
2-PMAP Ameliorates Cerebral Vasospasm and Brain Injury after Subarachnoid Hemorrhage by Regulating Neuro-Inflammation in Rats

Abstract
:1. Introduction
2. Materials and Methods
2.1. Ethics Statement
2.2. Animal Preparation
2.3. SAH Model
2.4. Neurological Scores
2.5. Tissue Processing
2.6. BA Morphometric Studies
2.7. Immunofluorescence Staining
2.8. Western Blotting Analysis
2.9. Quantitative Real-Time Polymerase Chain Reaction (qRT-PCR)
2.10. Enzyme-Linked Immunosorbent Assay (ELISA)
2.11. Measurement of Reactive Oxygen Species (ROS)
2.12. Statistical Analyses
3. Results
3.1. Mortality and Neurological Deficit in SAH Rats
3.2. 2-PMAP Reduced Vasospasm Severity in SAH Rats
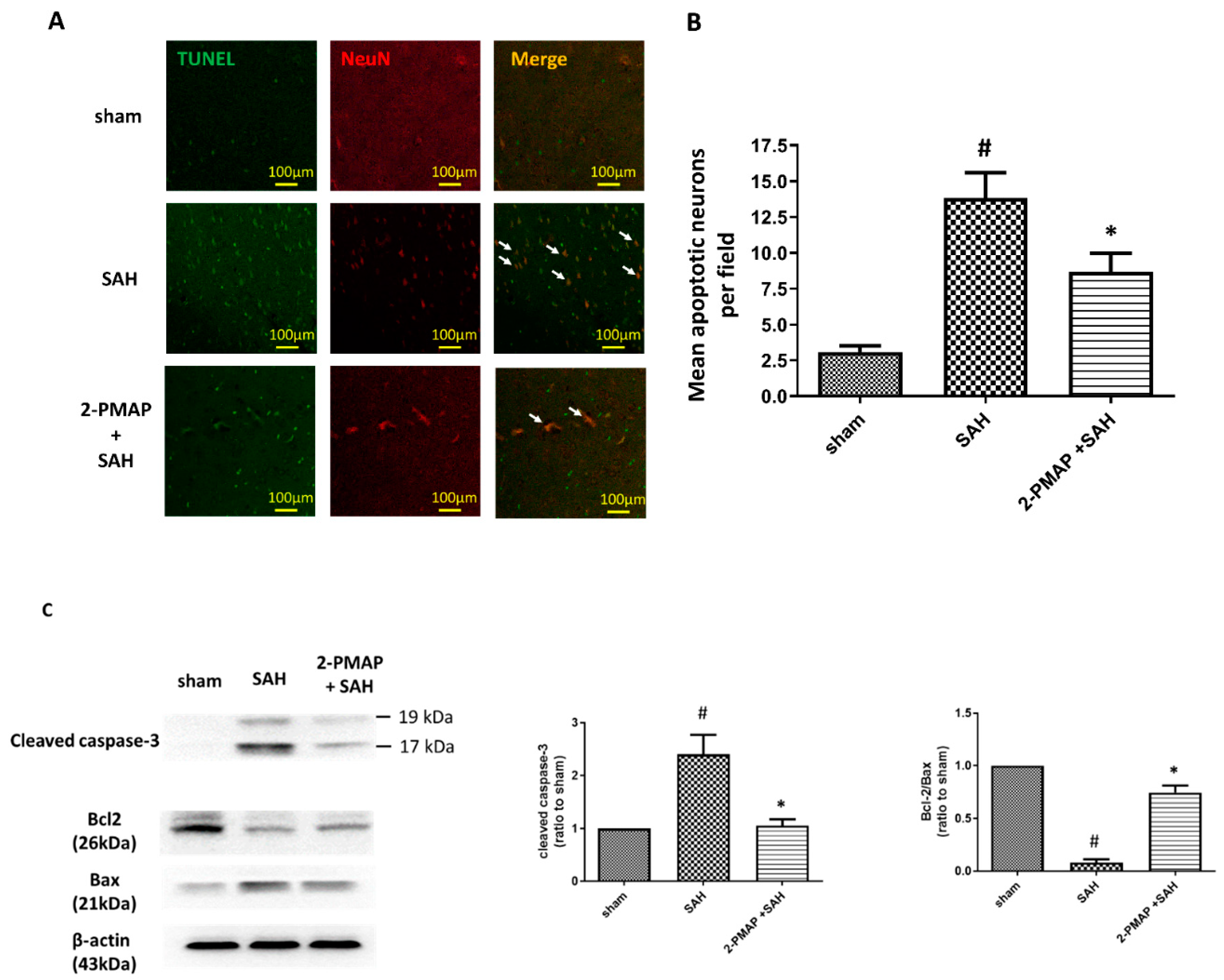
3.3. 2-PMAP Decreased Apoptotic Neurons in SAH Rats
3.4. 2-PMAP Decreased Inflammatory Cytokines in SAH Rats
3.5. 2-PMAP Inhibited Microglia Activation in SAH Rats
3.6. 2-PMAP Decreased Astrocyte Activation in SAH Rats
3.7. 2-PMAP Decreased ROS in SAH Rats
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Macdonald, R.L. Delayed neurological deterioration after subarachnoid haemorrhage. Nat. Rev. Neurol. 2014, 10, 44–58. [Google Scholar] [CrossRef]
- de Rooij, N.K.; Linn, F.H.; van der Plas, J.A.; Algra, A.; Rinkel, G.J. Incidence of subarachnoid haemorrhage: A systematic review with emphasis on region, age, gender and time trends. J. Neurol. Neurosurg. Psychiatry 2007, 78, 1365–1372. [Google Scholar] [CrossRef] [PubMed]
- Goldberg, J.; Schoeni, D.; Mordasini, P.; Z’Graggen, W.; Gralla, J.; Raabe, A.; Beck, J.; Fung, C. Survival and outcome after poor-grade aneurysmal subarachnoid hemorrhage in elderly patients. Stroke 2018, 49, 2883–2889. [Google Scholar] [CrossRef] [PubMed]
- Tseng, M.Y.; Czosnyka, M.; Richards, H.; Pickard, J.D.; Kirkpatrick, P.J. Effects of acute treatment with pravastatin on cerebral vasospasm, autoregulation, and delayed ischemic deficits after aneurysmal subarachnoid hemorrhage: A phase II randomized placebo-controlled trial. Stroke 2005, 36, 1627–1632. [Google Scholar] [CrossRef] [PubMed]
- Kassell, N.F.; Sasaki, T.; Colohan, A.R.; Nazar, G. Cerebral vasospasm following aneurysmal subarachnoid hemorrhage. Stroke. 1985, 16, 562–572. [Google Scholar] [CrossRef] [Green Version]
- Hankey, G.J. Potential new risk factors for ischemic stroke: What is their potential? Stroke 2006, 37, 2181–2188. [Google Scholar] [CrossRef] [Green Version]
- Rinkel, G.J.; Algra, A. Long-term outcomes of patients with aneurysmal subarachnoid haemorrhage. Lancet Neurol. 2011, 10, 349–356. [Google Scholar] [CrossRef]
