
Mesenchymal Stem Cell Therapy in Diabetic Cardiomyopathy

 , , , ,
, , , ,  , ,
, ,
Abstract
1. Introduction
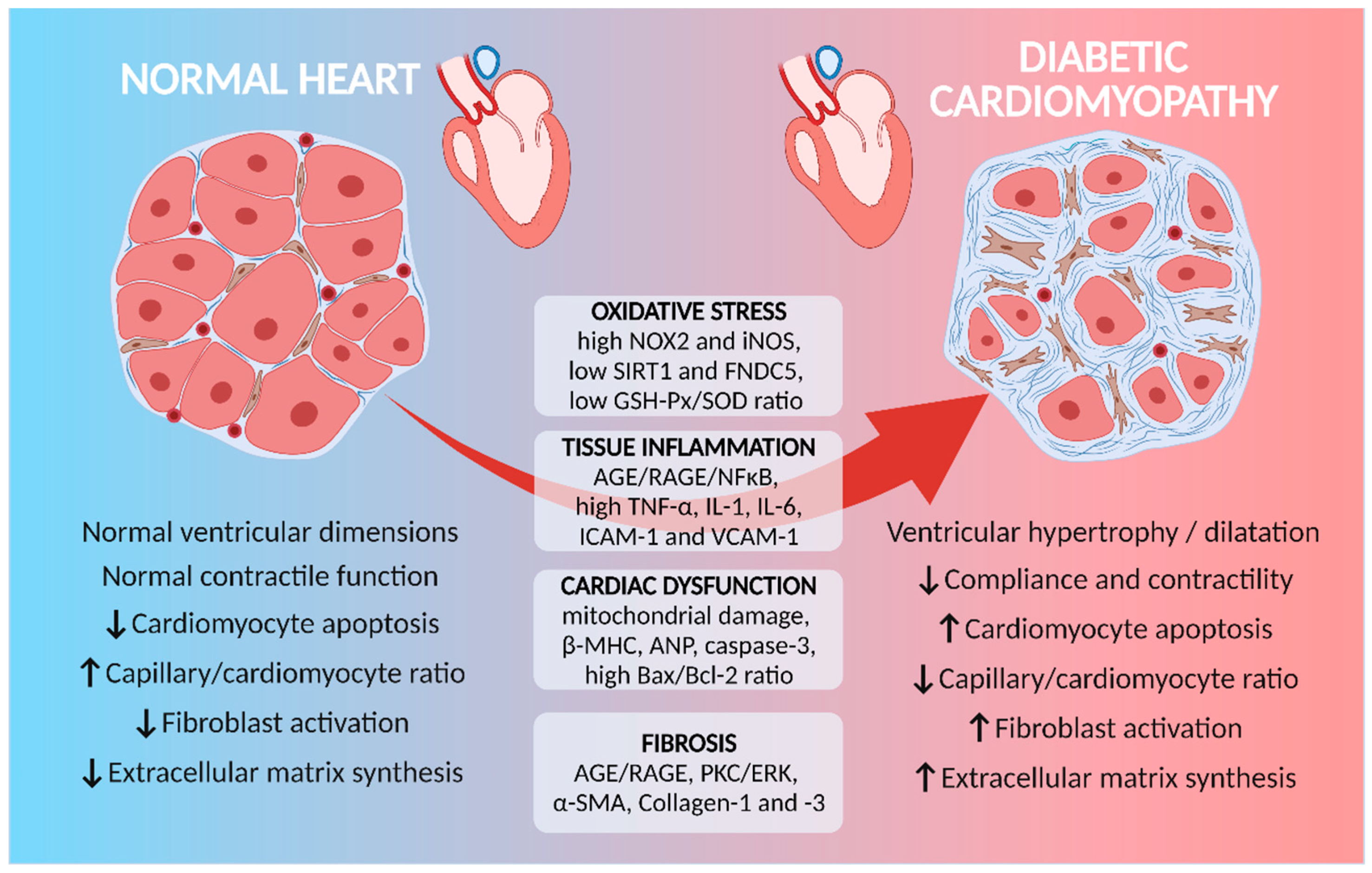
2. Molecular Mechanisms Involved in Diabetes-Induced Cardiac Remodeling
3. Myocardial Inflammation and Diabetes-Induced Cardiac Remodeling
4. Epigenetic Involvement in Diabetic Cardiomyopathy
5. Preclinical and Clinical Approaches to MSC in Diabetes-Induced Cardiac Complications
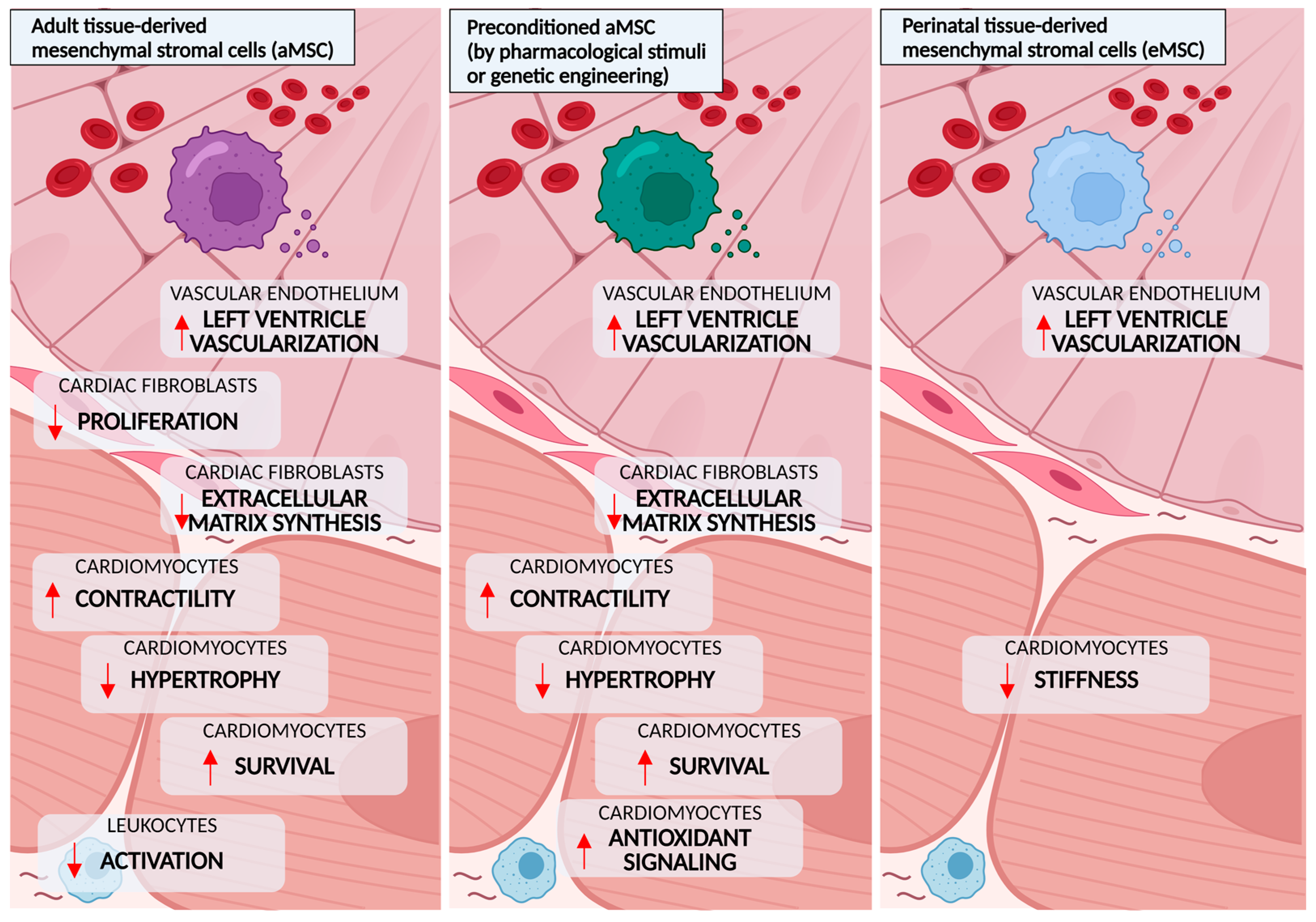
5.1. Adult Tissue-Derived MSCs
5.2. Preconditioning and Genetic Modification of Adult Tissue-Derived MSCs
5.3. Perinatal Tissue-Derived MSCs
5.4. Clinical Studies
6. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Sinclair, A.; Saeedi, P.; Kaundal, A.; Karuranga, S.; Malanda, B.; Williams, R. Diabetes and global ageing among 65–99-year-old adults: Findings from the International Diabetes Federation Diabetes Atlas, 9th edition. Diabetes Res. Clin. Pract. 2020, 162, 108078. [Google Scholar] [CrossRef]
- Saeedi, P.; Petersohn, I.; Salpea, P.; Malanda, B.; Karuranga, S.; Unwin, N.; Colagiuri, S.; Guariguata, L.; Motala, A.A.; Ogurtsova, K.; et al. Global and regional diabetes prevalence estimates for 2019 and projections for 2030 and 2045: Results from the International Diabetes Federation Diabetes Atlas, 9th edition. Diabetes Res. Clin. Pract. 2019, 157, 107843. [Google Scholar] [CrossRef] [PubMed]
- Kaur, R.; Kaur, M.; Singh, J. Endothelial dysfunction and platelet hyperactivity in type 2 diabetes mellitus: Molecular insights and therapeutic strategies. Cardiovasc. Diabetol. 2018, 17, 121. [Google Scholar] [CrossRef] [PubMed]
- El Hayek, M.S.; Ernande, L.; Benitah, J.-P.; Gomez, A.-M.; Pereira, L. The role of hyperglycaemia in the development of diabetic cardiomyopathy. Arch. Cardiovasc. Dis. 2021, 114, 748–760. [Google Scholar] [CrossRef]
- Azevedo, P.S.; Polegato, B.F.; Minicucci, M.F.; Paiva, S.A.R.; Zornoff, L.A.M. Cardiac Remodeling: Concepts, Clinical Impact, Pathophysiological Mechanisms and Pharmacologic Treatment. Arq. Bras. Cardiol. 2016, 106, 62–69. [Google Scholar] [CrossRef] [PubMed]
- Chen, W.; Ju, J.; Yang, Y.; Wang, H.; Chen, W.; Zhao, X.; Ye, H.; Zhang, Y. Astragalus polysaccharides protect cardiac stem and progenitor cells by the inhibition of oxidative stress-mediated apoptosis in diabetic hearts. Drug Des. Dev. Ther. 2018, 12, 943–954. [Google Scholar] [CrossRef] [PubMed]
- Yang, F.; Yu, X.; Li, T.; Wu, J.; Zhao, Y.; Liu, J.; Sun, A.; Dong, S.; Wu, J.; Zhong, X.; et al. Exogenous H2S regulates endoplasmic reticulum-mitochondria cross-talk to inhibit apoptotic pathways in STZ-induced type I diabetes. Am. J. Physiol. Endocrinol. Metab. 2017, 312, E190–E203. [Google Scholar] [CrossRef] [PubMed]
- Lin, C.; Guo, Y.; Xia, Y.; Li, C.; Xu, X.; Qi, T.; Zhang, F.; Fan, M.; Hu, G.; Zhao, H.; et al. FNDC5/Irisin attenuates diabetic cardiomyopathy in a type 2 diabetes mouse model by activation of integrin αV/β5-AKT signaling and reduction of oxidative/nitrosative stress. J. Mol. Cell. Cardiol. 2021, 160, 27–41. [Google Scholar] [CrossRef] [PubMed]
- Tavares, A.M.; Silva, J.H.; de Oliveira Bensusan, C.; Ferreira, A.C.F.; de Lima Matos, L.P.; de Araujo e Souza, K.L.; de Carvalho Cardoso-Weide, L.; Taboada, G.F. Altered superoxide dismutase-1 activity and intercellular adhesion molecule 1 (ICAM-1) levels in patients with type 2 diabetes mellitus. PLoS ONE 2019, 14, e0216256. [Google Scholar] [CrossRef]
- Papanas, N.; Tziakas, D.; Chalikias, G.; Floros, D.; Trypsianis, G.; Papadopoulou, E.; Kortsaris, A.; Symeonidis, G.; Souliou, E.; Maltezos, E.; et al. Gliclazide treatment lowers serum ICAM-1 levels in poorly controlled type 2 diabetic patients. Diabetes Metab. 2006, 32, 344–349. [Google Scholar] [CrossRef]
- Chen, F.; Song, J. Cardioprotective Action of Glycyrrhizin on Diabetic Rats with Myocardial Remodeling. J. Healthc. Eng. 2021, 2021, 6343677. [Google Scholar] [CrossRef] [PubMed]
- Mishra, V.; Nayak, P.; Sharma, M.; Albutti, A.; Alwashmi, A.S.S.; Aljasir, M.A.; Alsowayeh, N.; Tambuwala, M.M. Emerging Treatment Strategies for Diabetes Mellitus and Associated Complications: An Update. Pharmaceutics 2021, 13, 1568. [Google Scholar] [CrossRef] [PubMed]
- Naji, A.; Eitoku, M.; Favier, B.; Deschaseaux, F.; Rouas-Freiss, N.; Suganuma, N. Biological functions of mesenchymal stem cells and clinical implications. Cell. Mol. Life Sci. 2019, 76, 3323–3348. [Google Scholar] [CrossRef] [PubMed]
- Ocansey, D.K.W.; Pei, B.; Yan, Y.; Qian, H.; Zhang, X.; Xu, W.; Mao, F. Improved therapeutics of modified mesenchymal stem cells: An update. J. Transl. Med. 2020, 18, 42. [Google Scholar] [CrossRef] [PubMed]
- Tan, Y.; Zhang, Z.; Zheng, C.; Wintergerst, K.A.; Keller, B.B.; Cai, L. Mechanisms of diabetic cardiomyopathy and potential therapeutic strategies: Preclinical and clinical evidence. Nat. Rev. Cardiol. 2020, 17, 585–607. [Google Scholar] [CrossRef] [PubMed]
- Li, K.; Zhai, M.; Jiang, L.; Song, F.; Zhang, B.; Li, J.; Li, H.; Li, B.; Xia, L.; Xu, L.; et al. Tetrahydrocurcumin Ameliorates Diabetic Cardiomyopathy by Attenuating High Glucose-Induced Oxidative Stress and Fibrosis via Activating the SIRT1 Pathway. Oxid. Med. Cell. Longev. 2019, 2019, 6746907. [Google Scholar] [CrossRef]
