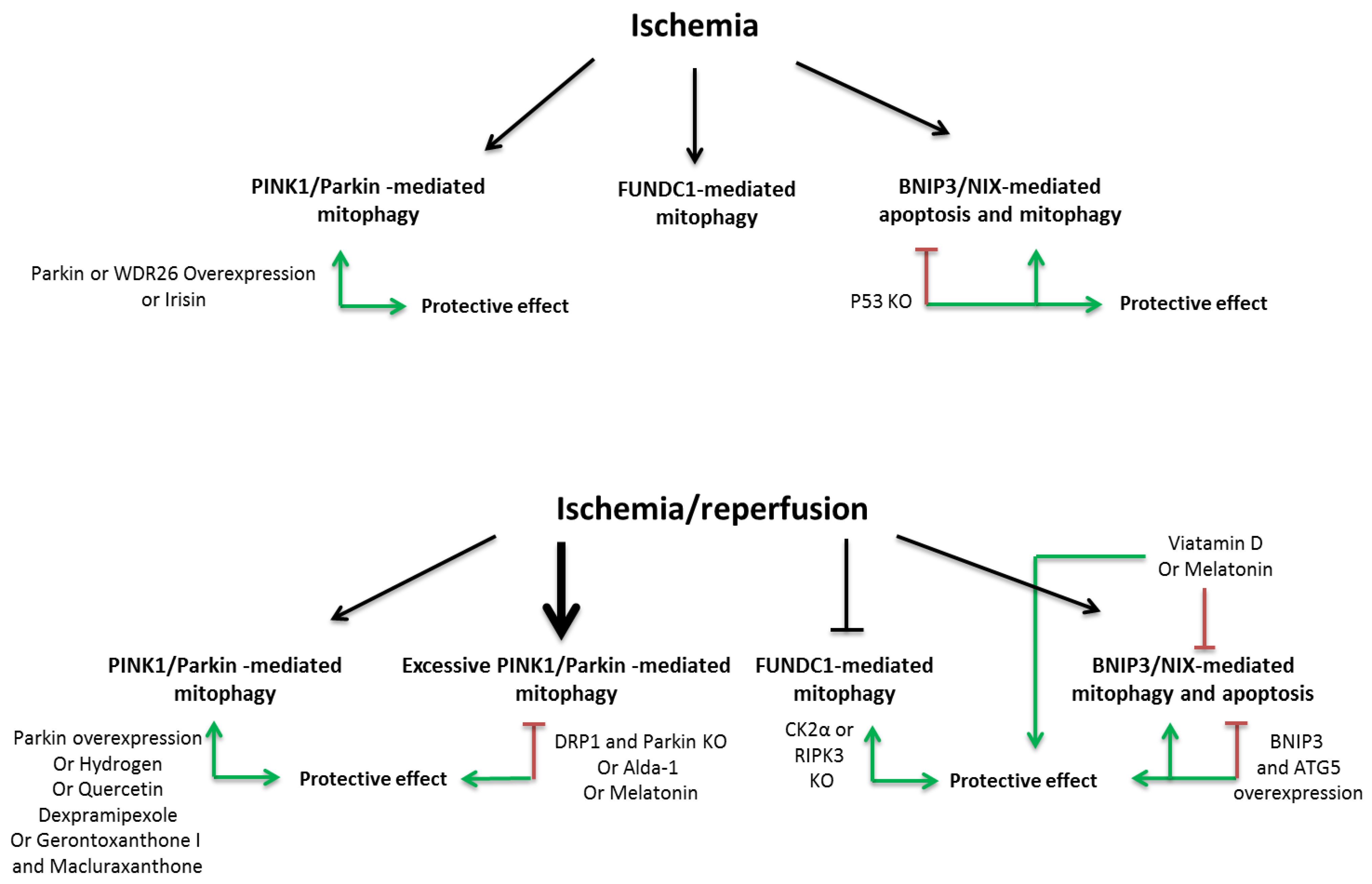
3. Role of Mitophagy during and Following Myocardial Infarction (MI)
3.1. PINK1/Parkin Pathway
3.2. FUNDC1 Pathway
3.3. BNIP3 Pathway
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Benjamin, E.J.; Blaha, M.J.; Chiuve, S.E.; Cushman, M.; Das, S.R.; Deo, R.; de Ferranti, S.D.; Floyd, J.; Fornage, M.; Gillespie, C.; et al. Heart Disease and Stroke Statistics-2017 Update: A Report From the American Heart Association. Circulation 2017, 135, e146–e603. [Google Scholar] [CrossRef]
- Pfeffer, M.A.; Braunwald, E. Ventricular remodeling after myocardial infarction. Experimental observations and clinical implications. Circulation 1990, 81, 1161–1172. [Google Scholar] [CrossRef] [Green Version]
- St John Sutton, M.; Pfeffer, M.A.; Plappert, T.; Rouleau, J.L.; Moyé, L.A.; Dagenais, G.R.; Lamas, G.A.; Klein, M.; Sussex, B.; Goldman, S. Quantitative two-dimensional echocardiographic measurements are major predictors of adverse cardiovascular events after acute myocardial infarction. The protective effects of captopril. Circulation 1994, 89, 68–75. [Google Scholar] [CrossRef] [Green Version]
- Bauters, C.; Dubois, E.; Porouchani, S.; Saloux, E.; Fertin, M.; de Groote, P.; Lamblin, N.; Pinet, F. Long-term prognostic impact of left ventricular remodeling after a first myocardial infarction in modern clinical practice. PLoS ONE 2017, 12, e0188884. [Google Scholar] [CrossRef] [Green Version]
- de Groote, P.; Fertin, M.; Duva Pentiah, A.; Goéminne, C.; Lamblin, N.; Bauters, C. Long-term functional and clinical follow-up of patients with heart failure with recovered left ventricular ejection fraction after β-blocker therapy. Circ. Heart Fail. 2014, 7, 434–439. [Google Scholar] [CrossRef] [Green Version]
- Richard, V.; Murry, C.; Jennings, R.; Reimer, K. Oxygen-derived free radicals and postischemic myocardial reperfusion: Therapeutic implications. Fundam. Clin. Pharmacol. 1990, 4, 85–103. [Google Scholar] [CrossRef]
- Zhou, H.; Zhang, Y.; Hu, S.; Shi, C.; Zhu, P.; Ma, Q.; Jin, Q.; Cao, F.; Tian, F.; Chen, Y. Melatonin protects cardiac microvasculature against ischemia/reperfusion injury via suppression of mitochondrial fission-VDAC1-HK2-mPTP-mitophagy axis. J. Pineal Res. 2017, 63, e12413. [Google Scholar] [CrossRef]
- Ohsumi, Y. Historical landmarks of autophagy research. Cell Res. 2014, 24, 9–23. [Google Scholar] [CrossRef] [Green Version]
- Clark, S.L. Cellular differentiation in the kidneys of newborn mice studies with the electron microscope. J. Biophys. Biochem. Cytol. 1957, 3, 349–362. [Google Scholar] [CrossRef] [Green Version]
- Deter, R.L.; De Duve, C. Influence of glucagon, an inducer of cellular autophagy, on some physical properties of rat liver lysosomes. J. Cell Biol. 1967, 33, 437–449. [Google Scholar] [CrossRef] [Green Version]
- Nakai, A.; Yamaguchi, O.; Takeda, T.; Higuchi, Y.; Hikoso, S.; Taniike, M.; Omiya, S.; Mizote, I.; Matsumura, Y.; Asahi, M.; et al. The role of autophagy in cardiomyocytes in the basal state and in response to hemodynamic stress. Nat. Med. 2007, 13, 619–624. [Google Scholar] [CrossRef]
- Gan, B.; Peng, X.; Nagy, T.; Alcaraz, A.; Gu, H.; Guan, J.-L. Role of FIP200 in cardiac and liver development and its regulation of TNFalpha and TSC-mTOR signaling pathways. J. Cell Biol. 2006, 175, 121–133. [Google Scholar] [CrossRef]
- Kaizuka, T.; Mizushima, N. Atg13 Is Essential for Autophagy and Cardiac Development in Mice. Mol. Cell. Biol. 2016, 36, 585–595. [Google Scholar] [CrossRef] [Green Version]
- Geisler, S.; Holmström, K.M.; Skujat, D.; Fiesel, F.C.; Rothfuss, O.C.; Kahle, P.J.; Springer, W. PINK1/Parkin-mediated mitophagy is dependent on VDAC1 and p62/SQSTM1. Nat. Cell Biol. 2010, 12, 119–131. [Google Scholar] [CrossRef]
- Matsuda, N.; Sato, S.; Shiba, K.; Okatsu, K.; Saisho, K.; Gautier, C.A.; Sou, Y.S.; Saiki, S.; Kawajiri, S.; Sato, F.; et al. PINK1 stabilized by mitochondrial depolarization recruits Parkin to damaged mitochondria and activates latent Parkin for mitophagy. J. Cell Biol. 2010, 189, 211–221. [Google Scholar] [CrossRef]