- Hara, A.; Yoshimi, N.; Mori, H. Evidence for apoptosis in human intracranial aneurysms. Neurol. Res. 1998, 20, 127–130. [Google Scholar] [CrossRef]
- Wu, C.H.; Tsai, Y.C.; Tsai, T.H.; Kuo, K.L.; Su, Y.F.; Chang, C.H.; Lin, C.L. Valproic acid reduces vasospasm through modulation of akt phosphorylation and attenuates neuronal apoptosis in subarachnoid hemorrhage rats. Int. J. Mol. Sci. 2021, 22, 5975. [Google Scholar] [CrossRef]
- Sehba, F.A.; Hou, J.; Pluta, R.M.; Zhang, J.H. The importance of early brain injury after subarachnoid hemorrhage. Prog. Neurobiol. 2012, 97, 14–37. [Google Scholar] [CrossRef] [Green Version]
- Provencio, J.J. Inflammation in subarachnoid hemorrhage and delayed deterioration associated with vasospasm: A review. Acta Neurochir. Suppl. 2013, 115, 233–238. [Google Scholar]
- Aragon, M.J.; Topper, L.; Tyler, C.R.; Sanchez, B.; Zychowski, K.; Young, T.; Herbert, G.; Hall, P.; Erdely, A.; Eye, T.; et al. Serum-borne bioactivity caused by pulmonary multiwalled carbon nanotubes induces neuroinflammation via blood-brain barrier impairment. Proc. Natl. Acad. Sci. USA 2017, 114, E1968–E1976. [Google Scholar] [CrossRef] [Green Version]
- Bulters, D.; Gaastra, B.; Zolnourian, A.; Alexander, S.; Ren, D.; Blackburn, S.L.; Borsody, M.; Dore, S.; Galea, J.; Iihara, K.; et al. Haemoglobin scavenging in intracranial bleeding: Biology and clinical implications. Nat. Rev. Neurol. 2018, 14, 416–443. [Google Scholar] [CrossRef] [PubMed]
- Hyzak, L.; Moos, R.; von Rath, F.; Wulf, V.; Wirtz, M.; Melchior, D.; Kling, H.W.; Kohler, M.; Gab, S.; Schmitz, O.J. Quantitative matrix-assisted laser desorption ionization-time-of-flight mass spectrometry analysis of synthetic polymers and peptides. Anal. Chem. 2011, 83, 9467–9471. [Google Scholar] [CrossRef] [PubMed]
- Pradilla, G.; Chaichana, K.L.; Hoang, S.; Huang, J.; Tamargo, R.J. Inflammation and cerebral vasospasm after subarachnoid hemorrhage. Neurosurg. Clin. N. Am. 2010, 21, 365–379. [Google Scholar] [CrossRef]
- Frosen, J.; Cebral, J.; Robertson, A.M.; Aoki, T. Flow-induced, inflammation-mediated arterial wall remodeling in the formation and progression of intracranial aneurysms. Neurosurg. Focus. 2019, 47, E21. [Google Scholar] [CrossRef] [Green Version]
- Stratoulias, V.; Venero, J.L.; Tremblay, M.E.; Joseph, B. Microglial subtypes: Diversity within the microglial community. EMBO J. 2019, 38, e101997. [Google Scholar] [CrossRef]
- Hanafy, K.A. The role of microglia and the TLR4 pathway in neuronal apoptosis and vasospasm after subarachnoid hemorrhage. J. Neuroinflammation 2013, 10, 83. [Google Scholar] [CrossRef] [Green Version]
- Lawrence, T. The nuclear factor NF-kappaB pathway in inflammation. Cold Spring Harb Perspect Biol. 2009, 1, a001651. [Google Scholar] [CrossRef] [Green Version]