- Li, L.; Zhao, Q.; Kong, W. Extracellular matrix remodeling and cardiac fibrosis. Matrix Biol. 2018, 68–69, 490–506. [Google Scholar] [CrossRef]
- Jia, G.; Hill, M.A.; Sowers, J.R. Diabetic Cardiomyopathy: An Update of Mechanisms Contributing to This Clinical Entity. Circ. Res. 2018, 122, 624–638. [Google Scholar] [CrossRef]
- Ren, J.; Bi, Y.; Sowers, J.R.; Hetz, C.; Zhang, Y. Endoplasmic reticulum stress and unfolded protein response in cardiovascular diseases. Nat. Rev. Cardiol. 2021, 18, 499–521. [Google Scholar] [CrossRef]
- Tao, A.; Song, J.; Lan, T.; Xu, X.; Kvietys, P.; Kao, R.; Martin, C.; Rui, T. Cardiomyocyte-fibroblast interaction contributes to diabetic cardiomyopathy in mice: Role of HMGB1/TLR4/IL-33 axis. Biochim. Biophys. Acta 2015, 1852, 2075–2085. [Google Scholar] [CrossRef]
- Dozio, E.; Massaccesi, L.; Corsi Romanelli, M.M. Glycation and Glycosylation in Cardiovascular Remodeling: Focus on Advanced Glycation End Products and O-Linked Glycosylations as Glucose-Related Pathogenetic Factors and Disease Markers. J. Clin. Med. 2021, 10, 4792. [Google Scholar] [CrossRef] [PubMed]
- Russo, I.; Frangogiannis, N.G. Diabetes-associated cardiac fibrosis: Cellular effectors, molecular mechanisms and therapeutic opportunities. J. Mol. Cell. Cardiol. 2016, 90, 84–93. [Google Scholar] [CrossRef] [PubMed]
- Katare, R.; Oikawa, A.; Cesselli, D.; Beltrami, A.P.; Avolio, E.; Muthukrishnan, D.; Munasinghe, P.E.; Angelini, G.; Emanueli, C.; Madeddu, P. Boosting the pentose phosphate pathway restores cardiac progenitor cell availability in diabetes. Cardiovasc. Res. 2013, 97, 55–65. [Google Scholar] [CrossRef] [PubMed]
- Leonardini, A.; Avogaro, A. Abnormalities of the cardiac stem and progenitor cell compartment in experimental and human diabetes. Arch. Physiol. Biochem. 2013, 119, 179–187. [Google Scholar] [CrossRef]
- Feng, W.; Lei, T.; Wang, Y.; Feng, R.; Yuan, J.; Shen, X.; Wu, Y.; Gao, J.; Ding, W.; Lu, Z. GCN2 deficiency ameliorates cardiac dysfunction in diabetic mice by reducing lipotoxicity and oxidative stress. Free Radic. Biol. Med. 2019, 130, 128–139. [Google Scholar] [CrossRef]
- Li, C.; Zhang, J.; Xue, M.; Li, X.; Han, F.; Liu, X.; Xu, L.; Lu, Y.; Cheng, Y.; Li, T.; et al. SGLT2 inhibition with empagliflozin attenuates myocardial oxidative stress and fibrosis in diabetic mice heart. Cardiovasc. Diabetol. 2019, 18, 15. [Google Scholar] [CrossRef]
- Zhu, H.Z.; Zhang, L.Y.; Zhai, M.E.; Xia, L.; Cao, Y.; Xu, L.; Li, K.F.; Jiang, L.Q.; Shi, H.; Li, X.; et al. GDF11 Alleviates Pathological Myocardial Remodeling in Diabetic Cardiomyopathy Through SIRT1-Dependent Regulation of Oxidative Stress and Apoptosis. Front. Cell Dev. Biol. 2021, 9, 686848. [Google Scholar] [CrossRef]
- Bindu, S.; Pillai, V.B.; Gupta, M.P. Role of Sirtuins in Regulating Pathophysiology of the Heart. Trends Endocrinol. Metab. 2016, 27, 563–573. [Google Scholar] [CrossRef]
- Han, Y.; Sun, W.; Ren, D.; Zhang, J.; He, Z.; Fedorova, J.; Sun, X.; Han, F.; Li, J. SIRT1 agonism modulates cardiac NLRP3 inflammasome through pyruvate dehydrogenase during ischemia and reperfusion. Redox Biol. 2020, 34, 101538. [Google Scholar] [CrossRef]
- Ren, B.; Zhang, Y.; Liu, S.; Cheng, X.; Yang, X.; Cui, X.; Zhao, X.; Zhao, H.; Hao, M.; Li, M.; et al. Curcumin alleviates oxidative stress and inhibits apoptosis in diabetic cardiomyopathy via Sirt1-Foxo1 and PI3K-Akt signalling pathways. J. Cell. Mol. Med. 2020, 24, 12355–12367. [Google Scholar] [CrossRef]
- Waldman, M.; Nudelman, V.; Shainberg, A.; Zemel, R.; Kornwoski, R.; Aravot, D.; Peterson, S.J.; Arad, M.; Hochhauser, E. The Role of Heme Oxygenase 1 in the Protective Effect of Caloric Restriction against Diabetic Cardiomyopathy. Int. J. Mol. Sci. 2019, 20, 2427. [Google Scholar] [CrossRef]
- Liu, J.-J.; Wong, M.D.S.; Toy, W.C.; Tan, C.S.H.; Liu, S.; Ng, X.W.; Tavintharan, S.; Sum, C.F.; Lim, S.C. Lower circulating irisin is associated with type 2 diabetes mellitus. J. Diabetes Complicat. 2013, 27, 365–369. [Google Scholar] [CrossRef]
- Shen, C.-Y.; Lu, C.-H.; Wu, C.-H.; Li, K.-J.; Kuo, Y.-M.; Hsieh, S.-C.; Yu, C.-L. The Development of Maillard Reaction, and Advanced Glycation End Product (AGE)-Receptor for AGE (RAGE) Signaling Inhibitors as Novel Therapeutic Strategies for Patients with AGE-Related Diseases. Molecules 2020, 25, 5591. [Google Scholar] [CrossRef]
- Zhao, J.; Randive, R.; Stewart, J.A. Molecular mechanisms of AGE/RAGE-mediated fibrosis in the diabetic heart. World J. Diabetes 2014, 5, 860–867. [Google Scholar] [CrossRef] [PubMed]
- Bansode, S.B.; Gacche, R.N. Glycation-induced modification of tissue-specific ECM proteins: A pathophysiological mechanism in degenerative diseases. Biochim. Biophys. Acta Gen. Subj. 2019, 1863, 129411. [Google Scholar] [CrossRef] [PubMed]
- Monroe, T.B.; Anderson, E.J. A Catecholaldehyde Metabolite of Norepinephrine Induces Myo fi broblast Activation and Toxicity via the Receptor for Advanced Glycation Endproducts: Mitigating Role of l-Carnosine. Chem. Res. Toxicol. 2021, 34, 2194–2201. [Google Scholar] [CrossRef]
- Fowlkes, V.; Clark, J.; Fix, C.; Law, B.A.; Morales, M.O.; Qiao, X.; Ako-Asare, K.; Goldsmith, J.G.; Carver, W.; Murray, D.B.; et al. Type II diabetes promotes a myofibroblast phenotype in cardiac fibroblasts. Life Sci. 2013, 92, 669–676. [Google Scholar] [CrossRef] [PubMed]