- Narendra, D.; Tanaka, A.; Suen, D.F.; Youle, R.J. Parkin is recruited selectively to impaired mitochondria and promotes their autophagy. J. Cell Biol. 2008, 183, 795–803. [Google Scholar] [CrossRef] [Green Version]
- Ji, H.; Wu, D.; Kimberlee, O.; Li, R.; Qian, G. Molecular Perspectives of Mitophagy in Myocardial Stress: Pathophysiology and Therapeutic Targets. Front. Physiol. 2021, 12, 700585. [Google Scholar] [CrossRef]
- Moyzis, A.; Gustafsson, Å.B. Multiple recycling routes: Canonical vs. non-canonical mitophagy in the heart. Biochim. Biophys. Acta-Mol. Basis Dis. 2019, 1865, 797–809. [Google Scholar] [CrossRef]
- Liu, L.; Sakakibara, K.; Chen, Q.; Okamoto, K. Receptor-mediated mitophagy in yeast and mammalian systems. Cell Res. 2014, 24, 787–795. [Google Scholar] [CrossRef] [Green Version]
- Kabeya, Y.; Kamada, Y.; Baba, M.; Takikawa, H.; Sasaki, M.; Ohsumi, Y. Atg17 functions in cooperation with Atg1 and Atg13 in yeast autophagy. Mol. Biol. Cell 2005, 16, 2544–2553. [Google Scholar] [CrossRef] [Green Version]
- Narendra, D.P.; Jin, S.M.; Tanaka, A.; Suen, D.-F.; Gautier, C.A. Is Selectively Stabilized on Impaired Mitochondria to Activate Parkin. PLoS Biol. 2010, 8, 1000298. [Google Scholar] [CrossRef] [Green Version]
- Vives-Bauza, C.; Zhou, C.; Huang, Y.; Cui, M.; De Vries, R.L.A.; Kim, J.; May, J.; Tocilescu, M.A.; Liu, W.; Ko, H.S.; et al. PINK1-dependent recruitment of Parkin to mitochondria in mitophagy. Proc. Natl. Acad. Sci. USA 2010, 107, 378–383. [Google Scholar] [CrossRef] [Green Version]
- Lin, W.; Kang, U.J. Characterization of PINK1 processing, stability, and subcellular localization. J. Neurochem. 2008, 106, 464–474. [Google Scholar] [CrossRef] [Green Version]
- Jin, S.M.; Lazarou, M.; Wang, C.; Kane, L.A.; Narendra, D.P.; Youle, R.J. Mitochondrial membrane potential regulates PINK1 import and proteolytic destabilization by PARL. J. Cell Biol. 2010, 191, 933–942. [Google Scholar] [CrossRef] [Green Version]
- Deas, E.; Plun-Favreau, H.; Gandhi, S.; Desmond, H.; Kjaer, S.; Loh, S.H.Y.; Renton, A.E.M.; Harvey, R.J.; Whitworth, A.J.; Martins, L.M.; et al. PINK1 cleavage at position A103 by the mitochondrial protease PARL. Hum. Mol. Genet. 2011, 20, 867–879. [Google Scholar] [CrossRef]
- Yamano, K.; Youle, R.J. PINK1 is degraded through the N-end rule pathway. Autophagy 2013, 9, 1758–1769. [Google Scholar] [CrossRef] [Green Version]
- Sekine, S.; Youle, R.J. PINK1 import regulation; a fine system to convey mitochondrial stress to the cytosol. BMC Biol. 2018, 16, 2. [Google Scholar] [CrossRef] [Green Version]
- Xiong, W.; Hua, J.; Liu, Z.; Cai, W.; Bai, Y.; Zhan, Q.; Lai, W.; Zeng, Q.; Ren, H.; Xu, D. PTEN induced putative kinase 1 (PINK1) alleviates angiotensin II-induced cardiac injury by ameliorating mitochondrial dysfunction. Int. J. Cardiol. 2018, 266, 198–205. [Google Scholar] [CrossRef]
- Shiba-Fukushima, K.; Imai, Y.; Yoshida, S.; Ishihama, Y.; Kanao, T.; Sato, S.; Hattori, N. PINK1-mediated phosphorylation of the Parkin ubiquitin-like domain primes mitochondrial translocation of Parkin and regulates mitophagy. Sci. Rep. 2012, 2, 1002. [Google Scholar] [CrossRef]
- Zhang, R.; Krigman, J.; Luo, H.; Ozgen, S.; Yang, M.; Sun, N. Mitophagy in cardiovascular homeostasis. Mech. Ageing Dev. 2020, 188, 111245. [Google Scholar] [CrossRef]
- Morales, P.E.; Arias-Durán, C.; Ávalos-Guajardo, Y.; Aedo, G.; Verdejo, H.E.; Parra, V.; Lavandero, S. Emerging role of mitophagy in cardiovascular physiology and pathology. Mol. Aspects Med. 2020, 71, 100822. [Google Scholar] [CrossRef]
- Yang, M.; Linn, B.S.; Zhang, Y.; Ren, J. Mitophagy and mitochondrial integrity in cardiac ischemia-reperfusion injury. Biochim. Biophys. Acta-Mol. Basis Dis. 2019, 1865, 2293–2302. [Google Scholar] [CrossRef]
- Luo, H.; Zhang, R.; Krigman, J.; McAdams, A.; Ozgen, S.; Sun, N. A Healthy Heart and a Healthy Brain: Looking at Mitophagy. Front. Cell Dev. Biol. 2020, 8, 294. [Google Scholar] [CrossRef]