- Sehba, F.A.; Pluta, R.M.; Zhang, J.H. Metamorphosis of subarachnoid hemorrhage research: From delayed vasospasm to early brain injury. Mol. Neurobiol. 2011, 43, 27–40. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Asuni, A.A.; Guridi, M.; Pankiewicz, J.E.; Sanchez, S.; Sadowski, M.J. Modulation of amyloid precursor protein expression reduces beta-amyloid deposition in a mouse model. Ann. Neurol. 2014, 75, 684–699. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Querfurth, H.W.; LaFerla, F.M. Alzheimer’s disease. N. Engl. J. Med. 2010, 362, 329–344. [Google Scholar] [CrossRef] [Green Version]
- Kakinohana, M.; Nakamura, S.; Fuchigami, T.; Davison, K.J.; Marsala, M.; Sugahara, K. Mu and delta, but not kappa, opioid agonists induce spastic paraparesis after a short period of spinal cord ischaemia in rats. Br. J. Anaesth. 2006, 96, 88–94. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shimokawa, H.; Rashid, M. Development of Rho-kinase inhibitors for cardiovascular medicine. Trends Pharmacol. Sci. 2007, 28, 296–302. [Google Scholar] [CrossRef]
- Kamat, P.K.; Ahmad, A.S.; Doré, S. Carbon monoxide attenuates vasospasm and improves neurobehavioral function after subarachnoid hemorrhage. Arch. Biochem. Biophys. 2019, 676, 108117. [Google Scholar] [CrossRef]
- Echigo, R.; Shimohata, N.; Karatsu, K.; Yano, F.; Kayasuga-Kariya, Y.; Fujisawa, A.; Ohto, T.; Kita, Y.; Nakamura, M.; Suzuki, S.; et al. Trehalose treatment suppresses inflammation, oxidative stress, and vasospasm induced by experimental subarachnoid hemorrhage. J. Transl. Med. 2012, 10, 80. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, Y.; Cai, H.; Wang, Z.; Li, J.; Wang, K.; Yu, Z.; Chen, G. Induction of autophagy by cystatin C: A potential mechanism for prevention of cerebral vasospasm after experimental subarachnoid hemorrhage. Eur. J. Med. Res. 2013, 18, 21. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yuksel, S.; Tosun, Y.B.; Cahill, J.; Solaroglu, I. Early brain injury following aneurysmal subarachnoid hemorrhage: Emphasis on cellular apoptosis. Turk. Neurosurg. 2012, 22, 529–533. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Biswas, S.; Bachay, G.; Chu, J.; Hunter, D.D.; Brunken, W.J. Laminin-dependent interaction between astrocytes and microglia: A role in retinal angiogenesis. Am. J. Pathol. 2017, 187, 2112–2127. [Google Scholar] [CrossRef] [Green Version]
- Chou, S.H.; Lan, J.; Esposito, E.; Ning, M.; Balaj, L.; Ji, X.; Lo, E.H.; Hayakawa, K. Extracellular mitochondria in cerebrospinal fluid and neurological recovery after subarachnoid hemorrhage. Stroke 2017, 48, 2231–2237. [Google Scholar] [CrossRef]