- Burr, S.D.; Stewart, J.A. Extracellular matrix components isolated from diabetic mice alter cardiac fibroblast function through the AGE/RAGE signaling cascade. Life Sci. 2020, 250, 117569. [Google Scholar] [CrossRef] [PubMed]
- Huang, S.; Wang, W.; Li, L.; Wang, T.; Zhao, Y.; Lin, Y.; Huang, W.; Wang, Y.; Huang, Z. P2X7 Receptor Deficiency Ameliorates STZ-induced Cardiac Damage and Remodeling through PKCβ and ERK. Front. Cell Dev. Biol. 2021, 9, 692028. [Google Scholar] [CrossRef]
- Burnstock, G. Purinergic Signaling in the Cardiovascular System. Circ. Res. 2017, 120, 207–228. [Google Scholar] [CrossRef]
- Sathanoori, R.; Swärd, K.; Olde, B.; Erlinge, D. The ATP Receptors P2X7 and P2X4 Modulate High Glucose and Palmitate-Induced Inflammatory Responses in Endothelial Cells. PLoS ONE 2015, 10, e0125111. [Google Scholar] [CrossRef]
- Martinez, C.G.; Zamith-Miranda, D.; da Silva, M.G.; Ribeiro, K.C.; Brandão, I.T.; Silva, C.L.; Diaz, B.L.; Bellio, M.; Persechini, P.M.; Kurtenbach, E. P2×7 purinergic signaling in dilated cardiomyopathy induced by auto-immunity against muscarinic M2 receptors: Autoantibody levels, heart functionality and cytokine expression. Sci. Rep. 2015, 5, srep16940. [Google Scholar] [CrossRef]
- Wenzl, F.A.; Ambrosini, S.; Mohammed, S.A.; Kraler, S.; Lüscher, T.F.; Costantino, S.; Paneni, F. Inflammation in Metabolic Cardiomyopathy. Front. Cardiovasc. Med. 2021, 8, 742178. [Google Scholar] [CrossRef] [PubMed]
- Mishra, S.; Kass, D.A. Cellular and molecular pathobiology of heart failure with preserved ejection fraction. Nat. Rev. Cardiol. 2021, 18, 400–423. [Google Scholar] [CrossRef] [PubMed]
- Nishida, K.; Otsu, K. Inflammation and metabolic cardiomyopathy. Cardiovasc. Res. 2017, 113, 389–398. [Google Scholar] [CrossRef] [PubMed]
- Frati, G.; Schirone, L.; Chimenti, I.; Yee, D.; Biondi-Zoccai, G.; Volpe, M.; Sciarretta, S. An overview of the inflammatory signalling mechanisms in the myocardium underlying the development of diabetic cardiomyopathy. Cardiovasc. Res. 2017, 113, 378–388. [Google Scholar] [CrossRef]
- Urbina, P.; Singla, D.K. BMP-7 attenuates adverse cardiac remodeling mediated through M2 macrophages in prediabetic cardiomyopathy. Am. J. Physiol. Heart Circ. Physiol. 2014, 307, H762–H772. [Google Scholar] [CrossRef]
- Abd El Motteleb, D.M.; Abd El Aleem, D.I. Renoprotective effect of Hypericum perforatum against diabetic nephropathy in rats: Insights in the underlying mechanisms. Clin. Exp. Pharmacol. Physiol. 2017, 44, 509–521. [Google Scholar] [CrossRef]
- Volz, H.C.; Kaya, Z.; Katus, H.A.; Andrassy, M. The role of HMGB1/RAGE in inflammatory cardiomyopathy. Semin. Thromb. Hemost. 2010, 36, 185–194. [Google Scholar] [CrossRef]
- Jin, X.; Yao, T.; Zhou, Z.; Zhu, J.; Zhang, S.; Hu, W.; Shen, C. Advanced Glycation End Products Enhance Macrophages Polarization into M1 Phenotype through Activating RAGE/NF-κB Pathway. Biomed. Res. Int. 2015, 2015, 732450. [Google Scholar] [CrossRef]
- Mouton, A.J.; Li, X.; Hall, M.E.; Hall, J.E. Obesity, Hypertension, and Cardiac Dysfunction: Novel Roles of Immunometabolism in Macrophage Activation and Inflammation. Circ. Res. 2020, 126, 789–806. [Google Scholar] [CrossRef] [PubMed]
- Zayas, J.; Amaia, A.; Ainhoa, A.; De Yurre, R.; Echeazarra, L.; Fernández, V. Kv1. 3 Channel Blockade Improves Inflammatory Profile, Reduces Cardiac Electrical Remodeling, and Prevents Arrhythmia in Type 2 Diabetic Rats. Cardiovasc. Drugs Ther. 2021, 2021, 1–11. [Google Scholar] [CrossRef]
- Zayas-Arrabal, J.; Alquiza, A.; Tuncay, E.; Turan, B.; Gallego, M.; Casis, O. Molecular and electrophysiological role of diabetes-associated circulating inflammatory factors in cardiac arrhythmia remodeling in a metabolic-induced model of type 2 diabetic rat. Int. J. Mol. Sci. 2021, 22, 6827. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Q.; Liu, T.; Ng, C.Y.; Li, G. Diabetes Mellitus and Atrial Remodeling: Mechanisms and Potential Upstream Therapies. Cardiovasc. Ther. 2014, 32, 233–241. [Google Scholar] [CrossRef] [PubMed]
- Xiao, L.; Dudley, A.C. Fine-tuning vascular fate during endothelial-mesenchymal transition. J. Pathol. 2017, 241, 25–35. [Google Scholar] [CrossRef]
- Yue, Y.; Meng, K.; Pu, Y.; Zhang, X. Transforming growth factor beta (TGF-β) mediates cardiac fibrosis and induces diabetic cardiomyopathy. Diabetes Res. Clin. Pract. 2017, 133, 124–130. [Google Scholar] [CrossRef]
- Kovacic, J.C. The Endothelial-Metabolic Axis: A Novel Cardiometabolic Disease Target. Trends Endocrinol. Metab. 2018, 29, 527–529. [Google Scholar] [CrossRef]
- Okayama, K.; Azuma, J.; Dosaka, N.; Iekushi, K.; Sanada, F.; Kusunoki, H.; Iwabayashi, M.; Rakugi, H.; Taniyama, Y.; Morishita, R. Hepatocyte growth factor reduces cardiac fibrosis by inhibiting endothelial-mesenchymal transition. Hypertens 2012, 59, 958–965. [Google Scholar] [CrossRef]
- Tian, J.; Zhang, M.; Suo, M.; Liu, D.; Wang, X.; Liu, M.; Pan, J.; Jin, T.; An, F. Dapagliflozin alleviates cardiac fibrosis through suppressing EndMT and fibroblast activation via AMPKα/TGF-β/Smad signalling in type 2 diabetic rats. J. Cell. Mol. Med. 2021, 25, 7642–7659. [Google Scholar] [CrossRef]
- Hu, X.; Bai, T.; Xu, Z.; Liu, Q.; Zheng, Y.; Cai, L. Pathophysiological Fundamentals of Diabetic Cardiomyopathy. Compr. Physiol. 2017, 7, 693–711. [Google Scholar] [CrossRef]