- Lazarou, M.; Sliter, D.A.; Kane, L.A.; Sarraf, S.A.; Wang, C.; Burman, J.L.; Sideris, D.P.; Fogel, A.I.; Youle, R.J. The ubiquitin kinase PINK1 recruits autophagy receptors to induce mitophagy. Nature 2015, 524, 309–314. [Google Scholar] [CrossRef] [Green Version]
- Chen, Y.; Dorn, G.W. PINK1-phosphorylated mitofusin 2 is a parkin receptor for culling damaged mitochondria. Science 2013, 340, 471–475. [Google Scholar] [CrossRef] [Green Version]
- Kane, L.A.; Lazarou, M.; Fogel, A.I.; Li, Y.; Yamano, K.; Sarraf, S.A.; Banerjee, S.; Youle, R.J. PINK1 phosphorylates ubiquitin to activate parkin E3 ubiquitin ligase activity. J. Cell Biol. 2014, 205, 143–153. [Google Scholar] [CrossRef]
- Kazlauskaite, A.; Kondapalli, C.; Gourlay, R.; Campbell, D.G.; Ritorto, M.S.; Hofmann, K.; Alessi, D.R.; Knebel, A.; Trost, M.; Muqit, M.M.K. Accelerated publication: Parkin is activated by PINK1-dependent phosphorylation of ubiquitin at Ser65. Biochem. J. 2014, 460, 127–139. [Google Scholar] [CrossRef] [Green Version]
- Kazlauskaite, A.; Martínez-Torres, R.J.; Wilkie, S.; Kumar, A.; Peltier, J.; Gonzalez, A.; Johnson, C.; Zhang, J.; Hope, A.G.; Peggie, M.; et al. Binding to serine 65-phosphorylated ubiquitin primes Parkin for optimal PINK 1-dependent phosphorylation and activation. EMBO Rep. 2015, 16, 939–954. [Google Scholar] [CrossRef]
- Wang, B.; Nie, J.; Wu, L.; Hu, Y.; Wen, Z.; Dong, L.; Zou, M.-H.; Chen, C.; Wang, D.W. AMPKα2 Protects Against the Development of Heart Failure by Enhancing Mitophagy via PINK1 Phosphorylation. Circ. Res. 2018, 122, 712–729. [Google Scholar] [CrossRef]
- Zhou, H.; Zhu, P.; Wang, J.; Zhu, H.; Ren, J.; Chen, Y. Pathogenesis of cardiac ischemia reperfusion injury is associated with CK2α-disturbed mitochondrial homeostasis via suppression of FUNDC1-related mitophagy. Cell Death Differ. 2018, 25, 1080–1093. [Google Scholar] [CrossRef] [Green Version]
- Egan, D.F.; Shackelford, D.B.; Mihaylova, M.M.; Gelino, S.; Kohnz, R.A.; Mair, W.; Vasquez, D.S.; Joshi, A.; Gwinn, D.M.; Taylor, R.; et al. Phosphorylation of ULK1 (hATG1) by AMP-Activated Protein Kinase Connects Energy Sensing to Mitophagy. Science 2011, 331, 456–461. [Google Scholar] [CrossRef] [Green Version]
- Quinsay, M.N.; Thomas, R.L.; Lee, Y.; Gustafsson, Å.B. Bnip3-mediated mitochondrial autophagy is independent of the mitochondrial permeability transition pore. Autophagy 2010, 6, 855–862. [Google Scholar] [CrossRef] [Green Version]
- Sandoval, H.; Thiagarajan, P.; Dasgupta, S.K.; Schumacher, A.; Prchal, J.T.; Chen, M.; Wang, J. Essential role for Nix in autophagic maturation of erythroid cells. Nature 2008, 454, 232–235. [Google Scholar] [CrossRef]
- Schweers, R.L.; Zhang, J.; Randall, M.S.; Loyd, M.R.; Li, W.; Dorsey, F.C.; Kundu, M.; Opferman, J.T.; Cleveland, J.L.; Miller, J.L.; et al. NIX is required for programmed mitochondrial clearance during reticulocyte maturation. Proc. Natl. Acad. Sci. USA 2007, 104, 19500–19505. [Google Scholar] [CrossRef] [Green Version]
- Marinković, M.; Šprung, M.; Novak, I. Dimerization of mitophagy receptor BNIP3L/NIX is essential for recruitment of autophagic machinery. Autophagy 2021, 17, 1232–1243. [Google Scholar] [CrossRef]
- Kubasiak, L.A.; Hernandez, O.M.; Bishopric, N.H.; Webster, K.A. Hypoxia and acidosis activate cardiac myocyte death through the Bcl-2 family protein BNIP3. Proc. Natl. Acad. Sci. USA 2002, 99, 12825–12830. [Google Scholar] [CrossRef] [Green Version]
- Dorn, G.W.; Kirshenbaum, L.A. Cardiac reanimation: Targeting cardiomyocyte death by BNIP3 and NIX/BNIP3L. Oncogene 2008, 27, S158–S167. [Google Scholar] [CrossRef] [Green Version]
- Tong, M.; Zablocki, D.; Sadoshima, J. The role of Drp1 in mitophagy and cell death in the heart. J. Mol. Cell. Cardiol. 2020, 142, 138–145. [Google Scholar] [CrossRef]
- Lee, Y.; Lee, H.-Y.; Hanna, R.A.; Gustafsson, Å.B. Mitochondrial autophagy by Bnip3 involves Drp1-mediated mitochondrial fission and recruitment of Parkin in cardiac myocytes. Am. J. Physiol. Circ. Physiol. 2011, 301, H1924–H1931. [Google Scholar] [CrossRef]