- Zhang, Z.; Zhang, A.; Liu, Y.; Hu, X.; Fang, Y.; Wang, X.; Luo, Y.; Lenahan, C.; Chen, S. New mechanisms and targets of subarachnoid hemorrhage: A focus on mitochondria. Curr. Neuropharmacol. 2021. [Google Scholar] [CrossRef] [PubMed]
- Linn, F.H.; Rinkel, G.J.; Algra, A.; van Gijn, J. Incidence of subarachnoid hemorrhage: Role of region, year, and rate of computed tomography: A meta-analysis. Stroke 1996, 27, 625–629. [Google Scholar] [CrossRef] [PubMed]
- Trimble, J.L.; Kockler, D.R. Statin treatment of cerebral vasospasm after aneurysmal subarachnoid hemorrhage. Ann. Pharmacother. 2007, 41, 2019–2023. [Google Scholar] [CrossRef]
- Dumont, A.S.; Dumont, R.J.; Chow, M.M.; Lin, C.L.; Calisaneller, T.; Ley, K.F.; Kassell, N.F.; Lee, K.S. Cerebral vasospasm after subarachnoid hemorrhage: Putative role of inflammation. Neurosurgery 2003, 53, 123–133. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Borel, C.O.; McKee, A.; Parra, A.; Haglund, M.M.; Solan, A.; Prabhakar, V.; Sheng, H.; Warner, D.S.; Niklason, L. Possible role for vascular cell proliferation in cerebral vasospasm after subarachnoid hemorrhage. Stroke. 2003, 34, 427–433. [Google Scholar] [CrossRef] [Green Version]
- Durand, E.; Mallat, Z.; Addad, F.; Vilde, F.; Desnos, M.; Guerot, C.; Tedgui, A.; Lafont, A. Time courses of apoptosis and cell proliferation and their relationship to arterial remodeling and restenosis after angioplasty in an atherosclerotic rabbit model. J. Am. Coll. Cardiol. 2002, 39, 1680–1685. [Google Scholar] [CrossRef] [Green Version]
- Ogihara, K.; Zubkov, A.Y.; Bernanke, D.H.; Lewis, A.I.; Parent, A.D.; Zhang, J.H. Oxyhemoglobin-induced apoptosis in cultured endothelial cells. J. Neurosurg. 1999, 91, 459–465. [Google Scholar] [CrossRef]
- Peterson, E.W.; Searle, R.; Mandy, F.F.; Leblanc, R. Ther reversal of experimental vasospasm by dibutyryl-3’,5’-adenosine monophosphate. J. Neurosurg. 1973, 39, 730–734. [Google Scholar] [CrossRef]
- Sobey, C.G.; Quan, L. Impaired cerebral vasodilator responses to NO and PDE V inhibition after subarachnoid hemorrhage. Am. J. Physiol. 1999, 277, H1718–H1724. [Google Scholar] [CrossRef]
- Zubkov, A.Y.; Ogihara, K.; Bernanke, D.H.; Parent, A.D.; Zhang, J. Apoptosis of endothelial cells in vessels affected by cerebral vasospasm. Surg. Neurol. 2000, 53, 260–266. [Google Scholar] [CrossRef]
- Raymond, M.A.; Desormeaux, A.; Laplante, P.; Vigneault, N.; Filep, J.G.; Landry, K.; Pshezhetsky, A.V.; Hebert, M.J. Apoptosis of endothelial cells triggers a caspase-dependent anti-apoptotic paracrine loop active on VSMC. FASEB J. 2004, 18, 705–707. [Google Scholar] [CrossRef] [PubMed]