- Salvatore, T.; Pafundi, P.C.; Galiero, R.; Albanese, G.; Di Martino, A.; Caturano, A.; Vetrano, E.; Rinaldi, L.; Sasso, F.C. The Diabetic Cardiomyopathy: The Contributing Pathophysiological Mechanisms. Front. Med. 2021, 8, 695792. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Wang, S.; Wang, J.; Xiao, M.; Guo, Y.; Tang, Y.; Zhang, J.; Gu, J. Epigenetic Regulation Associated with Sirtuin 1 in Complications of Diabetes Mellitus. Front. Endocrinol. (Lausanne) 2021, 11, 598012. [Google Scholar] [CrossRef] [PubMed]
- Fernandez-Twinn, D.S.; Hjort, L.; Novakovic, B.; Ozanne, S.E.; Saffery, R. Intrauterine programming of obesity and type 2 diabetes. Diabetologia 2019, 62, 1789–1801. [Google Scholar] [CrossRef] [PubMed]
- Mönkemann, H.; De Vriese, A.S.; Blom, H.J.; Kluijtmans, L.A.J.; Heil, S.G.; Schild, H.H.; Golubnitschaja, O. Early molecular events in the development of the diabetic cardiomyopathy. Amino Acids 2002, 23, 331–336. [Google Scholar] [CrossRef] [PubMed]
- Tao, H.; Shi, P.; Xuan, H.-Y.; Ding, X.-S. DNA methyltransferase-1 inactivation of androgen receptor axis triggers homocysteine induced cardiac fibroblast autophagy in diabetic cardiac fibrosis. Arch. Biochem. Biophys. 2020, 692, 108521. [Google Scholar] [CrossRef] [PubMed]
- Ghosh, A.K. p300 in Cardiac Development and Accelerated Cardiac Aging. Aging Dis. 2020, 11, 916. [Google Scholar] [CrossRef]
- Bugyei-Twum, A.; Advani, A.; Advani, S.L.; Zhang, Y.; Thai, K.; Kelly, D.J.; Connelly, K.A. High glucose induces Smad activation via the transcriptional coregulator p300 and contributes to cardiac fibrosis and hypertrophy. Cardiovasc. Diabetol. 2014, 13, 89. [Google Scholar] [CrossRef]
- Nicholls, S.J.; Schwartz, G.G.; Buhr, K.A.; Ginsberg, H.N.; Johansson, J.O.; Kalantar-Zadeh, K.; Kulikowski, E.; Toth, P.P.; Wong, N.; Sweeney, M.; et al. Apabetalone and hospitalization for heart failure in patients following an acute coronary syndrome: A prespecified analysis of the BETonMACE study. Cardiovasc. Diabetol. 2021, 20, 13. [Google Scholar] [CrossRef]
- Kalantar-Zadeh, K.; Schwartz, G.G.; Nicholls, S.J.; Buhr, K.A.; Ginsberg, H.N.; Johansson, J.O.; Kulikowski, E.; Lebioda, K.; Toth, P.P.; Wong, N.; et al. Effect of Apabetalone on Cardiovascular Events in Diabetes, CKD, and Recent Acute Coronary Syndrome. Clin. J. Am. Soc. Nephrol. 2021, 16, 705–716. [Google Scholar] [CrossRef]
- Mu, J.; Zhang, D.; Tian, Y.; Xie, Z.; Zou, M. BRD4 inhibition by JQ1 prevents high-fat diet-induced diabetic cardiomyopathy by activating PINK1/Parkin-mediated mitophagy in vivo. J. Mol. Cell. Cardiol. 2020, 149, 1–14. [Google Scholar] [CrossRef]
- Wang, Q.; Sun, Y.; Li, T.; Liu, L.; Zhao, Y.; Li, L.; Zhang, L.; Meng, Y. Function of BRD4 in the pathogenesis of high glucose-induced cardiac hypertrophy. Mol. Med. Rep. 2018, 16, 705–716. [Google Scholar] [CrossRef] [PubMed]
- Palomer, X.; Román-Azcona, M.S.; Pizarro-Delgado, J.; Planavila, A.; Villarroya, F.; Valenzuela-Alcaraz, B.; Crispi, F.; Sepúlveda-Martínez, Á.; Miguel-Escalada, I.; Ferrer, J.; et al. SIRT3-mediated inhibition of FOS through histone H3 deacetylation prevents cardiac fibrosis and inflammation. Signal Transduct. Target. Ther. 2020, 5, 14. [Google Scholar] [CrossRef] [PubMed]
- Ma, W.; Guo, W.; Shang, F.; Li, Y.; Li, W.; Liu, J.; Ma, C.; Teng, J. Bakuchiol Alleviates Hyperglycemia-Induced Diabetic Cardiomyopathy by Reducing Myocardial Oxidative Stress via Activating the SIRT1/Nrf2 Signaling Pathway. Oxid. Med. Cell. Longev. 2020, 2020, 3732718. [Google Scholar] [CrossRef] [PubMed]
- Costantino, S.; Mohammed, S.A.; Ambrosini, S.; Paneni, F. Epigenetic processing in cardiometabolic disease. Atherosclerosis 2019, 281, 150–158. [Google Scholar] [CrossRef] [PubMed]
- Yu, W.; Gao, B.; Li, N.; Wang, J.; Qiu, C.; Zhang, G.; Liu, M.; Zhang, R.; Li, C.; Ji, G.; et al. Sirt3 deficiency exacerbates diabetic cardiac dysfunction: Role of Foxo3A-Parkin-mediated mitophagy. Biochim. Biophys. Acta Mol. Basis Dis. 2017, 1863, 1973–1983. [Google Scholar] [CrossRef] [PubMed]
- Bagul, P.K.; Deepthi, N.; Sultana, R.; Banerjee, S.K. Resveratrol ameliorates cardiac oxidative stress in diabetes through deacetylation of NFkB-p65 and histone 3. J. Nutr. Biochem. 2015, 26, 1298–1307. [Google Scholar] [CrossRef] [PubMed]
- Wang, B.; Yang, Q.; Sun, Y.; Xing, Y.; Wang, Y.; Lu, X.; Bai, W.; Liu, X.; Zhao, Y. Resveratrol-enhanced autophagic flux ameliorates myocardial oxidative stress injury in diabetic mice. J. Cell. Mol. Med. 2014, 18, 1599–1611. [Google Scholar] [CrossRef]
- Olaniyi, K.S.; Amusa, O.A.; Areola, E.D.; Olatunji, L.A. Suppression of HDAC by sodium acetate rectifies cardiac metabolic disturbance in streptozotocin–nicotinamide-induced diabetic rats. Exp. Biol. Med. 2020, 245, 667–676. [Google Scholar] [CrossRef]
- Malek, V.; Sharma, N.; Gaikwad, A.B. Histone Acetylation Regulates Natriuretic Peptides and Neprilysin Gene Expressions in Diabetic Cardiomyopathy and Nephropathy. Curr. Mol. Pharmacol. 2019, 12, 61–71. [Google Scholar] [CrossRef]
- Xu, Z.; Tong, Q.; Zhang, Z.; Wang, S.; Zheng, Y.; Liu, Q.; Qian, L.; Chen, S.; Sun, J.; Cai, L. Inhibition of HDAC3 prevents diabetic cardiomyopathy in OVE26 mice via epigenetic regulation of DUSP5-ERK1/2 pathway. Clin. Sci. 2017, 131, 1841–1857. [Google Scholar] [CrossRef]