- Zhang, H.; Bosch-Marce, M.; Shimoda, L.A.; Tan, Y.S.; Baek, J.H.; Wesley, J.B.; Gonzalez, F.J.; Semenza, G.L. Mitochondrial Autophagy Is an HIF-1-dependent Adaptive Metabolic Response to Hypoxia. J. Biol. Chem. 2008, 283, 10892–10903. [Google Scholar] [CrossRef] [Green Version]
- Birse-Archbold, J.L.A.; Kerr, L.E.; Jones, P.A.; McCulloch, J.; Sharkey, J. Differential profile of Nix upregulation and translocation during hypoxia/ischaemia in vivo versus in vitro. J. Cereb. Blood Flow Metab. 2005, 25, 1356–1365. [Google Scholar] [CrossRef] [Green Version]
- Pattingre, S.; Tassa, A.; Qu, X.; Garuti, R.; Xiao, H.L.; Mizushima, N.; Packer, M.; Schneider, M.D.; Levine, B. Bcl-2 antiapoptotic proteins inhibit Beclin 1-dependent autophagy. Cell 2005, 122, 927–939. [Google Scholar] [CrossRef] [Green Version]
- Russell, R.C.; Tian, Y.; Yuan, H.; Park, H.W.; Chang, Y.Y.; Kim, J.; Kim, H.; Neufeld, T.P.; Dillin, A.; Guan, K.L. ULK1 induces autophagy by phosphorylating Beclin-1 and activating VPS34 lipid kinase. Nat. Cell Biol. 2013, 15, 741–750. [Google Scholar] [CrossRef] [Green Version]
- Park, J.M.; Seo, M.; Jung, C.H.; Grunwald, D.; Stone, M.; Otto, N.M.; Toso, E.; Ahn, Y.; Kyba, M.; Griffin, T.J.; et al. ULK1 phosphorylates Ser30 of BECN1 in association with ATG14 to stimulate autophagy induction. Autophagy 2018, 14, 584–597. [Google Scholar] [CrossRef] [Green Version]
- Wu, W.; Tian, W.; Hu, Z.; Chen, G.; Huang, L.; Li, W.; Zhang, X.; Xue, P.; Zhou, C.; Liu, L.; et al. ULK1 translocates to mitochondria and phosphorylates FUNDC1 to regulate mitophagy. EMBO Rep. 2014, 15, 566–575. [Google Scholar] [CrossRef] [Green Version]
- Turkieh, A.; Charrier, H.; Dubois-Deruy, E.; Porouchani, S.; Bouvet, M.; Pinet, F. Noncoding RNAs in Cardiac Autophagy following Myocardial Infarction. Oxid. Med. Cell. Longev. 2019, 2019, 8438650. [Google Scholar] [CrossRef]
- Wang, L.; Li, Q.; Diao, J.; Lin, L.; Wei, J. MiR-23a Is Involved in Myocardial Ischemia/Reperfusion Injury by Directly Targeting CX43 and Regulating Mitophagy. Inflammation 2021, 44, 1581–1591. [Google Scholar] [CrossRef]
- Lv, W.; Jiang, J.; Li, Y.; Fu, L.; Meng, F.; Li, J. MiR-302a-3p aggravates myocardial ischemia-reperfusion injury by suppressing mitophagy via targeting FOXO3. Exp. Mol. Pathol. 2020, 117, 104522. [Google Scholar] [CrossRef]
- Yang, F.; Li, T.; Dong, Z.; Mi, R. MicroRNA-410 is involved in mitophagy after cardiac ischemia/reperfusion injury by targeting high-mobility group box 1 protein. J. Cell. Biochem. 2018, 119, 2427–2439. [Google Scholar] [CrossRef]
- Wang, K.; Zhou, L.-Y.; Wang, J.-X.; Wang, Y.; Sun, T.; Zhao, B.; Yang, Y.-J.; An, T.; Long, B.; Li, N.; et al. E2F1-dependent miR-421 regulates mitochondrial fragmentation and myocardial infarction by targeting Pink1. Nat. Commun. 2015, 6, 7619. [Google Scholar] [CrossRef] [Green Version]
- Hu, Q.; Yuan, Y.; Wu, Y.; Huang, Y.; Zhao, Z.; Xiao, C. MicroRNA-137 exerts protective effects on hypoxia-induced cell injury by inhibiting autophagy/mitophagy and maintaining mitochondrial function in breast cancer stem-like cells. Oncol. Rep. 2020, 44, 1627–1637. [Google Scholar] [CrossRef]
- Li, W.; Zhang, X.; Zhuang, H.; Chen, H.G.; Chen, Y.; Tian, W.; Wu, W.; Li, Y.; Wang, S.; Zhang, L.; et al. MicroRNA-137 is a novel hypoxia-responsive MicroRNA that inhibits mitophagy via regulation of two mitophagy receptors FUNDC1 and NIX. J. Biol. Chem. 2014, 289, 10691–10701. [Google Scholar] [CrossRef] [Green Version]
- Li, X.; Luo, S.; Zhang, J.; Yuan, Y.; Jiang, W.; Zhu, H.; Ding, X.; Zhan, L.; Wu, H.; Xie, Y.; et al. lncRNA H19 Alleviated Myocardial I/RI via Suppressing miR-877-3p/Bcl-2-Mediated Mitochondrial Apoptosis. Mol. Ther.-Nucleic Acids 2019, 17, 297–309. [Google Scholar] [CrossRef] [Green Version]