- Leclerc, J.L.; Garcia, J.M.; Diller, M.A.; Carpenter, A.M.; Kamat, P.K.; Hoh, B.L.; Dore, S. A Comparison of pathophysiology in humans and rodent models of subarachnoid hemorrhage. Front. Mol. Neurosci. 2018, 11, 71. [Google Scholar] [CrossRef]
- Yu, J.; Zheng, J.; Lu, J.; Sun, Z.; Wang, Z.; Zhang, J. AdipoRon protects against secondary brain injury after intracerebral hemorrhage via alleviating mitochondrial dysfunction: Possible involvement of AdipoR1-AMPK-PGC1alpha pathway. Neurochem. Res. 2019, 44, 1678–1689. [Google Scholar] [CrossRef]
- Prunell, G.F.; Svendgaard, N.A.; Alkass, K.; Mathiesen, T. Inflammation in the brain after experimental subarachnoid hemorrhage. Neurosurgery 2005, 56, 1082–1092. [Google Scholar]
- Haruma, J.; Teshigawara, K.; Hishikawa, T.; Wang, D.; Liu, K.; Wake, H.; Mori, S.; Takahashi, H.K.; Sugiu, K.; Date, I.; et al. Anti-high mobility group box-1 (HMGB1) antibody attenuates delayed cerebral vasospasm and brain injury after subarachnoid hemorrhage in rats. Sci. Rep. 2016, 6, 37755. [Google Scholar] [CrossRef]
- Meguro, T.; Chen, B.; Parent, A.D.; Zhang, J.H. Caspase inhibitors attenuate oxyhemoglobin-induced apoptosis in endothelial cells. Stroke 2001, 32, 561–566. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhou, C.; Yamaguchi, M.; Colohan, A.R.; Zhang, J.H. Role of p53 and apoptosis in cerebral vasospasm after experimental subarachnoid hemorrhage. J. Cereb. Blood Flow Metab. 2005, 25, 572–582. [Google Scholar] [CrossRef] [Green Version]
- Niwa, A.; Osuka, K.; Nakura, T.; Matsuo, N.; Watabe, T.; Takayasu, M. Interleukin-6, MCP-1, IP-10, and MIG are sequentially expressed in cerebrospinal fluid after subarachnoid hemorrhage. J. Neuroinflammation 2016, 13, 217. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mathiesen, T.; Andersson, B.; Loftenius, A.; von Holst, H. Increased interleukin-6 levels in cerebrospinal fluid following subarachnoid hemorrhage. J. Neurosurg. 1993, 78, 562–567. [Google Scholar] [CrossRef] [Green Version]
- Mathiesen, T.; Edner, G.; Ulfarsson, E.; Andersson, B. Cerebrospinal fluid interleukin-1 receptor antagonist and tumor necrosis factor-alpha following subarachnoid hemorrhage. J. Neurosurg. 1997, 87, 215–220. [Google Scholar] [CrossRef]
- Hanafy, K.A.; Grobelny, B.; Fernandez, L.; Kurtz, P.; Connolly, E.S.; Mayer, S.A.; Schindler, C.; Badjatia, N. Brain interstitial fluid TNF-alpha after subarachnoid hemorrhage. J. Neurol. Sci. 2010, 291, 69–73. [Google Scholar] [CrossRef] [Green Version]