- Chen, Y.; Du, J.; Zhao, Y.T.; Zhang, L.; Lv, G.; Zhuang, S.; Qin, G.; Zhao, T.C. Histone deacetylase (HDAC) inhibition improves myocardial function and prevents cardiac remodeling in diabetic mice. Cardiovasc. Diabetol. 2015, 14, 99. [Google Scholar] [CrossRef] [PubMed]
- Gaikwad, A.B.; Gupta, J.; Tikoo, K. Epigenetic changes and alteration of Fbn1 and Col3A1 gene expression under hyperglycaemic and hyperinsulinaemic conditions. Biochem. J. 2010, 432, 333–341. [Google Scholar] [CrossRef] [PubMed]
- Yu, X.-Y.; Geng, Y.-J.; Liang, J.-L.; Lin, Q.-X.; Lin, S.-G.; Zhang, S.; Li, Y. High levels of glucose induce apoptosis in cardiomyocyte via epigenetic regulation of the insulin-like growth factor receptor. Exp. Cell Res. 2010, 316, 2903–2909. [Google Scholar] [CrossRef] [PubMed]
- Thakur, V.; Alcoreza, N.; Cazares, J.; Chattopadhyay, M. Changes in Stress-Mediated Markers in a Human Cardiomyocyte Cell Line under Hyperglycemia. Int. J. Mol. Sci. 2021, 22, 10802. [Google Scholar] [CrossRef]
- Shepherd, D.L.; Hathaway, Q.A.; Nichols, C.E.; Durr, A.J.; Pinti, M.V.; Hughes, K.M.; Kunovac, A.; Stine, S.M.; Hollander, J.M. Mitochondrial proteome disruption in the diabetic heart through targeted epigenetic regulation at the mitochondrial heat shock protein 70 (mtHsp70) nuclear locus. J. Mol. Cell. Cardiol. 2018, 119, 104–115. [Google Scholar] [CrossRef]
- Yu, X.-Y.; Geng, Y.-J.; Liang, J.-L.; Zhang, S.; Lei, H.-P.; Zhong, S.-L.; Lin, Q.-X.; Shan, Z.-X.; Lin, S.-G.; Li, Y. High levels of glucose induce “metabolic memory” in cardiomyocyte via epigenetic histone H3 lysine 9 methylation. Mol. Biol. Rep. 2012, 39, 8891–8898. [Google Scholar] [CrossRef]
- Guo, Y.; Feng, X.; Wang, D.; Kang, X.; Zhang, L.; Ren, H.; Yuan, G. Long Non-coding RNA: A Key Regulator in the Pathogenesis of Diabetic Cardiomyopathy. Front. Cardiovasc. Med. 2021, 8, 8891–8898. [Google Scholar] [CrossRef]
- Jakubik, D.; Fitas, A.; Eyileten, C.; Jarosz-Popek, J.; Nowak, A.; Czajka, P.; Wicik, Z.; Sourij, H.; Siller-Matula, J.M.; De Rosa, S.; et al. MicroRNAs and long non-coding RNAs in the pathophysiological processes of diabetic cardiomyopathy: Emerging biomarkers and potential therapeutics. Cardiovasc. Diabetol. 2021, 20, 55. [Google Scholar] [CrossRef]
- Rai, A.K.; Lee, B.; Gomez, R.; Rajendran, D.; Khan, M.; Garikipati, V.N.S. Current Status and Potential Therapeutic Strategies for Using Non-coding RNA to Treat Diabetic Cardiomyopathy. Front. Physiol. 2021, 11, 55. [Google Scholar] [CrossRef]
- Ahmed, U.; Khaliq, S.; Ahmad, H.U.; Ahmad, I.; Ashfaq, U.A.; Qasim, M.; Masoud, M.S. Pathogenesis of Diabetic Cardiomyopathy and Role of miRNA. Crit. Rev. Eukaryot. Gene Expr. 2021, 31, 79–92. [Google Scholar] [CrossRef]
- Hussain, S.; Khan, A.W.; Akhmedov, A.; Suades, R.; Costantino, S.; Paneni, F.; Caidahl, K.; Mohammed, S.A.; Hage, C.; Gkolfos, C.; et al. Hyperglycemia Induces Myocardial Dysfunction via Epigenetic Regulation of JunD. Circ. Res. 2020, 127, 1261–1273. [Google Scholar] [CrossRef] [PubMed]
- Costantino, S.; Paneni, F.; Mitchell, K.; Mohammed, S.A.; Hussain, S.; Gkolfos, C.; Berrino, L.; Volpe, M.; Schwarzwald, C.; Lüscher, T.F.; et al. Hyperglycaemia-induced epigenetic changes drive persistent cardiac dysfunction via the adaptor p66 Shc. Int. J. Cardiol. 2018, 268, 179–186. [Google Scholar] [CrossRef] [PubMed]
- Vecellio, M.; Spallotta, F.; Nanni, S.; Colussi, C.; Cencioni, C.; Derlet, A.; Bassetti, B.; Tilenni, M.; Carena, M.C.; Farsetti, A.; et al. The Histone Acetylase Activator Pentadecylidenemalonate 1b Rescues Proliferation and Differentiation in the Human Cardiac Mesenchymal Cells of Type 2 Diabetic Patients. Diabetes 2014, 63, 2132–2147. [Google Scholar] [CrossRef]
- Friedenstein, A.J.; Chailakhjan, R.K.; Lalykina, K.S. The Development of Fibroblast Colonies in Monolayer Cultures of Guinea-Pig Bone Marrow and Spleen Cells. Cell Prolif. 1970, 3, 393–403. [Google Scholar] [CrossRef]
- Kabat, M.; Bobkov, I.; Kumar, S.; Grumet, M. Trends in mesenchymal stem cell clinical trials 2004-2018: Is efficacy optimal in a narrow dose range? Stem Cells Transl. Med. 2020, 9, 17–27. [Google Scholar] [CrossRef]
- Dominici, M.; Le Blanc, K.; Mueller, I.; Slaper-Cortenbach, I.; Marini, F.; Krause, D.S.; Deans, R.J.; Keating, A.; Prockop, D.J.; Horwitz, E.M. Minimal criteria for defining multipotent mesenchymal stromal cells. The International Society for Cellular Therapy position statement. Cytotherapy 2006, 8, 315–317. [Google Scholar] [CrossRef] [PubMed]
- Harrell, C.R.; Jankovic, M.G.; Fellabaum, C.; Volarevic, A.; Djonov, V.; Arsenijevic, A.; Volarevic, V. Molecular Mechanisms Responsible for Anti-Inflammatory and Immunosuppressive Effects of Mesenchymal Stem Cell-Derived Factors. Cells 2019, 8, 1605. [Google Scholar] [CrossRef]
- Ranjbaran, H.; Abediankenari, S.; Khalilian, A.; Rahmani, Z.; Momeninezhad Amiri, M.; Hosseini Khah, Z. Differentiation of Wharton’s Jelly Derived Mesenchymal Stem Cells into Insulin Producing Cells. Int. J. Hematol. Stem Cell Res. 2018, 12, 220–229. [Google Scholar]
- Xin, Y.; Jiang, X.; Wang, Y.; Su, X.; Sun, M.; Zhang, L.; Tan, Y.; Wintergerst, K.A.; Li, Y.; Li, Y. Insulin-Producing Cells Differentiated from Human Bone Marrow Mesenchymal Stem Cells In Vitro Ameliorate Streptozotocin-Induced Diabetic Hyperglycemia. PLoS ONE 2016, 11, e0145838. [Google Scholar] [CrossRef]