- Wang, S.-H.; Zhu, X.-L.; Wang, F.; Chen, S.-X.; Chen, Z.-T.; Qiu, Q.; Liu, W.-H.; Wu, M.-X.; Deng, B.-Q.; Xie, Y.; et al. LncRNA H19 governs mitophagy and restores mitochondrial respiration in the heart through Pink1/Parkin signaling during obesity. Cell Death Dis. 2021, 12, 557. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Zhou, L.; Li, H.; Sun, T.; Wen, X.; Li, X.; Meng, Y.; Li, Y.; Liu, M.; Liu, S.; et al. Nuclear-Encoded lncRNA MALAT1 Epigenetically Controls Metabolic Reprogramming in HCC Cells through the Mitophagy Pathway. Mol. Ther.-Nucleic Acids 2021, 23, 264–276. [Google Scholar] [CrossRef] [PubMed]
- Hu, H.; Wu, J.; Li, D.; Zhou, J.; Yu, H.; Ma, L. Knockdown of lncRNA MALAT1 attenuates acute myocardial infarction through miR-320-Pten axis. Biomed. Pharmacother. 2018, 106, 738–746. [Google Scholar] [CrossRef]
- Huang, J.; Jiang, R.; Chu, X.; Wang, F.; Sun, X.; Wang, Y.; Pang, L. Overexpression of microRNA-23a-5p induces myocardial infarction by promoting cardiomyocyte apoptosis through inhibited of PI3K/AKT signalling pathway. Cell Biochem. Funct. 2020, 38, 1047–1055. [Google Scholar] [CrossRef]
- Song, Y.; Wang, B.; Zhu, X.; Hu, J.; Sun, J.; Xuan, J.; Ge, Z. Human umbilical cord blood–derived MSCs exosome attenuate myocardial injury by inhibiting ferroptosis in acute myocardial infarction mice. Cell Biol. Toxicol. 2021, 37, 51–64. [Google Scholar] [CrossRef]
- Mao, J.; Lv, Z.; Zhuang, Y. MicroRNA-23a is involved in tumor necrosis factor-α induced apoptosis in mesenchymal stem cells and myocardial infarction. Exp. Mol. Pathol. 2014, 97, 23–30. [Google Scholar] [CrossRef]
- Clark, A.L.; Maruyama, S.; Sano, S.; Accorsi, A.; Girgenrath, M.; Walsh, K.; Naya, F.J. miR-410 and miR-495 Are Dynamically Regulated in Diverse Cardiomyopathies and Their Inhibition Attenuates Pathological Hypertrophy. PLoS ONE 2016, 11, e0151515. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhao, T.; Qiu, Z.; Gao, Y. MiR-137-3p exacerbates the ischemia-reperfusion injured cardiomyocyte apoptosis by targeting KLF15. Naunyn. Schmiedebergs. Arch. Pharmacol. 2020, 393, 1013–1024. [Google Scholar] [CrossRef]
- Zhou, H.; Wang, B.; Yang, Y.; Jia, Q.; Zhang, A.; Qi, Z.; Zhang, J. Long Noncoding RNAs in Pathological Cardiac Remodeling: A Review of the Update Literature. Biomed. Res. Int. 2019, 2019, 7159592. [Google Scholar] [CrossRef]
- Dubois-Deruy, E.; Cuvelliez, M.; Fiedler, J.; Charrier, H.; Mulder, P.; Hebbar, E.; Pfanne, A.; Beseme, O.; Chwastyniak, M.; Amouyel, P.; et al. MicroRNAs regulating superoxide dismutase 2 are new circulating biomarkers of heart failure. Sci. Rep. 2017, 7, 14747. [Google Scholar] [CrossRef] [Green Version]
- Vausort, M.; Wagner, D.R.; Devaux, Y. Long noncoding RNAs in patients with acute myocardial infarction. Circ. Res. 2014, 115, 668–677. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhao, Z.-H.; Hao, W.; Meng, Q.-T.; Du, X.-B.; Lei, S.-Q.; Xia, Z.-Y. Long non-coding RNA MALAT1 functions as a mediator in cardioprotective effects of fentanyl in myocardial ischemia-reperfusion injury. Cell Biol. Int. 2017, 41, 62–70. [Google Scholar] [CrossRef] [PubMed]
- Shao, G.; Zhao, Z.; Zhao, W.; Hu, G.; Zhang, L.; Li, W.; Xing, C.; Zhang, X. Long non-coding RNA MALAT1 activates autophagy and promotes cell proliferation by downregulating microRNA-204 expression in gastric cancer. Oncol. Lett. 2020, 19, 805–812. [Google Scholar] [CrossRef] [PubMed]
- Xiao, J.; Zhu, X.; He, B.; Zhang, Y.; Kang, B.; Wang, Z.; Ni, X. MiR-204 regulates cardiomyocyte autophagy induced by ischemia-reperfusion through LC3-II. J. Biomed. Sci. 2011, 18, 35. [Google Scholar] [CrossRef] [Green Version]
- Zhang, L.; Fang, Y.; Zhao, X.; Zheng, Y.; Ma, Y.; Li, S.; Huang, Z.; Li, L. miR-204 silencing reduces mitochondrial autophagy and ROS production in a murine AD model via the TRPML1-activated STAT3 pathway. Mol. Ther.-Nucleic Acids 2021, 24, 822–831. [Google Scholar] [CrossRef]
- Kang, R.; Livesey, K.M.; Zeh, H.J.; Lotze, M.T.; Tang, D. Metabolic regulation by HMGB1-mediated autophagy and mitophagy. Autophagy 2011, 7, 1256–1258. [Google Scholar] [CrossRef] [Green Version]
- Gong, G.; Song, M.; Csordas, G.; Kelly, D.P.; Matkovich, S.J.; Dorn, G.W. Parkin-mediated mitophagy directs perinatal cardiac metabolic maturation in mice. Science 2015, 350, 33–48. [Google Scholar] [CrossRef] [Green Version]