- Chou, S.H.; Feske, S.K.; Atherton, J.; Konigsberg, R.G.; De Jager, P.L.; Du, R.; Ogilvy, C.S.; Lo, E.H.; Ning, M. Early elevation of serum tumor necrosis factor-alpha is associated with poor outcome in subarachnoid hemorrhage. J. Investig. Med. 2012, 60, 1054–1058. [Google Scholar] [CrossRef] [PubMed]
- Kawakita, F.; Fujimoto, M.; Liu, L.; Nakano, F.; Nakatsuka, Y.; Suzuki, H. Effects of toll-like receptor 4 antagonists against cerebral vasospasm after experimental subarachnoid hemorrhage in mice. Mol. Neurobiol. 2017, 54, 6624–6633. [Google Scholar] [CrossRef] [PubMed]
- Ma, C.; Zhou, W.; Yan, Z.; Qu, M.; Bu, X. Toll-like receptor 4 (TLR4) is Associated with cerebral vasospasm and delayed cerebral ischemia in aneurysmal subarachnoid hemorrhage. Neurol. Med. Chir. 2015, 55, 878–884. [Google Scholar] [CrossRef] [Green Version]
- Buchanan, M.M.; Hutchinson, M.; Watkins, L.R.; Yin, H. Toll-like receptor 4 in CNS pathologies. J. Neurochem. 2010, 114, 13–27. [Google Scholar] [CrossRef] [PubMed]
- Patel, H.; McIntire, J.; Ryan, S.; Dunah, A.; Loring, R. Anti-inflammatory effects of astroglial alpha7 nicotinic acetylcholine receptors are mediated by inhibition of the NF-kappaB pathway and activation of the Nrf2 pathway. J. Neuroinflamm. 2017, 14, 192. [Google Scholar] [CrossRef] [PubMed]
- Nowak, D.E.; Tian, B.; Jamaluddin, M.; Boldogh, I.; Vergara, L.A.; Choudhary, S.; Brasier, A.R. RelA Ser276 phosphorylation is required for activation of a subset of NF-kappaB-dependent genes by recruiting cyclin-dependent kinase 9/cyclin T1 complexes. Mol. Cell Biol. 2008, 28, 3623–3638. [Google Scholar] [CrossRef] [Green Version]
- Nicodeme, E.; Jeffrey, K.L.; Schaefer, U.; Beinke, S.; Dewell, S.; Chung, C.W.; Chandwani, R.; Marazzi, I.; Wilson, P.; Coste, H.; et al. Suppression of inflammation by a synthetic histone mimic. Nature 2010, 468, 1119–1123. [Google Scholar] [CrossRef]
- Arora, S.; Dev, K.; Agarwal, B.; Das, P.; Syed, M.A. Macrophages: Their role, activation and polarization in pulmonary diseases. Immunobiology 2018, 223, 383–396. [Google Scholar] [CrossRef]
- Mastroeni, D.; Sekar, S.; Nolz, J.; Delvaux, E.; Lunnon, K.; Mill, J.; Liang, W.S.; Coleman, P.D. ANK1 is up-regulated in laser captured microglia in Alzheimer’s brain; the importance of addressing cellular heterogeneity. PLoS ONE 2017, 12, e0177814. [Google Scholar]
- Borea, P.A.; Gessi, S.; Merighi, S.; Vincenzi, F.; Varani, K. Pharmacology of adenosine receptors: The state of the art. Physiol. Rev. 2018, 98, 1591–1625. [Google Scholar] [CrossRef]
- Novak, M.L.; Koh, T.J. Macrophage phenotypes during tissue repair. J. Leukoc. Biol. 2013, 93, 875–881. [Google Scholar] [CrossRef] [Green Version]