- Wartchow, K.M.; Rodrigues, L.; Suardi, L.Z.; Federhen, B.C.; Selistre, N.G.; Gonçalves, C.-A.; Sesterheim, P. Short-Term Protocols to Obtain Insulin-Producing Cells from Rat Adipose Tissue: Signaling Pathways and In Vivo Effect. Int. J. Mol. Sci. 2019, 20, 2458. [Google Scholar] [CrossRef]
- Hsiao, C.-Y.; Chen, T.-H.; Huang, B.-S.; Chen, P.-H.; Su, C.-H.; Shyu, J.-F.; Tsai, P.-J. Comparison between the therapeutic effects of differentiated and undifferentiated Wharton’s jelly mesenchymal stem cells in rats with streptozotocin-induced diabetes. World J. Stem Cells 2020, 12, 139–151. [Google Scholar] [CrossRef] [PubMed]
- Huang, Q.; Huang, Y.; Liu, J. Mesenchymal Stem Cells: An Excellent Candidate for the Treatment of Diabetes Mellitus. Int. J. Endocrinol. 2021, 2021, 9938658. [Google Scholar] [CrossRef] [PubMed]
- Gubert, F.; da Silva, J.S.; Vasques, J.F.; de Jesus Gonçalves, R.G.; Martins, R.S.; de Sá, M.P.L.; Mendez-Otero, R.; Zapata-Sudo, G. Mesenchymal Stem Cells Therapies on Fibrotic Heart Diseases. Int. J. Mol. Sci. 2021, 22, 7447. [Google Scholar] [CrossRef] [PubMed]
- Quevedo, H.C.; Hatzistergos, K.E.; Oskouei, B.N.; Feigenbaum, G.S.; Rodriguez, J.E.; Valdes, D.; Pattany, P.M.; Zambrano, J.P.; Hu, Q.; McNiece, I.; et al. Allogeneic mesenchymal stem cells restore cardiac function in chronic ischemic cardiomyopathy via trilineage differentiating capacity. Proc. Natl. Acad. Sci. USA 2009, 106, 14022–14027. [Google Scholar] [CrossRef] [PubMed]
- Gallina, C.; Turinetto, V.; Giachino, C. A New Paradigm in Cardiac Regeneration: The Mesenchymal Stem Cell Secretome. Stem Cells Int. 2015, 2015, 765846. [Google Scholar] [CrossRef]
- Zhang, N.; Li, J.; Luo, R.; Jiang, J.; Wang, J.-A. Bone Marrow Mesenchymal Stem Cells Induce Angiogenesis and Attenuate the Remodeling of Diabetic Cardiomyopathy. Exp. Clin. Endocrinol. Diabetes 2008, 116, 104–111. [Google Scholar] [CrossRef]
- Calligaris, S.D.; Conget, P. Intravenous administration of bone marrow-derived multipotent mesenchymal stromal cells has a neutral effect on obesity-induced diabetic cardiomyopathy. Biol. Res. 2013, 46, 251–255. [Google Scholar] [CrossRef][Green Version]
- Dong, X.; Zhu, F.; Liu, Q.; Zhang, Y.; Wu, J.; Jiang, W.; Zhang, L.; Dong, S. Transplanted bone marrow mesenchymal stem cells protects myocardium by regulating 14-3-3 protein in a rat model of diabetic cardiomyopathy. Int. J. Clin. Exp. Pathol. 2014, 7, 3714–3723. [Google Scholar]
- Lin, Y.; Zhang, F.; Lian, X.-F.; Peng, W.-Q.; Yin, C.-Y. Mesenchymal stem cell-derived exosomes improve diabetes mellitus-induced myocardial injury and fibrosis via inhibition of TGF-β1/Smad2 signaling pathway. Cell. Mol. Biol. (Noisy-le-Grand) 2019, 65, 123–126. [Google Scholar] [CrossRef]
- Ammar, H.I.; Shamseldeen, A.M.; Shoukry, H.S.; Ashour, H.; Kamar, S.S.; Rashed, L.A.; Fadel, M.; Srivastava, A.; Dhingra, S. Metformin impairs homing ability and efficacy of mesenchymal stem cells for cardiac repair in streptozotocin-induced diabetic cardiomyopathy in rats. Am. J. Physiol. Circ. Physiol. 2021, 320, H1290–H1302. [Google Scholar] [CrossRef]
- Pappritz, K.; Dong, F.; Miteva, K.; Kovacs, A.; El-Shafeey, M.; Kerim, B.; O’Flynn, L.; Elliman, S.J.; O’Brien, T.; Hamdani, N.; et al. Impact of Syndecan-2-Selected Mesenchymal Stromal Cells on the Early Onset of Diabetic Cardiomyopathy in Diabetic db/db Mice. Front. Cardiovasc. Med. 2021, 8, 632728. [Google Scholar] [CrossRef] [PubMed]
- Chu, D.-T.; Nguyen Thi Phuong, T.; Tien, N.L.B.; Tran, D.K.; Minh, L.B.; Van Thanh, V.; Gia Anh, P.; Pham, V.H.; Thi Nga, V. Adipose Tissue Stem Cells for Therapy: An Update on the Progress of Isolation, Culture, Storage, and Clinical Application. J. Clin. Med. 2019, 8, 917. [Google Scholar] [CrossRef] [PubMed]
- Jin, L.; Deng, Z.; Zhang, J.; Yang, C.; Liu, J.; Han, W.; Ye, P.; Si, Y.; Chen, G. Mesenchymal stem cells promote type 2 macrophage polarization to ameliorate the myocardial injury caused by diabetic cardiomyopathy. J. Transl. Med. 2019, 17, 251. [Google Scholar] [CrossRef] [PubMed]
- Jin, L.; Zhang, J.; Deng, Z.; Liu, J.; Han, W.; Chen, G.; Si, Y.; Ye, P. Mesenchymal stem cells ameliorate myocardial fibrosis in diabetic cardiomyopathy via the secretion of prostaglandin E2. Stem Cell Res. Ther. 2020, 11, 122. [Google Scholar] [CrossRef]
- Chen, T.; Chuang, S.; Shen, C.; Ho, T.; Chang, R.; Yeh, Y.; Kuo, C.; Mahalakshmi, B.; Kuo, W.; Huang, C. Antioxidant Sirt1/Akt axis expression in resveratrol pretreated adipose-derived stem cells increases regenerative capability in a rat model with cardiomyopathy induced by diabetes mellitus. J. Cell. Physiol. 2021, 236, 4290–4302. [Google Scholar] [CrossRef]
- Xu, X.; Tenth, S.; Tao, L. Adipose-derived mesenchymal stem cells (ADMSCs) protect against hyperglycemia and hyperlipidemia-induced heart failure by inhibiting autophagy-related apoptosis. Res. Sq. 2021, 2021, 1–22. [Google Scholar] [CrossRef]
- Khan, M.; Ali, F.; Mohsin, S.; Akhtar, S.; Mehmood, A.; Choudhery, M.S.; Khan, S.N.; Riazuddin, S. Preconditioning diabetic mesenchymal stem cells with myogenic medium increases their ability to repair diabetic heart. Stem Cell Res. Ther. 2013, 4, 58. [Google Scholar] [CrossRef]