- Vásquez-Trincado, C.; García-Carvajal, I.; Pennanen, C.; Parra, V.; Hill, J.A.; Rothermel, B.A.; Lavandero, S. Mitochondrial dynamics, mitophagy and cardiovascular disease. J. Physiol. 2016, 594, 509–525. [Google Scholar] [CrossRef] [PubMed]
- Lampert, M.A.; Orogo, A.M.; Najor, R.H.; Hammerling, B.C.; Leon, L.J.; Wang, B.J.; Kim, T.; Sussman, M.A.; Gustafsson, Å.B. BNIP3L/NIX and FUNDC1-mediated mitophagy is required for mitochondrial network remodeling during cardiac progenitor cell differentiation. Autophagy 2019, 15, 1182–1198. [Google Scholar] [CrossRef]
- Billia, F.; Hauck, L.; Konecny, F.; Rao, V.; Shen, J.; Mak, T.W. PTEN-inducible kinase 1 (PINK1)/Park6 is indispensable for normal heart function. Proc. Natl. Acad. Sci. USA 2011, 108, 9572–9577. [Google Scholar] [CrossRef] [Green Version]
- Kubli, D.A.; Zhang, X.; Lee, Y.; Hanna, R.A.; Quinsay, M.N.; Nguyen, C.K.; Jimenez, R.; Petrosyans, S.; Murphy, A.N.; Gustafsson, Å.B. Parkin protein deficiency exacerbates cardiac injury and reduces survival following myocardial infarction. J. Biol. Chem. 2013, 288, 915–926. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, Y.; Ruggeri, Z.M.; Du, X. 14-3-3 proteins in platelet biology and glycoprotein Ib-IX signaling. Blood 2018, 131, 2436–2448. [Google Scholar] [CrossRef] [Green Version]
- Durrant, T.N.; Van Den Bosch, M.T.; Hers, I. Integrin αIIbβ3 outside-in signaling. Blood 2017, 130, 1607–1619. [Google Scholar] [CrossRef] [Green Version]
- Metharom, P.; Berndt, M.C.; Baker, R.I.; Andrews, R.K. Current state and novel approaches of antiplatelet therapy. Arterioscler. Thromb. Vasc. Biol. 2015, 35, 1327–1338. [Google Scholar] [CrossRef] [Green Version]
- Xu, X.R.; Wang, Y.; Adili, R.; Ju, L.; Spring, C.M.; Jin, J.W.; Yang, H.; Neves, M.A.D.; Chen, P.; Yang, Y.; et al. Apolipoprotein A-IV binds αIIbβ3 integrin and inhibits thrombosis. Nat. Commun. 2018, 9, 3608. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zharikov, S.; Shiva, S. Platelet mitochondrial function: From regulation of thrombosis to biomarker of disease. Biochem. Soc. Trans. 2013, 41, 118–123. [Google Scholar] [CrossRef]
- Zhang, W.; Ren, H.; Xu, C.; Zhu, C.; Wu, H.; Liu, D.; Wang, J.; Liu, L.; Li, W.; Ma, Q.; et al. Hypoxic mitophagy regulates mitochondrial quality and platelet activation and determines severity of I/R heart injury. eLife 2016, 5, e21407. [Google Scholar] [CrossRef]
- Zhang, W.; Chen, C.; Wang, J.; Liu, L.; He, Y.; Chen, Q. Mitophagy in cardiomyocytes and in platelets: A major mechanism of cardioprotection against ischemia/reperfusion injury. Physiology 2018, 33, 86–98. [Google Scholar] [CrossRef]
- Lee, S.H.; Du, J.; Stitham, J.; Atteya, G.; Lee, S.; Xiang, Y.; Wang, D.; Jin, Y.; Leslie, K.L.; Spollett, G.; et al. Inducing mitophagy in diabetic platelets protects against severe oxidative stress. EMBO Mol. Med. 2016, 8, 779–795. [Google Scholar] [CrossRef]
- Lee, S.H.; Lee, S.; Du, J.; Jain, K.; Ding, M.; Kadado, A.J.; Atteya, G.; Jaji, Z.; Tyagi, T.; Kim, W.; et al. Mitochondrial MsrB2 serves as a switch and transducer for mitophagy. EMBO Mol. Med. 2019, 11, e10409. [Google Scholar] [CrossRef]
- Zhang, W.; Ma, Q.; Siraj, S.; Ney, P.A.; Liu, J.; Liao, X.; Yuan, Y.; Li, W.; Liu, L.; Chen, Q. Nix-mediated mitophagy regulates platelet activation and life span. Blood Adv. 2019, 3, 2342–2354. [Google Scholar] [CrossRef] [Green Version]
- Yao, L.; Chen, H.; Wu, Q.; Xie, K. Hydrogen-rich saline alleviates inflammation and apoptosis in myocardial I/R injury via PINK-mediated autophagy. Int. J. Mol. Med. 2019, 44, 1048–1062. [Google Scholar] [CrossRef] [Green Version]
- Jiang, J.; Liang, S.; Zhang, J.; Du, Z.; Xu, Q.; Duan, J.; Sun, Z. Melatonin ameliorates PM 2.5 -induced cardiac perivascular fibrosis through regulating mitochondrial redox homeostasis. J. Pineal Res. 2021, 70, e12686. [Google Scholar] [CrossRef]
- Hamacher-Brady, A.; Brady, N.R.; Logue, S.E.; Sayen, M.R.; Jinno, M.; Kirshenbaum, L.A.; Gottlieb, R.A.; Gustafsson, Å.B. Response to myocardial ischemia/reperfusion injury involves Bnip3 and autophagy. Cell Death Differ. 2007, 14, 146–157. [Google Scholar] [CrossRef] [Green Version]