- Mantovani, A.; Biswas, S.K.; Galdiero, M.R.; Sica, A.; Locati, M. Macrophage plasticity and polarization in tissue repair and remodelling. J. Pathol. 2013, 229, 176–185. [Google Scholar] [CrossRef] [PubMed]
- Kumar, A.; Loane, D.J. Neuroinflammation after traumatic brain injury: Opportunities for therapeutic intervention. Brain Behav. Immun. 2012, 26, 1191–1201. [Google Scholar] [CrossRef]
- Helbok, R.; Schiefecker, A.J.; Beer, R.; Dietmann, A.; Antunes, A.P.; Sohm, F.; Fischer, M.; Hackl, W.O.; Rhomberg, P.; Lackner, P.; et al. Early brain injury after aneurysmal subarachnoid hemorrhage: A multimodal neuromonitoring study. Crit. Care 2015, 19, 75. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Murakami, K.; Koide, M.; Dumont, T.M.; Russell, S.R.; Tranmer, B.I.; Wellman, G.C. Subarachnoid hemorrhage induces gliosis and increased expression of the pro-inflammatory cytokine high mobility group box 1 protein. Transl. Stroke Res. 2011, 2, 72–79. [Google Scholar] [CrossRef] [Green Version]
- Nau, R.; Haase, S.; Bunkowski, S.; Bruck, W. Neuronal apoptosis in the dentate gyrus in humans with subarachnoid hemorrhage and cerebral hypoxia. Brain Pathol. 2002, 12, 329–336. [Google Scholar]
- Han, Y.; Zhang, T.; Su, J.; Zhao, Y.; Chenchen; Wang; Li, X. Apigenin attenuates oxidative stress and neuronal apoptosis in early brain injury following subarachnoid hemorrhage. J. Clin. Neurosci. 2017, 40, 157–162. [Google Scholar] [CrossRef]
- Lu, Y.Y.; Wu, C.H.; Hong, C.H.; Chang, K.L.; Lee, C.H. Glut-1 enhances glycolysis, oxidative stress, and fibroblast proliferation in keloid. Life 2021, 11, 505. [Google Scholar] [CrossRef] [PubMed]
- Hong, J.Y.; Kim, H.; Jeon, W.J.; Lee, J.; Ha, I.H. Elevated mitochondrial reactive oxygen species within cerebrospinal fluid as new index in the early detection of lumbar spinal stenosis. Diagnostics 2021, 11, 748. [Google Scholar] [CrossRef]
- Al Shahrani, M.; Heales, S.; Hargreaves, I.; Orford, M. Oxidative stress: Mechanistic insights into inherited mitochondrial disorders and parkinson’s disease. J. Clin. Med. 2017, 6, 100. [Google Scholar] [CrossRef]
- Rong, Y.; Distelhorst, C.W. Bcl-2 protein family members: Versatile regulators of calcium signaling in cell survival and apoptosis. Annu. Rev. Physiol. 2008, 70, 73–91. [Google Scholar] [CrossRef] [PubMed]
- Guttenplan, K.A.; Liddelow, S.A. Astrocytes and microglia: Models and tools. J. Exp. Med. 2019, 216, 71–83. [Google Scholar] [CrossRef]
- Koyama, Y. Signaling molecules regulating phenotypic conversions of astrocytes and glial scar formation in damaged nerve tissues. Neurochem. Int. 2014, 78, 35–42. [Google Scholar] [CrossRef]