- De Paula, D.R.M.; Capuano, V.; Filho, D.M.; Carneiro, A.C.D.M.; de Oliveira Crema, V.; de Oliveira, L.F.; Rodrigues, A.R.A.; Montano, N.; da Silva, V.J.D. Biological properties of cardiac mesenchymal stem cells in rats with diabetic cardiomyopathy. Life Sci. 2017, 188, 45–52. [Google Scholar] [CrossRef]
- Jin, P.; Zhang, X.; Wu, Y.; Li, L.; Yin, Q.; Zheng, L.; Zhang, H.; Sun, C. Streptozotocin-Induced Diabetic Rat–Derived Bone Marrow Mesenchymal Stem Cells Have Impaired Abilities in Proliferation, Paracrine, Antiapoptosis, and Myogenic Differentiation. Transplant. Proc. 2010, 42, 2745–2752. [Google Scholar] [CrossRef]
- Li, J.H.; Zhang, N.; Wang, J.A. Improved anti-apoptotic and anti-remodeling potency of bone marrow mesenchymal stem cells by anoxic pre-conditioning in diabetic cardiomyopathy. J. Endocrinol. Investig. 2008, 31, 103–110. [Google Scholar] [CrossRef]
- Delucchi, F.; Berni, R.; Frati, C.; Cavalli, S.; Graiani, G.; Sala, R.; Chaponnier, C.; Gabbiani, G.; Calani, L.; Del Rio, D.; et al. Resveratrol Treatment Reduces Cardiac Progenitor Cell Dysfunction and Prevents Morpho-Functional Ventricular Remodeling in Type-1 Diabetic Rats. PLoS ONE 2012, 7, e39836. [Google Scholar] [CrossRef]
- ShamsEldeen, A.M.; Ashour, H.; Shoukry, H.S.; Fadel, M.; Kamar, S.S.; Aabdelbaset, M.; Rashed, L.A.; Ammar, H.I. Combined treatment with systemic resveratrol and resveratrol preconditioned mesenchymal stem cells, maximizes antifibrotic action in diabetic cardiomyopathy. J. Cell. Physiol. 2019, 234, 10942–10963. [Google Scholar] [CrossRef]
- Chen, T.S.; Liou, S.Y.; Lin, H.H.; Hung, M.Y.; Lin, C.C.; Lin, Y.M.; Lin, K.H.; Padma, V.V.; Yao, C.H.; Kuo, W.W.; et al. Oral administration of green tea Epigallocatechin-3-gallate reduces oxidative stress and enhances restoration of cardiac function in diabetic rats receiving autologous transplantation of adipose-derived stem cells. Arch. Physiol. Biochem. 2021, 127, 82–89. [Google Scholar] [CrossRef] [PubMed]
- Meng, K.; Cai, H.; Cai, S.; Hong, Y.; Zhang, X. Adiponectin Modified BMSCs Alleviate Heart Fibrosis via Inhibition TGF-beta1/Smad in Diabetic Rats. Front. Cell Dev. Biol. 2021, 9, 644160. [Google Scholar] [CrossRef] [PubMed]
- Nagamura-Inoue, T. Umbilical cord-derived mesenchymal stem cells: Their advantages and potential clinical utility. World J. Stem Cells 2014, 6, 195. [Google Scholar] [CrossRef] [PubMed]
- Van Linthout, S.; Hamdani, N.; Miteva, K.; Koschel, A.; Müller, I.; Pinzur, L.; Aberman, Z.; Pappritz, K.; Linke, W.A.; Tschöpe, C. Placenta-Derived Adherent Stromal Cells Improve Diabetes Mellitus-Associated Left Ventricular Diastolic Performance. Stem Cells Transl. Med. 2017, 6, 2135–2145. [Google Scholar] [CrossRef] [PubMed]
- Butler, J.; Epstein, S.E.; Greene, S.J.; Quyyumi, A.A.; Sikora, S.; Kim, R.J.; Anderson, A.S.; Wilcox, J.E.; Tankovich, N.I.; Lipinski, M.J.; et al. Intravenous Allogeneic Mesenchymal Stem Cells for Nonischemic Cardiomyopathy. Circ. Res. 2017, 120, 332–340. [Google Scholar] [CrossRef]
- Xiao, W.; Guo, S.; Gao, C.; Dai, G.; Gao, Y.; Li, M.; Wang, X.; Hu, D. A Randomized Comparative Study on the Efficacy of Intracoronary Infusion of Autologous Bone Marrow Mononuclear Cells and Mesenchymal Stem Cells in Patients with Dilated Cardiomyopathy. Int. Heart J. 2017, 58, 238–244. [Google Scholar] [CrossRef]
- Tompkins, B.A.; Rieger, A.C.; Florea, V.; Banerjee, M.N.; Natsumeda, M.; Nigh, E.D.; Landin, A.M.; Rodriguez, G.M.; Hatzistergos, K.E.; Schulman, I.H.; et al. Comparison of Mesenchymal Stem Cell Efficacy in Ischemic Versus Nonischemic Dilated Cardiomyopathy. J. Am. Heart Assoc. 2018, 7, e008460. [Google Scholar] [CrossRef]
- Hare, J.M.; DiFede, D.L.; Rieger, A.C.; Florea, V.; Landin, A.M.; El-Khorazaty, J.; Khan, A.; Mushtaq, M.; Lowery, M.H.; Byrnes, J.J.; et al. Randomized Comparison of Allogeneic Versus Autologous Mesenchymal Stem Cells for Nonischemic Dilated Cardiomyopathy. J. Am. Coll. Cardiol. 2017, 69, 526–537. [Google Scholar] [CrossRef]
- Premer, C.; Wanschel, A.; Porras, V.; Balkan, W.; Legendre-Hyldig, T.; Saltzman, R.G.; Dong, C.; Schulman, I.H.; Hare, J.M. Mesenchymal Stem Cell Secretion of SDF-1α Modulates Endothelial Function in Dilated Cardiomyopathy. Front. Physiol. 2019, 10, 1182. [Google Scholar] [CrossRef] [PubMed]
- Golpanian, S.; El-Khorazaty, J.; Mendizabal, A.; DiFede, D.L.; Suncion, V.Y.; Karantalis, V.; Fishman, J.E.; Ghersin, E.; Balkan, W.; Hare, J.M. Effect of Aging on Human Mesenchymal Stem Cell Therapy in Ischemic Cardiomyopathy Patients. J. Am. Coll. Cardiol. 2015, 65, 125–132. [Google Scholar] [CrossRef] [PubMed]
- Florea, V.; Rieger, A.C.; Natsumeda, M.; Tompkins, B.A.; Banerjee, M.N.; Schulman, I.H.; Premer, C.; Khan, A.; Valasaki, K.; Heidecker, B.; et al. The impact of patient sex on the response to intramyocardial mesenchymal stem cell administration in patients with non-ischaemic dilated cardiomyopathy. Cardiovasc. Res. 2020, 116, 2131–2141. [Google Scholar] [CrossRef] [PubMed]
- Cho, J.; D’Antuono, M.; Glicksman, M.; Wang, J.; Jonklaas, J. A review of clinical trials: Mesenchymal stem cell transplant therapy in type 1 and type 2 diabetes mellitus. Am. J. Stem Cells 2018, 7, 82–93. [Google Scholar] [PubMed]



{kind=link}
{kind=link}
| Agent/Cells | Preconditioning | Model | Effect and/or Mechanism | Ref. |