- Ji, W.; Wei, S.; Hao, P.; Xing, J.; Yuan, Q.; Wang, J.; Xu, F.; Chen, Y. Aldehyde dehydrogenase 2 has cardioprotective effects on myocardial ischaemia/reperfusion injury via suppressing mitophagy. Front. Pharmacol. 2016, 7, 101. [Google Scholar] [CrossRef] [Green Version]
- Feng, Y.; Zhao, J.; Hou, H.; Zhang, H.; Jiao, Y.; Wang, J.; Wang, Y.; Sun, Y. WDR26 promotes mitophagy of cardiomyocytes induced by hypoxia through Parkin translocation. Acta Biochim. Biophys. Sin. (Shanghai) 2016, 48, 1075–1084. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xin, T.; Lu, C. Irisin activates Opa1-induced mitophagy to protect cardiomyocytes against apoptosis following myocardial infarction. Aging (Albany NY) 2020, 12, 4474–4488. [Google Scholar] [CrossRef] [PubMed]
- Siddall, H.K.; Yellon, D.M.; Ong, S.B.S.-B.; Mukherjee, U.A.; Burke, N.; Hall, A.R.; Angelova, P.R.; Ludtmann, M.H.R.; Deas, E.; Davidson, S.M.; et al. Loss of PINK1 Increases the Heart’s Vulnerability to Ischemia-Reperfusion Injury. PLoS ONE 2013, 8, 62400. [Google Scholar] [CrossRef]
- Song, M.; Gong, G.; Burelle, Y.; Gustafsson, Å.B.; Kitsis, R.N.; Matkovich, S.J.; Dorn, G.W. Interdependence of parkin-mediated mitophagy and mitochondrial fission in adult mouse hearts. Circ. Res. 2015, 117, 346–351. [Google Scholar] [CrossRef] [Green Version]
- Chang, X.; Zhang, T.; Meng, Q.; Wang, S.; Yan, P.; Wang, X.; Luo, D.; Zhou, X.; Ji, R. Quercetin Improves Cardiomyocyte Vulnerability to Hypoxia by Regulating SIRT1/TMBIM6-Related Mitophagy and Endoplasmic Reticulum Stress. Oxid. Med. Cell. Longev. 2021, 2021, 5529913. [Google Scholar] [CrossRef]
- Tang, L.; Li, Y.-P.; Hu, J.; Chen, A.-H.; Mo, Y. Dexpramipexole attenuates myocardial ischemia/reperfusion injury through upregulation of mitophagy. Eur. J. Pharmacol. 2021, 899, 173962. [Google Scholar] [CrossRef]
- Xiang, Q.; Wu, M.; Zhang, L.; Fu, W.; Yang, J.; Zhang, B.; Zheng, Z.; Zhang, H.; Lao, Y.; Xu, H. Gerontoxanthone I and Macluraxanthone Induce Mitophagy and Attenuate Ischemia/Reperfusion Injury. Front. Pharmacol. 2020, 11, 452. [Google Scholar] [CrossRef] [Green Version]
- Wu, J.; Yang, Y.; Gao, Y.; Wang, Z.; Ma, J. Melatonin attenuates anoxia/reoxygenation injury by inhibiting excessive mitophagy through the MT2/SIRT3/FOXO3A signaling pathway in H9C2 cells. Drug Des. Dev. Ther. 2020, 14, 2047–2060. [Google Scholar] [CrossRef] [PubMed]
- Huang, C.; Andres, A.M.; Ratliff, E.P.; Hernandez, G.; Lee, P.; Gottlieb, R.A. Preconditioning Involves Selective Mitophagy Mediated by Parkin and p62/SQSTM1. PLoS ONE 2011, 6, e20975. [Google Scholar] [CrossRef] [PubMed]
- Chen, G.; Han, Z.; Feng, D.; Chen, Y.; Chen, L.; Wu, H.; Huang, L.; Zhou, C.; Cai, X.; Fu, C.; et al. A Regulatory Signaling Loop Comprising the PGAM5 Phosphatase and CK2 Controls Receptor-Mediated Mitophagy. Mol. Cell 2014, 54, 362–377. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhou, H.; Zhu, P.; Guo, J.; Hu, N.; Wang, S.; Li, D.; Hu, S.; Ren, J.; Cao, F.; Chen, Y. Ripk3 induces mitochondrial apoptosis via inhibition of FUNDC1 mitophagy in cardiac IR injury. Redox Biol. 2017, 13, 498–507. [Google Scholar] [CrossRef] [PubMed]
- Syed, F.; Odley, A.; Hahn, H.S.; Brunskill, E.W.; Lynch, R.A.; Marreez, Y.; Sanbe, A.; Robbins, J.; Dorn, G.W. Physiological growth synergizes with pathological genes in experimental cardiomyopathy. Circ. Res. 2004, 95, 1200–1206. [Google Scholar] [CrossRef] [Green Version]
- Dorn, G.W. Mitochondrial Pruning by Nix and BNip3: An Essential Function for Cardiac-Expressed Death Factors. J. Cardiovasc. Transl. Res. 2010, 3, 374–383. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hoshino, A.; Matoba, S.; Iwai-Kanai, E.; Nakamura, H.; Kimata, M.; Nakaoka, M.; Katamura, M.; Okawa, Y.; Ariyoshi, M.; Mita, Y.; et al. p53-TIGAR axis attenuates mitophagy to exacerbate cardiac damage after ischemia. J. Mol. Cell. Cardiol. 2012, 52, 175–184. [Google Scholar] [CrossRef] [PubMed]