- Pekny, M.; Nilsson, M. Astrocyte activation and reactive gliosis. Glia 2005, 50, 427–434. [Google Scholar] [CrossRef]
- Sofroniew, M.V. Reactive astrocytes in neural repair and protection. Neuroscientist 2005, 11, 400–407. [Google Scholar] [CrossRef]
- Nylen, K.; Csajbok, L.Z.; Ost, M.; Rashid, A.; Blennow, K.; Nellgard, B.; Rosengren, L. Serum glial fibrillary acidic protein is related to focal brain injury and outcome after aneurysmal subarachnoid hemorrhage. Stroke 2007, 38, 1489–1494. [Google Scholar] [CrossRef] [Green Version]
- Petzold, A.; Keir, G.; Kerr, M.; Kay, A.; Kitchen, N.; Smith, M.; Thompson, E.J. Early identification of secondary brain damage in subarachnoid hemorrhage: A role for glial fibrillary acidic protein. J. Neurotrauma 2006, 23, 1179–1184. [Google Scholar] [CrossRef] [PubMed]
- Pereira, A.R.; Sanchez-Pena, P.; Biondi, A.; Sourour, N.; Boch, A.L.; Colonne, C.; Lejean, L.; Abdennour, L.; Puybasset, L. Predictors of 1-year outcome after coiling for poor-grade subarachnoid aneurysmal hemorrhage. Neurocrit. Care 2007, 7, 18–26. [Google Scholar] [CrossRef] [PubMed]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Motor | Behavior | Score |
|---|---|---|
| Ambulation | Normal (symmetric and coordinated) | 0 |
| Toes flat under the body while walking with ataxia | 1 | |
| Knuckle walking | 2 | |
| Movement in lower extremities but unable to knuckle walk | 3 | |
| No movement, dragging lower extremities | 4 | |
| Placing/stepping reflex | Normal (coordinated lifting and placing response) | 0 |
| Weak response | 1 | |
| No stepping | 2 |
| Treatment | Ambulation | Placing/Stepping Reflex | MDI |
|---|---|---|---|
| Sham | 0 | 0 | 0 |
| SAH | 2.56 ± 0.88 # | 1.78 ± 0.44 # | 4.33 ± 1 # |
| 2-PMAP + SAH | 1.44 ± 0.53 * | 1.22 ± 0.49 * | 2.67 ± 0.71 * |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wu, C.-H.; Tsai, H.-P.; Su, Y.-F.; Tsai, C.-Y.; Lu, Y.-Y.; Lin, C.-L. 2-PMAP Ameliorates Cerebral Vasospasm and Brain Injury after Subarachnoid Hemorrhage by Regulating Neuro-Inflammation in Rats. Cells 2022, 11, 242. https://doi.org/10.3390/cells11020242
Wu C-H, Tsai H-P, Su Y-F, Tsai C-Y, Lu Y-Y, Lin C-L. 2-PMAP Ameliorates Cerebral Vasospasm and Brain Injury after Subarachnoid Hemorrhage by Regulating Neuro-Inflammation in Rats. Cells. 2022; 11(2):242. https://doi.org/10.3390/cells11020242
Chicago/Turabian StyleWu, Chieh-Hsin, Hung-Pei Tsai, Yu-Feng Su, Cheng-Yu Tsai, Ying-Yi Lu, and Chih-Lung Lin. 2022. "2-PMAP Ameliorates Cerebral Vasospasm and Brain Injury after Subarachnoid Hemorrhage by Regulating Neuro-Inflammation in Rats" Cells 11, no. 2: 242. https://doi.org/10.3390/cells11020242