|---|---|---|---|---|
| BM-MSCs | - | STZ rats | ↓ Cardiac hypertrophy (LV posterior wall thickness and relative wall thickness); | [106] |
| ↑ Myocardial arteriole density; | ||||
| ↑ LV systolic function and FS; | ||||
| ↓ LV collagen content; | ||||
| ↓ Cardiac expression of MMP-9; | ||||
| BM-MSCs | - | HF diet mice | ↑/↓ Cardiac contractility (+dP/dt) and relaxation (-dP/dt); | [107] |
| BM-MSCs | - | STZ+HF/HS diet rats | ↓ Cardiac expression of caspase-3; | [108] |
| ↑ Cardiac expression of 14-3-3, p-Ask1; | ||||
| BM-MSCs | - | STZ rats treated with RSV | ↓ Cardiac apoptosis (Bax/Bcl2 ratio); | [122] |
| ↓ Cardiac expression of Wnt3 and β-catenin; | ||||
| ↓ Cardiomyocyte hypertrophy; | ||||
| ↑ Myocardial capillary density; | ||||
| ↑ Cardiac antioxidant defenses (TAC, SOD); | ||||
| BM-MSCs | - | STZ rats treated with MET | Attenuated reduction in blood glucose; | [110] |
| Attenuated cardiac angiogenesis; | ||||
| Attenuated reduction in LV collagen content; | ||||
| AT-MSCs | - | STZ rats | ↓ LV wall thinning and dilation; | [113] |
| ↓ Diastolic dysfunction; | ||||
| ↓ Cardiac collagen content and fibrosis; | ||||
| ↓ Proliferation of cardiac fibroblasts; | ||||
| ↓ Cardiac expression of IL-6, TNF-α, TGF-β; | ||||
| ↑ Macrophage polarization to M2 phenotype; | ||||
| AT-MSCs | - | STZ+HF diet mice | ↓ Blood glucose and cholesterol; | [116] |
| ↑ LV systolic function (FS and EF); | ||||
| ↓ Cardiomyocyte hypertrophy; | ||||
| ↓ Cardiac collagen content; | ||||
| ↓ Cardiac macrophage number; | ||||
| ↓ Cardiac TNF, CXCL15, IL6 mRNA levels; | ||||
| ↓ Cardiac expression of IL-1β; | ||||
| AT-MSCs (autologous) | - | STZ rats treated with EGCG | ↓ Cardiac expression of TGF-β, MMP-9, p-NFκB, COX-2; | [123] |
| Syndecan-2+ BM-MSCs | - | db/db mice | Attenuated cardiac angiogenesis; | [111] |
| Attenuated reduction in cardiomyocyte stiffness; | ||||
| BM-MSC exosomes | - | STZ rats | ↓ LV collagen content; | [109] |
| ↓ Cardiac TGF-β, Smad2 mRNA levels; | ||||
| BM-MSCs | Anoxia | STZ rats | ↓ Cardiac hypertrophy (heart weight); | [120] |
| ↑ LV systolic function (FS); | ||||
| ↑ Myocardial capillary density; | ||||
| BM-MSCs | RSV | STZ ratstreated with RSV | ↓ Cardiac apoptosis (Bax/Bcl2 ratio); | [121,122] |
| ↓ Cardiac expression of Wnt3, β-catenin and sFRP2; | ||||
| ↓ Cardiac collagen content; | ||||
| ↓ Cardiomyocyte hypertrophy; | ||||
| ↑ Myocardial capillary density; | ||||
| ↑ Cardiac antioxidant defenses (TAC, SOD); | ||||
| AT-MSCs (autologous) | RSV | STZ rats | ↓ Blood glucose; | [115] |
| ↑ LV systolic function (EF and FS); | ||||
| ↑ Cardiac expression of p-IGF1R, p-PI3K, p-Akt, p-AMPK, Sirt1, PGF1α, SOD2; | ||||
| ↓ Cardiac expression of ANP, BNP; | ||||
| ↓ Cardiac expression of p-Bad, Bcl2, caspase-3; | ||||
| ↓ Cardiomyocyte apoptosis (TUNEL); | ||||
| BM-MSCs | Adiponectin overexpression | HG-stimulated H9c2 cells | ↓ Expression of TGF-β, Smad2/3 | [124] |
| BM-MSCs | Adiponectin overexpression | STZ+HF diet rats | ↓ Cardiac hypertrophy; | [124] |
| ↑ LV systolic function (FS); | ||||
| ↓ LV collagen content; | ||||
| ↓ Cardiac expression of TGF-β, Smad2/3; | ||||
| dm-BM-MSCs | Conditioned medium from HG+H2O2-stimulated-primary neonatal rat cardiomyocytes | STZ mice | ↑ LV systolic function (EF, +dP/dt); | [117] |
| ↑ LV diastolic function (LVEDP, -dP/dt); | ||||
| ↑ Cardiac MEF2c, NKX2.5, GATA-4 mRNA levels; | ||||
| ↓ Cardiac NFκB mRNA levels; | ||||
| ↓ Cardiac expression of caspase-3; | ||||
| ↑ Cardiac VEGF, ANG-1 mRNA levels; | ||||
| ↑ Myocardial capillary density; | ||||
| ↓ Cardiac collagen content; | ||||
| PLX | - | STZ mice | ↓ Diastolic dysfunction (-dP/dt, tau); | [126] |
| ↓ Cardiomyocyte stiffness (p-titin); | ||||
| ↑ Cardiac PKA and PKG activities; | ||||
| ↑ Cardiac VEGF mRNA levels | ||||
| ↑ Myocardial arteriole density; | ||||
| ↓ Cardiac IFN-γ and VCAM-1 mRNA levels; | ||||
| ↑ Circulating Treg cells; | ||||
| PLX | - | HG-stimulated cardiac fibroblasts | ↓ Collagen production | [126] |
| ↓ Myofibroblast transdifferentiation (α-SMA) |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
da Silva, J.S.; Gonçalves, R.G.J.; Vasques, J.F.; Rocha, B.S.; Nascimento-Carlos, B.; Montagnoli, T.L.; Mendez-Otero, R.; de Sá, M.P.L.; Zapata-Sudo, G. Mesenchymal Stem Cell Therapy in Diabetic Cardiomyopathy. Cells 2022, 11, 240. https://doi.org/10.3390/cells11020240
da Silva JS, Gonçalves RGJ, Vasques JF, Rocha BS, Nascimento-Carlos B, Montagnoli TL, Mendez-Otero R, de Sá MPL, Zapata-Sudo G. Mesenchymal Stem Cell Therapy in Diabetic Cardiomyopathy. Cells. 2022; 11(2):240. https://doi.org/10.3390/cells11020240
Chicago/Turabian Styleda Silva, Jaqueline S., Renata G. J. Gonçalves, Juliana F. Vasques, Bruna S. Rocha, Bianca Nascimento-Carlos, Tadeu L. Montagnoli, Rosália Mendez-Otero, Mauro P. L. de Sá, and Gisele Zapata-Sudo. 2022. "Mesenchymal Stem Cell Therapy in Diabetic Cardiomyopathy" Cells 11, no. 2: 240. https://doi.org/10.3390/cells11020240
APA Styleda Silva, J. S., Gonçalves, R. G. J., Vasques, J. F., Rocha, B. S., Nascimento-Carlos, B., Montagnoli, T. L., Mendez-Otero, R., de Sá, M. P. L., & Zapata-Sudo, G. (2022). Mesenchymal Stem Cell Therapy in Diabetic Cardiomyopathy. Cells, 11(2), 240. https://doi.org/10.3390/cells11020240