- Lee, T.L.; Lee, M.H.; Chen, Y.C.; Lee, Y.C.; Lai, T.C.; Lin, H.Y.H.; Hsu, L.F.; Sung, H.C.; Lee, C.W.; Chen, Y.L. Vitamin D Attenuates Ischemia/Reperfusion-Induced Cardiac Injury by Reducing Mitochondrial Fission and Mitophagy. Front. Pharmacol. 2020, 11, 604700. [Google Scholar] [CrossRef] [PubMed]
- Georgakopoulos, N.D.; Wells, G.; Campanella, M. The pharmacological regulation of cellular mitophagy. Nat. Chem. Biol. 2017, 13, 136–146. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
{kind=link}
| Non-Coding RNAs | Role on Mitophagy | Mitochondrial Targets | Consequences | References |
|---|---|---|---|---|
| miR-23a | Induces PINK-1/parkin dependent mitophagy | CX43 | Increases cardiac cells apoptosis | [57] |
| miR-302-3p | Inhibits mitophagy | FOXO3 | Increases mitochondrial dysfunction and apoptosis | [58] |
| miR-410 | Inhibits mitophagy | HMGB-1 | Increases mitochondrial dysfunction and apoptosis | [59] |
| miR-421 | Not shown | PINK-1 | Increases mitochondrial fragmentation and apoptosis | [60] |
| miR-137 | Unknown in cardiac cells Inhibits mitophagy in cancer cells and brain | Unknown in cardiac cells FUNDC-1 BNIP3L/NIX | Unknown in cardiac cells Restores mitochondrial functions and decreases apoptosis in breast cancer cells. | [61,62] |
| LncRNA H19 | Unknown in I/R Decreases excessive mitophagy in palmitate treated-H9c2. | miR-877-3p/Bcl-2 pathway Hinder the binding of eIF4A2-PINK1 mRNA | Decreases apoptosis in I/R Decreases apoptosis in palmitate-treated-H9c2 | [63,64] |
| LncRNA MALAT1 | Unknown in cardiac cells Increases mitophagy in cancer cells | miR-320/PTEN miR-145/BNIP3 Unknow in cancer cells | Decreases cardiac cells apoptosis Improves mitochondrial structure and function in cancer cells | [65,66] |
| Non-Coding RNAs | Models | Regulation during and Post-MI | Consequences on Cardiac Function | References |
|---|---|---|---|---|
| miR-23a | Rat: 30 min I/R Rat primary cardiomyocytes 4 h H/2 h R LAD coronary artery 1 day | Increased | Exosomes derived from HUCB-MSC containing miR-23a decreases infarct size overexpression of miR-23a in BM-MSC decreases infarct size and LVESD, and increases EF, FS and IVS | [57,67,68,69] |
| miR-302-3p | Mice: 45 min I/2 h R Adult mice cardiomyocytes 3 h H/6 h R | Increased | Not described | [58] |
| miR-410 | MI mice: LAD coronary artery 1-3-7 days Mice: 45 min I/6-72 h R Adult human cardiomyocytes 8 h H/16 h R | Increased | Not described in the heart but decreases cell area and ANP, BNP expressions in cardiomyocytes treated with AngII | [59,70] |
| miR-421 | Mice: 45 min I/3 h R or 1 week | Increased | Overexpression increases infarct size but has no effect on FS. | [60] |
| miR-137 | Rat: 45 min I/2 h R H9c2: 6 h H/18 h R | Increased | Inhibition of miR-137-3p improves EF and FS | [71] |
| LncRNA H19 | Mice: 45 min I/24 h R MI rat: LAD coronary artery 4 weeks | Decreased | Overexpression of LncRNA H19 decreases infarct area and improves cardiac function: increased EF and FS, decreased ANP, BNP and fibrosis markers. | [63,72] |
| LncRNA MALAT1 | MI mice: LAD coronary artery 3 days Neonatal mice cardiomyocytes 12, 24, 48 h H | Increased | Inhibition of MALAT1 decreases infarct area, LVEDD and LVESD, and increases EF and FS | [66] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Turkieh, A.; El Masri, Y.; Pinet, F.; Dubois-Deruy, E. Mitophagy Regulation Following Myocardial Infarction. Cells 2022, 11, 199. https://doi.org/10.3390/cells11020199
Turkieh A, El Masri Y, Pinet F, Dubois-Deruy E. Mitophagy Regulation Following Myocardial Infarction. Cells. 2022; 11(2):199. https://doi.org/10.3390/cells11020199
Chicago/Turabian StyleTurkieh, Annie, Yara El Masri, Florence Pinet, and Emilie Dubois-Deruy. 2022. "Mitophagy Regulation Following Myocardial Infarction" Cells 11, no. 2: 199. https://doi.org/10.3390/cells11020199
APA StyleTurkieh, A., El Masri, Y., Pinet, F., & Dubois-Deruy, E. (2022). Mitophagy Regulation Following Myocardial Infarction. Cells, 11(2), 199. https://doi.org/10.3390/cells11020199